

Abstract
CD4+CD25+ regulatory T (TR) cells can inhibit a variety of autoimmune and inflammatory diseases, but the precise mechanisms by which they suppress immune responses in vivo remain unresolved. Here, we have used Helicobacter hepaticus infection of T cell–reconstituted recombination-activating gene (RAG)−/− mice as a model to study the ability of CD4+CD25+ TR cells to inhibit bacterially triggered intestinal inflammation. H. hepaticus infection elicited both T cell-mediated and T cell–independent intestinal inflammation, both of which were inhibited by adoptively transferred CD4+CD25+ TR cells. T cell–independent pathology was accompanied by activation of the innate immune system that was also inhibited by CD4+CD25+ TR cells. Suppression of innate immune pathology was dependent on T cell–derived interleukin 10 and also on the production of transforming growth factor β. Thus, CD4+CD25+ TR cells do not only suppress adaptive T cell responses, but are also able to control pathology mediated by innate immune mechanisms.
Keywords: regulatory T cells, Helicobacter, immune tolerance, mucosal immunity, IL-10
Introduction
The key role that the immune system plays in regulating intestinal integrity has been demonstrated by the development of multiple rodent models of mucosal inflammation that are associated with immunological defects, particularly those that affect T cells (1). Growing evidence indicates that T cell responses toward intestinal antigens are normally of a tolerogenic nature and that dysregulation of mucosal T cell responses may lead to a loss of tolerance and the development of harmful intestinal inflammation similar to that observed in human inflammatory bowel disease (IBD)* (1–3).
A well characterized murine IBD model is the intestinal inflammation that occurs after adoptive transfusion of naive CD4+CD45RBhigh T cells from normal mice into immunodeficient recipients (4, 5). The resultant pathology is due to an excessive Th1-driven inflammatory response in the intestine and cotransfer of CD4+CD45RBlowCD25+ T cells from normal mice inhibits the colitis (6, 7). Additional studies in a variety of experimental models of autoimmune and inflammatory diseases have confirmed that regulatory T (TR) cells capable of controlling immune pathology are an integral part of the T cell repertoire in normal animals (for reviews, see references 8 and 9) and that their activity is enriched within the minor subpopulation of CD4+CD45RBlow T cells that express the CD25 marker (10).
Although the precise cellular and molecular pathways involved remain to be defined, current evidence indicates that TR cells are able to utilize multiple mechanisms to suppress immune responses. In vitro studies have demonstrated that TR cells can inhibit the activation of other T cells either directly via a cell contact–dependent mechanism (11–13), or indirectly by down-modulating the activity of antigen-presenting cells (14, 15). In addition, studies in animal models have provided strong evidence of a role for cytokines in the effector function of TR cells in vivo, and the inhibition of intestinal inflammation appeared to be crucially dependent on the presence of the immunosuppressive cytokines IL-10 and TGF-β (7, 16, 17). These molecules can mediate multiple suppressive effects on both T cells and on cells of the innate immune system (18, 19). Although these studies raise the possibility that TR cells also inhibit innate immune activation in vivo, the ability of TR cells to suppress non-T cell–mediated immune pathology has not yet been investigated.
The intestinal microflora are essential for the development of intestinal inflammation and a number of different bacterial species have been identified as being able to trigger disease in rodent models of IBD (20–23). One such organism is Helicobacter hepaticus, a Gram negative spiral bacterium that colonizes the intestinal crypts of the cecum and colon, establishing a life-long infection (24). Although H. hepaticus does not usually cause disease in immunocompetent mice, infection is associated with hepatitis in susceptible inbred strains (25) and it can cause colitis and typhlitis in immunodeficient mice (26, 27). H. hepaticus infection has also been shown to exacerbate colitis in specific pathogen-free (SPF) IL-10−/− mice and in RAG−/− mice reconstituted with naive CD45RBhigh CD4+ T cells (22, 23). In this study, we have used H. hepaticus infection of immunodeficient RAG−/− mice as a model to study the ability of CD4+CD25+ TR cells to inhibit bacterially driven T cell–dependent and T cell–independent intestinal inflammation.
Materials and Methods
Mice.
129SvEv, 129SvEvRAG2−/−, and 129SvEvIL-10−/− mice were bred under SPF conditions and maintained in micro-isolator cages with filtered air in the Pathology Services Building at the University of Oxford. Mice were routinely screened for the presence of Helicobacter spp. and were 6–10 wk old when first used.
Bacteria.
Helicobacter hepaticus was isolated from infected 129SvEvRAG2−/− mice as described previously (24), by culturing cecal scrapings from infected mice on blood agar plates containing trimethoprim, vancomycin and polymixin B (all from Oxoid) under microaerophilic conditions (CampyPak; Oxoid). H. hepaticus were characterized morphologically using phase-contrast microscopy and were Gram negative, and urease, catalase, and oxidase positive. Identity was confirmed by PCRs specific for H. hepaticus 16sRNA (27), urease (28), and cytolethal distending toxin (CDT; reference 29). Cultures were expanded for 3–4 d in TSB (Oxoid) containing 10% FCS (GIBCO BRL) and Helicobacter-free 129SvEvRAG2−/− mice were inoculated by oral gavage of 2 × 108 cfu H. hepaticus.
Purification of T Cell Subsets.
CD4+ T cell subsets were isolated from spleen cells from Helicobacter-free 129SvEv mice using magnetic bead based separation techniques and FACS® sorting, essentially as described previously (7, 30). Briefly, single cell suspensions were depleted of CD8+, MHC class II+, Mac-1+, and B220+ cells by negative selection using a panel of rat mAb, followed by sheep anti–rat–coated Dynabeads (Dynal). CD4+CD45RBlowCD25+ T cells were purified by staining with biotinylated-anti-CD25 (BD Biosciences) followed by streptavidin-conjugated microbeads (Miltenyi Biotec) and positively selected using AutoMACS (Miltenyi Biotec), according to the manufacturer's instructions. CD4+CD45RBhigh T cells were similarly isolated using AutoMACS, following staining with FITC-anti-CD45RB (BD Biosciences), followed by anti-FITC-conjugated microbeads (Miltenyi Biotec). Alternatively, after staining with Cy-Chrome-conjugated anti-CD4, PE-conjugated anti-CD25 and FITC-anti-CD45RB (all from BD Biosciences), CD4+ T cell subsets were purified by cell sorting, using a FACSVantage™ (BD Biosciences). Purity of the CD4+ T cell subsets was assessed using flow cytometry and ranged from 92–99%.
T Cell Reconstitution and Antibody Treatments.
129SvEv RAG2−/− mice were reconstituted by intraperitoneal injection of 4 × 105 CD4+CD45RBhigh T cells and/or 105 CD4+CD45RBlowCD25+ T cells and infected with H. hepaticus 24 h later. Antibody treatments were commenced the day after T cell reconstitution. Where indicated, mice were treated with 1 mg/wk anti–IL-12p40 (C17.8.20) for the first 2 wk after infection, or weekly throughout the duration of the experiment with 2 mg anti-TNF-α (XT22), 2 mg anti-TGF-β (1D11), or 0.5 mg anti-IL10R (1B1.2) or isotype control by intraperitoneal injection.
Assessment of Intestinal Inflammation.
Mice were killed 8–12 wk after H. hepaticus infection and samples of cecum, proximal, mid, and distal colon were isolated and fixed in buffered 10% formalin. 5 μm paraffin-embedded sections were cut and stained with hematoxylin and eosin. Inflammation was assessed as previously described (7, 17), each sample was graded semiquantitatively from 0–4 and typical features of each grade are: 0 = normal; 1 = mild epithelial hyperplasia; 2 = pronounced hyperplasia and significant inflammatory infiltrates; 3 = severe hyperplasia and infiltration with significant decrease in goblet cells; 4 = severe hyperplasia, severe transmural inflammation, ulceration, crypt abscesses, and substantial depletion of goblet cells. Ceca and colons were assessed separately and at least three separate sections from each sample were scored blind.
Flow Cytometry.
Aliquots of 1–5 × 105 cells were stained in FACS® buffer (PBS, 0.1% BSA, 5 mM EDTA; both from Sigma-Aldrich) using the following panel of mAb to murine cell surface molecules (all from BD Biosciences); biotinylated anti-NK cells (DX5), biotinylated anti-Gr1 (Ly6G), allophycocyanin (APC)-conjugated anti-CD11b, PE-anti-CD11c, peridinine chlorophyll protein (PerCP)-conjugated anti-CD4, FITC-conjugated anti-class II MHC, PE-anti–mouse TCRβ. Biotinylated antibodies were detected using PE-, APC-, or FITC-conjugated streptavidin (SA; all from BD Biosciences). Staining steps were performed for 30 min on ice and, after washing with FACS® buffer, samples were fixed in FACS® buffer containing 2% paraformaldehyde (BDH) and analyzed using a FACSCalibur™ with CELLQuest™ software (Becton Dickinson). For intracellular cytokine analysis, spleen cells (2.5 × 106 cells/ml) were incubated in complete RPMI 1640 (10% heat-inactivated FCS, 2 mM l-glutamine, 100 U/ml penicillin and streptomycin [GIBCO BRL], and 0.05 mM 2-mercaptoethanol [Sigma-Aldrich]) supplemented with 1,000 U/ml rhIL-2 (Sigma), for 18 h at 37°C in a humidified incubator with 5% CO2. Brefeldin A (BFA, 10 mg/ml; Sigma-Aldrich) was added for the final 4 h of incubation. Cell surface staining was performed as described above and after fixing, the samples were incubated in permeabilization (Perm) buffer (PBS, 0.1% BSA, 0.5% saponin; Sigma-Aldrich) containing PE-anti–mouse IFN-γ or isotype control (BD Biosciences) for 30 min at room temperature, washed in Perm buffer, and analyzed as above.
Isolation of Lamina Propria Leukocytes.
Lamina propria leukocytes (LPLs) were purified from ceca as described (6). Briefly, ceca were pooled from 4–6 mice per group, contents were removed, and ceca were rinsed in PBS. Ceca were cut into 0.3–0.5-cm pieces and repeatedly incubated in Ca- and Mg-free PBS containing 10% FCS and 5 mM EDTA to release intestinal epithelial cells. The remaining tissue was further digested with collagenase/dispase (100 U/ml; Sigma-Aldrich) and the LPL were then layered on a discontinuous Percoll gradient (Amersham Biosciences). After centrifugation at 600 g for 20 min, the LPL population was recovered at the 40/75% interface, washed twice in PBS/BSA, and analyzed using flow cytometry as detailed above.
Quantitation of H. hepaticus Using Real-time PCR.
DNA was purified from cecal or fecal scrapings taken from H. hepaticus–infected mice using the DNA Stool kit (QIAGEN). H. hepaticus DNA was quantified as described (29) using a real-time PCR method based on the cdtB gene, performed with the ABI prism Taqman 7700 sequence detection system (PE Biosystems). Standard curves were constructed using H. hepaticus DNA that was purified from bacterial cultures using the DNeasy kit (QIAGEN).
Statistics.
The nonparametric Mann-Whitney test was used for comparing pathology scores and cell counts and differences were considered statistically significant when P < 0.05.
Results
CD4+CD25+ TR Cells Protect Against both T Cell–dependent and T Cell–independent Intestinal Inflammation Triggered by Infection with H. hepaticus.
To examine whether TR cells could prevent intestinal inflammation induced by the bacterial pathogen H. hepaticus, we isolated CD4+ CD45RBlowCD25+ T cells and CD4+CD45RBlow CD25− T cells from normal mice and coinjected these subpopulations with CD4+CD45RBhigh T cells into 129SvEvRAG2−/− recipients that were then infected with H. hepaticus. Although CD4+CD45RBhigh T cell reconstitution of RAG2−/− mice induced only minimal changes in intestinal architecture (Figs. 1 and 2 D), concomitant infection of these mice with H. hepaticus led to the development of severe intestinal inflammation, characterized by marked epithelial cell hyperplasia, extensive inflammatory infiltrates, and goblet cell depletion (Figs. 1 and 2 E). While cotransfer of CD4+CD45RBlowCD25+ T cells completely inhibited the development of disease, CD4+CD45RBlowCD25− T cells were unable to prevent H. hepaticus–triggered intestinal inflammation (Figs. 1 and 2, F and G).
Figure 1.

CD4+CD25+ T cells prevent both T cell–dependent and T cell–independent intestinal inflammation. 129SvEvRAG2−/− mice were reconstituted with purified CD4+ T cell subsets as indicated (4 × 105 CD45RBhigh T cells and/or 105 CD4+CD45RBlowCD25+ T cells or CD4+CD45RBlowCD25− T cells) and infected 24 h later by oral gavage with 2 × 108 cfu H. hepaticus. Mice were killed 8–12 wk later and pathology in the colon (A) and cecum (B) was assessed histologically. Each symbol represents a single animal and results are representative of two similar experiments. **P < 0.01 vs. RBhi + Hh; *P < 0.05 vs. Hh.
Figure 2.
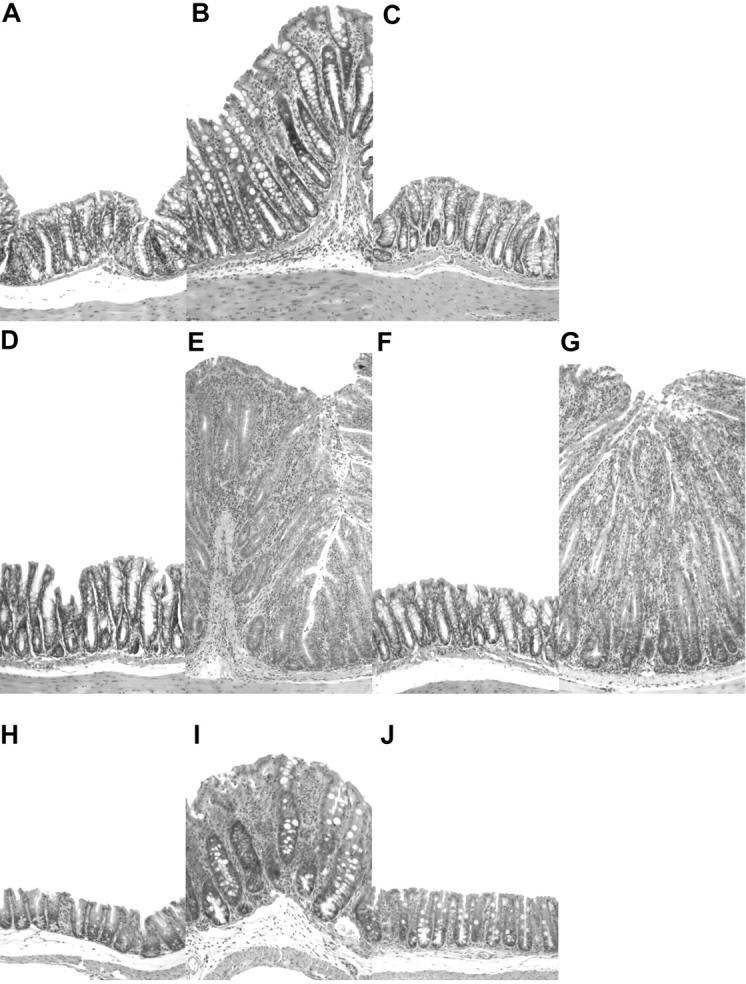
Representative photomicrographs of hematoxylin and eosin stained intestinal tissues from 129SvEvRAG2−/− mice that were reconstituted with purified CD4+ T cell subsets and/or infected with H. hepaticus as outlined in Fig. 1. (A–G) Colon sections from a control RAG2−/− mouse (A) and from mice that received; H. hepaticus only (B), H. hepaticus and CD4+CD45RBlowCD25+ T cells (C), CD4+CD45RBhigh T cells only (D), CD4+CD45RBhigh T cells and H. hepaticus (E), CD45RBhighCD4+ T cells plus CD4+CD45RBlowCD25+ T cells and H. hepaticus (F), or CD45RBhighCD4+ T cells plus CD4+CD45RBlowCD25− T cells and H. hepaticus (G). (H–J) Cecum sections from a control RAG2−/− mouse (H) and from mice that received; H. hepaticus only (I), or H. hepaticus and CD4+CD45RBlowCD25+ T cells (J). Original magnification: ×50.
H. hepaticus infection of RAG2−/− mice led to the development of significant intestinal inflammation, even in the absence of T cell reconstitution (Figs. 1 and 2 B). This T cell–independent pathology was generally less severe than the T cell–mediated disease, again involving marked epithelial cell hyperplasia, but with little depletion of mucin-producing cells (compare Fig. 2, B and E). Inflammatory infiltrates comprised of both polymorphonuclear cells and mononuclear cells were also present, although markedly less dense than those present in the T cell-mediated form of disease (compare Fig. 2, B and E). Consistent with previous reports of H. hepaticus–induced intestinal disease (23, 27), the most severe T cell–independent inflammation was observed in the cecum (Figs. 1 and 2 I). Importantly, reconstitution of RAG2−/− mice with CD4+ CD45RBlowCD25+ T cells also inhibited the development of H. hepaticus–induced T cell–independent intestinal inflammation in both the cecum and colon (Figs. 1 and 2, C and J). Conversely, reconstitution with CD4+CD45RBlow CD25− T cells did not prevent H. hepaticus–triggered T cell–independent intestinal pathology (unpublished data). Together, these results demonstrate that H. hepaticus is able to induce significant intestinal inflammation in RAG2−/− mice, that this inflammation is exacerbated when the RAG2−/− mice are reconstituted with CD4+CD45RBhigh T cells and, most importantly, that both the T cell–dependent and T cell–independent forms of disease are inhibited by CD4+CD45RBlowCD25+ TR cells.
T Cell–independent Immune Pathology Triggered by Infection with H. hepaticus Is Driven by Proinflammatory Cytokines and Is Associated with Local and Systemic Activation of Innate Immunity that Is Inhibited by CD4+CD25+ TR Cells.
To examine the role of proinflammatory cytokines in the T cell–independent intestinal inflammation we treated H. hepaticus–infected RAG2−/− mice with monoclonal antibodies to IL-12p40 or TNF-α, from the onset of infection. As shown in Fig. 3 , treatment with anti–IL-12p40 or anti–TNF-α significantly inhibited the development of intestinal inflammation, suggesting that the intestinal pathology was driven by the activation of host innate immune responses.
Figure 3.
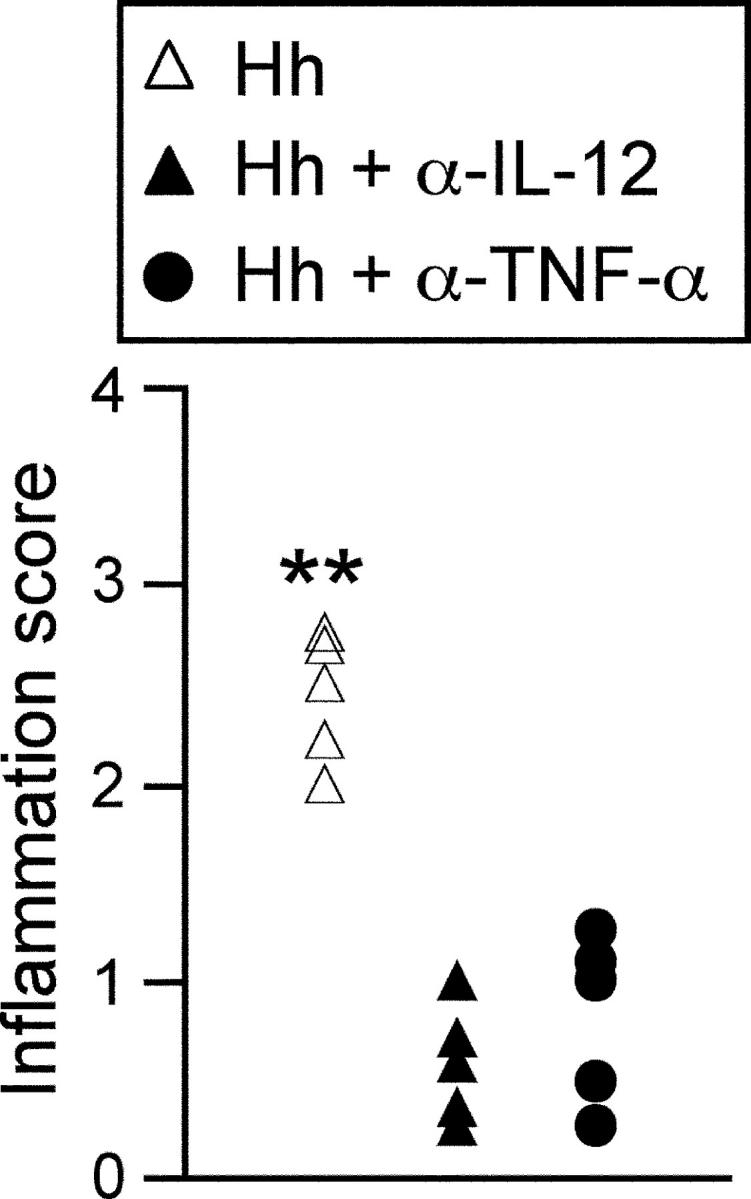
H. hepaticus–triggered T cell–independent intestinal inflammation is driven by proinflammatory cytokines. 129SvEvRAG2−/− mice were infected by oral gavage with 2 × 108 cfu H. hepaticus. Where indicated, mice received 1 mg/wk anti–IL-12p40 for the first 2 wk after infection or 2 mg/wk anti-TNF-α throughout the course of the infection. Mice were killed 8–12 wk later and pathology in the cecum was assessed histologically. Each symbol represents a single animal and results are representative of two similar experiments. **P < 0.01 vs. anti–IL-12p40 or anti-TNF-α.
In addition to the marked intestinal changes, we also observed significant splenomegaly in RAG2−/− mice that were infected with H. hepaticus (Fig. 4 A). Flow cytometric analyses (31) revealed that this was primarily due to substantial increases in the numbers of neutrophils (Gr1highCD11bhigh, 10 to 20-fold; Fig. 4 B) and monocytes/macrophages (Gr1lowCD11bhigh, 5 to 10-fold; Fig. 4 C) present in the spleens of H. hepaticus–infected RAG2−/− mice, with only modest increases in the number of dendritic cells (CD11chighMHC IIhigh, twofold; Fig. 4 D). Although there were only similarly modest increases in the total number of NK cells (twofold; Fig. 4 E), there was also a twofold increase in the proportion of NK cells producing IFN-γ (control 5.6 ± 1.3%; Hh 10.4 ± 2.8%; Hh + CD25+T 4.1 ± 1.9%), which together resulted in substantial increases in the number of IFN-γ+ NK cells (fourfold; Fig. 4 F), confirming that H. hepaticus infection led to extensive activation of innate immune cells in RAG2−/− mice. Importantly, adoptive transfer of CD4+ CD45RBlowCD25+ TR cells significantly reduced splenomegaly, granulocyte and monocyte recruitment and NK cell activation observed in RAG2−/− mice after H. hepaticus infection (Fig. 4).
Figure 4.

H. hepaticus infection of RAG2−/− mice induces systemic activation of innate immunity that is inhibited by CD4+CD25+ T cells. 129SvEvRAG2−/− mice were reconstituted with 105 CD4+ CD45RBlowCD25+ T cells and/or infected by oral gavage with 2 × 108 cfu H. hepaticus. Mice were killed 8–12 wk later and spleen cell populations were enumerated using FACS® analyses. Graphs shown represent numbers of total spleen cells (A), neutrophils (B), monocytes/macrophages (C), dendritic cells (D), NK cells (E), and IFN-γ–producing NK cells (F). Each symbol represents a single animal and results are representative of two similar experiments. **P < 0.01 vs. Hh; *P < 0.05 vs. Hh.
To assess innate immune activation at the site of infection, we isolated leukocyte populations from the lamina propria of the cecum (LPL) and from the mesenteric lymph node (MLN) and characterized their composition using flow cytometry. Due to the small numbers of leukocytes present in these tissues, we analyzed pooled LPL or MLN populations from the different groups of mice (Table I). H. hepaticus infection of RAG−/− mice elicited a marked increase in the number of LPL isolated from cecal tissue (eightfold, Table I). Strikingly, although they were not present in LPL populations isolated from control RAG−/− mice, neutrophils constituted almost 20% of LPL isolated from inflamed ceca in H. hepaticus–infected RAG−/− mice (Table I). By contrast, no marked differences in the relative proportions of monocytes or NK cells were observed in LPL populations isolated from control or H. hepaticus–infected RAG−/− mice (Table I). Importantly, reconstitution of H. hepaticus–infected RAG−/− mice with CD4+CD25+ TR cells prevented the accumulation of neutrophils in the cecal lamina propria and reduced LPL numbers to those found in control RAG mice (Table I). Interestingly, CD4+ T cells accounted for around 15% of the LPL isolated from H. hepaticus–infected RAG−/− mice that had received CD4+CD25+ TR cells (Table I). Although H. hepaticus infection also slightly increased the number of MLN cells when compared with control RAG−/− mice (2–3-fold), this was primarily due to an increased proportion of monocytes, with no significant increase in the proportion of neutrophils and a marked decrease in the proportion of NK cells (Table I). However, in contrast to the spleen and LPL populations, fivefold higher numbers of MLN cells were present in H. hepaticus–infected RAG−/− mice that had received CD4+CD25+ TR cells, and this was primarily due to the high proportion (22%) of CD4+ T cells found in the MLN populations isolated from these mice (Table I).
Table I.
Composition of Mucosal Lymphoid Populations in H. hepaticus–infected RAG−/− Mice
| Tissue | Group | Cell no. | Neuts | Monos | DC | NK | CD4+
T |
|---|---|---|---|---|---|---|---|
| % | % | % | % | % | |||
| LPL | Control | 2.8 × 104 | 0.6 | 18.2 | 8.8 | 3.0 | 0.3 |
| Hh | 2.1 × 105 | 17.5 | 13.0 | 6.0 | 8.1 | 0.3 | |
| Hh + 25+ | 2.5 × 104 | 0.4 | 14.6 | 7.3 | 5.8 | 15.2 | |
| MLN | Control | 1.4 × 105 | 1.8 | 15.1 | 4.5 | 47.7 | 0.2 |
| Hh | 3.6 × 105 | 4.0 | 31.7 | 4.7 | 25.2 | 0.3 | |
| Hh + 25+ | 7.7 × 105 | 0.5 | 10.2 | 5.6 | 15.8 | 22.1 |
Groups of 129SvEvRAG2−/− mice were left untreated (Control), or infected by oral gavage with 2 × 108 cfu H. hepaticus (Hh), or infected with H. hepaticus and reconstituted with 1 × 105 CD4+ CD45RBlowCD25+ T cells (Hh + 25+). Mice were sacrificed 8–12 wk later and LPL and MLN populations (pooled from 4–6 mice per group) were characterized using FACS® analyses. Mean cell number for each tissue was calculated by dividing the total number of cells isolated by the number of mice pooled. Results shown are representative of two similar experiments.
Together, the results described above indicate that CD4+CD25+ TR cells do not mediate their effects only on other T cells, but are also able to control chronic immune pathology mediated by sustained activation of innate immune mechanisms. In addition, they show that CD4+ CD25+ TR cells efficiently reconstitute both mucosal and systemic lymphoid compartments, suggesting that they may act at multiple sites to inhibit harmful immune pathology caused by the accumulation and activation of innate immune cells.
CD4+CD25+ TR Cells Do Not Influence H. hepaticus Colonization Levels.
To assess whether CD4+CD25+ TR cells might have inhibited intestinal pathology by mediating protective immunity against H. hepaticus, we used a real time PCR assay (29) to quantitate H. hepaticus levels in cecum and colon scrapings from infected RAG2−/− mice, in the presence or absence of CD4+CD25+ TR cells. In infected RAG2−/− mice we detected the highest levels of H. hepaticus in the cecum, with 5 to 10-fold lower levels present in the colon (Fig. 5) . Crucially, reconstitution of RAG2−/− mice with CD4+CD25+ TR cells had no measurable effect on the levels of H. hepaticus DNA detected in the cecum or colon (Fig. 5), indicating that the protection from H. hepaticus–triggered intestinal inflammation was not due to eradication of the bacteria or decreased levels of colonization.
Figure 5.

CD4+CD25+ T cell reconstitution does not affect H. hepaticus colonization levels in RAG2−/− mice. 129SvEvRAG2−/− mice were reconstituted with 105 CD4+ CD45RBlowCD25+ T cells and/or infected by oral gavage with 2 × 108 cfu H. hepaticus. Mice were killed 10 wk later, DNA was isolated from cecum and colon samples and H. hepaticus DNA was quantified using a real-time PCR assay. Each symbol represents a single animal and results are representative of two similar experiments.
CD4+CD25+ TR Cell–mediated Protection against T Cell-independent Intestinal Inflammation Is Cytokine Dependent.
Previous studies have highlighted the important roles of IL-10 and TGF-β in the regulation of intestinal inflammation (7, 16, 17, 32). In addition, H. hepaticus has also been shown to be able to trigger severe intestinal inflammation in IL-10−/− mice (23, 33). We therefore examined whether these cytokines also played a role in the inhibition of T cell–independent intestinal inflammation induced by H. hepaticus. Thus, RAG2−/− mice were reconstituted with CD4+CD25+ TR cells, infected with H. hepaticus and treated throughout the course of infection with monoclonal antibodies reactive with IL-10R or TGF-β. As shown in Fig. 6 , treatment with anti–IL-10R or anti–TGF-β completely ablated the ability of CD4+CD25+ TR cells to inhibit H. hepaticus–induced intestinal inflammation. Furthermore, we also observed that CD4+CD25+ T cells from IL-10−/− mice were unable to inhibit H. hepaticus–induced intestinal inflammation (Fig. 6), suggesting that T cell–derived IL-10 plays a crucial role in the control of innate immune pathology by CD4+CD25+ TR cells. The inability to protect from T cell–independent intestinal inflammation was not due to ineffective T cell reconstitution, as approximately twofold more CD4+T cells were recovered from RAG2−/− mice that received IL-10−/− CD4+CD25+ T cells or anti–IL-10R or anti–TGF-β treatment, when compared with those that received wild-type CD4+CD25+ T cells only (unpublished data).
Figure 6.
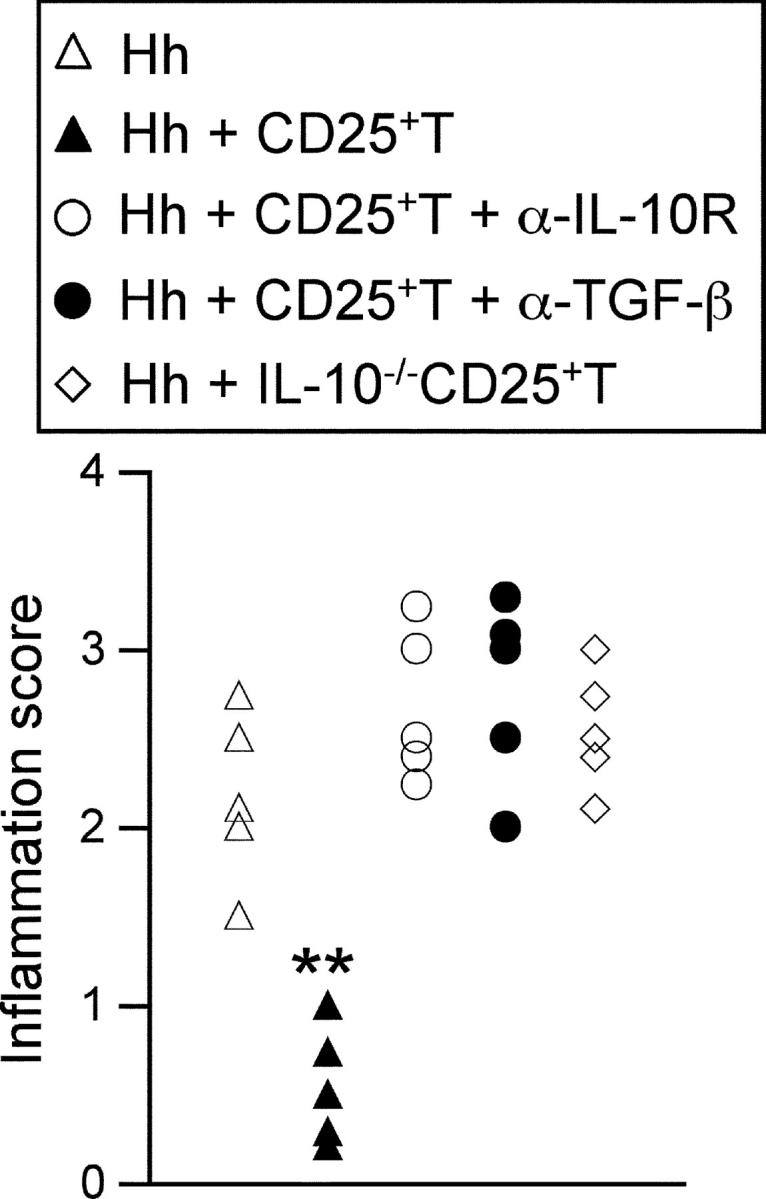
CD4+CD25+ T cell–mediated protection against T cell–independent intestinal inflammation is dependent on IL-10 and TGF-β. 129SvEvRAG2−/− mice were reconstituted with 105 CD4+CD45RBlowCD25+ T cells from wild-type or IL-10−/− mice and/or infected by oral gavage with 2 × 108 cfu H. hepaticus. Where indicated mice received 0.5 mg/wk anti–IL-10R or 2 mg/wk anti-TGF-β. Mice were killed 8–12 wk later, ceca samples were taken for histological examination. Each symbol represents a single animal and results are representative of two similar experiments. **P < 0.01 vs. Hh.
Discussion
Although the role of T cells in the induction of intestinal inflammation has received much attention, these results emphasize that innate immune mechanisms are also able to mediate significant intestinal inflammation upon stimulation with pathogenic organisms like H. hepaticus (26, 27, 34, 35). In addition, we show here that T cell–independent intestinal inflammation triggered by H. hepaticus infection of RAG2−/− mice was driven by proinflammatory cytokines, was accompanied by local and systemic innate immune activation and was completely inhibited by cotransfer of CD4+CD25+ TR cells through cytokine-dependent mechanisms. These results represent the first clear evidence showing that the immune suppressive properties of CD4+CD25+ TR cells are not limited to effects on other T cell responses, but also include inhibition of immune pathology mediated by cells of the innate immune system.
The mechanisms by which TR cells mediate their suppressive functions are the subject of intensive debate. Different in vivo models of disease suppression show different patterns of dependency on various immune suppressive cytokines and there is good evidence that a cell contact–dependent mechanism is involved in the suppression of T cell responses in vitro (for reviews, see references 8 and 36). An alternative explanation that has been proposed for TR cell phenomena is that regulatory activity is not the sole property of a defined T cell subpopulation, but rather can be accomplished by any kind of T cell that is able to compete for ‘space’ and survival factors with potentially pathogenic T cells (37). However, suppression of H. hepaticus–triggered innate immune pathology was a unique property of CD4+CD25+ TR cells. In addition, our results illustrate that CD4+CD25+ TR cells do not exclusively mediate their suppressive functions by inhibiting other T cell responses. Since in many inflammatory and autoimmune diseases pathology is associated with the activation and accumulation of innate immune effector cells, the inhibition of such responses by TR cells extends the therapeutic potential of manipulations of TR cell function. Nevertheless, it is important to emphasize the delicate balance that TR cells must maintain between allowing protective immune responses to proceed efficiently, while inhibiting harmful immune pathology. This raises the possibility that excessive TR cell activity may actually impede protective immunity. One such instance was recently reported in scid mice infected with Pneumocystis carinii, where the elimination of P. carinii mediated by adoptively transferred CD4+CD25− T cells was suppressed when CD4+CD25+ T cells were cotransferred (38).
The suppression of T cell–independent immune pathology by CD4+CD25+ TR cells was dependent on the actions of IL-10 and TGF-β, as treatment with anti–IL-10R or anti–TGF-β completely abrogated protection against H. hepaticus–induced intestinal inflammation. This observation further confirms the central role of these immunosuppressive cytokines in preventing intestinal inflammation, highlighted by their key role in the suppression of T cell–mediated colitis by CD4+CD45RBlow T cells (6, 16, 17) and by the development of colitis in IL-10−/− mice (32). Our findings are also consistent with a previous study showing that the development of T cell–independent intestinal inflammation in RAG2−/− mice exposed to a flora that included H. hepaticus could be inhibited by treatment with rIL-10 (39). Importantly, our results suggest a crucial role for T cell–derived IL-10 in this process, as CD4+CD25+ T cells from IL-10−/− mice were unable to inhibit the development of intestinal inflammation. There is clear evidence that IL-10 has inhibitory effects on cells of the innate immune system, including the suppression of pro-inflammatory cytokine and chemokine production by activated monocytes/macrophages and neutrophils (for a review, see reference 19). An excellent example highlighting the importance of the actions of IL-10 in controlling innate immune cells in the intestine is the observation that transgenic mice with a cell-type deletion in Stat-3, in which macrophages and neutrophils are unresponsive to IL-10, spontaneously develop intestinal inflammation (40).
We observed that H. hepaticus infection of 129SvEv RAG2−/− mice triggered intestinal inflammation that was characterized by epithelial cell hyperplasia and by marked infiltration of both granulocytic and mononuclear cells. Immune pathology was most prominent in the cecum, where the highest colonization by H. hepaticus occurred. The immune pathology found in H. hepaticus-infected RAG2−/− mice was driven by proinflammatory cytokines, as it was significantly inhibited by treatment with anti–IL-12p40 or anti–TNF-α antibodies. In addition, Helicobacter-triggered intestinal inflammation is associated with increased production of proinflammatory molecules, such as IL-1, TNF-α, iNOS, and IFN-γ (33, 35). The induction of innate immune pathology in the gastrointestinal tract has also been observed in immunodeficient mice infected with other bacteria, including Campylobacter fetus (41) and Citrobacter rodentium (42), suggesting that potentially pathogenic innate immune responses may occur commonly in the intestine after colonization with pathogenic organisms.
The present study demonstrates that the development of T cell–mediated colitis in 129SvEvRAG2−/− mice requires H. hepaticus infection, whereas we have previously found that adoptive transfer of naive syngeneic CD4+ T cells into CB17 scid or C57BL/6 RAG−/− mice leads to severe colitis, even in the absence of H. hepaticus (7). While the reasons for these differences are not yet clear, they highlight the fact that the incidence and severity of colitis observed in immune deficient mice after T cell adoptive transfer will be heavily dependent on both environmental factors, especially the intestinal micro-flora, as well as genetic factors, such as mouse strain differences. Similarly, different immune deficient strains in our animal facilities exhibit different susceptibilities to T cell–independent intestinal inflammation induced by H. hepaticus infection, with C57BL6 RAG−/− mice and CB17 scid mice exhibiting minimal intestinal inflammation after infection with H. hepaticus (unpublished data). In our opinion, similar host environmental and genetic influences, in combination with bacterial strain differences, may account for the wide range of susceptibilities, kinetics, and severity of disease observed in different immunodeficient mouse strains upon infection with H. hepaticus (26, 27, 34, 35, 43).
The inflammatory changes found in H. hepaticus–infected 129SvEvRAG2−/− mice were not limited to the intestine, they were accompanied by systemic innate immune activation as evidenced by pronounced splenomegaly that was associated with accumulation of neutrophils and monocytes and by the enhanced activation of NK cells. Importantly, we found that adoptively transferred CD4+CD25+ TR cells efficiently reconstituted both systemic and mucosal lymphoid compartments, including the lamina propria of the large intestine and the mesenteric lymph nodes. Reconstitution with CD4+CD25+ TR cells completely inhibited the accumulation and activation of innate immune cells observed in H. hepaticus–infected RAG−/− mice, both in the intestinal lamina propria and also in the spleen. Our results show that, in addition to inhibiting innate immune pathology in the intestine, CD4+CD25+ TR cells can also suppress the activation and accumulation of innate immune cells in systemic lymphoid tissues, suggesting that CD4+CD25+ TR cells may operate both locally at the inflammatory site and systemically to prevent immune pathology.
It is not yet clear whether CD4+CD25+ TR cells recognize self-peptides, peptides derived from foreign antigens or both. In this study we isolated CD4+CD25+ TR cells from normal 129SvEv mice that were free from Helicobacter infection and demonstrated that they could inhibit intestinal inflammation triggered by H. hepaticus. These results are consistent with the finding that CD4+CD25+ TR cells isolated from germ-free mice were able to inhibit intestinal pathology mediated by CD4+CD45RBhigh T cells (44), confirming that TR cells can suppress harmful immune pathology triggered by organisms to which they have not been previously exposed. The nature of CD4+ T cell responses directed against H. hepaticus has been examined by Kullberg and colleagues in the IL-10−/− murine model of IBD (23, 33). H. hepaticus–infected IL-10−/− mice developed severe colitis that was characterized by the presence of H. hepaticus-specific CD4+ Th1 cell responses in the MLN, while H. hepaticus infected wild-type mice did not develop intestinal pathology and mounted an IL-10 dominated response (23, 33). Recently, Kullberg et al. have extended these findings using H. hepaticus infection of naive T cell-reconstituted C57BL10 RAG−/− mice as a model of bacterially driven T cell–mediated colitis (43). This study found that both CD25+ and CD25−CD4+CD45RBlow T cell populations from wild-type C57BL10 mice could inhibit the development of colitis, but only when these cells were purified from H. hepaticus-infected donors (43). Protection from colitis was associated with IL-10 production by the CD4+CD45RBlow T cells, but was not dependent on TGF-β. These results suggest H. hepaticus infection of normal immune competent mice induces CD4+ TR cells that prevent bacterially induced colitis. While it is difficult to reconcile the discrepancies between these findings and our own observations, we believe that a number of factors may contribute. First, as noted above, both genetic strain differences and environmental factors influence susceptibility to H. hepaticus–induced intestinal inflammation and it is possible that different H. hepaticus isolates may also exhibit differing pathogenic potentials. Second, the intestinal inflammation triggered by H. hepaticus in T cell–reconstituted C57BL10 RAG−/− mice is clearly more severe than the T cell–independent pathology we observed in 129SvEv RAG−/− mice, therefore it is not surprising that the inhibition of T cell–mediated colitis may well require higher numbers of bacteria-specific TR cells. Lastly, as CD25− CD4+ T cell populations have been shown to possess TR cell activity in other experimental models (45–47) and because expression of CD25 by TR cells is highly dynamic in vivo (48), it is not entirely surprising that H. hepaticus infection of normal immune competent mice may give rise to a heterogenous population of CD45RBlowCD4+ TR cells capable of inhibiting H. hepaticus–driven intestinal inflammation. Further studies will be required to more clearly define the roles of H. hepaticus–specific TR cells, and of TR cells recognizing self-antigens or other components of the intestinal flora, in the regulation of pathological innate and adaptive immune responses triggered after infection with H. hepaticus.
Acknowledgments
We would like to thank Liz Darley for performing the histology, Nigel Rust for assistance with the FACS® sorting, staff at the PSB for animal care and O. Annacker, L. Stephens, and H. Uhlig for critical review of the manuscript.
This project was supported by a Wellcome Trust Project grant (F. Powrie) and by Wellcome Trust Fellowships (F. Powrie and N.J. Saunders).
Footnotes
Abbreviations used in this paper: IBD, inflammatory bowel disease; LPL, lamina propria leukocyte; MLN, mesenteric lymph node; TR, regulatory T.
References
- 1.Strober, W., I.J. Fuss, and R.S. Blumberg. 2002. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 20:495–549. [DOI] [PubMed] [Google Scholar]
- 2.Duchmann, R., I. Kaiser, E. Hermann, W. Mayet, K. Ewe, and K.H. Meyer zum Buschenfelde. 1995. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 102:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khoo, U.Y., I.E. Proctor, and A.J. Macpherson. 1997. CD4+ T cell down-regulation in human intestinal mucosa: evidence for intestinal tolerance to luminal bacterial antigens. J. Immunol. 158:3626–3634. [PubMed] [Google Scholar]
- 4.Powrie, F., M.W. Leach, S. Mauze, L.B. Caddle, and R.L. Coffman. 1993. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5:1461–1471. [DOI] [PubMed] [Google Scholar]
- 5.Morrissey, P.J., K. Charrier, S. Braddy, D. Liggitt, and J.D. Watson. 1993. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 178:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powrie, F., M.W. Leach, S. Mauze, S. Menon, L.B. Caddle, and R.L. Coffman. 1994. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1:553–562. [DOI] [PubMed] [Google Scholar]
- 7.Read, S., V. Malmstrom, and F. Powrie. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 9.Shevach, E.M. 2000. Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 18:423–449. [DOI] [PubMed] [Google Scholar]
- 10.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 11.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10:1969–1980. [DOI] [PubMed] [Google Scholar]
- 12.Thornton, A.E., and E.M. Shevach. 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thornton, A.M., and E.M. Shevach. 2000. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164:183–190. [DOI] [PubMed] [Google Scholar]
- 14.Cederbom, L., H. Hall, and F. Ivars. 2000. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 30:1538–1543. [DOI] [PubMed] [Google Scholar]
- 15.Vendetti, S., J.G. Chai, J. Dyson, E. Simpson, G. Lombardi, and R. Lechler. 2000. Anergic T cells inhibit the antigen-presenting function of dendritic cells. J. Immunol. 165:1175–1181. [DOI] [PubMed] [Google Scholar]
- 16.Powrie, F., J. Carlino, M.W. Leach, S. Mauze, and R.L. Coffman. 1996. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1–mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 183:2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asseman, C., S. Mauze, M.W. Leach, R.L. Coffman, and F. Powrie. 1999. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Letterio, J.J., and A.B. Roberts. 1998. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 16:137–161. [DOI] [PubMed] [Google Scholar]
- 19.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 20.Rath, H.C., H.H. Herfarth, J.S. Ikeda, W.B. Grenther, T.E. Hamm, Jr., E. Balish, J.D. Taurog, R.E. Hammer, K.H. Wilson, and R.B. Sartor. 1996. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest. 98:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellon, R.K., S. Tonkonogy, M. Schultz, L.A. Dieleman, W. Grenther, E. Balish, D.M. Rennick, and R.B. Sartor. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66:5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahill, R.J., C.J. Foltz, J.G. Fox, C.A. Dangler, F. Powrie, and D.B. Schauer. 1997. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect. Immun. 65:3126–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kullberg, M.C., J.M. Ward, P.L. Gorelick, P. Caspar, S. Hieny, A. Cheever, D. Jankovic, and A. Sher. 1998. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66:5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox, J.G., F.E. Dewhirst, J.G. Tully, B.J. Paster, L. Yan, N.S. Taylor, M.J. Collins, Jr., P.L. Gorelick, and J.M. Ward. 1994. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32:1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox, J.G., L. Yan, B. Shames, J. Campbell, J.C. Murphy, and X. Li. 1996. Persistent hepatitis and enterocolitis in germfree mice infected with Helicobacter hepaticus. Infect. Immun. 64:3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward, J.M., M.R. Anver, D.C. Haines, J.M. Melhorn, P. Gorelick, L. Yan, and J.G. Fox. 1996. Inflammatory large bowel disease in immunodeficient mice naturally infected with Helicobacter hepaticus. Lab. Anim. Sci. 46:15–20. [PubMed] [Google Scholar]
- 27.Li, X., J.G. Fox, M.T. Whary, L. Yan, B. Shames, and Z. Zhao. 1998. SCID/NCr mice naturally infected with Helicobacter hepaticus develop progressive hepatitis, proliferative typhlitis, and colitis. Infect. Immun. 66:5477–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckwith, C.S., D.J. McGee, H.L. Mobley, and L.K. Riley. 2001. Cloning, expression, and catalytic activity of Helicobacter hepaticus urease. Infect. Immun. 69:5914–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge, Z., D.A. White, M.T. Whary, and J.G. Fox. 2001. Fluorogenic PCR-based quantitative detection of a murine pathogen, Helicobacter hepaticus. J. Clin. Microbiol. 39:2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malmstrom, V., D. Shipton, B. Singh, A. Al-Shamkhani, M.J. Puklavec, A.N. Barclay, and F. Powrie. 2001. Cd134l expression on dendritic cells in the mesenteric lymph nodes drives colitis in t cell-restored scid mice. J. Immunol. 166:6972–6981. [DOI] [PubMed] [Google Scholar]
- 31.Lagasse, E., and I.L. Weissman. 1996. Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods. 197:139–150. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 33.Kullberg, M.C., A.G. Rothfuchs, D. Jankovic, P. Caspar, T.A. Wynn, P.L. Gorelick, A.W. Cheever, and A. Sher. 2001. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 69:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chin, E.Y., C.A. Dangler, J.G. Fox, and D.B. Schauer. 2000. Helicobacter hepaticus infection triggers inflammatory bowel disease in T cell receptor alphabeta mutant mice. Comp. Med. 50:586–594. [PubMed] [Google Scholar]
- 35.Burich, A., R. Hershberg, K. Waggie, W. Zeng, T. Brabb, G. Westrich, J.L. Viney, and L. Maggio-Price. 2001. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G764–G778. [DOI] [PubMed] [Google Scholar]
- 36.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 37.Stockinger, G., T. Barthlott, and G. Kassiotis. 2001. T cell regulation: a special job or everyone's responsibility? Nat. Immunol. 2:757–758. [DOI] [PubMed] [Google Scholar]
- 38.Hori, S., T.L. Carvalho, and J. Demengeot. 2002. CD25+CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur. J. Immunol. 32:1282–1291. [DOI] [PubMed] [Google Scholar]
- 39.von Freeden-Jeffry, U., N. Davidson, R. Wiler, M. Fort, S. Burdach, and R. Murray. 1998. IL-7 deficiency prevents development of a non-T cell non-B cell-mediated colitis. J. Immunol. 161:5673–5680. [PubMed] [Google Scholar]
- 40.Takeda, K., B.E. Clausen, T. Kaisho, T. Tsujimura, N. Terada, I. Forster, and S. Akira. 1999. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 10:39–49. [DOI] [PubMed] [Google Scholar]
- 41.Young, V.B., C.A. Dangler, J.G. Fox, and D.B. Schauer. 2000. Chronic atrophic gastritis in SCID mice experimentally infected with Campylobacter fetus. Infect. Immun. 68:2110–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vallance, B.A., W. Deng, L.A. Knodler, and B.B. Finlay. 2002. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 70:2070–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kullberg, M.C., D. Jankovic, P.L. Gorelick, P. Caspar, J.J. Letterio, A.W. Cheever, and A. Sher. 2002. Bacteria-triggered CD4+ T regulatory cells suppress Helicobacter hepaticus-induced colitis. J. Exp. Med. 196:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh, B., S. Read, C. Asseman, V. Malmstrom, C. Mottet, L.A. Stephens, R. Stepankova, H. Tlaskalova, and F. Powrie. 2001. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 182:190–200. [DOI] [PubMed] [Google Scholar]
- 45.Olivares-Villagomez, D., A.K. Wensky, Y. Wang, and J.J. Lafaille. 2000. Repertoire requirements of CD4+ T cells that prevent spontaneous autoimmune encephalomyelitis. J. Immunol. 164:5499–5507. [DOI] [PubMed] [Google Scholar]
- 46.Stephens, L.A., and D. Mason. 2000. CD25 is a marker for CD4+ thymocytes that prevent autoimmune diabetes in rats, but peripheral T cells with this function are found in both CD25+ and CD25− subpopulations. J. Immunol. 165:3105–3110. [DOI] [PubMed] [Google Scholar]
- 47.Annacker, O., R. Pimenta-Araujo, O. Burlen-Defranoux, T.C. Barbosa, A. Cumano, and A. Bandeira. 2001. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J. Immunol. 166:3008–3018. [DOI] [PubMed] [Google Scholar]
- 48.Annacker, O., R. Pimenta-Araujo, O. Burlen-Defranoux, and A. Bandeira. 2001. On the ontogeny and physiology of regulatory T cells. Immunol. Rev. 182:5–17. [DOI] [PubMed] [Google Scholar]