

Abstract
Early interactions between lung dendritic cells (LDCs) and Mycobacterium tuberculosis, the etiological agent of tuberculosis, are thought to be critical for mounting a protective anti-mycobacterial immune response and for determining the outcome of infection. However, these interactions are poorly understood, at least at the molecular level. Here we show that M. tuberculosis enters human monocyte-derived DCs after binding to the recently identified lectin DC-specific intercellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN). By contrast, complement receptor (CR)3 and mannose receptor (MR), which are the main M. tuberculosis receptors on macrophages (Mφs), appeared to play a minor role, if any, in mycobacterial binding to DCs. The mycobacteria-specific lipoglycan lipoarabinomannan (LAM) was identified as a key ligand of DC-SIGN. Freshly isolated human LDCs were found to express DC-SIGN, and M. tuberculosis–derived material was detected in CD14−HLA-DR+DC-SIGN+ cells in lymph nodes (LNs) from patients with tuberculosis. Thus, as for human immunodeficiency virus (HIV), which is captured by the same receptor, DC-SIGN–mediated entry of M. tuberculosis in DCs in vivo is likely to influence bacterial persistence and host immunity.
Keywords: Mycobacteria, tuberculosis, dendritic cell, DC-SIGN, lipoarabinomannan
Introduction
M. tuberculosis infections are responsible for 1.5 to 2 million deaths annually. Such a dramatic situation is due, at least in part, to the ability of the airborne bacillus to resist killing by, and to parasitize host alveolar macrophages (Mφs; reference 1). Protective anti-mycobacterial immune response involves mainly T lymphocytes that activate the Mφ microbicidal functions through the release of interferon γ (2, 3). Priming of naive T lymphocytes against mycobacterial antigens is thought to occur in the proximal LNs and to rely on a particular subset of phagocytic cells, the dendritic cells (DCs). Indeed, DCs exhibit the unique ability to activate naive lymphocytes after migration from infection sites, where they capture antigens, to the LNs where they express high amounts of presentation molecules, such as MHC-II, and costimulatory molecules, such as CD80 and CD86 (4). The early interaction between the DCs present as a dense network in the airway mucosa (5) and M. tuberculosis is thus likely to be critical for mounting a protective anti-mycobacterial immune response (3, 6–9). However, M. tuberculosis interactions with DCs are poorly understood at the molecular level. In particular, the ability of M. tuberculosis to replicate in DCs, relative to Mφs, remains controversial (7, 9, 10), and the receptor(s) used by M. tuberculosis to bind and to enter DCs are still unknown, whereas those involved in the parasitism of Mφs have been well characterized in vitro. Mycobacterial binding to Mφs occurs in cholesterol-rich domains of the host cell plasma membrane (11) and involves CR3, together with other molecules like MR, CR1, CR4, CD14, surfactant protein (SP)-A receptors, as well as scavenger receptors (12, 13). Other surface molecules, such as Toll-like receptors (TLRs), are also essential for mycobacterial interactions with phagocytic cells (14), though their role in mycobacterial entry remains to be evaluated. Some of these receptors (e.g., CR3, MR) are present on DCs and may be involved in the binding and entry of mycobacteria into these cells. However, DCs express additional receptors that are dedicated to capture of antigens. These additional receptors include the recently identified DC-SIGN (15), a calcium-dependent (C-type) lectin, containing a carbohydrate recognition domain (CRD) at its extracellular COOH-terminal end, that recognizes mannose-rich molecules (16). DC-SIGN was initially described as a receptor for ICAM-3 at the surface of T cells, triggering the formation of the immunological synapse between DCs and naive T lymphocytes. Interestingly, DC-SIGN binds to HIV and simian immunodeficiency viruses, and is involved in the trans-infection of CD4+ T lymphocytes by HIV- or SIV-infected DCs (17). DC-SIGN has also been recently involved in Leishmania pifanoi binding to DCs (18).
Here we show that M. tuberculosis infects DCs via ligation of DC-SIGN by the mycobacterial surface-exposed lipoglycan lipoarabinomannan (LAM). Freshly isolated LDCs were found to express DC-SIGN, and M. tuberculosis–derived material was detected in DC-SIGN+CD14−HLA-DR+ cells in LNs from patients with tuberculosis.
Materials and Methods
Cells and Bacteria.
HeLa-derived cells expressing or not DC-SIGN (P4-DC and P4, respectively) (19), were cultured and infected in RPMI-1640 (GIBCO BRL/Invitrogen) supplemented with 10% heat-inactivated FCS (Dutscher). Mononuclear cells were isolated from the blood of healthy volunteers (Etablissement Français du Sang) by Ficoll-Paque centrifugation. T, B, and NK cells were depleted using M-450 Pan T/CD2 and M-450 Pan B/CD19 Dynabeads (Dynal). The recovered cells, referred to as monocytes, were seeded in 6-well plates at 2 × 106 cells/well in 3 ml RPMI-1640, 10% FCS, l-glutamine, granulocyte/Mφ-colony stimulating factor (10 ng/ml), and interleukin 4 (20 ng/ml; both from R&D Systems). This resulted in DCs after 5 d of culture. Cultures were fed every 2 d with fresh medium containing full doses of cytokines. GFP-expressing strains of M. tuberculosis H37Rv and M. bovis BCG were generated by transformation with the GFP-encoding plasmid pEGFP and propagated in medium containing 50 μg/ml hygromycin B (Boehringer). Human lung DCs (LDCs) were isolated as described (20, 21). Lung samples were from surgical specimen distant from primary carcinoma, obtained with the patients' consent, and used according to institutional guidelines. In brief, after treating the lung fragments with collagenase, cells were separated on a Ficoll-Paque gradient to obtain pulmonary mononuclear cells, which were cultured in Petri dishes for 1 h before removing nonadherent cells. Adherent cells were further incubated for 16 h in medium. Loosely adherent mononuclear cells released after three rinses in saline were separated into LDCs and autofluorescent alveolar Mφs with a FACStar™ (Becton Dickinson) according to the presence or absence of autofluorescent inclusions, using a 488 nm wavelength for excitation and a 588 nm filter for emission. Gates were set to remove cell debris and to select LDCs. In contrast to alveolar Mφs, the latter cells are potent stimulators of allogeneic T lymphocytes (data not depicted, and reference 20). LDCs represented 0.3 to 0.8% of the total cells.
Lymph Node Samples.
Lymph nodes were referred to the Laboratory of Pathology at the Saint-Louis Hospital (Paris, France) for the purpose of tuberculosis diagnosis with the patients' consent and used according to institutional guidelines. Human tissues were previously fixed in AFA (Carlo Erba), a mix of 2% formalin (vol/vol), 5% acetic acid (vol/vol), 75% ethanol (vol/vol), and 18% water (vol/vol), and then embedded in paraffin for histopathological diagnosis. Smear positive and/or culture positive (in less than 12 d) biopsies were selected from the collection between May 1995 and December 2001, and ∼10 sections per biopsy were used for immunostaining.
Binding Assay.
Cells were infected at the indicated multiplicity of infection (MOI) for 4 h at 4°C in RPMI-1640, 10% FCS, extensively washed in RPMI-1640, and analyzed by flow cytometry. Fluorescence was assessed on a total of 2 × 104 cells per sample using a FACSCalibur™ and CELLQuest™ software (Becton Dickinson). In some experiments, the same samples were also plated out onto agar medium and CFUs were scored after 3 wk at 37°C. Alternatively, infections were performed in the presence of 10% complete human serum, in order to opsonize bacteria with complement, as indicated (22).
Antibodies.
The following mAbs were used in binding inhibition experiments: anti-CR3/CD11b (clone M1/70; BD Biosciences), -CR3 (2LPM19c; Dako), -MR (clone 15/2; HyCult Biotechnology), -CD40 (clone 5C3; BD Biosciences), and -DC-SIGN (clones 120507; R&D Systems). 1B10 (IgG2a-κ) were produced as follows: Balb/c mice were immunized with 293T cells transfected with cDNA encoding DC-SIGN (cloned from human monocyte-derived DCs). Hybridoma supernatants were screened for the ability to recognize DC-SIGN–expressing HeLa cells, and were purified from bulk cultures. 1B10 neutralizes HIV gp120 binding to DC-SIGN and prevents trans-infection of CD4+ T cells by HIV-pulsed DC-SIGN+ cell lines (A. Amara, personal communication). Binding inhibition experiments were performed by preincubating the cells with the indicated mAbs at different concentrations (see legends to figures) for 1 h at 4°C before the binding assay. For confocal microscopy, DC-SIGN was detected using clone 120507 mAb and a Cy3-conjugated secondary mAb (Amersham Biosciences). For flow cytometry, DC-SIGN, CR3/CD11b, and MR were detected using phycoerythrin-conjugated mAbs from clones 120507, M1/70, and 3.29B1.10 (Immunotech), respectively; CD14 was detected using an allophycocyanin-conjugated mAb (clone M5E2, BD Biosciences); HLA-DR was detected using fluorescein isothiocyanate-conjugated mAb (clone H279; Immunotech). For immunohistochemistry, CD3, CD20, DC-SIGN, CD14, HLA-DR, and M. tuberculosis were detected using polyclonal rabbit serum (Dako), mAbs from clone H1(FB1; Dako), from clone 1B10, from clone 7 (Novocastra), from clone CR3/43 (Dako), and an anti–M. bovis BCG polyclonal rabbit serum, respectively.
Results and Discussion
Given the unique richness of the mycobacterial envelope in poly-mannosylated materials (23), we asked whether M. tuberculosis interacts with DC-SIGN on the surface of DCs. We first compared the binding of virulent GFP-expressing M. tuberculosis H37Rv, the most commonly used reference M. tuberculosis strain, to HeLa-derived cells expressing or not DC-SIGN (P4-DC and P4, respectively; reference 19). We found that M. tuberculosis bound to P4-DC cells up to 25 times more than to P4 cells (Fig. 1 A). Similar results were obtained with M. tuberculosis clinical isolate MT103 (data not shown), indicating that binding to DC-SIGN was not restricted to laboratory mycobacterial strains. We then studied the binding of M. tuberculosis to DCs, and the role of DC-SIGN in this process, as compared with CR3 and MR. As reported (24), human monocyte-derived DCs (MDDCs; reference 25) expressed high levels of DC-SIGN together with CR3 and MR. We performed a binding assay with MDDCs that had been preincubated or not with different mAbs. Preincubation of MDDCs with two different anti–DC-SIGN mAbs inhibited attachment of M. tuberculosis up to 90% (Fig. 1 B). Interestingly, preincubation with two anti-CR3 mAbs, used in combination, an anti-MR mAb, or an irrelevant (i.e., directed against a non-mycobacteria-binding protein) anti-CD40 mAb, had only minor effects on the binding of M. tuberculosis to MDDCs (Fig. 1 B). Under the same conditions anti-CR3 and -MR mAbs inhibited M. tuberculosis binding to monocyte-derived Mφs (MDMφs) by ∼50 and ∼45%, respectively (data not shown), thus confirming previous reports that CRs and MR are major M. tuberculosis receptors on MDMφs (22). It was also important to assess whether DC-SIGN was still the predominant M. tuberculosis receptor on DCs in the presence of a complement source, a condition that might be expected in vivo. To this end, we performed a binding inhibition experiment in the presence of complete human serum. Anti-DC-SIGN mAbs were then still able to inhibit up to 90% mycobacterial binding to DCs (Fig. 1 B). Under the same conditions, anti-CR3 and -MR mAbs inhibited mycobacterial binding to MDMφs by ∼60 and ∼20%, respectively (data not shown). Thus, our results indicate that DC-SIGN acts as the major M. tuberculosis receptor on human MDDCs, even in the presence of complement.
Figure 1.

M. tuberculosis binds to DC-SIGN. (A) Epithelial HeLa-derived P4 cells expressing or not DC-SIGN (P4-DC and P4, respectively) were infected with GFP expressing (GFP+) M. tuberculosis H37Rv at different MOIs. Bacteria binding was evaluated by flow cytometry (left panel) and CFU counts (right panel). Data represent means (±SD) of three separate experiments. (B) MDDCs were infected with GFP-M. tuberculosis H37Rv at an MOI of 1 bacterium per cell in the presence or not of 10% complete human serum, either directly (control) or after preincubation with 10 μg/ml of mAbs directed against CR3/CD11b, MR, CD40, or DC-SIGN. Bacteria binding was assessed by flow cytometry. Preincubation with the corresponding isotype controls led to no significant inhibition of mycobacteria binding (not shown). Data were expressed as percentages of binding relative to control values (100%, no mAb), and means (±SD) of three independent experiments are shown. (C) MDDCs were infected with GFP-M. bovis BCG, Salmonella typhimurium (clinical isolate), or Listeria monocytogenes (clinical isolate) and subjected to the binding assay. In experiments using S. typhimurium and L. monocytogenes, infected cells were plated out onto agar medium and CFUs were scored after 24 h at 37°C. Data are expressed as in B. (D) M. tuberculosis binding to DC-SIGN is inhibited by LAM. Cells were pretreated for 1 h at 4°C with 10 μg/ml mannan as control, or with 10 μg/ml LAM, and subjected to the binding assay. Data are expressed as in B.
We next examined whether DC-SIGN contributes to the attachment of other intracellular bacterial species to MDDCs. Binding of the vaccine strain Mycobacterium bovis bacillus Calmette-Guérin (BCG), which belongs to the tuberculosis complex, was also found to be mediated by DC-SIGN (Fig. 1 C). However, pretreatment with anti–DC-SIGN mAbs had no effect on the binding of either the Gram-positive Listeria monocytogenes or the Gram-negative Salmonella typhimurium species (Fig. 1 C). Major structural differences exist at the surface of mycobacteria compared with Gram+ and Gram− species, which may explain differences in binding to DC-SIGN. In particular, LAM is an abundant poly-mannosylated lipoglycan, specific to the mycobacterial envelope (26), and it has been shown to bind to various human C-type lectins on Mφs, such as surfactant protein D (27). Inasmuch as DC-SIGN is a C-type lectin that recognizes mannose-rich molecules (16), we investigated whether M. tuberculosis binding to the lectin was mannosyl-defined. Binding of M. tuberculosis to both P4-DC and MDDCs was inhibited up to 90% by yeast mannan as well as by M. tuberculosis H37Rv-derived LAM (Fig. 1 D). By contrast, preincubation of cells with LPS derived from Escherichia coli or with dextran had no effect on the binding process (data not shown). These findings suggest that LAM may constitute a privileged mycobacterial ligand for DC-SIGN, even though other mycobacterial components may also bind to the lectin. Interestingly, Mycobacterium smegmatis–derived LAM, that is devoid of mannose capping residues, was found to only moderately inhibit M. tuberculosis binding to DC-SIGN (unpublished data).
After attaching to the cell surface, pathogenic mycobacteria are taken up by phagocytic cells and reside in phagosomes that do not fuse with host cell late endosomes and lysosomes, but take part in the recycling pathway (1). To investigate DC-SIGN trafficking in M. tuberculosis-infected cells, we performed confocal microscopy analysis of MDDCs infected with GFP-expressing mycobacteria. During the first hour of phagocytosis, most bacilli were detected as either attached extracellularly to the cells (Fig. 2 A, top panel), or colocalized with DC-SIGN in nascent phagosomes (Fig. 2 A, middle panel). However, DC-SIGN staining was not detected on phagosomes that had detached from the plasma membrane, indicating that it was excluded from the vacuoles very soon after phagocytosis (Fig. 2 A, bottom panel). These data indicate that DC-SIGN is present in M. tuberculosis vacuoles during the early steps of bacterial uptake, and is then rapidly expelled from the phagosome (Fig. 2 B), possibly as a result of recycling to the cell plasma membrane. We also examined whether M. tuberculosis infection could modify DC-SIGN expression at the surface of infected cells. As reported (6, 7), infection was found to induce cell maturation, as illustrated by up-regulation of CD83 (Fig. 2 C), CD86, and HLA-DR (data not shown). DC-SIGN expression was only slightly down-modulated in mature infected cells, even 48 h after infection (Fig. 2 C).
Figure 2.


DC-SIGN is transiently present onto the M. tuberculosis phagosome. Cells were pulsed at 4°C for 3 h with GFP-M. tuberculosis, washed extensively in RPMI-1640, and chased at 37°C for the indicated periods of time. (A) The two top panels shows cells representative of early phagocytosis events; DC-SIGN was detected both at the cell surface and in intracellular vesicles, but due to the strong surface staining, the red signal had to be reduced. Each panel shows a representative cell. (B) Kinetics of DC-SIGN colocalization with M. tuberculosis (M. tb): a minimum of 100 bacteria were scored at each time point. Extracellular bacteria that were found attached to the cells (early time points, 15 and 30 min) were scored as colocalizing with DC-SIGN. Results are means (±SD) of three separate experiments. (C) Surface expression of CD83 (left panel) and DC-SIGN (right panel) on MDDCs was assessed by flow cytometry 48 h after infection with GFP-M. tuberculosis H37Rv. For analysis, cells were gated on GFP-M. tuberculosis–infected MDDCs. Dotted line: isotype control labeling; plain lines: uninfected cells; bold lines: infected cells.
It was then important to determine whether M. tuberculosis could interact with DC-SIGN+ cells in vivo. We first evaluated the presence of the lectin on human interstitial LDCs as compared with in vitro–generated MDDCs. LDCs were isolated from surgical specimens in tissues distant from limited primary lung carcinomas. As reported (20), all LDCs were HLA-DR+ (Fig. 3 A) and CD14− (data not shown), a phenotype shared by MDDCs. Like MDDCs, LDCs expressed surface DC-SIGN, CR3, and MR (Fig. 3 A). Although we cannot formally rule out the possibility that LDC preparations were devoid of contaminant cells of other type(s) (e.g., activated macrophages), it is tempting to suggest that M. tuberculosis may encounter and interact with DC-SIGN+ DCs during the natural course of infection. However, too few cells could be recovered from surgical samples to allow us to perform binding experiments to test this hypothesis. To further investigate DC-SIGN possible involvement in the interactions of M. tuberculosis with DCs in vivo, we reasoned that if DC-SIGN+ DCs take up M. tuberculosis in the lungs, and since DC-SIGN expression is only slightly reduced by infection-associated maturation of the cells, then we should be able to detect mycobacteria-derived material in DC-SIGN+ DCs in LNs from patients with tuberculosis. LN paraffin-embedded sections from seven patients were stained with anti-mycobacterial and anti–DC-SIGN mAbs (Fig. 3 B). Samples containing viable bacilli were selected based on smear and culture positivity. The selected LNs were rich in granulomas, characterized by caseous centers surrounded by rings of T and B lymphocytes (Fig. 3 B, left panel). Granulomas are typical of mycobacterial infections. They develop after recruitment and accumulation of effector lymphocytes around infected foci. As reported (15, 28), DC-SIGN was detected mostly in intergerminal T cell zones and not in germinal centers. In addition, DC-SIGN+ cells were located in granulomatous structures, and were CD14− and HLA-DR+ (Fig. 3 B, middle and right panels, respectively). Mycobacteria and mycobacteria-derived antigens were immunodetected in samples from 5 out of 7 patients, in both granulomatous (Fig. 3 C, left and right panels), and nongranulomatous regions of the LNs, including subcapsular sinuses (Fig. 3 C, middle panel). In most cases (∼80–100%), mycobacteria-specific signal was detected within DC-SIGN+ cells (Fig. 3 C). These findings indicate that M. tuberculosis interacts with DC-SIGN in vivo and that DC-SIGN+ cells, possibly DCs, may carry mycobacteria or mycobacteria-derived material from the lungs to the LNs during their maturation process.
Figure 3.

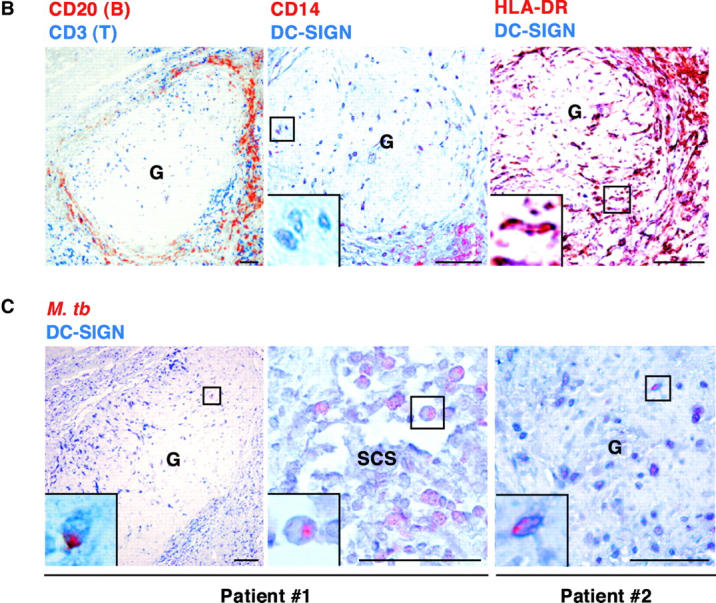
DC-SIGN expression on lung DCs (LDCs) and in lymph nodes (LNs). (A) LDCs are HLA-DR+ and express DC-SIGN, CR3, and MR. Surface expression of HLA-DR, DC-SIGN, CR3/CD11b, and MR on LDCs from a noninfected patient was assessed by flow cytometry using the appropriate cytochrome-conjugated mAbs. (B) DC-SIGN expression in the LN from a patient with tuberculosis (G, granuloma). Left panel, CD3 (blue) and CD20 (red); middle panel, DC-SIGN (blue) and CD14 (red); right panel, DC-SIGN (blue) and HLA-DR (red). (C) Localization of M. tuberculosis–derived antigens in DC-SIGN+ cells in LNs from two patients with tuberculosis. DC-SIGN (blue) and M. tuberculosis (red) were immunodetected both in granulomas (G; left and right panels) and in nongranulomatous regions, including subcapsular sinuses (SCS; middle panel). In B and C, bars represent 0.5 mm and squares represent areas shown at higher magnification at the single cell level in the insets. Staining of the samples with IgG2a (1B10 isotype control) or with a naive rabbit serum led to no detectable signal (data not depicted).
Altogether, our results demonstrate that DC-SIGN/CD209 is the predominant M. tuberculosis receptor on human DCs, whereas the mycobacterial Mφ receptors, CR3 and MR, appear to play a minor role, if any, in this binding. This exclusivity may be due to DC-SIGN abundance on DCs relative to CR3 and MR, and/or to the affinity of the lectin for its ligand(s). Affinity for DC-SIGN seems to be fairly restrictive among bacteria, as neither Gram+ L. monocytogenes nor Gram− S. typhimurium species bound to the lectin. This is not surprising, as the envelopes of Gram+ and Gram− bacteria are very poor in poly-mannosylated material. Unique characteristics of the mycobacterial cell wall, which is the most complex of all bacterial cell surfaces (23), might thus account for the affinity for DC-SIGN. Our finding that LAM, like mannan, which contain common mannosyl motifs, can block DC-SIGN–mediated attachment of M. tuberculosis to DCs and to P4-DC cells is consistent with the high affinity of the lectin for mannose-rich molecules (16), and suggests that LAM may be one of its major mycobacterial ligands.
Ligation of DC-SIGN by mycobacteria is likely to have important effects on the immunological and pathological events associated with M. tuberculosis infection. Differential receptor usage by M. tuberculosis on DCs and Mφs may account for the different survival ability and trafficking patterns of mycobacteria in the two cell types, which is still a matter of debate (7, 9, 10). DC-SIGN has been detected on alveolar Mφs (28, 29), that constitute the privileged cell targets of M. tuberculosis during the early steps of infection. It will be important to evaluate whether the lectin is also a predominant mycobacterial receptor on this cell population. It will also be of great interest to investigate what type of pro- or antiinflammatory cytokines are induced or repressed upon DC-SIGN ligation by mycobacteria (30), as compared with ligation of other signal transducers, such as the TLRs (14). DCs are also involved in the early activation of non-MHC–restricted and CD1-restricted T cells specific for various mycobacterial glycolipids, including LAM (31). The intracellular trafficking pattern of DC-SIGN in M. tuberculosis-infected DCs suggests that DC-SIGN may carry mycobacterial glycolipids from the bacterial vacuole to the cell plasma membrane and/or to various subcellular compartments, where glycolipids could be loaded onto CD1 molecules for presentation to CD1-restricted lymphocytes (32).
In the lungs, submucosal and interstitial LDCs are thought to play a key role in immune surveillance of the respiratory tract (5). In particular, interactions of DCs present in the alveolar septal walls with M. tuberculosis could be crucial for initiating an efficient anti-mycobacterial immune response. Our finding that LDCs express DC-SIGN suggests that DC-SIGN is likely to interact with M. tuberculosis in vivo. This is strengthened by detection of mycobacteria-derived material in DC-SIGN+ DCs in LNs from patients with tuberculosis. It is interesting that both M. tuberculosis and HIV can bind to DC-SIGN. DC-SIGN–expressing cells may carry either intracellular or surface-attached M. tuberculosis during their migration from the site of infection to the LNs, and could thus constitute a mycobacterial reservoir. This has also been suggested for HIV (17), and might account for several pathological and immunological aspects of M. tuberculosis infection, e.g., mediastinal adenitis, the formation of secondary granulomas in the LNs, and the chronic stimulation of the immune system that is required to maintain the latency period of the disease.
Acknowledgments
We thank S. Wain-Hobson, L. Quintana-Murci, C. Petit, V. Rosas Magalanes, N. Winter, and P. Davis for reading the manuscript and helpful discussions. We acknowledge F. Amira for excellent technical assistance in histology, and A. Janin for helping to obtain LN samples. We thank G. Stewart for providing pEGFP, and F.X. Weill for providing GFP-expressing M. bovis BCG. We acknowledge the Colorado State University and the National Institutes of Health, National Institute of Allergy and Infectious Diseases contract NO1 AI-75320 “Tuberculosis Research Materials and Vaccine Testing,” for the gift of purified LAM.
O. Neyrolles and L. Tailleux are fellows from the Fondation pour la Recherche Médicale. This work was supported by grants from Sidaction, Agence Nationale de Recherche sur le SIDA, Association pour la Recherche sur le Cancer, Ligue contre le Cancer, European “Cluster for tuberculosis vaccine development”, and Institut Pasteur.
References
- 1.Russell, D.G. 2001. Mycobacterium tuberculosis: here today, and here tomorrow. Nat. Rev. Mol. Cell Biol. 2:569–577. [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann, S.H. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1:20–30. [DOI] [PubMed] [Google Scholar]
- 3.Flynn, J.L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129. [DOI] [PubMed] [Google Scholar]
- 4.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 5.Holt, P.G. 2000. Antigen presentation in the lung. Am. J. Respir. Crit. Care Med. 162:S151–156. [DOI] [PubMed] [Google Scholar]
- 6.Henderson, R.A., S.C. Watkins, and J.L. Flynn. 1997. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J. Immunol. 159:635–643. [PubMed] [Google Scholar]
- 7.Fortsch, D., M. Rollinghoff, and S. Stenger. 2000. IL-10 converts human dendritic cells into macrophage-like cells with increased antibacterial activity against virulent Mycobacterium tuberculosis. J. Immunol. 165:978–987. [DOI] [PubMed] [Google Scholar]
- 8.Inaba, K., M. Inaba, M. Naito, and R.M. Steinman. 1993. Dendritic cell progenitors phagocytose particulates, including bacillus Calmette-Guerin organisms, and sensitize mice to mycobacterial antigens in vivo. J. Exp. Med. 178:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao, X., R. Lo-Man, P. Guermonprez, L. Fiette, E. Deriaud, S. Burgaud, B. Gicquel, N. Winter, and C. Leclerc. 2002. Dendritic cells are host cells for mycobacteria in vivo that trigger innate and acquired immunity. J. Immunol. 168:1294–1301. [DOI] [PubMed] [Google Scholar]
- 10.Bodnar, K.A., N.V. Serbina, and J.L. Flynn. 2001. Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect. Immun. 69:800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatfield, J., and J. Pieters. 2000. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 288:1647–1650. [DOI] [PubMed] [Google Scholar]
- 12.Schorey, J.S., M.C. Carroll, and E.J. Brown. 1997. A macrophage invasion mechanism of pathogenic mycobacteria. Science. 277:1091–1093. [DOI] [PubMed] [Google Scholar]
- 13.Ernst, J.D. 1998. Macrophage receptors for Mycobacterium tuberculosis. Infect. Immun. 66:1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brightbill, H.D., D.H. Libraty, S.R. Krutzik, R.B. Yang, J.T. Belisle, J.R. Bleharski, M. Maitland, M.V. Norgard, S.E. Plevy, S.T. Smale, et al. 1999. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 285:732–736. [DOI] [PubMed] [Google Scholar]
- 15.Geijtenbeek, T.B., R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, G.J. Adema, Y. van Kooyk, and C.G. Figdor. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 100:575–585. [DOI] [PubMed] [Google Scholar]
- 16.Feinberg, H., D.A. Mitchell, K. Drickamer, and W.I. Weis. 2001. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science. 294:2163–2166. [DOI] [PubMed] [Google Scholar]
- 17.Geijtenbeek, T.B., D.S. Kwon, R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, J. Middel, I.L. Cornelissen, H.S. Nottet, V.N. KewalRamani, D.R. Littman, et al. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 100:587–597. [DOI] [PubMed] [Google Scholar]
- 18.Colmenares, M., A. Puig-Kroger, O. Muniz Pello, A.L. Corbi, and L. Rivas. 2002. Dendritic-cell specific ICAM-3 grabbing nonintegrin (DC-SIGN, CD209), a C-type surface lectin in human dendritic cells, is a receptor for Leishmania amastigotes. J. Biol. Chem. 277:36766–36769. [DOI] [PubMed] [Google Scholar]
- 19.Sol-Foulon, N., A. Moris, C. Nobile, C. Boccaccio, A. Engering, J.P. Abastado, J.M. Heard, Y. van Kooyk, and O. Schwartz. 2002. HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity. 16:145–155. [DOI] [PubMed] [Google Scholar]
- 20.Cochand, L., P. Isler, F. Songeon, and L.P. Nicod. 1999. Human lung dendritic cells have an immature phenotype with efficient mannose receptors. Am. J. Respir. Cell Mol. Biol. 21:547–554. [DOI] [PubMed] [Google Scholar]
- 21.van den Heuvel, M.M., C.E.G. Havenith, and R.H.J. Beelen. 2001. Isolation of human lung dendritic cells. Dendritic Cell Protocols. S.P. Robinson and A.J. Stagg, editors. Humana Press Inc., Totowa, NJ. 163–173. [DOI] [PubMed]
- 22.Schlesinger, L.S. 1993. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J. Immunol. 150:2920–2930. [PubMed] [Google Scholar]
- 23.Ehlers, M.R., and M. Daffe. 1998. Interactions between Mycobacterium tuberculosis and host cells: are mycobacterial sugars the key? Trends Microbiol. 6:328–335. [DOI] [PubMed] [Google Scholar]
- 24.Relloso, M., A. Puig-Kroger, O.M. Pello, J.L. Rodriguez-Fernandez, G. de la Rosa, N. Longo, J. Navarro, M.A. Munoz-Fernandez, P. Sanchez-Mateos, and A.L. Corbi. 2002. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-beta, and anti-inflammatory agents. J. Immunol. 168:2634–2643. [DOI] [PubMed] [Google Scholar]
- 25.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 26.Chatterjee, D., K. Lowell, B. Rivoire, M.R. McNeil, and P.J. Brennan. 1992. Lipoarabinomannan of Mycobacterium tuberculosis. Capping with mannosyl residues in some strains. J. Biol. Chem. 267:6234–6239. [PubMed] [Google Scholar]
- 27.Ferguson, J.S., D.R. Voelker, F.X. McCormack, and L.S. Schlesinger. 1999. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163:312–321. [PubMed] [Google Scholar]
- 28.Soilleux, E.J., L.S. Morris, G. Leslie, J. Chehimi, Q. Luo, E. Levroney, J. Trowsdale, L.J. Montaner, R.W. Doms, D. Weissman, et al. 2002. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J. Leukoc. Biol. 71:445–457. [PubMed] [Google Scholar]
- 29.Lee, B., G. Leslie, E. Soilleux, U. O'Doherty, S. Baik, E. Levroney, K. Flummerfelt, W. Swiggard, N. Coleman, M. Malim, and R.W. Doms. 2001. cis expression of DC-SIGN allows for more efficient entry of human and simian immunodeficiency viruses via CD4 and a coreceptor. J. Virol. 75:12028–12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figdor, C.G., Y. van Kooyk, and G.J. Adema. 2002. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2:77–84. [DOI] [PubMed] [Google Scholar]
- 31.Ernst, W.A., J. Maher, S. Cho, K.R. Niazi, D. Chatterjee, D.B. Moody, G.S. Besra, Y. Watanabe, P.E. Jensen, S.A. Porcelli, et al. 1998. Molecular interaction of CD1b with lipoglycan antigens. Immunity. 8:331–340. [DOI] [PubMed] [Google Scholar]
- 32.Schaible, U.E., K. Hagens, K. Fischer, H.L. Collins, and S.H. Kaufmann. 2000. Intersection of group I CD1 molecules and mycobacteria in different intracellular compartments of dendritic cells. J. Immunol. 164:4843–4852. [DOI] [PubMed] [Google Scholar]