

Abstract
A number of diseases are due to lysosomal destabilization, which results in damaging cell loss. To investigate the mechanisms of lysosomal cell death, we characterized the cytotoxic action of two widely used quinolone antibiotics: ciprofloxacin (CPX) or norfloxacin (NFX). CPX or NFX plus UV light (NFX*) induce lysosomal membrane permeabilization (LMP), as detected by the release of cathepsins from lysosomes. Inhibition of the lysosomal accumulation of CPX or NFX suppresses their capacity to induce LMP and to kill cells. CPX- or NFX-triggered LMP results in caspase-independent cell death, with hallmarks of apoptosis such as chromatin condensation and phosphatidylserine exposure on the plasma membrane. LMP triggers mitochondrial membrane permeabilization (MMP), as detected by the release of cytochrome c. Both CPX and NFX* cause Bax and Bak to adopt their apoptotic conformation and to insert into mitochondrial membranes. Bax−/− Bak−/− double knockout cells fail to undergo MMP and cell death in response to CPX- or NFX-induced LMP. The single knockout of Bax or Bak (but not Bid) or the transfection-enforced expression of mitochondrion-targeted (but not endoplasmic reticulum–targeted) Bcl-2 conferred protection against CPX (but not NFX*)-induced MMP and death. Altogether, our data indicate that mitochondria are indispensable for cell death initiated by lysosomal destabilization.
Keywords: Bax, Bcl-2, apoptosis, autophagy, caspases
Introduction
The ultimate morphological and biochemical features of apoptotic cells are largely independent of the death-initiating pathway, a fact that led to the concept of the “central executioner,” a self-amplifying device that involves caspase activation cascades and/or mitochondrial membrane permeabilization (MMP;* references 1, 2). Caspase activation and MMP are intimately linked because MMP stimulates caspase activation through the mitochondrial release of several caspase-activating proteins, in particular cytochrome c (3). Conversely, caspase activation culminates in the proteolytic maturation of proteins (e.g., truncated Bid, Bad, Bcl-XL, etc.), which trigger MMP (2, 4). MMP manifests at the level of the outer membrane, which allows for the release of cytochrome c, as well as at the level of the inner membrane as a loss of the mitochondrial transmembrane potential (ΔΨm; references 1, 2). MMP-mediated control of cell death has been firmly established by the fact that mouse embryonic fibroblasts (MEF) lacking both Bax and Bak, two proteins required for MMP induction, are protected against a plethora of different apoptotic stimuli (5). Moreover, Bcl-2 and Bcl-XL, two proteins inserted into the outer mitochondrial membrane as well as other intracellular membranes, inhibit apoptosis, at least in part, by preventing MMP (1, 2, 6). Thus far, it has been established that MMP is a rate-limiting step for cell death induction when apoptosis is triggered, in an organelle-specific fashion, by the nuclear DNA damage response, by the unfolded stress response at the level of the endoplasmic reticulum (ER), or by mitochondrial dysfunction (4).
Lysosomes, which have been referred to as “suicide bags,” notoriously contribute to autophagic cell death (also called “type II” cell death; references 7–9). Recent observations suggest that p53 can trigger a primary lysosomal destabilization that contributes to cell death via apoptosis (10). In pro-oxidant–induced apoptosis, the release of cathepsin D from lysosomes reportedly precedes the release of cytochrome c from mitochondria (11). Ceramide generated by the lysosomal sphingomyelinase can trigger the proteolytic autoactivation of cathepsin D (12). Hepatocytes from cathepsin B knockout mice are relatively resistant to TNF-α–induced apoptosis (13), and cathepsin B participates in TNF-α-induced, caspase-independent cell death of WEHI-S cells (14). Enzymatic defects resulting in lysosomal destabilization can cause unwarranted (possibly apoptotic) cell loss, in particular in the central nervous system. This applies to clinically important diseases such as Sandhoff disease, Gaucher disease, Farber's disease, Niemann Picks' disease, and neural ceroid lipofuscinosis (4).
In spite of this wealth of information, it is currently unknown to what extent MMP is required for cell death initiated at the lysosomal level. On theoretical grounds, two possibilities could be envisioned. On the one hand, MMP could be a rate-limiting step of lysosome-initiated death, which would be in line with the idea that MMP would be part of the “central executer” (1) or “gateway to death” (5). On the other hand, some data suggest that lysosomal destabilization and cathepsins might trigger cell death via novel, MMP-independent pathways, e.g., via direct cathepsin effects on the nucleus (9, 15, 16).
Lysosomotropic agents are lipophilic bases that accumulate in the lysosomal lumen and can exert detergent-like or local phototoxic effects on lysosomal membranes. Two quinolone antibiotics, norfloxacin (NFX) and ciprofloxacin (CFX), which are used on millions of patients each year, are lysosomotropic and can induce apoptosis either in the presence (NFX) or absence (CFX) of a low dose of UV light (17–19). This property may explain the unwarranted cytotoxicity of such compounds (and, in particular, the deleterious effect of NFX medication plus sunlight exposure) and lead to the proposal that quinolone antibiotics could be used for cancer therapy (19).
Here, we show that NFX and CFX kill cells through a lysosomal mechanism. Using NFX and CFX as specific tools for the organelle-specific initiation of cell death, we addressed the functional hierarchy between lysosomes, MMP, and caspases. Our results indicate that MMP mediated by Bax and/or Bak is strictly required for the induction of cell death through a caspase-independent pathway initially triggered at the lysosomal level.
Materials and Methods
Cell Lines, Culture Conditions, and Transfection.
HeLa cells were stably transfected with pcDNA3.1 control vector (Neo), with human Bcl-2 (Bcl-2) or the cytomegalovirus UL37 exon 1 gene (vMIA, provided by Dr. V. Goldmacher, ImmunoGen, Cambridge, MA; reference 20), or a histone H2B–green fluorescent protein (GFP) fusion construct (H2B-GFP, a gift from Dr. G.M. Wahl, Salk Institute, La Jolla, CA; reference 21). Rat-1/myc fibroblasts (provided by Dr. D. Andrews, McMaster University, Hamilton, Canada) were stably transfected with pRc/CMV-based plasmids (Neo) and constitutively express human Bcl-2, mitochondrion-targeted Bcl-2 acta, and ER-targeted Bcl2-cb5 (22, 23). Cells were cultured in DMEM supplemented with 10% FCS, 1 mM of pyruvate, 10 mM Hepes, and 100 U/ml penicillin/streptomycin at 37°C under 5% CO2. SV40-transformed MEF, whose genotype was either wild type, Bax−/−, Bak−/−, Bax−/− Bak−/− double knockout (DKO), Bid+/+, or Bid−/− (provided by Dr. S. Korsmeyer, Harvard Medical School, Boston, MA), were cultivated in IMDM (Life Technologies) supplemented with 20% FCS, 1× NEAA (Sigma-Aldrich), and 100 U/ml penicillin/streptomycin at 37°C under 5% CO2 (5). MEF from wild-type (WT), cathepsin B–deficient (24), cathepsin L–deficient (25), cathepsin S–deficient (26), and cathepsin D–deficient mice (provided by Dr. M. Boes, Harvard Medical School, Boston, MA; reference 24) were immortalized with SV40 (27). Transfection with pcDNA3.1 vector only or p53-responsive enhanced GFP plasmid (a gift from K. Wiman, Karolinska Cancer Center, Stockholm, Sweden) was performed by electroporation 24 h before induction of cell death. The p53-responsive GFP plasmid was generated by replacing the luciferase gene in PG13PY Luc (a luciferase construct containing 13 repeats of the p53-binding oligonucleotide 5′-CCTGCCCTGGGACTTGCCTGG-3′, a gift from Dr. B. Vogelstein, Johns Hopkins University, Baltimore, MD) with an EcoA7III–MluI fragment from pEGFP-C1. DAP kinase cDNA and its mutants were a gift from A. Kimchi (Weizmann Institute, Rehovot, Israel).
Cell Death Induction and Inhibition.
Cells were cultured with CPX (standard dose 350 μg/ml, Bayer), chloroquine (Sigma-Aldrich), 1 μM staurosporine, 100 μM etoposide, 200 μM cisplatin (Sigma-Aldrich), or a combination of anti-CD95 (clone 7C11, 100 ng/ml for 15 h; Immunotech) and 35 μM cycloheximide. Serum withdrawal was performed for 24 h. Alternatively, cells were incubated with NFX (standard dose 10 μg/ml; Sigma-Aldrich) for 1 h and irradiated with a UV lamp (365 ± 5 nm) at 12 J/cm2. 0.1 μM Bafilomycin A1 (Baf A1, Sigma-Aldrich), 100 μM z-FA-fmk, N-benzyloxycarbonyl-Phe-Ala-fluoromethylketone; 100 μM z-VAD-fmk, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Bachem), 10 μM CA-074-Me (Peptides International), 100 μM of pepstatin A (Sigma-Aldrich), 10 μM of cyclic pifthrin-α (Calbiochem), 10 mM GSH ethyl ester (Sigma-Aldrich), or 100 μM MnTBAP (Calbiochem) were added 1 h before induction of cell death. MEF (107 in 150 μl RPMI 1640) were electroporated (with a Bio-Rad gene pulser set at 250 V and 960 μF) with 10 μg vector only (pcDNA3.1), WT DAP kinase or its constitutively active or inactive mutants (DAP kinase ΔCaM or DAP kinase ΔDD, respectively; reference 28), together with 10 μg pDS-Red1-mito vector (CLONTECH Laboratories, Inc.), for the identification of transfected (∼50%) cells.
Lysosomal Stability Assessment.
To determine the intracellular localization of NFX and CPX, cells cultured on coverslips were loaded with 10 μg/ml of the antibiotics and visualized under fluorescence microscopy. To label lysosomes, cells were incubated with 0.5 μM of LysoTracker red (Molecular Probes) for 15 min at 37°C and visualized by fluorescence microscopy (10, 29) or with acridine orange (AO, 5 μM, 30 min, 37°C), followed by determination of the red AO fluorescence by flow cytometry (30).
Flow Cytometry.
We used 40 nM 3, 3′ dihexyloxacarbocyanine iodide (DiOC6(3)) for ΔΨm quantification, 1 μg/ml propidium iodide (PI) for determination of cell viability, 10 μM hydroethidine ( Molecular Probes) for the determination of superoxide anion generation, and Annexin V conjugated with fluorescein isothiocyanate kit (Bender Medsystems) for the assessment of phosphatidylserine (PS) exposure (31). Quantification of DNA content was performed on ethanol-fixed cells which were stained with 4′,6-diamidino-2-phenylindole (DAPI; 2.5 μg/ml; Molecular Probes) for 30 min at 37°C.
Light Microscopy and Immunofluorescence.
Cells cultured on coverslips were stained with 3 μM 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Molecular Probes) and 2 μM Hoechst 33342 (Sigma-Aldrich), followed by fluorescence microscopic assessment. Alternatively, cells were fixed with paraformaldehyde (4% wt/vol) and picric acid (0.19% vol/vol) for immunofluorescence assays (32). Cells were stained for the detection of cytochrome c (mAb 6H2.B4; BD Biosciences), activated Bax (mAb 6A7; BD Biosciences), activated Bak (mAb Ab-1 specific for the NH2 terminus, Oncogene Research Products), cathepsin B (Calbiochem), and monoclonal anti–cathepsin D (clone CTD-19; Sigma-Aldrich); all were detected by a goat anti–mouse or goat anti–rabbit IgG conjugated with Alexa® Fluor (Molecular Probes; reference 33). Giemsa stainings were performed with a kit from Sigma-Aldrich.
Electron Microscopy.
Cells were fixed for 1 h at 4°C in 2.5% glutaraldehyde in phosphate buffer (pH 7.4), washed, and fixed again in 2% osmium tetroxide before embedding in Epon. Electron microscopy was performed with an transmission electron microscope (model EM902; Carl Zeiss MicroImaging, Inc.), at 80 kV, on ultrathin sections (80 nm) stained with uranyl acetate and lead citrate.
Immunoblot.
After the different treatments, cells were lysed for 15 min in 50 mM Hepes, 150 mM NaCl, 5 mM EDTA, and 0.1% NP-40, supplemented with protease inhibitor cocktail (1 mM DTT and 1 mM PMSF; Roche), and centrifuged at 13,000 g for 10 min to remove cell debris. Total protein content was determined with the Bio-Rad DC kit. 40 μg of protein were loaded on a 15% SDS-PAGE. Anti-cleaved caspase-3 antibody (Cell Signaling Technology) was used to determine caspase-3 activation and anti–glyceraldehyde-3-phosphate dehydrogenase (Chemicon) was used as a loading control.
Results and Discussion
NFX and CPX Induce Permeabilization of Lysosomal Membranes.
Based on the intrinsic low-level fluorescence of NFX and CPX (34), we determined their subcellular localization. Both agents were found enriched in the nucleolus as well as in cytoplasmic vesicles (Fig. 1 A). Inhibition of the lysosomal vacuolar H+ ATPase with Baf A1 abolished the cytoplasmic (but not the nucleolar) accumulation of NFX and CPX, indicating that the NFX/CPX-containing vesicles are lysosomes (Fig. 1 A). Cells exposed to NFX or to a low dose of UV light alone exhibited normal staining, with two acidophilic dyes specifically labeling lysosomes: namely a punctate lysosomal staining with LysoTracker red (Fig. 1 B) and a marked red fluorescence with AO (Fig. 1 C). When NFX was combined with UV light (NFX*), an immediate phototoxic effect was noted at the level of lysosomes, leading to the loss of the punctate LysoTracker red staining pattern (Fig. 1 B) and a diminution of the red AO fluorescence (Fig. 1 C). NFX*, but not NFX or UV light alone, caused lysosomes to release cathepsins B and D into the cytosol (Fig. 1, D and E). Similarly, CPX reduced LysoTracker incorporation (Fig. 1 B), attenuated AO staining (Fig. 1 C), and led to the translocation of cathepsins B and D (Fig. 1, D and E). In conclusion, NFX mediates the UV-elicited (photodynamic) permeabilization of lysosomal membranes, and CPX alone exerts similar effects.
Figure 1.
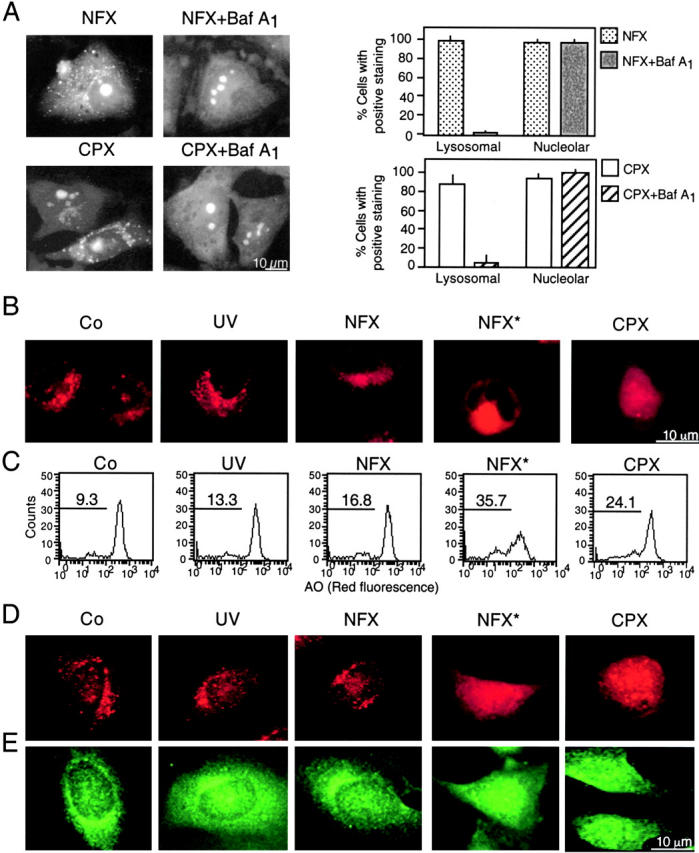
Specific lysosomal destabilization by NFX* and CPX. (A) Lysosomal localization of NFX and CPX. HeLa cells were incubated with 10 μg/ml NFX or CPX, and the CPX- or NFX-mediated fluorescence was determined in control cells or in cells pretreated with 100 nM Baf A1. The percentage of cells exhibiting a clear cytoplasmic punctate (lysosomal) or a nucleolar staining was determined. (B and C) NFX*- and CPX-mediated disruption of acidic vacuoles. Cells were stained with LysoTracker red (B) or AO (C) after treatment with UV, NFX alone, NFX*, or CPX, followed by fluorescence microscopy (B) or cytofluorometric analysis of the 630 ± 11-nm fluorescence (C). Numbers indicate the percentage of cells with decreased AO red fluorescence. (D and E) Translocation of cathepsins B and D from lysosomes. Cells treated for 8 h were fixed, permeabilized, and stained for the immunofluorescence of cathepsin B (D) and D (E). Results are representative of three to five independent determinations.
NFX* and CPX Induce Caspase-independent Cell Death with MMP.
The combination of NFX and UV light (NFX*), as well as CPX alone, induced several hallmarks of apoptosis, in particular the exposure of residues on the plasma membrane surface (Fig. 2, A and B) and the loss of nuclear DNA (Fig. 2 C). Although CPX caused proteolytic maturation of caspase-3, NFX* failed to induce caspase activation, as determined by immunoblots (Fig. 2 D). Importantly, treatment with the pan-caspase inhibitor z-VAD-fmk failed to prevent the induction of chromatin condensation by NFX* or CPX, as determined by Giemsa staining (Fig. 2 E) as well as in unfixed cells engineered to stably express a histone H2B-GFP chimera, allowing for the constant monitoring of chromatin structure (Fig. 2, F and H). This chromatin condensation was less pronounced as that induced by CD95 cross-linking (which, however, was fully inhibited by z-VAD-fmk) (Fig. 2, E, F, and H), and was of a more peripheral type, as determined by electron microscopy (Fig. 2 G). z-VAD-fmk failed to prevent PS exposure induced by NFX* or CPX (Fig. 2 I). NFX* and CPX induced signs of MMP such as the dissipation of ΔΨm, measured as a reduction of the DiOC6(3) incorporation (Fig. 3, A and B) or a spectral red–green shift in the fluorescence of JC-1 (Fig. 3 C), and the mitochondrial release of cytochrome c (Fig. 3 D). Again, z-VAD-fmk failed to affect the signs of MMP induced by NFX* or CPX (Fig. 3 E). The population of cells having released cytochrome c from mitochondria constituted a subensemble of NFX*- or CPX-treated cells with diffuse cathepsin B staining (Fig. 3 F), indicating that MMP occurs after lysosomal membrane permeabilization (LMP). Baf A1 inhibited all hallmarks of cell death induced by NFX* or CPX: loss of the ΔΨm (Fig. 4 A), loss of viability (Fig. 4 B), mitochondrial release of cytochrome c (Fig. 4 C), and lysosomal release of cathepsin B (Fig. 4 D). In conclusion, NFX* and CPX induce MMP and z-VAD-fmk–resistant cell death via a pathway that strictly depends on their Baf A1–inhibitable accumulation in lysosomes.
Figure 2.
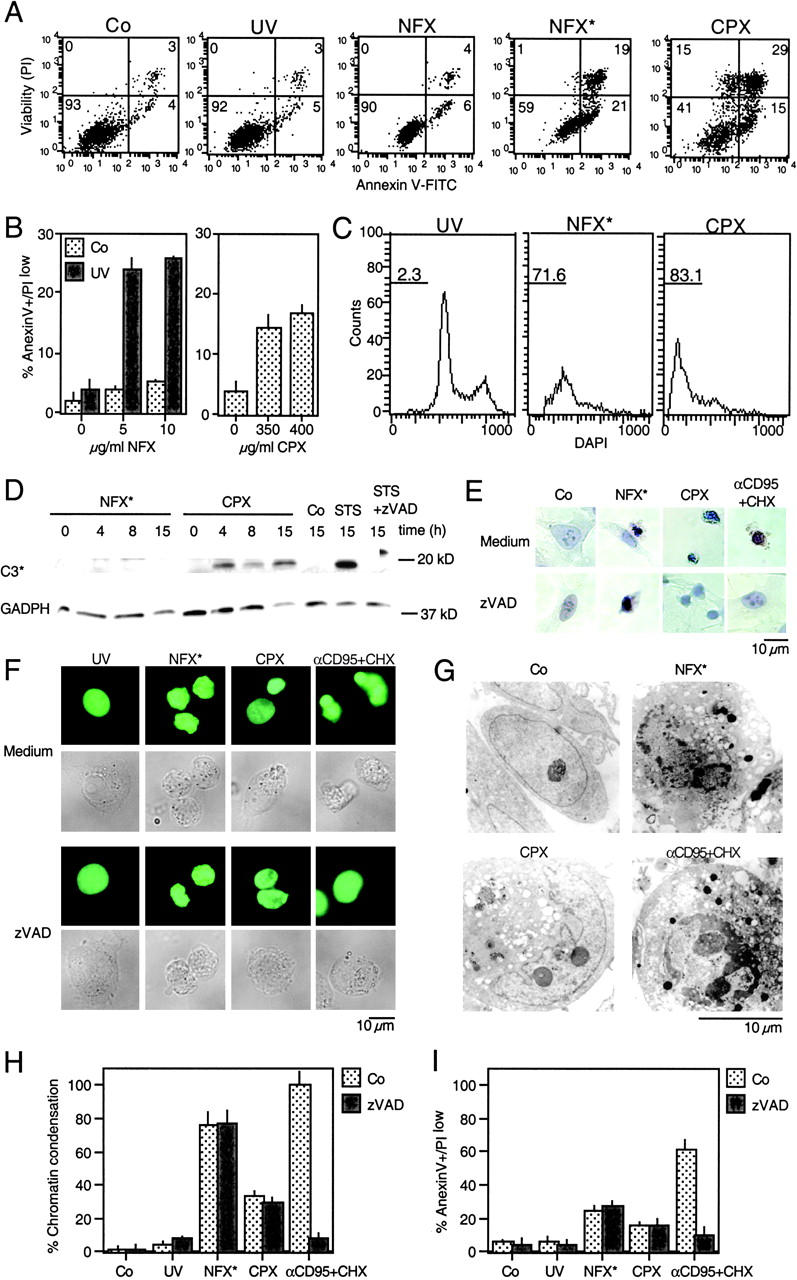
Caspase-independent cell death induction by NFX* and CPX. (A) PS exposure resulting from lysosomal destabilization. After the indicated treatment, cells were stained for PS exposure (with annexin V–FITC) or plasma membrane permeabilization (with PI) and subjected to cytofluorometric analysis. Numbers in each quadrant refer to the percentage of cells. (B) Quantitation of data obtained in A. (C) DNA hypodiploidy induced by NFX* and CPX. After 24 h, the nuclear DNA content was determined with DAPI by cytofluorometric analysis in fixed-permeabilized cells. Numbers indicate the percentage of cells exhibiting a subdiploid (apoptotic) DNA content. (D) Immunoblot determination of caspase-3 maturation. Cell extracts obtained after the indicated treatment and interval were subjected to immunoblotting with antibodies specific for cleaved caspase-3 (17-kD fragment) and glyceraldehyde-3-phosphate dehydrogenase (as a loading control). (E) Giemsa staining of HeLa cells treated with NFX*, CPX, or αCD95, in the absence or presence of 100 μM z-VAD-fmk. (F) Chromatin structure of living HeLa cells stably expressing a histone H2B-GFP fusion protein after treatment with NFX* or CPX. Representative micrographs of the GFP fluorescence and phase-contrast pictures are shown. (G) Ultrastructure of NFX*- or CPX-treated cells exhibiting nuclear chromatin condensation and vacuolization, as compared with apoptosis induced by αCD95. (H) Chromatin condensation induced by NFX* or CPX, both in the absence or presence of 100 μM z-VAD-fmk, as determined in F. (I) Failure of z-VAD-fmk (100 μM) to inhibit PS exposure in NFX*- and CPX-treated cells. Data are shown as mean values ± SD of three independent experiments.
Figure 3.
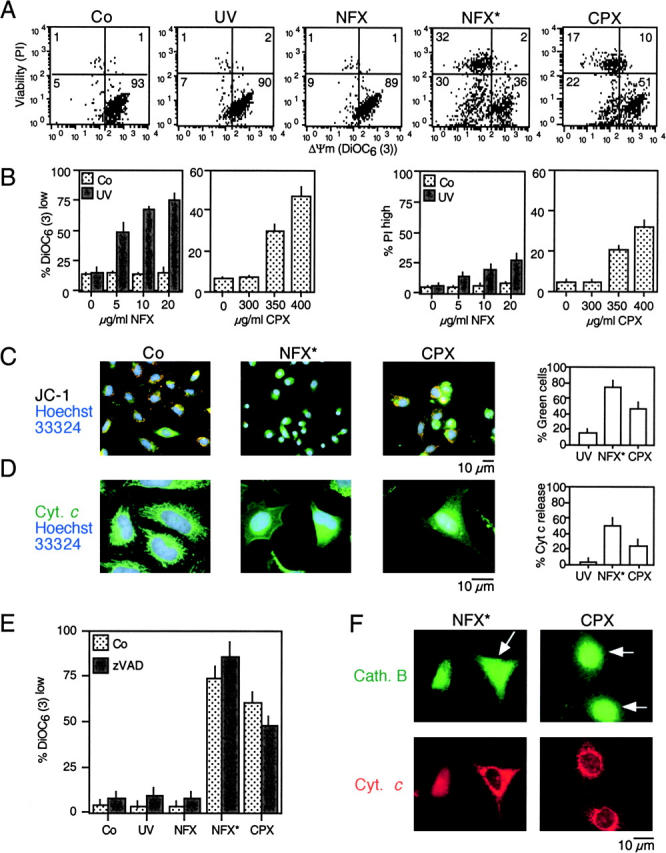
MMP induced by NFX* and CPX. (A) Dissipation of the mitochondrial transmembrane potential (ΔΨm). After NFX* (or UV and NFX alone) or CPX treatment, cells were stained simultaneously with DiOC6(3), which incorporates into cells driven by the ΔΨm, and the vital marker propidium iodide (PI). (B) Dose response of the ΔΨm dissipation and viability loss induced by NFX* and CPX, determined as in A. (C) In situ determination of ΔΨm loss, as indicated by the spectral red–green shift of JC-1–stained cells. (D) In situ evidence for outer mitochondrial membrane permeabilization. After an 8-h treatment and fixation, cells were stained for the immunodetection of cytochrome c (green fluorescence) and counterstained with Hoechst 33324. (E) Caspase independence of mitochondrial changes. Cells were pretreated (60 min) with subtoxic doses of z-VAD-fmk (100 μM), exposed overnight to NFX* or CPX, followed by detection of ΔΨm reduction. (F) Cytochrome c release occurs after cathepsin B translocation. Cells were treated for 8 h with NFX* or CPX and subjected to simultaneous immunofluorescence staining for cytochrome c (red) and cathepsin B (green). Note that some cells (arrows) have a diffuse cathepsin B distribution, yet still retain cytochrome c in a punctate pattern.
Figure 4.
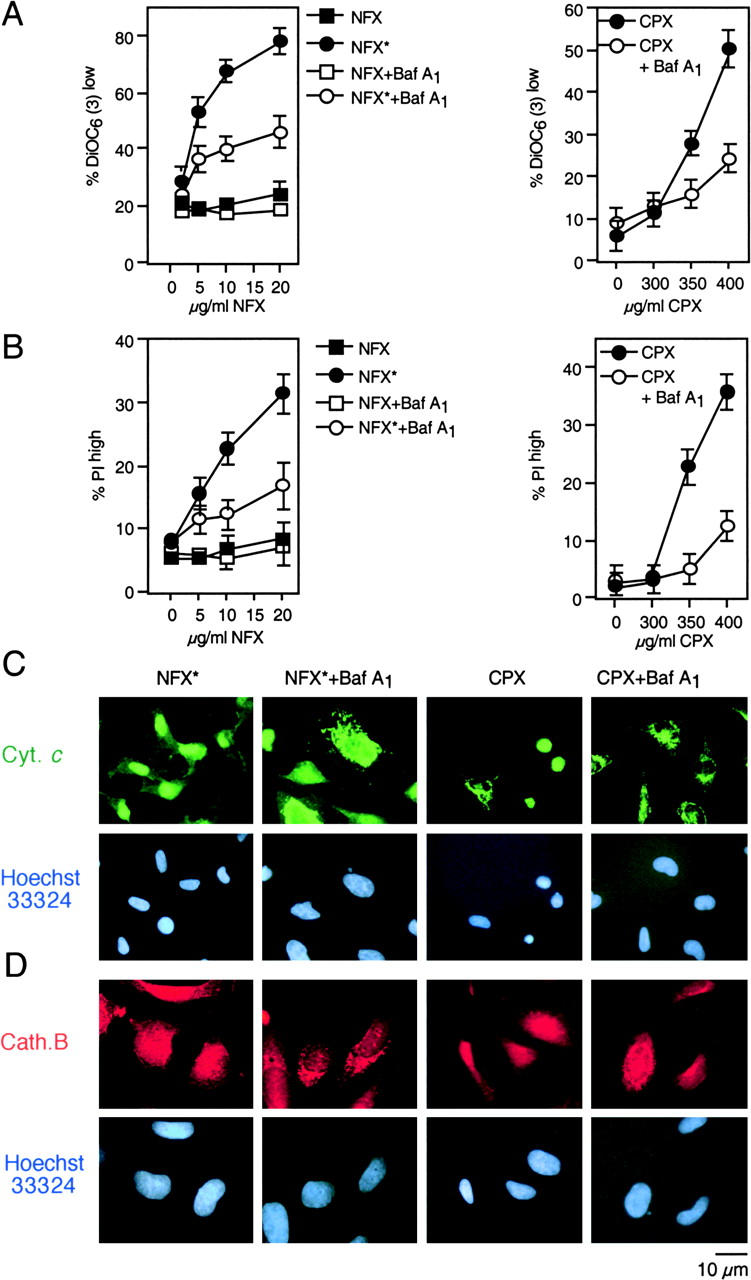
NFX* and CPX exert their mitochondriotoxic and cytotoxic effect via lysosomes. (A and B) Effect of the vacuolar ATPase inhibitor Baf A1 on ΔΨm loss and cell death. Cells were pretreated for 1 h with Baf A1 and exposed overnight to NFX* or CPX, followed by staining with DiOC6(3) (A) and PI (B). (C and D) Baf A1–mediated protection against lysosomal and mitochondrial permeabilization. Cells treated as in A and B were subjected to immunofluorescence detection of cytochrome c (C) or cathepsin B (D). Representative cells showing that Baf A1 pretreatment maintains cytochrome c and cathepsin B in cytoplasmic organelles are shown.
Lysosomal Damage Triggers MMP and Cell Death via Bax and Bak.
Both NFX* and CPX induced a conformational change of Bax and Bak (detectable with monoclonal antibodies specific for the NH2 terminus of these molecules), which are exposed only in the “apoptotic conformation” (35, 36). Both NFX* and CPX treatment resulted in a punctate cytoplasmic staining of apoptotic Bax and Bak (Fig. 5 A), which were found to colocalize with the mitochondrial marker Hsp60 (not depicted). The activation of Bax and Bak occurs after LMP (as assessed by measuring the LysoTracker red incorporation and cathepsin B release), but before the mitochondrial release of cytochrome c and before caspase-3–activation (Fig. 5, B and C). These alterations were not due to transcriptional activation of p53 because NFX* and CPX (in contrast to the positive control etoposide) failed to induce a p53-inducible GFP reporter gene (Fig. 5 D). Moreover, NFX*- and CPX-induced MMP and cell death were not inhibited by pifithrin-α, a chemical inhibitor of p53-mediated transactivation (Fig. 5 E) or cycloheximide, a general inhibitor of protein synthesis (not depicted). To directly address the contribution of Bax and Bak to cell death induced by lysomotropic agents, we took advantage of SV40–immortalized MEF lacking Bax, Bak, Bid, or both Bax and Bak. Bax−/−Bak−/− DKO were found to be profoundly resistant to the induction of MMP and PS exposure by NFX* or CPX (Fig. 6 A). Only the DKO of Bax and Bak led to cytoprotection against NFX* (Fig. 6 B). In contrast, the absence of either Bax or Bak (but not Bid) was sufficient to prevent cell death induction by CPX (Fig. 6 B). WT and Bax−/−Bak−/− cells exposed to CPX or NFX* underwent a similar degree of lysosomal disruption, as measured with cathepsin B immunostaining (Fig. 6 C). However, Bax−/− Bak−/− cells manifested much less diffuse cytochrome c staining induced by CPX or NFX*, as compared with controls (Fig. 6 C). The knockout of the genes coding for cathepsin B, D, L, or S and several cathepsin inhibitors failed to prevent CPX- or NFX-induced MMP and cell death (Fig. 6, D and E). Altogether, these data demonstrate that Bax and/or Bak, but not cathepsins, control cell death induced by lysosomotropic agents.
Figure 5.
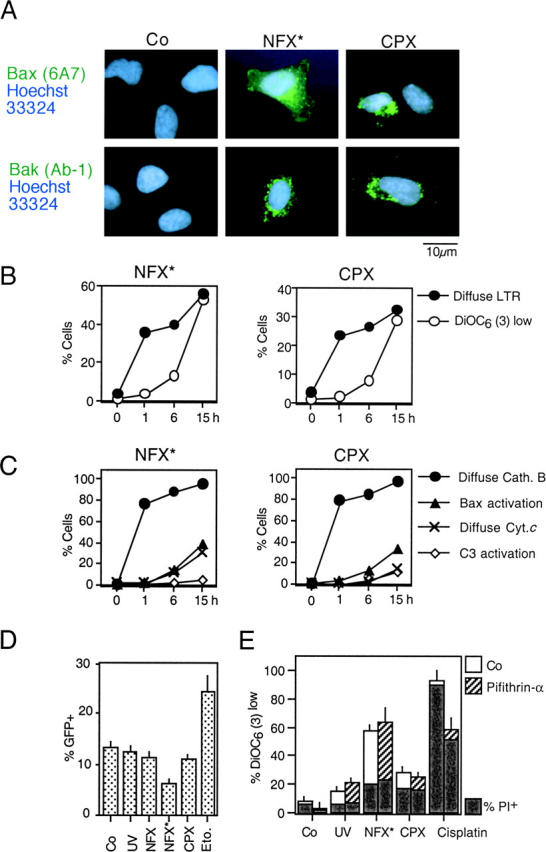
Activation of Bax and Bak by NFX* and CPX through a p53-independent mechanism. (A) Conformational change of Bax and Bak. Cells were stained with monoclonal antibodies recognizing the apoptotic conformation of Bax (6A7) and Bak (Ab-1) 8 h after treatment with NFX* or CPX. (B) Time course of lysosomal and mitochondrial permeabilization induced by NFX* and CPX, as determined by staining with LysoTracker Red and DiOC6(3) in unfixed cells. (C) Kinetics of NFX*- and CPX-induced lysosomal cathepsin B release, Bax activation (determined as in A), mitochondrial cytochrome c release, and caspase-3 activation, as determined by immunofluorescence of fixed and permeabilized cells. Results are representative of three experiments. (D) Failure of NFX* and CPX to activate p53-dependent transcription. Cells were transfected with a p53-inducible GFP reporter construct, and the percentage of cells expressing GFP was determined by FACS® analysis. DNA damage by etoposide served as positive control. (E) Failure of pifithrin-α to inhibit NFX*- and CPX-induced cell death. Cells were pretreated for 60 min with the p53 inhibitor pifithrin-α, and the frequency of 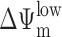 (DiOC6(3)low) and death (PIhigh) was determined by cytofluorometry.
(DiOC6(3)low) and death (PIhigh) was determined by cytofluorometry.
Figure 6.
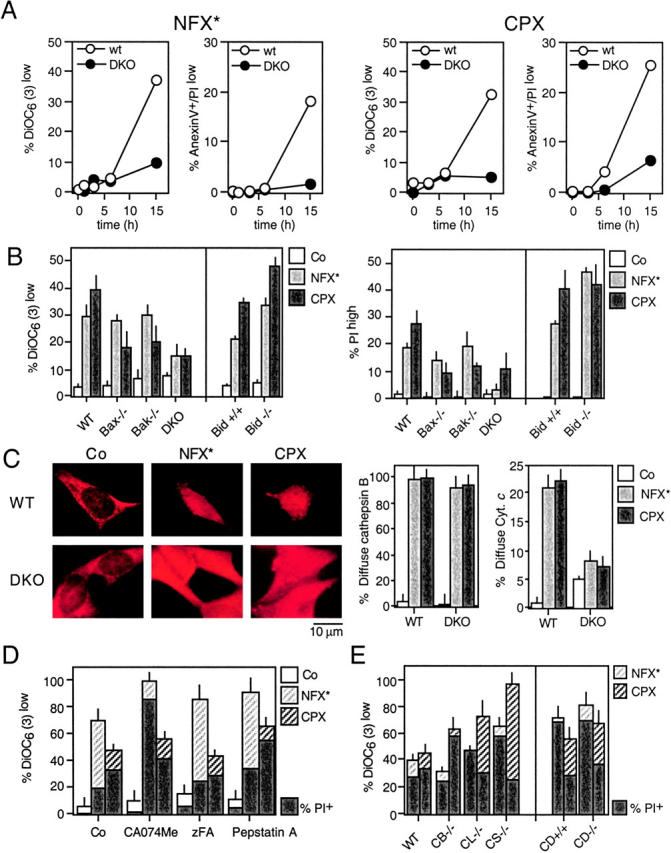
Requirement of Bax and/or Bak for NFX*- or CPX-induced cell death. (A) SV40-transformed WT MEF or Bax−/− Bak−/− DKO cells were exposed to NFX* and CPX for the indicated period, followed by determination of the frequency of DiOC6(3)low and annexin V–positive cells. (B) MEF lacking Bax, Bak, Bax and Bak (DKO), or Bid were exposed to either NFX* or CPX and the indicated parameters were measured. (C) NFX*- and CPX-induced cathepsin B release in control and Bax−/− Bak−/− DKO cells, as determined by immunostaining. The percentage of cells exhibiting diffuse cathepsin B staining as well as those with diffuse cytochrome c staining has been quantified. (D) Failure of chemical cathepsin inhibition to prevent NFX*- or CPX-induced cell death. HeLa cells were pretreated with the indicated cathepsin inhibitors, followed by determination of the frequency of DiOC6(3)low and PIhigh cells. (E) Cathepsins are dispensable for NFX*- or CPX-induced cell death. MEF with the indicated genotypes were treated with NFX* or CPX, and apoptotic parameters were measured as in D. Results are means ± SD of three independent determinations.
Mitochondrial Bcl-2 and vMIA Selectively Inhibit CPX-mediated MMP and Cell Death.
HeLa cells stably transfected with Bcl-2 or vMIA, a cytomegalovirus-encoded protein that interacts with the mitochondrial adenine nucleotide translocase (20), exhibited a relative resistance to the induction of ΔΨm loss and cell death by CPX but not by NFX* (Fig. 7 A). Bcl-2 specifically targeted to the mitochondrial outer membrane (Bcl-2 acta) was as efficient as WT Bcl-2 in inhibiting CPX-induced MMP and cell killing (Fig. 7 B). In contrast, ER-targeted Bcl-2 (Bcl-2 cathepsin B5; references 22, 23) failed to antagonize CPX (Fig. 7 B). Bcl-2 and vMIA failed to antagonize the dissipation of LysoTracker red staining by CPX (Fig. 7 C). Thus, Bcl-2 and vMIA inhibit CPX but not NFX*-induced cell death at the mitochondrial level.
Figure 7.
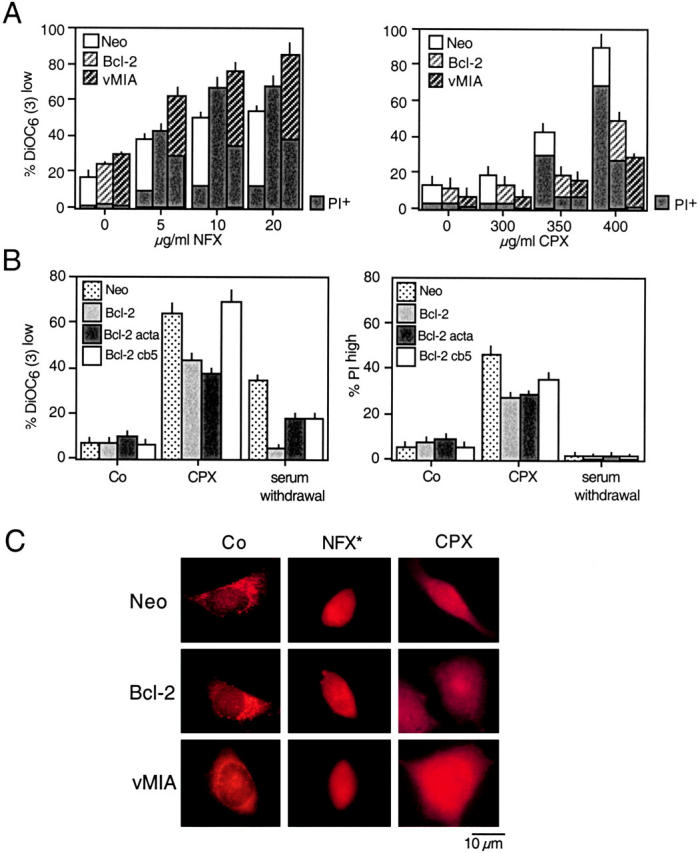
Mitochondrial membrane stabilization by Bcl-2 or vMIA prevent CPX-induced cell death. (A) HeLa cells stably transfected with vector only (Neo), or cDNAs coding for human Bcl-2 or cytomegalovirus-derived vMIA were treated with NFX* or CPX, and the frequency of DiOC6(3)low or PIhigh cells was assessed. Note that Bcl-2– and vMIA-expressing cells are more susceptible to cell death induction by NFX*, yet relatively resistant to CPX. (B) Mitochondrial, not ER-localized, Bcl-2 prevents CPX-induced cell death. Rat1 cells stably transfected with vector only (Neo), WT Bcl-2, mitochondrion-targeted Bcl-2 (Bcl-2 acta), and ER-targeted Bcl-2 (Bcl-2 cathepsin B5), were subjected to the indicated treatment, and the loss of mitochondrial and plasma membrane integrity was assessed with DiOC6(3) and PI, respectively. (C) Failure of Bcl-2 or vMIA to confer lysosomal stabilization. HeLa cells expressing Neo, Bcl-2, or vMIA were treated with NFX* or CPX, fixed, and immunostained for cathepsin B. Note that all cells exhibit a diffuse cathepsin B staining, although Bcl-2 and vMIA do inhibit CPX-induced MMP (A) and cell death (B) as an internal control of their efficacy.
Involvement of Reactive Oxygen Species (ROS) as Mediators of MMP in NFX*-induced Cell Death.
The NFX*-induced ΔΨm loss was preceded by an increase in ROS production (as quantified by the ROS-mediated conversion of hydroethidine into the fluorescent product ethidium), whereas CPX-induced ΔΨm loss occurred before ROS production increased (Fig. 8 A). Furthermore, the antioxidants glutathione ethyl ester and MnTBAP prevented NFX*-induced MMP, ROS generation, PS exposure, and cell death, yet had no effect on CPX-induced cell death (Fig. 8, B and C). MnTBAP also inhibited the NFX-induced apoptotic conformation of Bax, yet had no significant effect on CPX-induced Bax activation (Fig. 8 D). The antioxidant partially (∼60%) inhibited the NFX* (but not the CPX)-induced cathepsin B relocalization (Fig. 8 E). Thus, NFX* induces cell death through a photodynamic effect involving ROS upstream of MMP, whereas ROS are not involved in CPX-induced MMP.
Figure 8.

Differential involvement of ROS in CPX- and NFX-induced cell death. (A) Relationship between ROS production and ΔΨm loss triggered by NFX* and CPX. HeLa cells were exposed to NFX* or CPX for the indicated interval, and stained simultaneously with the ΔΨm-sensitive dye DiOC6(3) and the ROS-sensitive probe hydroethidine (HE), whose oxidation by superoxide anions yields the red fluorescent product ethidium (Eth). Note that NFX*treatment causes ROS production before the ΔΨm drops, whereas after CPX treatment, ROS production is only observed in a subpopulation of DiOC6(3)low cells. (B–E) Effects of antioxidants on CPX- and NFX-induced cell death. HeLa cells were pretreated (60 min) with the cell-permeable GSH ethyl ester (10 mM) or the superoxide dismutase analogue MnTBAP (100 μM), followed by overnight treatment with NFX* or CPX and determination of the frequency of 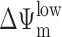 , ROS-overproducing (B), PS-exposing, dead (C) cells, and cells exhibiting Bax in the apoptotic conformation (D, determined with the conformation-specific 6A7 mAB as in Fig. 5 A) and diffuse cathepsin B staining (E, determined as in Fig. 1 D).
, ROS-overproducing (B), PS-exposing, dead (C) cells, and cells exhibiting Bax in the apoptotic conformation (D, determined with the conformation-specific 6A7 mAB as in Fig. 5 A) and diffuse cathepsin B staining (E, determined as in Fig. 1 D).
MMP Is a General Requirement for Lysosome-initiated Cell Death.
As shown in Fig. 9 A, the cytotoxic effect of chloroquine, yet another lysosomotropic drug, is abolished by either Baf A1 or by the DKO of Bax and Bak. The absence of Bax and Bak also inhibited the ΔΨm loss (Fig. 9 A), the release of cytochrome c, and chromatin condensation induced by chloroquine, yet had no effect on the translocation of cathepsin B (Fig. 9 B). The constitutively active DAP kinase (DAP kinase ΔCaM) has been shown to stimulate a lysosomal/autophagic cell death pathway, when overexpressed in HeLa cells (28). DAP kinase ΔCaM (but not WT DAP kinase or the inactive DAP kinase ΔDD mutant) induces a ΔΨm loss and kills WT MEF, yet has no major effect on Bax−/− Bak−/− MEF. These findings confirm that Bax/Bak-dependent MMP is involved in lysosome-triggered cell death.
Figure 9.

Effect of the Bax/Bak DKO on apoptosis induced by two different lysomal stimuli. (A) Chloroquine-induced ΔΨm loss and cell death is inhibited by Baf A1 as well as by ablation of Bax and Bak. WT MEF or DKO MEF were exposed to 30 μg/ml chloroquine (24 h), followed by determination of DiOC6(3)/PI staining and flow cytometric analysis. (B) Chloroquine-induced cathepsin B translocation, cytochrome c release, and chromatin condensation are inhibited in DKO cells. Cells, treated as in A, were subjected to immunofluorescence analysis (anti–cathepsin B, anti–cytochrome c, and Hoechst 33324), and the cells exhibiting the indicated phenotype were determined. (C) DAP kinase-induced cell death and ΔΨm loss in WT and DKO MEF. Cells were transfected with the constitutively active DAP kinase construct ΔCaM or controls. After 72 h, the cells were stained with DiOC6(3) and DAPI, and the percentage of 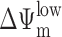 (DiOC6(3)low) or dead (DAPI+) cells was measured by cytofluorometry.
(DiOC6(3)low) or dead (DAPI+) cells was measured by cytofluorometry.
Concluding Remarks.
We report here the unexpected finding that MMP occurs downstream of LMP, as a down-stream rate-limiting step of cellular demise, based on several signs of evidence. First, LMP is detectable within a few hours after NFX* or CPX treatment, whereas MMP appears later (Figs. 1 and 3 and Fig. 5, B and C). Second, cells with signs of MMP (e.g., cytochrome c release) are a sub-ensemble of cells with signs of LMP (e.g., cathepsin B release), not vice versa (Fig. 3). Third, inhibition of LMP with Baf A1 suppresses MMP (Fig. 4), whereas prevention of MMP by the Bax/Bak DKO, Bcl-2, or vMIA does not block LMP (Figs. 6 C, 7 C, and 9 B). Fourth, when MMP is prevented (by deletion of Bax and/or Bak or overexpression of Bcl-2 or vMIA), LMP fails to cause late manifestations of cell death such as PS exposure, (partial) chromatin condensation, and loss of viability (Figs. 6 and 7). Importantly, the evidence placing LMP upstream of MMP has been obtained with three different clinically relevant drugs (NFX*, CPX, and chloroquine), as well as with DAP-kinase, a signal-transducing molecule known to be involved in physiological cell death induction by γ-interferon and TNF-α (Fig. 9; reference 28). Thus, the importance of MMP for LMP-induced cell death appears to be a general phenomenon.
Although these data imply a clear hierarchical relationship between LMP and MMP with regards to cell death induction (Fig. 10) , the molecular link between LMP and MMP remains elusive. The lysosomal cathepsins B, D, L, and S do not constitute such a link, as shown by the knockout of each of these cathepsins (Fig. 6 E), as well as by pharmacological studies involving cathepsin inhibitors (Fig. 6 D). Moreover, although the proapoptotic Bcl-2–like protein Bid could constitute a link between LMP and MMP (37, 38), Bid clearly does not contribute to MMP induced by CPX or NFX*, as demonstrated by experiments involving Bid−/− cells (Fig. 6 B). p53-mediated effects, which also have been postulated to participate in LMP-induced apoptosis (39, 40), clearly do not participate in CPX- or NFX-induced MMP, which is independent of the transcription of p53 target genes (Fig. 5, D and E). It has been suggested that cathepsin B would directly activate caspase-3 (41) and/or trigger apoptotic chromatin condensation and nuclear DNA loss (15). However, the presence of cathepsin B in the cytosol and in the nucleus clearly is not sufficient to induce cell death, as shown by the fact that Bax−/−Bak−/− cells can release cathepsin from lysosomes, yet fail to translocate cytochrome c and continue to manifest a normal chromatin structure (Figs. 6 C and 9 B).
Figure 10.

Hypothetical mechanisms through which CPX and NFX* kill cells. CPX causes death through lysosomal membrane permeabilization (LMP), upstream of MMP, caspase-3 activation, and activation of caspase-independent death effectors including ROS. NFX* causes death through ROS-mediated phototoxic effects, upstream of LMP, and MMP. Perhaps as a result of the primary effect on cellular redox potentials, MMP occurs in a Bcl-2–independent fashion and does not result in caspase-3 activation.
Clear differences appear in the mechanisms through which NFX* and CPX induce MMP and cell death, the first one involving ROS (as to be expected for a photosensitizer) and the second acting independently from ROS (Fig. 8). Moreover, the NFX-induced MMP was more difficult to be inhibited than CPX-induced MMP. Only the knockout of both Bax and Bak conferred resistance to NFX* (Fig. 6, A and B). The knockout of either Bax or Bak alone, or the overexpression of vMIA, natural Bcl-2, or mitochondrion-targeted Bcl-2 did not attenuate the NFX-triggered MMP and death, whereas all these manipulations did prevent CPX-mediated MMP (Figs. 6 and 7). Although NFX failed to cause the activation of caspase-3, CPX triggered the proteolytic maturation of caspase-3 (Fig. 2 D). However, caspase inhibition did not block cell death in CPX- or NFX-treated cells (Fig. 2, F and G), which is as in line with the notion that caspase-independent death effectors seal the cell's fate in response to lysosomotropic agents. The ROS-inducing property of NFX* may explain why it acts as a stronger MMP inducer, yet as a weaker caspase elicitor than CPX. Indeed, it is known that primary thiol oxidation of mitochondrial proteins (which may result from an oxidative shift in the cellular redox potential) can induce a Bcl-2–refractory MMP (42–44) and that Bcl-2 can even sensitize to (ROS-mediated) photodynamic therapy (45). Moreover, oxidation of critical thiols within the catalytic center of caspases annihilates their latent proteolytic potential and, thus, precludes their (auto)activation (46).
In conclusion, the data reported herein reveal that MMP may be indispensable for cell death initiated at the lysosomal level, thus reenforcing the concept that MMP constitutes a central event in the death-inducing signaling cascade. Based on these results, studies are underway to explore the possibility that prevention of MMP might diminish pathogenic cell loss triggered by primary lysosomal lesions in pathologies as diverse as Gaucher disease, Sandhoff disease, Farber disease, Niemann Pick disease, neuronal ceroid lipofuscinosis, and age-related macular degeneration.
Acknowledgments
We thank Drs. D. Andrews, M. Boes, S. Korsmeyer, A. Kimchi, V. Goldmacher, K. Wiman, and G.M. Wahl for reagents, and G. Pierron (UMR 1983, Villejuif, France) for electron microscopy.
This work has been supported by a special grant from Ligue Nationale contre le Cancer, as well as grants from the Agence National pour la Recherche sur le SIDA, Fondation pour la Recherche Médicale, and European Commission (QLG1-CT-1999-00739 and QLK3-CT-2002-01956 to G. Kroemer). P. Boya received a fellowship from the European Commission (MCFI-2000-00943). K. Andreau received a Sidaction fellowship.
P. Boya and K. Andreau contributed equally to this work.
Footnotes
Abbreviations used in this paper: AO, acridine orange; Baf A1, Bafilomycin A1; CPX, ciprofloxacin; ΔΨm, mitochondrial transmembrane potential; DAPI, 4′,6-diamidino-2-phenylindole; DiOC6(3), 3,3′ dihexyloxacarbocyanine iodide; DKO, double knockout; GFP, green fluorescent protein; JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; LMP, lysosomal membrane permeabilization; MEF, mouse embryonic fibroblasts; MMP, mitochondrial membrane permeabilization; NFX, norfloxacin; NFX*, NFX plus UV light; PI, propidium iodide; PS, phosphatidylserine; ROS, reactive oxygen species; z-FA-fmk, N-benzyloxycarbonyl-Phe-Ala-fluoromethylketone; z-VAD-fmk, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone.
References
- 1.Green, D.R., and G. Kroemer. 1998. The central executioner of apoptosis: mitochondria or caspases? Trends Cell Biol. 8:267–271. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer, G., and J.C. Reed. 2000. Mitochondrial control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- 3.Wang, X. 2002. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- 4.Ferri, K.F., and G.K. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- 5.Wei, M.C., W.-X. Zong, E.H.-Y. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer, G. 1997. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3:614–620. [DOI] [PubMed] [Google Scholar]
- 7.Bursch, W. 2001. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8:569–581. [DOI] [PubMed] [Google Scholar]
- 8.Lockshin, R.A., and Z. Zakeri. 2001. Programmed cell death and apoptosis: origins of the theory. Nat. Rev. Mol. Cell Biol. 2:545–550. [DOI] [PubMed] [Google Scholar]
- 9.Leist, M., and M. Jaattela. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:589–598. [DOI] [PubMed] [Google Scholar]
- 10.Yuan, X.M., W. Li, H. Dalen, J. Lotem, R. Kama, L. Sachs, and U.T. Brunk. 2002. Lysosomal destabilization in p53-induced apoptosis. Proc. Natl. Acad. Sci. USA. 99:6286–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberg, K. 2001. Relocalization of cathepsin D and cytochrome c early in apoptosis revealed by immunoelectron microscopy. Lab. Invest. 81:149–158. [DOI] [PubMed] [Google Scholar]
- 12.Heinrich, M., M. Wickel, W. Schneider-Brachert, C. Snadberg, J. Gahr, R. Schwandner, T. Weber, J. Brunner, M. Krönke, and S. Schütze. 1999. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 18:5252–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guicciardi, M.E., J. Deussing, H. Miyoshi, S.F. Bronk, P.A. Svingen, C. Peters, S.H. Kaufmann, and G.J. Gores. 2000. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106:1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foghsgaard, M., D. Wissing, D. Mauch, U. Lademann, L. Bastholm, M. Boes, F. Elling, M. Leist, and M. Jäättelä. 2001. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vancompernolle, K., F. Van Herrewghe, G. Pynaert, M. Van de Craen, K. De Vos, N. Totty, A. Sterling, W. Fiers, P. Vandenabeele, and J. Grooten. 1998. Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett. 6:150–158. [DOI] [PubMed] [Google Scholar]
- 16.Biggs, J.R., J. Yang, U. Gullberg, C. Muchardt, M. Yaniv, and A.S. Kraft. 2001. The human brm protein is cleaved during apoptosis: the role of cathepsin G. Proc. Natl. Acad. Sci. USA. 98:3814–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ouedraogo, G., P. Morliere, C. Maziere, J.C. Maziere, and R. Santus. 2000. Alteration of the endocytotic pathway by photosensitization with fluoroquinolones. Photochem. Photobiol. 72:458–463. [DOI] [PubMed] [Google Scholar]
- 18.El-Rayes, B.F., R. Grignon, N. Aslam, O. Aranha, and F.H. Sarkar. 2002. Ciprofloxacin inhibits cell growth and synergises the effect of etoposide in hormone resistant prostate cancer cells. Int. J. Oncol. 21:207–211. [PubMed] [Google Scholar]
- 19.Herold, C., M. Ocker, M. Ganslmayer, H. Gerauer, E.G. Hahn, and D. Schuppan. 2002. Ciprofloxacin induces apoptosis and inhibits proliferation of human colorectal carcinoma cells. Br. J. Cancer. 86:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldmacher, V.S., L.M. Bartle, S. Skletskaya, C.A. Dionne, N.L. Kedersha, C.A. Vater, J.W. Han, R.J. Lutz, S. Watanabe, E.D.C. McFarland, et al. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA. 96:12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanda, T., K.F. Sullivan, and G.M. Wahl. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. [DOI] [PubMed] [Google Scholar]
- 22.Zhu, W., A. Cowie, G.W. Wasfy, L.Z. Penn, B. Leber, and D.W. Andrews. 1996. Bcl-2 mutants with restricted subcellular localization reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 23.Annis, M.G., N. Zamzami, W. Zhu, L.Z. Penn, G. Kroemer, B. Leber, and D.W. Andrews. 2001. Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene. 20:1939–1952. [DOI] [PubMed] [Google Scholar]
- 24.Deussing, J., W. Roth, P. Saftig, C. Peters, H.L. Ploegh, and J.A. Villadangos. 1998. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc. Natl. Acad. Sci. USA. 95:4516–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth, W., J. Deussing, V.A. Botchkarev, M. Pauly-Evers, P. Saftig, A. Hafner, P. Schmidt, W. Schmahl, J. Scherer, I. Anton-Lamprecht, et al. 2000. Cathepsin L deficiency as molecular defect of furless: hyperproliferation of keratinocytes and pertubation of hair follicle cycling. FASEB J. 14:2075–2086. [DOI] [PubMed] [Google Scholar]
- 26.Shi, G.P., J.A. Villadangos, G. Dranoff, C. Small, L. Gu, K.J. Haley, R. Riese, H.L. Ploegh, and H.A. Chapman. 1999. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 10:197–206. [DOI] [PubMed] [Google Scholar]
- 27.Foghsgaard, L., U. Lademann, D. Wissing, B. Poulsen, and M. Jaattela. 2002. Cathepsin B mediates tumor necrosis factor-induced arachidonic acid release in tumor cells. J. Biol. Chem. 277:39499–39506. [DOI] [PubMed] [Google Scholar]
- 28.Inbal, B., S. Bialik, I. Sabanay, G. Shani, and A. Kimchi. 2002. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagedal, K., M. Zhao, I. Svensson, and U.T. Brunk. 2001. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 359:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao, M., J.W. Eaton, and U.T. Brunk. 2001. Bcl-2 phosphorylation is required for inhibition of oxidative stress-induced lysosomal leak and ensuing apoptosis. FEBS Lett. 509:405–412. [DOI] [PubMed] [Google Scholar]
- 31.Zamzami, N., P. Marchetti, M. Castedo, D. Decaudin, A. Macho, T. Hirsch, S.A. Susin, P.X. Petit, B. Mignotte, and G. Kroemer. 1995. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferri, K.F., E. Jacotot, J. Blanco, J.A. Esté, A. Zamzami, S.A. Susin, G. Brothers, J.C. Reed, J.M. Penninger, and G. Kroemer. 2000. Apoptosis control in syncytia induced by the HIV-1–envelope glycoprotein complex. Role of mitochondria and caspases. J. Exp. Med. 192:1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castedo, M., K.F. Ferri, J. Blanco, T. Roumier, N. Larochette, J. Barretina, A. Amendola, R. Nardacci, D. Metivier, J.A. Este, et al. 2001. HIV-1 envelope glycoprotein complex-induced apoptosis involves mammalian target of rapamycin/FKBP12-rapamycin–associated protein-mediated p53 phosphorylation. J. Exp. Med. 194:1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussain, M.S., V. Chukwumaeze-Obiajunwa, and R.G. Micetich. 1995. Sensitive high-performance liquid chromatographic assay for norfloxacin utilizing fluorescence detection. J. Chromatogr. B. Biomed. Appl. 663:379–384. [DOI] [PubMed] [Google Scholar]
- 35.Wolter, K.G., Y.-T. Hsu, C.L. Smith, A. Nechushtan, X.-G. Xi, and R.J. Youle. 1997. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths, G.J., L. Dubrez, C.P. Morgan, N.A. Jones, J. Whitehouse, B.M. Corfe, C. Dive, and J.A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoka, V., B. Turk, S.L. Schendel, T.H. Kil, T. Cirman, S.J. Snipas, L.M. Ellerby, D. Bredesen, H. Freeze, M. Abrahamson, et al. 2001. Lysosomal protease pathways to apoptosis: cleavage of bid, not pro-caspase, is the most likely route. J. Biol. Chem. 276:3149–3157. [DOI] [PubMed] [Google Scholar]
- 38.Reiners, J.J., Jr., J.A. Caruso, P. Mathieu, B. Chelladurai, X.M. Yin, and D. Kessel. 2002. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves bid cleavage. Cell Death Differ. 9:934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaidi, A.U., J.S. McDonough, B.J. Klocke, C.B. Latham, S.J. Korsmeyer, R.A. Flavell, R.E. Schmidt, and K.A. Roth. 2001. Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 60:937–945. [DOI] [PubMed] [Google Scholar]
- 40.Moriwaki, Y., N.A. Begum, M. Kobayashi, M. Matsumoto, K. Toyoshima, and T. Seya. 2001. Mycobacterium bovis Bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and estrogen-inducible gene, EET-1. J. Biol. Chem. 276:23065–23076. [DOI] [PubMed] [Google Scholar]
- 41.Hishita, T., S. Tada-Oikawa, K. Tohyama, Y. Miura, T. Nishihara, Y. Tohyama, Y. Yoshida, T. Uchiyama, and S. Kawanishi. 2001. Caspase-3 activation by lysosomal enzymes in cytochrome c-independent apoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res. 61:2878–2884. [PubMed] [Google Scholar]
- 42.Zamzami, N., I. Marzo, S.A. Susin, C. Brenner, N. Larochette, P. Marchetti, J. Reed, R. Kofler, and G. Kroemer. 1998. The thiol-crosslinking agent diamide overcomes the apoptosis-inhibitory effect of Bcl-2 by enforcing mitochondrial permeability transition. Oncogene. 16:1055–1063. [DOI] [PubMed] [Google Scholar]
- 43.Costantini, P., A.-S. Belzacq, H.L.A. Vieira, N. Larochette, M. de Pablo, N. Zamzami, S.A. Susin, C. Brenner, and G. Kroemer. 2000. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 19:307–314. [DOI] [PubMed] [Google Scholar]
- 44.Pallis, M., M. Grundy, J. Turzanski, R. Kofler, and N. Russell. 2001. Mitochondrial membrane sensitivity to depolarization in acute myeloblastic leukemia is associated with spontaneous in vitro apoptosis, wild-type TP53, and vicinal thiol/disulfide status. Blood. 98:405–413. [DOI] [PubMed] [Google Scholar]
- 45.Klein, S.D., H. Walt, S. Rocha, P. Ghafourifar, M. Pruschy, K.H. Winterhalter, and C. Richter. 2001. Overexpression of Bcl-2 enhances sensitivity of L929 cells to a lipophilic cationic photosensitiser. Cell Death Differ. 8:204–206. [DOI] [PubMed] [Google Scholar]
- 46.Mannick, J.B., C. Schonhoff, N. Papeta, P. Ghafourifar, M. Szibor, K. Fang, and B. Gaston. 2001. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 154:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]