

Abstract
Transduction of Tat-tagged fusion proteins confirmed a hypothesis based on pharmacologic inhibitors (Fuortes, M., M. Melchior, H. Han, G.J. Lyon, and C. Nathan. 1999. J. Clin. Invest. 104:327–335) that proline-rich tyrosine kinase (Pyk2) plays a critical role in the activation of adherent human neutrophils, and allowed an analysis of individual Pyk2 domains not possible with chemical inhibitors. Acting as a dominant negative, the COOH terminus of Pyk2 fused to a Tat peptide (Tat-CT), but not other regions of Pyk2, specifically inhibited the respiratory burst of cells responding to tumor necrosis factor (TNF), Salmonella, or Listeria, while sparing responses induced by phorbol ester. Tat-CT suppressed TNF-triggered cell spreading and the phosphorylation of endogenous Pyk2 and the associated tyrosine kinase Syk without blocking the ability of neutrophils to degranulate and kill bacteria. Thus, separate signals control the respiratory burst and degranulation, and a normal rate of killing of some bacteria can be sustained by granule products in conjunction with a minimal residual respiratory burst. Inhibition of select inflammatory functions without impairment of antibacterial activity may commend the Pyk2 pathway as a potential target for antiinflammatory therapy.
Keywords: neutrophil, Pyk2, Syk, Tat, TNF
Introduction
An inherent dilemma of antiinflammatory therapy, the risk of impairing host defense, is particularly problematic with neutrophils. Adherence of neutrophils to endothelium, their diapedesis from venules into tissues and their release of peptides, proteases, and reactive oxygen intermediates underlie both their killing of bacteria and their damage to tissues. The histotoxic impact of neutrophils is prominent in several inflammatory settings that are not thought to involve bacterial infection, such as rheumatoid arthritis, Crohn's disease, and ischemia-reperfusion syndrome, or in which neutrophil-mediated injury can occur at sites remote from invading bacteria, as in the acute respiratory distress and systemic inflammatory response syndromes. The stimuli that activate neutrophils in these settings are host-derived mediators rather than bacteria themselves. Pharmacologic agents might be able to target some inflammatory functions of neutrophils while sparing antimicrobial functions if some signaling steps involved in the activation of neutrophils by host-derived mediators were distinct from steps involved in the ingestion and killing of bacteria.
Full activation of neutrophils by soluble host products requires a binary signal. One part of the signal consists of integrin ligation during adherence to extracellular matrix. Simultaneously, a factor such as TNF, lymphotoxin, C5a, formylated peptides, macrophage inflammatory protein (MIP)-1,* CSF-G, or GM-CSF must engage its receptor(s) on the neutrophil (1–4). TNF has been studied intensively both with respect to the mechanisms by which it activates adherent neutrophils (2, 5–15) and the benefit of its neutralization in inflammatory disorders such as rheumatoid arthritis (16), ankylosing spondylitis (17), and Crohn's disease (18).
Proline-rich tyrosine kinase (Pyk2; reference 19), also called related adhesion focal tyrosine kinase (20), cell adhesion kinase-β (21), calcium-dependent tyrosine kinase (22), and focal adhesion kinase 2 (23), is a nonreceptor kinase expressed abundantly in hematopoietic cells. Pyk2 is distinguished by a lack of SH2 and SH3 domains. Pyk2's kinase domain is flanked instead by large N- and COOH-terminal extensions that are involved in binding diverse kinases and substrates in various cells (24–26). Previous studies identified Pyk2 in human neutrophils, localized it to focal adhesions and podosomes, and demonstrated its tyrosine phosphorylation, activation, and association with paxillin during stimulation of adherent neutrophils by TNF (27, 28). One tyrosine kinase inhibitor of 51 that was tested, tyrphostin A9, blocked both the activation of TNF-treated neutrophils and the tyrosine phosphorylation of Pyk2 (27). However, it was not established whether tyrphostin A9 targeted Pyk2 directly. Tyrphostin A9 may instead have inhibited another kinase for which Pyk2 was a target. Tyrphostin A9 inhibits the platelet-derived growth factor receptor (29) and as with most chemical inhibitors, the full range of its targets is unknown.
In the analysis of intracellular signaling, limited availability, specificity, or knowledge of the specificity of chemical inhibitors has directed attention to interventions based on DNA, RNA, or peptide sequences. However, the options for applying such approaches to neutrophils have been more restricted than with most other cell types. Gene disruption by homologous recombination has aided the study of neutrophils in the mouse, but the relevance for human neutrophils is uncertain. Although the respiratory burst of mouse neutrophils in response to phorbol esters is of the same magnitude as that of human neutrophils, the response of mouse neutrophils to soluble, physiologic agonists like TNF is 10-fold lower than for their human counterparts (15, 30) and in some laboratories is rarely demonstrable (unpublished data and P. Detmers, personal communication). Such species differences underscore the importance of studies in human neutrophils themselves. Transfection of cDNAs, antisense constructs, and inhibitory RNAs is not useful in primary neutrophils, because neutrophil activation depends on posttranslational modifications of preformed proteins whose half-life exceeds the lifetime of the cells under the conditions of study. Myeloid cell lines can be transfected with difficulty (31), but do not undergo a TNF-induced respiratory burst even after differentiative agents drive a minority of the population toward maturity.
With other approaches so constrained, protein transduction holds special interest for the analysis of signaling in primary neutrophils. Loading procedures such as osmotic shock, scraping, streptolysin O permeabilization, and electroporation tend to activate or damage neutrophils as evidenced when subsequent studies span several hours. Recently, transduction based on membrane-permeant sequences in the HIV tat gene product (32) has been described in neutrophils (33, 34) and eosinophils (35–38), but questions remain that appear to have restricted the use of this approach.
In this work, we studied the transduction of Tat-tagged, Pyk2-derived polypeptides into human neutrophils. This allowed both a comparison to and an extension of earlier work with kinase inhibitors (27). The approaches used for purification of the fusion proteins were critical. The vast majority of Tat-tagged proteins taken up appeared to reside in endosomes. However, enough reached the cytosol to demonstrate that the carboxyl terminus of Pyk2 is critical for TNF or bacteria to trigger the respiratory burst. At the same time, the Pyk2 carboxyl terminus does not appear to control degranulation. Finally, it appears that the Pyk2 pathway can be targeted without impairing bacterial killing in vitro.
Materials and Methods
Cells.
Neutrophils were isolated to >95% purity from 10 U/ml heparinized blood of normal donors using Polymorphprep™ (Axis-Shield PoC) according to the manufacturer's instructions. Contaminating erythrocytes were lysed by hypotonic shock for 45 s with 0.2% saline. Neutrophils were resuspended in Krebs Ringer phosphate with glucose (KRPG) formulated as previously described (39).
Tat Fusion Proteins.
The vector pTatHA (32) was provided by S. Dowdy, Washington University, St. Louis, MO. Domains of Pyk2 were amplified from pRK5-Pyk2 (provided by J. Schlessinger, New York University, New York, NY) as a template using the following primer sets: autophosphorylation site (AP, nt 1102–1557): GTGAATTCGGATGGTGAGAAGCGGAACAGC and CTGAATTCTTCTTGTTCCGCTCCAGGTAGT; phosphatidyl inositol 3′ phosphate (PI3) kinase binding domain (PBM, nt 1741–2099): GTGAATTCGAAAGCCTCTGTGACTCGT and CAGAATTCGTTCGGTAGCGAGCATTCCT; Grb2 binding domain (GBM, nt 2617–2986): GTGAATTCAGACCGGACCGATGACCTG and GTGAATTC-TCCACAGCGTCGAGCAGGTT; and proline-rich region (PR, nt 2041–3030): CAGAATTCGGACGTTTATCAGATGGAGAA and GCGAATTCTCACTCTGCAGGTGGGTG. Full-length Rac2 was amplified using pCR-rac2 wt (provided by D. Ambruso, University of Colorado, Denver, CO) as template with primers GTGAATTCGATGCAGGCCATCAAG and CTGAATTCCTAGAGGAGGCTGCAG. PCR products and the pTatHA vector were digested with EcoRI, gel purified, ligated (Rapid Ligation Kit; Roche), and used for the transformation of Escherichia coli XL10 (Stratagene). Transformed E. coli BL21 DE 3 (Novagene) served as host for recombinant protein expression. BL21 cells were resuspended in buffer Z (8 M urea, 20 mM Hepes, 100 mM NaCl, pH 8.0), sonicated, and centrifuged at 17,500 g for 20 min. The supernatant was loaded on a Ni-NTA column (QIAGEN), which was washed with 50 column volumes of buffer Z to remove contaminating proteins, followed by 100 column volumes of 60% isopropanol in 20 mM Hepes and 50 column volumes of 20 mM Hepes to remove endotoxin (40). Residual LPS was measured by the chromogenic Limulus amebocyte assay (Biowhittaker). As previously reported (2), LPS at concentrations ranging from 10 to 100 ng/ml had no effect on the cell functions we analyzed either when tested alone or in the presence of TNF, nor did LPS affect results seen in the additional presence of Tat constructs (unpublished data). Recombinant Tat fusion proteins with NH2-terminal hexa-His tags were eluted with an imidazole gradient in buffer Z. Fractions containing >95% pure recombinant proteins were pooled, diluted 10-fold in 20 mM Hepes, pH 8.0, filtered six times on YM ultrafiltration discs (Millipore) using Amicon Stirred Cells (model 8050) to remove urea and imidazole, and stored at 4°C until use. Protein aggregates forming during storage were removed by ultracentrifugation at 100,000 g at 4°C for 15 min immediately before each experiment and protein concentration was measured by Dc protein assay (Bio-Rad Laboratories).
Flow Cytometry and Confocal Microscopy.
Tat fusion proteins were labeled with Alexa fluor 488 (Molecular Probes) according to the manufacturer's instructions. Neutrophils were incubated with labeled proteins for the indicated times at 37°C in 5-ml polypropylene round-bottom tubes (Falcon), precoated with FBS (Hyclone Laboratories), washed three times with cold washing buffer (0.1% Tween-20 in PBS), fixed with 2% paraformaldehyde in PBS, washed four times with cold washing buffer, resuspended in 2 ml 1% BSA in PBS, and analyzed on a FACScan™ (Becton Dickinson). Aliquots of the same cell suspensions were permeabilized with 3.3% paraformaldehyde, 0.05% glutaraldehyde, and 0.25 mg/ml saponin in PBS for 4 min at room temperature. The reactions were stopped with the same volume of 20 mM glycine buffer. The cells were washed three times with PBS, incubated in 25% goat serum in PBS at room temperature for 30 min, incubated with anti–lysosomal-associated membrane protein (LAMP)-1 mAb (provided by R. Silverstein, Weill Cornell Medical College, New York, NY) at 4°C overnight, washed, incubated with goat anti–mouse Ab conjugated with Alexa fluor 546 at room temperature for 30 min, and observed by confocal microscopy (LSM 510; Carl Zeiss MicroImaging, Inc.).
H2O2 Release and Degranulation.
The respiratory burst was measured as previously described (39). In brief, 96-well flat-bottomed plates (Primaria; Falcon) were coated with 25 μl/well FBS in 5% CO2 at 37°C for at least 1 h and washed three times with 0.9% saline. 1.5 × 104 neutrophils were added to triplicate wells containing 100 μl reaction mixture (2.4 nmol scopoletin, 0.5 μg horseradish peroxidase, and 1 mM NaN3) and either buffer control, TNF (Genentech, Inc. or PreproTech) or PMA, each at 100 ng/ml. The reduction of scopoletin by H2O2 was recorded every 15 min for 2 h on a plate-reading fluorometer and the amount of H2O2 released was calculated as previously described (39). The supernate from the H2O2 release assay with 1.0 × 105 neutrophils was used to measure degranulation using lactoferrin (LF) and myeloperoxidase (MPO) ELISA kits (Oxis International, Inc.), or to measure spontaneous cell death using Cytotoxicity Detection Kit (Roche). The total cell content of LF, MPO, and lactate dehydrogenase (LDH) was determined after lysing cells with 1% Triton X-100. The percent of cell death was determined by dividing the amount of LDH released spontaneously or in response to test agents by the amount of LDH released in response to detergent. Similarly, the percent of degranulation was determined by dividing the amount of LF or MPO released spontaneously or in response to test agents by the amount released in response to detergent, correcting for the amount of LF or MPO that may have been released in association with cell death.
Bacterial Killing.
Neutrophils were preincubated with or without 500 nM recombinant Tat fusion protein for 30 min at 37°C and then exposed to 10% autologous serum-opsonized Salmonella typhimurium (American Type Culture Collection [ATCC] 14028s) or Listeria monocytogenes (ATCC 15323) at a multiplicity of infection of 0.5 bacteria per neutrophil. Neutrophils were lysed at the indicated times with 1% sodium deoxycholate for assays with Salmonella or 1% Triton X-100 for assays with Listeria. Bacteria were grown in Luria-Bertani medium containing 10% Alamar Blue™ (Trek Diagnostic Systems, Inc.). The time for half-maximal growth was monitored and converted to CFUs using a standard curve as previously described (41).
Immunoprecipitation and Immunoblot.
15 × 106 neutrophils in KRPG were plated in 10-cm diameter dishes (Primaria) that had been precoated with FBS. After 15 min at 37°C, the cells were stimulated with 100 ng/ml TNF. When the cells were fully spread (40–60 min), they were treated with 5 mM diisopropylfluorophosphate to inhibit serine proteases and lysed with either 125 μl SDS lysis buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% SDS, 1 mM PMSF, 1 mM Na pyrophosphate, 1 mM NaF, 1 mM vanadate, 5 μg/ml each of aprotinin, leupeptin, chymostatin, and pepstatin A) for Western blot or with 200 μl nondenaturing modified RIPA buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, 1 mM Na pyrophosphate, 1 mM NaF, 1 mM vanadate, 5 μg/ml each of aprotinin, leupeptin, chymostatin, and pepstatin A) for immunoprecipitation. Cell lysates were separated by SDS-PAGE and transferred electrophoretically to nitrocellulose membranes (Schleicher & Schuell). The membranes were incubated with 5% milk in TBST (1 M Tris-HCl, pH 7.5, 9% NaCl, 0.1% Tween-20) for 1 h at 37°C and then overnight at 4°C with antiphospho-specific Pyk2 Abs (Biosource International), anti-Pyk2 Ab (Upstate Biotechnology), or anti-Syk Ab (Transduction Laboratories). Membranes were washed with TBST and incubated with secondary Ab conjugated with horseradish peroxidase in 5% milk in TBST for 1 h at 37°C. After additional washing with TBST, bound antibody was detected by enhanced chemiluminescence (Pierce Chemical Co.).
Results
Domains of Pyk2 Expressed as Tat Fusion Proteins.
Four recombinant Tat fusion proteins were designed based on homology to FAK and studies of Pyk2 in other cells (Fig. 1 ; reference 26). In Tat-AP, Tat peptide was fused to Pyk2's autophosphorylation domain (amino acid residues 365–518, including Y402, the residue that undergoes autophosphorylation). The fusion in Tat-PBM was to Pyk2's phosphatidylinositol-3 kinase (PI3K) binding motif (amino acid residues 581–700, including a sequence, YTLM, thought to be involved in binding the p85 SH2 domain of P13K) and in Tat-GBM was to the Grb2-binding motif (amino acids 873–995, including Y881). In Tat-CT, Tat peptide was fused to the NH2 terminus of Pyk2's proline-rich COOH-terminal domain (amino acid residues 680–1009). LPS was scrubbed to <5 ng/ml at 500 nM used after purification of the recombinant proteins from E. coli.
Figure 1.

Schematic representation of the Pyk2 regions expressed as Tat fusion proteins. Pro denotes the proline-rich regions critical for some of the protein–protein interactions associated with the COOH terminus of Pyk2. The ball and stick represent the Tat peptide, hexahistidine, and hemagglutinin tag. Amino acid residue numbers are shown in parentheses.
Uptake of Tat Fusion Proteins by Human Neutrophils.
Fluorescently labeled, Tat-tagged proteins were incubated with neutrophils and uptake was assessed by flow cytometry. Uptake varied strikingly with the method used to desalt the purified fusion proteins. Purification by ion exchange followed by gel permeation chromatography led to maximal uptake in 30 min, but factors eluting from the ion exchange column were toxic to the cells (not depicted). In contrast, preparations desalted by membrane filtration were nontoxic. These preparations were subject to increasing uptake with time over at least 120 min at 37°C, as shown for Tat-AP in Fig. 2 A and for Tat-CT in Fig. 2, B and C. The apparent extent of uptake was markedly augmented by chloroquine (Fig. 2 C), suggesting that lysosomal degradation may normally limit the accumulation and intracellular retention of the fluorescent marker. There was no detectable uptake at 4°C over the same time periods (not depicted). No saturation of uptake was achieved at concentrations up to 1 μM (Fig. 2, D and E). Fluorescence was almost entirely intracellular, as judged by the inability of trypan blue to quench it. As a control, trypan blue did quench fluorescence imparted by fluorescent antibody directed to a cell surface antigen, CD11c (not depicted). Optimal protein transduction required both Ca2+ and Mg2+ and was insensitive to pH over the range of 6.8–7.4 (not depicted). All additional work involved fusion proteins desalted by membrane filtration and a standard loading time of 30 min, because this sufficed to label ∼90% of the cells.
Figure 2.

Uptake of Tat fusion proteins by human PMNs, FACS® analysis. Flow cytometric analysis of the uptake of Tat fusion proteins by neutrophils. The cells were incubated with labeled Tat-AP (A and D) or Tat-CT (B, C, and E), either with 1 μM Tat construct for the indicated time at 37°C (A, B, and C), or for 60 min at 37°C with the indicated concentration of Tat construct (D and E). In C, cells were also preincubated with 100 μM chloroquine for 30 min at 37°C.
Intracellular Distribution of Tat Fusion Proteins.
Tat fusion proteins have been shown to enter polymorphonuclear leukocytes (34, 38), but it has not been assessed whether uptake is chiefly into the cytosol, as required to inhibit the signaling molecules targeted; into the nucleus, as expected from Tat's nuclear localization signal and as observed in several other types of cells (42–44); or into the endosome-lysosome system via absorptive endocytosis as also observed in various cells (43, 44). Confocal microscopy of neutrophils exposed to Tat-AP and Tat-CT confirmed that all detectable staining was intracellular (Fig. 3 , rows 1–3). The Tat fusion protein was excluded from rather than concentrated in the neutrophil nucleus (not depicted). Cytoplasmic staining was consistently visible although difficult to demonstrate in photomicrographs. However, the majority of staining was distributed in punctate collections throughout the cytoplasm that were almost entirely nonoverlapping with LAMP-1 (Fig. 3, rows 2 and 3). The variation in staining patterns illustrated in Fig. 3, rows 2 and 3, was not an intrinsic property of the Tat peptide studied. Both Tat-AP and Tat-Ct most often gave the pattern illustrated in row 2. Pretreatment of neutrophils with chloroquine before protein transduction led to dramatic intensification of both cytoplasmic and punctate staining (Fig. 3, row 1). Chloroquine elevates lysosomal pH (45) and blocks degradation of Tat in HeLa cells (42). The fluor used in these experiments is insensitive to pH over the range pH 4–10. Thus, a change in fluorescence efficiency does not account for the impact of chloroquine on signal intensity. A plausible interpretation of the findings in Figs. 2 C and 3 is that Tat constructs were taken up and largely retained in endosomes. Some passed into lysosomes, where degradation may have led to rapid removal of the fluor. With inhibition of lysosomal degradation, intact Tat fusion protein may have accumulated in lysosomes and endosomes. Biochemical evidence supported the conclusion that most of the Tat fusion proteins accumulating in neutrophils were in a degradative compartment. Although reagent Tat-CT was readily immunoprecipitated, no intact Tat-CT could be visualized on anti-hexahistidine immunoblots of immunoprecipitated lysates of neutrophils that had been incubated with Tat-CT. Instead, immunoblot displayed numerous His-tagged peptides of lower M r (unpublished data). In summary, large amounts of Tat proteins entered neutrophils, but the vast majority appeared to be degraded in the endosomal compartment with only traces reaching the cytosol.
Figure 3.
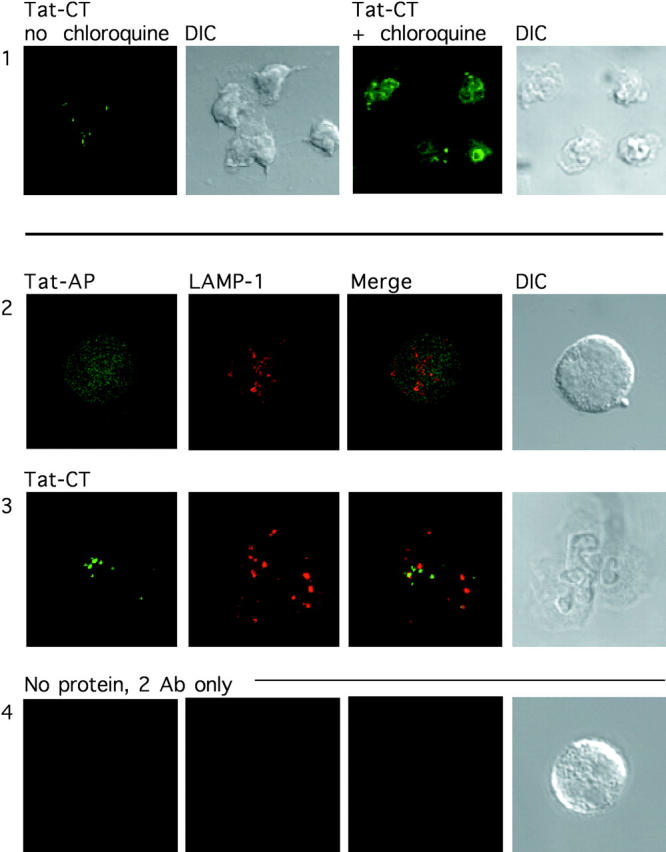
Distribution of Tat fusion proteins within human PMNs, confocal microscopy. Neutrophils were incubated with 1 μM fluorescent Tat-CT (rows 1 and 3), Tat-AP (row 2), or no protein (row 4), and stained for the lysosomal marker LAMP-1 (rows 2 and 3). Neutrophils were treated without (row 1, left) or with 100 μM chloroquine (row 1, right, and rows 2–4) along with the Tat fusion protein for 1 h at 37°C and then permeabilized and stained. Differential interference contrast microscopy (DIC) demonstrates the presence of cells. ×800 for row 1 and ×2,000 for rows 2–4.
Dominant Negative Pyk2 Constructs Inhibit the Respiratory Burst Triggered by TNF in Adherent Neutrophils.
Tat-CT selectively inhibited the TNF-triggered respiratory burst in a concentration-dependent manner (Fig. 4 A). Despite variation associated with the use of 18 different blood donors and 5 independently prepared batches of recombinant protein, 70–90% inhibition was consistently achieved by 500 nM Tat-CT and 50% inhibition required a mean of 250 nM (Fig. 4 B). Tat-CT had no effect on its own nor on the respiratory burst triggered by PMA. PMA was used as a control stimulus because it triggers the same spectrum of responses in adherent neutrophils as TNF, but bypasses surface receptors to act directly on protein kinase C. Sparing of the PMA-induced response indicated that the Tat transduction system was not toxic for neutrophils and did not interfere with the assay.
Figure 4.
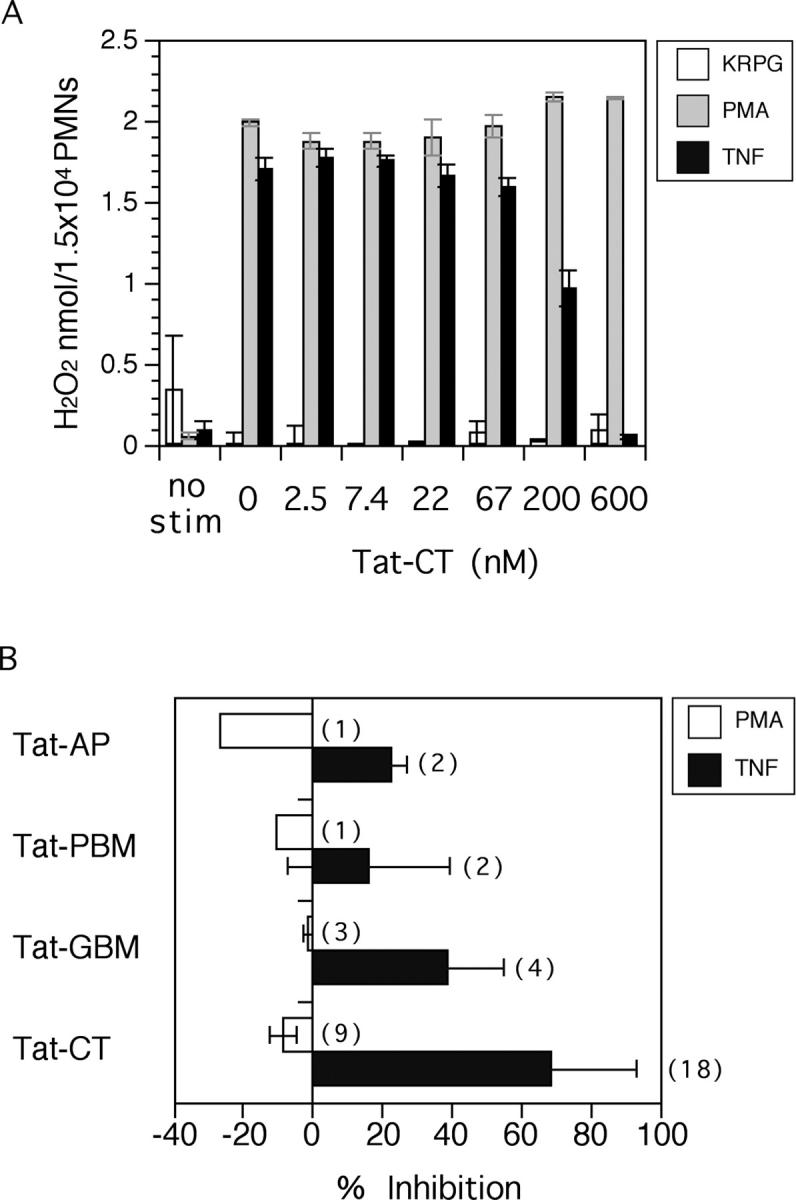
Effect of Pyk2 domains on the respiratory burst of adherent neutrophils. (A) Concentration-dependent effect of Tat-CT. Neutrophils were preincubated with the indicated concentrations of Tat-CT at 37°C for 30 min before stimulation with 100 ng/ml TNF, 100 ng/ml PMA, or an equivalent volume of KRPG buffer as a control. H2O2 release was measured at 15-min intervals. Results are displayed for 75 min and expressed as means ± SEM for triplicates in one representative experiment. (B) Summary of experiments with Tat-CT and other Pyk2 constructs. Results are means ± SEM for results at 100–120 min from the number of experiments with different blood donors shown in parentheses, each in triplicate. Percent inhibition was calculated in comparison to results for TNF without Tat fusion proteins in the same experiments.
Results of multiple experiments are summarized in Fig. 4 B. No constructs inhibited responses to PMA. With respect to responses to TNF, Tat-GBM afforded a mean of 38% inhibition, Tat-AP 23% inhibition, and Tat-PBM 16% inhibition when each was tested at 500 nM. The differences in the effect of individual domains of Pyk2 were not due to differences in uptake (Fig. 2). Another specificity control was provided by Tat-Rac2. Rac2 is a small GTP binding protein that constitutes a subunit of the phagocyte oxidase in human neutrophils (46). In three experiments, Tat-Rac2 afforded a mean of −15% inhibition of the respiratory burst induced by TNF and −4% of that induced by PMA (not depicted). However, the rapid aggregation of Tat-Rac2 upon storage required that a fresh purification be performed for every experiment. In subsequent experiments, Tat-CT was used as a test agent and ovalbumin as a control for added protein.
Inhibition of TNF-triggered Cell Spreading by Tat-CT.
The respiratory burst of adherent neutrophils triggered by soluble, physiologic agents depends on rearrangement of the actin-based cytoskeleton and cell spreading. Rearrangement of actin bundles from a dense, subcortical meshwork to interconnected focal adhesions and podosomes might be necessary to permit vesicular and cytoplasmic components of the phagocyte oxidase complex to be mobilized together to the plasma membrane. To determine whether Pyk2 helps mediate cytoskeletal rearrangement, we treated neutrophils with Tat-CT or buffer and then did or did not activate them with PMA or TNF. As shown in Fig. 5 , neutrophils adhered to serum-coated glass but did not spread unless activated. Tat-CT failed to impede cell spreading induced by PMA. Cells preincubated with Tat-CT and then exposed to TNF began to spread but were arrested before they had extended filopodia and completely flattened (Fig. 5). These findings suggested that Tat-CT did not interfere with the ability of TNF to bind to its receptors nor with the earliest steps in signal transduction. Instead, Tat-CT blocked an intermediate stage in the response to TNF.
Figure 5.
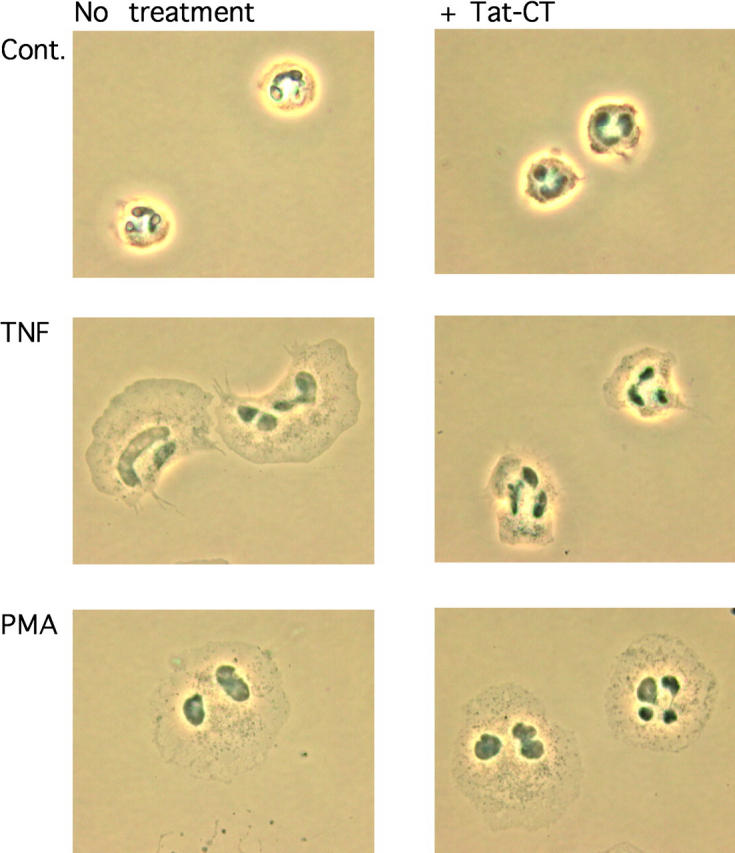
Inhibition of TNF-triggered neutrophil spreading by Tat-CT. Neutrophils were plated on FBS-coated glass coverslips and preincubated or not with 500 nM Tat-CT at 37°C for 30 min before stimulation with 100 ng/ml TNF, 100 ng/ml PMA, or an equal volume of KRPG (Cont.), and fixed and photographed with phase-contrast microscope. ×1,000.
Impact of Tat-CT on TNF-triggered Degranulation.
Any stimulus that triggers the respiratory burst in neutrophils generally triggers degranulation and vice versa. With extensive overlap in the signaling pathways inducing both processes, it is unclear whether there is any respect in which they are regulated differentially. To test the role of the carboxyl terminus of Pyk2 in degranulation, we did or did not activate neutrophils with TNF after preincubation with 500 nM Ova or Tat-CT for 30 min, harvested the supernatant at 60–90 min, and measured the concentration of the specific granule protein LF and of the azurophil granule protein MPO, correcting for cell death as reflected by release of the cytosolic enzyme LDH. Tat-CT had no effect on TNF-induced specific release of LF and MPO (Fig. 6) . In the same experiments, Tat-CT blocked the TNF-induced respiratory burst by ∼80%.
Figure 6.
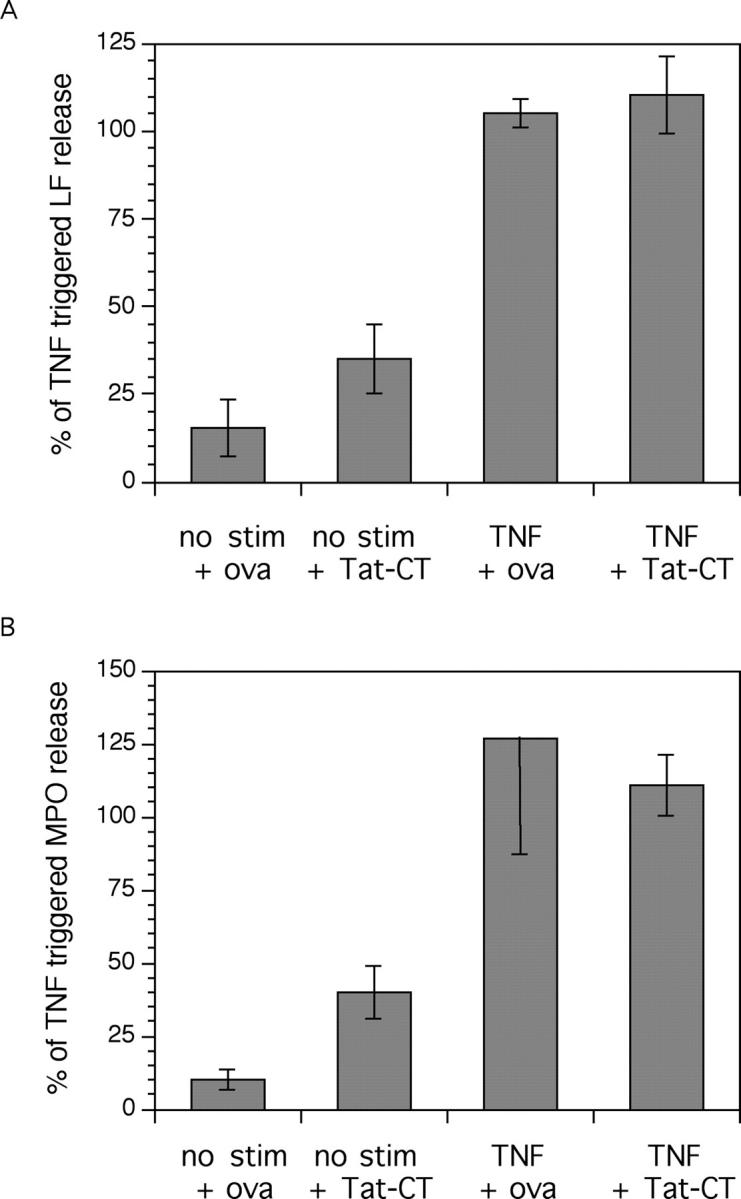
Effect of Tat-CT on TNF-triggered neutrophil degranulation. Neutrophils were incubated as indicated (test proteins were added at 500 nM) and their supernate was assayed for the presence of lactoferrin (LF) as a measure of specific granule release and myeloperoxidase (MPO) for azurophil granule release. LDH was measured as an indicator of spontaneous cell lysis. Specific LF (A) and MPO (B) release (corrected for cell lysis) are shown normalized to that seen with TNF alone in the absence of ovalbumin (ova) or Tat-CT. TNF alone led to the specific release of an average of 67 and 27% of total detergent-releasable LF and MPO content of the cells, respectively, over and above the mean 5% release that could be attributed to cell death. Results are the mean ± SEM in three independent experiments for LF release and two for MPO release.
Inhibition of TNF-dependent Tyrosine Phosphorylation of Endogenous Pyk2 in the Presence of Tat-CT.
To explore the biochemical basis for the inhibition of TNF-induced responses by Tat-CT, we first examined the most abundant tyrosine phosphoproteins by immunoblots after one-dimensional SDS-PAGE. As shown in Fig. 7 A, there was no gross alteration in the pattern of TNF-induced protein tyrosine phosphorylation in adherent neutrophils in the presence of Tat-CT, although the intensity of several bands was slightly diminished. Next, we immunoprecipitated Pyk2 and investigated the effect of Tat-CT on the phosphorylation of tyrosine residues 402, 570, and 881, which have been shown to play important roles in Pyk2's functions in other cell types. TNF-induced phosphorylation of all three tyrosines was inhibited in the presence of Tat-CT (Fig. 7 B). Thus, Tat-CT acted as a dominant negative in that it blocked the tyrosine phosphorylation of endogenous Pyk2. Because the tyrosine phosphorylation of residue 402 is essential for Pyk2's tyrosine kinase activity (26), it can be presumed that Tat-CT also blocked the enzyme activity of Pyk2. These findings implied that the COOH terminus of Pyk2 is involved in protein–protein interactions that are necessary for the activation of Pyk2.
Figure 7.

Inhibition of TNF-induced phosphorylation of endogenous Pyk2 by Tat-CT. Neutrophils were preincubated with Tat-CT or ovalbumin (ova; each 500 nM) at 37°C for 30 min and stimulated with TNF (T) or buffer alone as a control (C). Total cell lysates were separated by SDS-PAGE and western-blotted (WB) with (A) antiphosphotyrosine Ab or (B) Abs specific for individual phosphotyrosine (PY) residues of Pyk2. One membrane was stripped and reprobed with anti-Pyk2 Ab, demonstrating equal loading of the lanes.
Inhibition of TNF-dependent Association of Pyk2 and Syk by Tat-CT.
Syk, like Pyk2, is a tyrosine kinase that undergoes activation in TNF-treated human neutrophils (47). In earlier work, wortmannin, a PI3K inhibitor, appeared to block the TNF-induced tyrosine phosphorylation of only one protein in adherent neutrophils besides Pyk2, namely an unidentified 80-kD protein whose M r matched that of Syk (27). In mouse neutrophils, Syk associates with Pyk2 and is essential for the cells to respond to TNF, fMLF, MIP-2, and LPS (48). In our experiments, Pyk2 and Syk could indeed be coimmunoprecipitated from human neutrophils, but only after the cells were activated with TNF (Fig. 8 A). Wortmannin inhibited the TNF-induced tyrosine phosphorylation not only of Pyk2 (27) but also of Syk (Fig. 8 B). Finally, TNF-induced tyrosine phosphorylation of Syk was inhibited by Tat-CT (Fig. 8 C).
Figure 8.

TNF-induced association of Pyk2 and Syk, and the inhibition of tyrosine phosphorylation of Syk by wortmannin and Tat-CT. (A) Pyk2 was immunoprecipitated (IP) from neutrophils stimulated with TNF (T) or treated with buffer alone as a control (C) and Western blotted (WB) for Syk. (B) Syk was immunoprecipitated from neutrophils after preincubation with or without TNF (T) and/or 10 nM wortmannin and Western blotted for phosphotyrosine (PY; top) or Syk (bottom). (C) Syk was immunoprecipitated from neutrophils after preincubation with or without TNF (T) and/or with Tat-CT or ovalbumin (ova; each 500 nM) at 37°C for 30 min and probed with antiphosphotyrosine Ab (PY) or anti-Syk.
The Effect of Tat-CT on Bacteria-triggered Respiratory Burst and Killing.
Neutrophils underwent a robust respiratory burst upon encountering S. typhimurium (ATCC 14028s; a Gram-negative bacterium) and L. monocytogenes (ATCC 15323; a Gram-positive bacterium) that had been opsonized with autologous serum. Tat-CT inhibited the respiratory burst triggered by each pathogen by a mean of 81% for Salmonella and 72% for Listeria (Fig. 9 A). However, the ability of neutrophils to kill these organisms was unaffected by Tat-CT (Fig. 9 B).
Figure 9.

Effect of Tat-CT on interaction of neutrophils with bacteria. (A) Impact of Tat-CT or ovalbumin (ova; each 500 nM, 30 min preincubation) on the respiratory burst triggered by S. typhimurium or L. monocytogenes, measured at 150 min. Results are means ± SEM for triplicates in a representative experiment of two performed. (B) Survival of bacteria in culture only (open symbols) or in culture with neutrophils (closed symbols). Survival of bacteria in the presence of Ova (▪ and □) or Tat-CT (▴ and ▵) are shown in CFU/ml. Results are means ± SEM for triplicates in a representative experiment of three performed. Some of the error bars fall within the symbols.
Other Approaches to Protein Transduction.
Transducing peptides based on the homeodomain of Drosophila antennapedia were effective in epithelial cells but were not detectably taken up by human neutrophils (unpublished data, and D. Thomas, personal communication). A peptide carrier based on the signal sequence of the β3 integrin (49) was reported to be effective in myeloid cell lines (49, 50). The synthetic carrier (VTVLALGALAGVGVG) was coupled to biotin and attached via its COOH terminus to the Pyk2 autophosphorylation site SIESDIYAEIPDET (residues 396–409). Neutrophils incubated with this peptide at 100 μM showed intense cytoplasmic staining. The same cargo-carrier conjugate lacking biotin selectively blocked cell spreading and the respiratory burst triggered by TNF and formyl peptide, but not PMA. There was no inhibition by a control peptide whose cargo sequence (underlined) was scrambled (VTVLALGALAGVGVGATYEPSDISEIEID; unpublished data and R. Webber, personal communication). These results were dependent on the protocol used to purify the peptides and the factors responsible for producing consistently effective lots eluded identification. Thus, Tat-based transduction was the only approach that worked consistently with human neutrophils.
Discussion
These experiments appear to have afforded the first opportunity to compare Tat-based protein transduction with the use of a relatively specific pharmacologic inhibitor (27) for the study of signaling in neutrophils. That these two approaches gave consistent results helps to validate Tat-based transduction as a method of choice and increases confidence in additional conclusions regarding the role of specific protein domains that can only be addressed at present through protein transduction. Thus, the transduced COOH terminus of Pyk2 reproduced the known actions of tyrphostin A9 on neutrophils, blocking the TNF-induced tyrosine phosphorylation of Pyk2 and Syk, the release of hydrogen peroxide, and the spreading of the cells, while sparing PMA-induced responses, TNF-triggered degranulation, and bactericidal activity. Additional evidence for the validity of the Tat-based technique was provided by the specificity and selectivity of effects achieved with Tat-tagged constructs that incorporated other domains of Pyk2 or introduced a different protein, Rac2. By blocking tyrosine phosphorylation of endogenous Pyk2, the Tat-tagged Pyk2 COOH terminus acted as a dominant negative. Tat constructs afforded gain-of-function phenotypes in the two earlier reports with neutrophils (33, 34), illustrating the versatility of the approach.
Several protein transduction domains can confer the ability to carry attached molecules across biological membranes. The Tat protein from HIV-1, antennapaedia from Drosophila, the signal sequence from the β3 integrin and VP22 from HSV-1 have all been effective in some cells. Of the first three, only Tat was successful with neutrophils. The factors are unknown that confer a wider cell type range on Tat compared with other protein transduction domains. Nontoxicity of Tat constructs and consistency of their effects depended on how the proteins were purified. The bacterial expression system introduced LPS contamination. Not all constructs were stable enough for routine use. Thus, Tat-based transduction remains experimentally demanding.
Most applications of Tat-based transduction require and assume cytosolic localization. However, in work involving neutrophils or eosinophils, a predominant cytosolic localization for Tat-tagged proteins has apparently been documented in only one instance (34). In this study, the predominant localization of the Tat constructs appeared to be endosomal and the vast majority of the Tat construct taken up was rapidly degraded. Nonetheless, by confocal microscopy a very small fraction appeared to be localized in the cytosol. Tat constructs are deliberately denatured to foster their uptake (32). To function as a dominant negative, cytosolic Tat-CT presumably underwent sufficient renaturation to compete with endogenous Pyk2 for protein–protein interactions. The concentration of extracellular Tat-CT affording 50% inhibition was 250 nM. The concentration that was both cytosolic and renatured is unknown.
The assignment of critical functions to the COOH terminus of Pyk2 in neutrophils echoes the dominant negative effect of electroporating the Pyk2 COOH terminus into human monocytes to inhibit their spreading and motility (51). The COOH terminus of Pyk2 is a versatile partner for protein interactions in other cell types, for example with p130 Cas (52), Grb2 (52), Hic-5 (53), and Pap α/β (54). Immunoprecipitation of Pyk2 from neutrophils did not bring down p130 Cas or Grb2 (not depicted). Investigation is underway into the possible expression in neutrophils of Hic-5 and Pap α/β.
Among the proteins reported to interact with Pyk2, Syk is of particular interest because it was activated by TNF in adherent human neutrophils (47) and was essential for adherent mouse neutrophils to undergo activation in response to TNF, fMLF, MIP-2, and LPS (48). Syk was constitutively associated with β2 integrins in the myeloid cell line HL-60, where its activation required cell spreading (55). Because Pyk2 localized to focal adhesions and podosomes (27), of which β2 integrins form the core, Pyk2 might activate Syk directly or indirectly in focal adhesions and podosomes. Activated Syk phosphorylated the focal adhesion protein paxillin (56). Pharmacologic inhibition of Syk2 blocked tyrosine phosphorylation of Pyk2 (28). In this study, TNF triggered the association of Pyk2 and Syk, and inhibition of Pyk2 blocked the tyrosine phosphorylation of Syk. TNF also triggered the binding of PI3K to TNF receptors and activation of PI3K in adherent human neutrophils (57). In this study, inhibition of PI3K blocked the tyrosine phosphorylation of both Pyk2 and Syk. Thus, in response to TNF, Pyk2 and Syk both appear to control each other's activation, and PI3K appears to control the activation of both of them.
Despite the close interactions among these three enzymes, the consequences of inhibiting or deleting PI3K, Syk, and Pyk2 are not the same, either in isolated neutrophils or in the whole organism. Wortmannin-treated, TNF-stimulated neutrophils adhered but did not spread at all (27). In the presence of tyrphostin A9, TNF-stimulated neutrophils began to spread and succeeded in projecting filopodia, but spreading halted prematurely (27). Tat-CT allowed more spreading of TNF-treated neutrophils than wortmannin but less than tyrphostin A9, aborting their projection of filopodia. It appears that Tat-CT blocked TNF signaling in more ways, more completely, and/or at an earlier step than tyrphostin A9. Mice deficient in p85α subunit of PI3K die as embryos (58) and PI3Kγ-deficient mice displayed defects in thymocyte development, T cell activation, and neutrophil migration (59–61). Genetic deficiency of Syk (62, 63) led to perinatal lethality in mice. In contrast, Pyk2-deficient mice appeared to be normal except for impaired macrophage migration and a paucity of marginal zone B cells (64).
Tat-CT blocked the bacteria-induced respiratory burst but not the killing of Salmonella. This may seem paradoxical insofar as Salmonella can cause life-threatening infections in chronic granulomatous disease patients who lack the phagocyte oxidase (phox; reference 65) and in mice that are rendered phox-deficient through homologous recombination (66). However, in some chronic granulomatous disease patients with a regulatory rather than structural defect in a gene encoding a phox subunit, restoration of phox activity to 10% of normal by treatment with interferon-γ sufficed to endow their leukocytes with normal antibacterial activity (67). In our studies, inhibition of the respiratory burst by Tat-CT left the respiratory burst at ∼20% of normal, even though there appeared to be complete inhibition of the phosphorylation of two of the tyrosine residues of endogenous Pyk2. The combination of a small residual respiratory burst with a normal level of degranulation may account for the sparing of antibacterial activity in the presence of Tat-CT.
The tissue damaging potential of secreted reactive oxygen intermediates and lysosomal proteases has been demonstrated extensively (68–70). Critical for this discussion are the synergistic interactions between these two sets of neutrophil secretory products. First, neutrophil-derived oxidants contribute to the activation of latent neutrophil proteases, such as collagenase and gelatinase (71, 72). Second, neutrophil-derived oxidants can inactivate three of the major tissue antiproteinases: α1 antiproteinase, α2-macroglobulin, and secretory leukocyte protease inhibitor (69, 70, 73, 74). Thus, an intervention that markedly decreased the respiratory burst of neutrophils responding to inflammatory agonists might decrease tissue damage both directly and indirectly. Selective regulation of the respiratory burst might be an alternative to blocking all aspects of neutrophil activation, which would carry grave risk to host defense.
The cytochrome b558 components of phox (gp91 and gp22) are localized primarily in the membranes of specific granules in neutrophils (75) and assembly of the full oxidase complex involves their recruitment to plasma or phagosomal membranes (76). Thus, the activation of the respiratory burst depends on degranulation. Tat-CT blocked the respiratory burst but did not impair degranulation. It follows that the COOH terminus of Pyk2 is likely to mediate some aspect of phox assembly distal to the mobilization of gp91/gp22, a step that can be bypassed when PMA activates protein kinase C. Possible loci of action for Pyk2 (directly or indirectly) include the phosphorylation of phox47 and the recruitment of the small GTPase components of the oxidase complex.
The discovery that the neutrophil respiratory burst oxidase can be substantially inhibited without blocking degranulation or bacterial killing in vitro directs attention to a search for members of the Pyk2 pathway that might be more myeloid-specific than Pyk2 itself. Such proteins might be a suitable experimental target for antiinflammatory therapy.
Acknowledgments
We are grateful to D. Ambruso, S. Dowdy, J. Schlessinger, and R. Silverstein for gifts of plasmids or antibodies, Robert Webber (R&D Antibodies, Inc.) for extensive syntheses of peptides with the β3 integrin signal sequence, D. Thomas for trials of transduction with the antennapaedia carrier, M. Melchior for help with neutrophil preparation, and E. Block and A. Ding for advice.
This work was supported by National Institutes of Health (NIH) grant RO1-AI46382 and by an NIH predoctoral fellowship to H. Han via grant T32-AI07621.
Footnotes
Abbreviations used in this paper: GBM, Grb2 binding domain; KRPG, Krebs Ringer phosphate with glucose; LAMP, lysosomal-associated membrane protein; LDH, lactate dehydrogenase; LF, lactoferrin; MIP, macrophage inflammatory protein; MPO, myeloperoxidase; phox, phagocyte oxidase; PI3K, phosphatidylinositol-3 kinase; Pyk2, proline-rich tyrosine kinase.
References
- 1.Klebanoff, S.J., M.A. Vadas, J.M. Harlan, L.H. Sparks, J.R. Gamble, J.M. Agosti, and A.M. Waltersdorph. 1986. Stimulation of neutrophils by tumor necrosis factor. J. Immunol. 136:4220–4225. [PubMed] [Google Scholar]
- 2.Nathan, C.F. 1987. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J. Clin. Invest. 80:1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolpe, S.D., G. Davatelis, B. Sherry, B. Beutler, D.G. Hesse, H.T. Nguyen, L.L. Moldawer, C.F. Nathan, S.F. Lowry, and A. Cerami. 1988. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 167:570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan, C.F. 1989. Respiratory burst in adherent human neutrophils: triggering by colony-stimulating factors CSF-GM and CSF-G. Blood. 73:301–306. [PubMed] [Google Scholar]
- 5.De La Harpe, J., and C.F. Nathan. 1989. Adenosine regulates the respiratory burst of cytokine-triggered human neutrophils adherent to biologic surfaces. J. Immunol. 143:596–602. [PubMed] [Google Scholar]
- 6.Laudanna, C., S. Miron, G. Berton, and F. Rossi. 1990. Tumor necrosis factor-alpha/cachectin activates the O2 −-generating system of human neutrophils independently of the hydrolysis of phosphoinositides and the release of arachidonic acid. Biochem. Biophys. Res. Commun. 166:308–315. [DOI] [PubMed] [Google Scholar]
- 7.Nathan, C., and E. Sanchez. 1990. Tumor necrosis factor and CD11/CD18 (beta 2) integrins act synergistically to lower cAMP in human neutrophils. J. Cell Biol. 111:2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dri, P., R. Cramer, M. Romano, P. Spessotto, and P. Patriarca. 1991. Effect of biological surfaces on neutrophil O2 − production and its relationship to the CD11b/CD18 integrin-dependent adherence. Int. J. Tissue React. 13:193–201. [PubMed] [Google Scholar]
- 9.Dapino, P., F. Dallegri, L. Ottonello, and C. Sacchetti. 1993. Induction of neutrophil respiratory burst by tumour necrosis factor-alpha; priming effect of solid-phase fibronectin and intervention of CD11b-CD18 integrins. Clin. Exp. Immunol. 94:533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuortes, M., W.W. Jin, and C. Nathan. 1993. Adhesion-dependent protein tyrosine phosphorylation in neutrophils treated with tumor necrosis factor. J. Cell Biol. 120:777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan, C., Q.W. Xie, L. Halbwachs-Mecarelli, and W.W. Jin. 1993. Albumin inhibits neutrophil spreading and hydrogen peroxide release by blocking the shedding of CD43 (sialophorin, leukosialin). J. Cell Biol. 122:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liles, W.C., J.A. Ledbetter, A.W. Waltersdorph, and S.J. Klebanoff. 1995. Cross-linking of CD18 primes human neutrophils for activation of the respiratory burst in response to specific stimuli: implications for adhesion-dependent physiological responses in neutrophils. J. Leukoc. Biol. 58:690–697. [DOI] [PubMed] [Google Scholar]
- 13.Richter, J., U. Gullberg, and M. Lantz. 1995. TNF-induced superoxide anion production in adherent human neutrophils involves both the p55 and p75 TNF receptor. J. Immunol. 154:4142–4149. [PubMed] [Google Scholar]
- 14.Fuortes, M., W. Jin, and C. Nathan. 1996. Ceramide selectively inhibits early events in the response of human neutrophils to tumor necrosis factor. J. Leukoc. Biol. 59:451–460. [DOI] [PubMed] [Google Scholar]
- 15.Lowell, C.A., L. Fumagalli, and G. Berton. 1996. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell Biol. 133:895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldmann, M. 2002. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2:364–371. [DOI] [PubMed] [Google Scholar]
- 17.Braun, J., J. Brandt, J. Listing, A. Zink, R. Alten, W. Golder, E. Gromnica-Ihle, H. Kellner, A. Krause, M. Schneider, et al. 2002. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. 359:1187–1193. [DOI] [PubMed] [Google Scholar]
- 18.Sandborn, W.J., and S.B. Hanauer. 1999. Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm. Bowel Dis. 5:119–133. [DOI] [PubMed] [Google Scholar]
- 19.Lev, S., H. Moreno, R. Martinez, P. Canoll, E. Peles, J.M. Musacchio, G.D. Plowman, B. Rudy, and J. Schlessinger. 1995. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 376:737–745. [DOI] [PubMed] [Google Scholar]
- 20.Avraham, S., R. London, Y. Fu, S. Ota, D. Hiregowdara, J. Li, S. Jiang, L.M. Pasztor, R.A. White, J.E. Groopman, et al. 1995. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J. Biol. Chem. 270:27742–27751. [DOI] [PubMed] [Google Scholar]
- 21.Sasaki, H., K. Nagura, M. Ishino, H. Tobioka, K. Kotani, and T. Sasaki. 1995. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J. Biol. Chem. 270:21206–21219. [DOI] [PubMed] [Google Scholar]
- 22.Yu, H., X. Li, G.S. Marchetto, R. Dy, D. Hunter, B. Calvo, T.L. Dawson, M. Wilm, R.J. Anderegg, L.M. Graves, et al. 1996. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J. Biol. Chem. 271:29993–29998. [DOI] [PubMed] [Google Scholar]
- 23.Herzog, H., J. Nicholl, Y.J. Hort, G.R. Sutherland, and J. Shine. 1996. Molecular cloning and assignment of FAK2, a novel human focal adhesion kinase, to 8p11.2-p22 by nonisotopic in situ hybridization. Genomics. 32:484–486. [DOI] [PubMed] [Google Scholar]
- 24.Ganju, R.K., W.C. Hatch, H. Avraham, M.A. Ona, B. Druker, S. Avraham, and J.E. Groopman. 1997. RAFTK, a novel member of the focal adhesion kinase family, is phosphorylated and associates with signaling molecules upon activation of mature T lymphocytes. J. Exp. Med. 185:1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dikic, I., and J. Schlessinger. 1998. Identification of a new Pyk2 isoform implicated in chemokine and antigen receptor signaling. J. Biol. Chem. 273:14301–14308. [DOI] [PubMed] [Google Scholar]
- 26.Schlaepfer, D.D., and T. Hunter. 1998. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol. 8:151–157. [DOI] [PubMed] [Google Scholar]
- 27.Fuortes, M., M. Melchior, H. Han, G.J. Lyon, and C. Nathan. 1999. Role of the tyrosine kinase pyk2 in the integrin-dependent activation of human neutrophils by TNF. J. Clin. Invest. 104:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan, S.R., and M.J. Novak. 1999. Beta2 integrin-dependent phosphorylation of protein-tyrosine kinase Pyk2 stimulated by tumor necrosis factor alpha and fMLP in human neutrophils adherent to fibrinogen. FEBS Lett. 451:33–38. [DOI] [PubMed] [Google Scholar]
- 29.Bilder, G.E., J.A. Krawiec, K. Mcvety, A. Gazit, C. Gilon, R. Lyall, A. Zilberstein, A. Levitzki, M.H. Perrone, and A.B. Schreiber. 1991. Tyrphostins inhibit PDGF-induced DNA synthesis and associated early events in smooth muscle cells. Am. J. Physiol. 260:C721–C730. [DOI] [PubMed] [Google Scholar]
- 30.Yaffe, M.B., J. Xu, P.A. Burke, R.A. Forse, and G.E. Brown. 1999. Priming of the neutrophil respiratory burst is species-dependent and involves MAP kinase activation. Surgery. 126:248–254. [PubMed] [Google Scholar]
- 31.Downey, G.P., J.R. Butler, H. Tapper, L. Fialkow, A.R. Saltiel, B.B. Rubin, and S. Grinstein. 1998. Importance of MEK in neutrophil microbicidal responsiveness. J. Immunol. 160:434–443. [PubMed] [Google Scholar]
- 32.Nagahara, H., A.M. Vocero-Akbani, E.L. Snyder, A. Ho, D.G. Latham, N.A. Lissy, M. Becker-Hapak, S.A. Ezhevsky, and S.F. Dowdy. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4:1449–1452. [DOI] [PubMed] [Google Scholar]
- 33.Jones, S.L., J. Wang, C.W. Turck, and E.J. Brown. 1998. A role for the actin-bundling protein L-plastin in the regulation of leukocyte integrin function. Proc. Natl. Acad. Sci. USA. 95:9331–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruyninckx, W.J., K.M. Comerford, D.W. Lawrence, and S.P. Colgan. 2001. Phosphoinositide 3-kinase modulation of beta(3)-integrin represents an endogenous “braking” mechanism during neutrophil transmatrix migration. Blood. 97:3251–3258. [DOI] [PubMed] [Google Scholar]
- 35.Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Activation of RhoA and ROCK are essential for detachment of migrating leukocytes. Mol. Biol. Cell. 12:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capowski, E.E., S. Esnault, S. Bhattacharya, and J.S. Malter. 2001. Y box-binding factor promotes eosinophil survival by stabilizing granulocyte-macrophage colony-stimulating factor mRNA. J. Immunol. 167:5970–5976. [DOI] [PubMed] [Google Scholar]
- 37.Hall, D.J., J. Cui, M.E. Bates, B.A. Stout, L. Koenderman, P.J. Coffer, and P.J. Bertics. 2001. Transduction of a dominant-negative H-Ras into human eosinophils attenuates extracellular signal-regulated kinase activation and interleukin-5-mediated cell viability. Blood. 98:2014–2021. [DOI] [PubMed] [Google Scholar]
- 38.Myou, S., X. Zhu, E. Boetticher, S. Myo, A. Meliton, A. Lambertino, N.M. Munoz, and A.R. Leff. 2002. Blockade of focal clustering and active conformation in beta(2)-integrin-mediated adhesion of eosinophils to intercellular adhesion molecule-1 caused by transduction of HIV TAT-dominant negative Ras. J. Immunol. 169:2670–2676. [DOI] [PubMed] [Google Scholar]
- 39.De La Harpe, J., and C.F. Nathan. 1985. A semi-automated micro-assay for H2O2 release by human blood monocytes and mouse peritoneal macrophages. J. Immunol. Methods. 78:323–336. [DOI] [PubMed] [Google Scholar]
- 40.Franken, K.L., H.S. Hiemstra, K.E. Van Meijgaarden, Y. Subronto, J. Den Hartigh, T.H. Ottenhoff, and J.W. Drijfhout. 2000. Purification of his-tagged proteins by immobilized chelate affinity chromatography: the benefits from the use of organic solvent. Protein Expr. Purif. 18:95–99. [DOI] [PubMed] [Google Scholar]
- 41.Shiloh, M.U., J. Ruan, and C. Nathan. 1997. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect. Immun. 65:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frankel, A.D., and C.O. Pabo. 1988. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 55:1189–1193. [DOI] [PubMed] [Google Scholar]
- 43.Mann, D.A., and A.D. Frankel. 1991. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 10:1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fawell, S., J. Seery, Y. Daikh, C. Moore, L.L. Chen, B. Pepinsky, and J. Barsoum. 1994. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA. 91:664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lie, S.O., and B. Schofield. 1973. Inactivation of lysosomal function in normal cultured human fibroblasts by chloroquine. Biochem. Pharmacol. 22:3109–3114. [DOI] [PubMed] [Google Scholar]
- 46.Knaus, U.G., P.G. Heyworth, T. Evans, J.T. Curnutte, and G.M. Bokoch. 1991. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science. 254:1512–1515. [DOI] [PubMed] [Google Scholar]
- 47.Yan, S.R., M. Huang, and G. Berton. 1997. Signaling by adhesion in human neutrophils: activation of the p72syk tyrosine kinase and formation of protein complexes containing p72syk and Src family kinases in neutrophils spreading over fibrinogen. J. Immunol. 158:1902–1910. [PubMed] [Google Scholar]
- 48.Mocsai, A., M. Zhou, F. Meng, V.L. Tybulewicz, and C.A. Lowell. 2002. Syk is required for integrin signaling in neutrophils. Immunity. 16:547–558. [DOI] [PubMed] [Google Scholar]
- 49.Liu, K.Y., S. Timmons, Y.Z. Lin, and J. Hawiger. 1996. Identification of a functionally important sequence in the cytoplasmic tail of integrin beta 3 by using cell-permeable peptide analogs. Proc. Natl. Acad. Sci. USA. 93:11819–11824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawiger, J. 1997. Cellular import of functional peptides to block intracellular signaling. Curr. Opin. Immunol. 9:189–194. [DOI] [PubMed] [Google Scholar]
- 51.Watson, J.M., T.W. Harding, V. Golubovskaya, J.S. Morris, D. Hunter, X. Li, J.S. Haskill, and H.S. Earp. 2001. Inhibition of the calcium-dependent tyrosine kinase (CADTK) blocks monocyte spreading and motility. J. Biol. Chem. 276:3536–3542. [DOI] [PubMed] [Google Scholar]
- 52.Blaukat, A., I. Ivankovic-Dikic, E. Gronroos, F. Dolfi, G. Tokiwa, K. Vuori, and I. Dikic. 1999. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J. Biol. Chem. 274:14893–14901. [DOI] [PubMed] [Google Scholar]
- 53.Matsuya, M., H. Sasaki, H. Aoto, T. Mitaka, K. Nagura, T. Ohba, M. Ishino, S. Takahashi, R. Suzuki, and T. Sasaki. 1998. Cell adhesion kinase beta forms a complex with a new member, Hic-5, of proteins localized at focal adhesions. J. Biol. Chem. 273:1003–1014. [DOI] [PubMed] [Google Scholar]
- 54.Andreev, J., J.P. Simon, D.D. Sabatini, J. Kam, G. Plowman, P.A. Randazzo, and J. Schlessinger. 1999. Identification of a new Pyk2 target protein with Arf-GAP activity. Mol. Cell. Biol. 19:2338–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miura, Y., Y. Tohyama, T. Hishita, A. Lala, E. De Nardin, Y. Yoshida, H. Yamamura, T. Uchiyama, and K. Tohyama. 2000. Pyk2 and Syk participate in functional activation of granulocytic HL-60 cells in a different manner. Blood. 96:1733–1739. [PubMed] [Google Scholar]
- 56.Fernandez, R., and S.J. Suchard. 1998. Syk activation is required for spreading and H2O2 release in adherent human neutrophils. J. Immunol. 160:5154–5162. [PubMed] [Google Scholar]
- 57.Korchak, H.M., and L.E. Kilpatrick. 2001. TNFalpha elicits association of PI 3-kinase with the p60TNFR and activation of PI 3-kinase in adherent neutrophils. Biochem. Biophys. Res. Commun. 281:651–656. [DOI] [PubMed] [Google Scholar]
- 58.Fruman, D.A., S.B. Snapper, C.M. Yballe, L. Davidson, J.Y. Yu, F.W. Alt, and L.C. Cantley. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 283:393–397. [DOI] [PubMed] [Google Scholar]
- 59.Hirsch, E., V.L. Katanaev, C. Garlanda, O. Azzolino, L. Pirola, L. Silengo, S. Sozzani, A. Mantovani, F. Altruda, and M.P. Wymann. 2000. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 287:1049–1053. [DOI] [PubMed] [Google Scholar]
- 60.Li, Z., H. Jiang, W. Xie, Z. Zhang, A.V. Smrcka, and D. Wu. 2000. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 287:1046–1049. [DOI] [PubMed] [Google Scholar]
- 61.Sasaki, T., J. Irie-Sasaki, R.G. Jones, A.J. Oliveira-Dos-Santos, W.L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, et al. 2000. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046. [DOI] [PubMed] [Google Scholar]
- 62.Cheng, A.M., B. Rowley, W. Pao, A. Hayday, J.B. Bolen, and T. Pawson. 1995. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 378:303–306. [DOI] [PubMed] [Google Scholar]
- 63.Turner, M., P.J. Mee, P.S. Costello, O. Williams, A.A. Price, L.P. Duddy, M.T. Furlong, R.L. Geahlen, and V.L. Tybulewicz. 1995. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. 378:298–302. [DOI] [PubMed] [Google Scholar]
- 64.Guinamard, R., M. Okigaki, J. Schlessinger, and J.V. Ravetch. 2000. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 1:31–36. [DOI] [PubMed] [Google Scholar]
- 65.Segal, B.H., T.L. Leto, J.I. Gallin, H.L. Malech, and S.M. Holland. 2000. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 79:170–200. [DOI] [PubMed] [Google Scholar]
- 66.Mastroeni, P., A. Vazquez-Torres, F.C. Fang, Y. Xu, S. Khan, C.E. Hormaeche, and G. Dougan. 2000. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J. Exp. Med. 192:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ezekowitz, R.A., M.C. Dinauer, H.S. Jaffe, S.H. Orkin, and P.E. Newburger. 1988. Partial correction of the phagocyte defect in patients with X-linked chronic granulomatous disease by subcutaneous interferon gamma. N. Engl. J. Med. 319:146–151. [DOI] [PubMed] [Google Scholar]
- 68.Henson, P.M., and R.B. Johnston, Jr. 1987. Tissue injury in inflammation. Oxidants, proteinases, and cationic proteins. J. Clin. Invest. 79:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weiss, S.J. 1989. Tissue destruction by neutrophils. N. Engl. J. Med. 320:365–376. [DOI] [PubMed] [Google Scholar]
- 70.Leff, J.A., and J.E. Repine. 1993. Neutrphil-mediated tissue injury. The Neutrophil. J.S. Abramson, J.G. Wheeler, editors. Oxford University Press, Oxford, UK. 229–261.
- 71.Weiss, S.J., G. Peppin, X. Ortiz, C. Ragsdale, and S.T. Test. 1985. Oxidative autoactivation of latent collagenase by human neutrophils. Science. 227:747–749. [DOI] [PubMed] [Google Scholar]
- 72.Peppin, G.J., and S.J. Weiss. 1986. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc. Natl. Acad. Sci. USA. 83:4322–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pryor, W.A., M.M. Dooley, and D.F. Church. 1986. The inactivation of alpha-1-proteinase inhibitor by gas-phase cigarette smoke: protection by antioxidants and reducing species. Chem. Biol. Interact. 57:271–283. [DOI] [PubMed] [Google Scholar]
- 74.Stief, T.W., and N. Heimburger. 1988. Inactivation of serine proteinase inhibitors (serpins) in human plasma by reactive oxidants. Biol. Chem. Hoppe Seyler. 369:1337–1342. [DOI] [PubMed] [Google Scholar]
- 75.Bjerrum, O.W., and N. Borregaard. 1989. Dual granule localization of the dormant NADPH oxidase and cytochrome b559 in human neutrophils. Eur. J. Haematol. 43:67–77. [DOI] [PubMed] [Google Scholar]
- 76.Dewald, B., M. Baggiolini, J.T. Curnutte, and B.M. Babior. 1979. Subcellular localization of the superoxide-forming enzyme in human neutrophils. J. Clin. Invest. 63:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]