

Abstract
Toll-like receptors (TLRs) and members of the proinflammatory interleukin 1 receptor (IL-1R) family are dependent on the presence of MyD88 for efficient signal transduction. The bipartite nature of MyD88 (N-terminal death domain [DD] and COOH-terminal Toll/IL-1 receptor [TIR] domain) allows it to link the TIR domain of IL-1R/TLR with the DD of the Ser/Thr kinase termed IL-1R–associated kinase (IRAK)-1. This triggers IRAK-1 phosphorylation and in turn the activation of multiple signaling cascades such as activation of the transcription factor nuclear factor (NF)-κB. In contrast, expression of MyD88 short (MyD88s), an alternatively spliced form of MyD88 that lacks only the short intermediate domain separating the DD and TIR domains, leads to a shutdown of IL-1/lipopolysaccharide-induced NF-κB activation. Here, we provide the molecular explanation for this difference. MyD88 but not MyD88s strongly interacts with IRAK-4, a newly identified kinase essential for IL-1R/TLR signaling. In the presence of MyD88s, IRAK-1 is not phosphorylated and neither activates NF-κB nor is ubiquitinated. Thus, MyD88s acts as a negative regulator of IL-1R/TLR/MyD88-triggered signals, leading to a transcriptionally controlled negative regulation of innate immune responses.
Keywords: MyD88, IL-1R, TLR, NF-κB, IRAK
Introduction
IL-1R–associated kinase (IRAK)-1 was originally identified as a protein kinase that associates with the IL-1Rs (a heterocomplex composed of the IL-1RI and IL-1RAcP) after IL-1 stimulation (1, 2). It is now known to play an important role in the activation of NF-κB and mitogen-activated protein kinase signaling cascades initiated by IL-1β and Toll-like receptor (TLR) ligands (3, 4). Stimulation triggers the rapid recruitment of IRAK-1 to an activated receptor resulting in IRAK-1 hyperphosphorylation. Phosphorylation of IRAK-1 appears necessary for signal transduction. It is required for binding to TNFR-associated factor 6 and for the formation of a multiprotein signalosome that is crucial for driving the activation of the transcription factors activator protein 1 (AP1) and nuclear factor (NF)-κB (5–8). Phosphorylation of IRAK-1 also leads to its proteasome-mediated degradation, the function of which is unclear but might be a means of switching off or terminating the signal (9, 10).
The MyD88 short (MyD88s) protein (MyD88 without the intermediary domain (ID), amino acids 110–157) can be detected only after continuous stimulation with bacterial products (i.e., LPS) or proinflammatory cytokines (i.e., TNFα; reference 11). This inducible expression hints to its role as a negative regulator of Toll/IL-1 receptor (TIR) signaling that we previously proposed based on its ability to block IL-1/LPS-induced NF-κB activation. Like MyD88, MyD88s binds both the IL-1R and IRAK-1, but in contrast to MyD88, ectopic expression of MyD88s does not induce IRAK-1 phosphorylation (11). This suggested a potential mechanism for the inhibitory effect of MyD88s and that MyD88 via its ID might activate and/or bind a kinase required for IRAK-1 phosphorylation.
Materials and Methods
Biological Reagents and Cell Culture.
Recombinant mouse TNFα and IL-1β were provided by Apotech and Sigma-Aldrich, respectively. The source of the various antibodies used in this study is as follows: anti-Flag/M2 (Eastman Kodak Co.), anti–vesicular stomatitis virus (VSV; Sigma-Aldrich), anti–IRAK-1 (Qbiogene or Santa Cruz Biotechnology, Inc.), anti-MyD88 (ProSci Inc.), and anti-E (Amersham Biosciences) antibodies. 293T and the embryonic cell lines were maintained in DMEM Glutamax supplemented with 10% fetal calf serum and 100 μg penicillin/streptomycin.
Generation of MyD88−/− Mouse Embryo Fibroblasts (MEFs).
D14 MEFs from MyD88−/− were transformed by transfection with a vector encoding the SV40 T antigen (provided by F. Radtke, Institute Ludwig, Epalinges, Switzerland). These cells were then reconstituted with MyD88, MyD88s, or empty vector by retroviral infection. Populations of MyD88 or MyD88s reconstituted cells (called MEF+ MyD88 and MEF+ MyD88s, respectively) were selected with 1 μg/ml puromycin.
Expression Vectors and Yeast 2 Hybrid Constructs.
Expression vectors encoding IRAK-1, IRAK-1 D340N, MyD88, and MyD88s have been previously described (11, 12). IRAK-4 was PCR amplified from an expressed sequence tag (EST) clone and inserted into pCRIII containing an NH2-terminal Flag or a VSV tag, or inserted into pGAD10. Kinase-dead mutants of IRAK-4 (IRAK-4KK213AA or IRAK-4D311N) were generated by double PCR and inserted into a pCRIII vector with an NH2-terminal tag. pGBT9 MyD88, pGBT9 MyD88-N (amino acids 1–172), and pGBT9 MyD88-TIR (amino acids 161–296) expressing pGALDB (the GAL4 DNA-binding domain) fused to full-length MyD88 or the indicated deletion mutants have been described previously (13). pGBT9 MyD88-ID (amino acids 110–157) and pGBT9 MyD88-death domain (DD; amino acids 1–110) were prepared by inserting PCR-generated fragments into pGBT9. The sequence of all PCR-generated cDNAs were confirmed by DNA sequencing. pGAD10 IRAK-4–expressing Gal4AD-IRAK4 (a fusion protein of GAL4 transcription activation with full-length IRAK-4) was made by inserting IRAK-4 cDNA as an EcoRI fragment into pGAD10.
Yeast 2 Hybrid Interaction Studies.
Interaction of full-length IRAK-4 with different deletion mutants of MyD88 was evaluated by yeast 2 hybrid interaction studies, performed as previously described (14). In brief, yeast cells of the Saccharomyces cerevisiae strain HF7c were cotransformed with the pGAD10 IRAK-4 and pGBT9 MyD88 or pGBT9 fused to different MyD88 deletion mutants (schematically illustrated in Fig. 3 B). Transformation efficiency was verified by growth on appropriate synthetic media using Trp and Leu selection markers. Protein interaction was revealed by His auxotrophy and assessed by β galactosidase expression filter assays. ++ indicates strong color development within 60 min of the assay and − indicates no development of color within 24 h. All pGBT9 MyD88 fusion proteins were negative for autoactivation.
Figure 3.
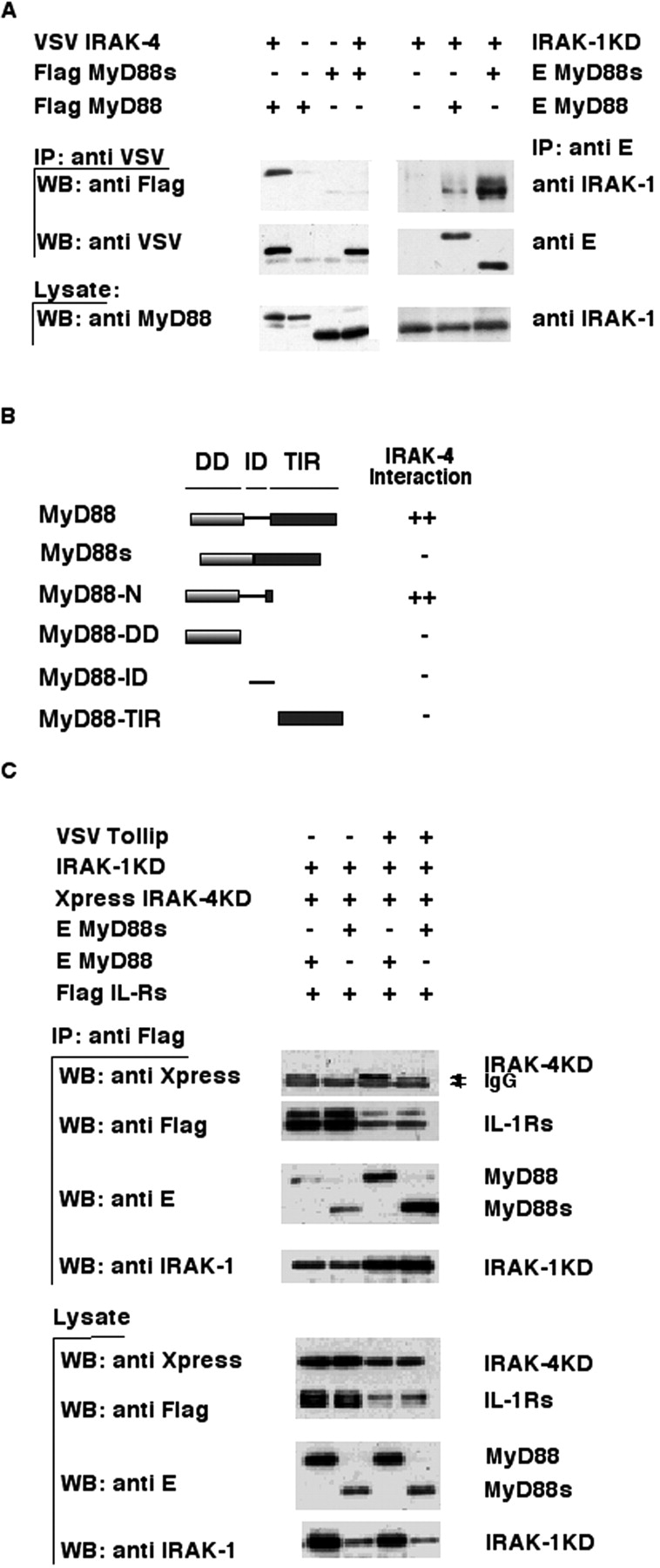
MyD88s does not bind IRAK-4 and blocks its recruitment to the IL-1Rs. (A) The left side of the top panel shows that IRAK-4 was coimmunoprecipitated using an anti-VSV antibody from lysates of 293T cells transfected with the indicated expression plasmids, and immuno-blotted with an anti-Flag antibody. The middle and bottom panels show Western blotting of immunoprecipitates and cell lysates, respectively, using anti-VSV or anti-MyD88 antibodies. The ride side of the top panel shows that MyD88 was coimmunoprecipitated using an anti-E antibody from lysates of 293T cells cotransfected with the indicated expression plasmids, and immunoblotted with an anti–IRAK-1 antibody. The middle and bottom panels show Western blotting of immunoprecipitates or cell lysates using anti-E (middle) or anti–IRAK-1 (bottom) antibodies. IP, immunoprecipitate; WB, Western blot. (B) HF7c yeast was cotransformed with expression vectors encoding the pGBT9 GAL4 DNA-binding domain fused to various MyD88 deletion mutants (schematically illustrated) and pGAD10 IRAK-4 expressing the GAL4 transcription activation domain fused to full-length IRAK-4. Interaction of the proteins was assessed by β galactosidase expression filter assays. ++ indicates strong color development within 60 min of the assay and − indicates no development of color within 24 h. All pGBT9 fusion proteins were checked for autoactivation and found to be negative. (C) Flag-tagged IL-1Rs were coimmunoprecipitated with anti-Flag antibodies from lysates of 293T cells cotransfected with the indicated expression plasmids and immunoblotted with an anti-Xpress antibody to detect IRAK-4KD association. The same blot was reprobed with anti-Flag, anti-E, and anti–IRAK-1 antibodies to monitor IL-1Rs, MyD88, and IRAK-1KD levels, respectively. Bottom panels show Western blotting of cell lysates using the indicated antibodies.
Immunoprecipitation and Kinase Assays.
Transfected 293T cells were lysed in lysis buffer (1% NP-40, 20 mM Hepes, pH 7.9, 250 mM NaCl, 20 mM β glycerophosphate, 10 mM NaF, 1 mM sodium orthovanadate, 2 mM dithiothreitol, 1 mM EDTA, and a protease inhibitor cocktail). After lysis, the cell extracts were incubated with one of the following antibodies for 2 h at 4°C: 1 μg anti-M2, anti-VSV, anti-IRAK-1, or anti-E preincubated with protein G Sepharose. After incubation the beads were washed six times with lysis buffer, separated by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblotting.
For the kinase assays, transiently transfected HEK 293T cells were lysed in 500 μl of 20 mM Tris, pH 7.5, 50 mM KCl, 5 mM MgCl2, 400 mM NaCl, 2 mM dithiothreitol, 1% Triton X-100, 20% glycerol, and protease and phosphatase inhibitors. IRAK-1KD was immunoprecipitated for 2 h at 4°C with an anti–IRAK-1 antibody (Qbiogene), followed by the addition of protein A trisacryl (Pierce Chemical Co.). Immune complexes were washed twice with lysis buffer and twice with kinase buffer containing 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 2 mM MgCl2, 2 mM MnCl2, 5% glycerol, and protease inhibitors. After the last wash, immune complexes were resuspended in 40 μl kinase buffer. For each kinase reaction 10 μl of the respective immune complexes were mixed with 5 μCi of [γ-32P] ATP (3,000 Ci/mmol) in a total volume of 25 μl. Reactions were allowed to proceed for 15 min at 30°C and then directly analyzed by SDS-PAGE and autoradiography. A reaction without ATP added was set up in parallel and analyzed by Western blot to estimate the input.
Results and Discussion
The ID of MyD88 Is Critical for IRAK-1 Phosphorylation.
Initially, to prove that MyD88's ID is indeed crucial for IRAK-1 phosphorylation, a MyD88−/− MEF cell line was generated and stably reconstituted with MyD88- or MyD88s-expressing vectors, referred to here as MEF+ MyD88 or MEF+ MyD88s. The cells were stimulated with IL-1β and IRAK-1 levels were monitored (as a readout for phosphorylation-induced degradation) in total cell extracts by Western blot analysis (Fig. 1 A). As previously reported, IRAK-1 was not activated in MyD88−/− cells (15, 16). However, activation of IRAK-1 was restored in the MEF+ MyD88 cells as evidenced by the disappearance of IRAK-1 in an IL-1β–dependent manner. Even in the unstimulated MEF+ MyD88 cells, very little IRAK-1 was detected demonstrating the effect that MyD88 overexpression alone has on IRAK-1 stability. In contrast, IRAK-1 levels in MEF+ MyD88s cells were comparable to those in control cells, confirming that expression of MyD88s does not trigger IRAK-1 phosphorylation and that the ID is essential for IRAK-1 phosphorylation. The ID of MyD88 is also essential for IL-1β–induced NF-κB activation, based on the absence of IκB degradation in MEF+ MyD88s cells stimulated with IL-1β (Fig. 1 B).
Figure 1.

The ID of MyD88 is critical for IRAK-1 phosphorylation. (A) MyD88−/−-deficient MEF cell line was reconstituted by retroviral infection with an empty vector, MyD88, or MyD88s expression vectors. Reconstituted populations of cells are referred to as MEF+ vector, MEF+ MyD88, and MEF+ MyD88s and were stimulated with 10 ng/ml IL-1β for 0.5 or 16 h. Total cell lysates were prepared and IRAK-1 levels were monitored by Western blotting with anti–IRAK-1 antibodies. The levels of MyD88 or MyD88s in MEF+ MyD88 and MEF+ MyD88s cells were confirmed by Western analysis with anti-MyD88 antibodies (middle). The loading control shows a nonspecific band detected by the anti-IRAK antibody. (B) MEF+ vector, MEF+ MyD88, and MEF+ MyD88s cells were stimulated with IL-1β or TNFα for the indicated times. Reconstitution of MyD88−/−-deficient MEFs with MyD88 but not MyD88s restores NF-κB signaling as evidenced by IκB degradation, which was monitored by Western analysis with anti-IκB antibodies. In both MEF+ vector and MEF+ MyD88s cells the IκB degradation machinery is intact as evidenced by treatment with TNFα.
MyD88 Is Required for IRAK-4–induced IRAK-1 Phosphorylation.
For sometime it was speculated that IRAK-1 was phosphorylated via its own kinase activity. However, this idea was challenged by the discovery that a kinase-dead mutant of IRAK-1 (subsequently referred to as IRAK-1KD) was phosphorylated in an IRAK-1–deficient cell line (17). A second kinase was postulated to phosphorylate IRAK-1 and perhaps to activate IRAK-1's own kinase activity. Recently, IRAK-4, so called for its homology to other members of the IRAK-1 family (other members include the kinase-inactive IRAK-2 and IRAK-M/3), was identified as a candidate for the IRAK-1 kinase (18–20). This was based on in vitro kinase assays and the observation that IL-1–induced degradation of IRAK-1 was partially blocked by overexpression of a kinase-inactive mutant of IRAK-4 (20). IRAK-4–deficient mice confirm that IRAK-4 is critical for signaling of IL-1 and TLR ligands (21) and in contrast to other IRAKs, IRAK-4 appears to require its kinase activity for signal transmission (20).
To obtain additional evidence that IRAK-4 is a kinase for IRAK-1, we developed a simple coexpression assay in HEK 293T cells. IRAK-4 was cotransfected with IRAK-1KD (IRAK-1D340N; used because it cannot self-phosphorylate like overexpressed wild-type IRAK-1) and phosphorylation monitored by the appearance of a slower migrating species in SDS-PAGE. As predicted, IRAK-4 induced phosphorylation of IRAK-1KD (Fig. 2 A). That phosphorylation was specifically induced by IRAK-4 was confirmed by the observation that coexpression of IRAK-1KD with two different IRAK-4 kinase-dead mutants, IRAK-4KD (IRAK-4KK213AA or IRAK-4D311N), did not similarly induce IRAK-1KD phosphorylation (Fig. 2 A and unpublished data). Although coexpression of IRAK-4 clearly induced IRAK-1 phosphorylation, only a partial conversion to the phosphorylated species was observed. The addition of MyD88, however, significantly enhanced IRAK-4–induced IRAK-1KD phosphorylation, suggesting that MyD88 stimulates IRAK-4's activity (Fig. 2, A and B). This was confirmed by an in vitro kinase assay (Fig. 2 B) performed on immunoprecipitated IRAK-1KD showing significant phosphorylation of IRAK-1KD when immunoprecipitated from cell extracts coexpressing MyD88 and IRAK-4, but not IRAK-4KD (compare lane 1 with lanes 3 and 5).
Figure 2.
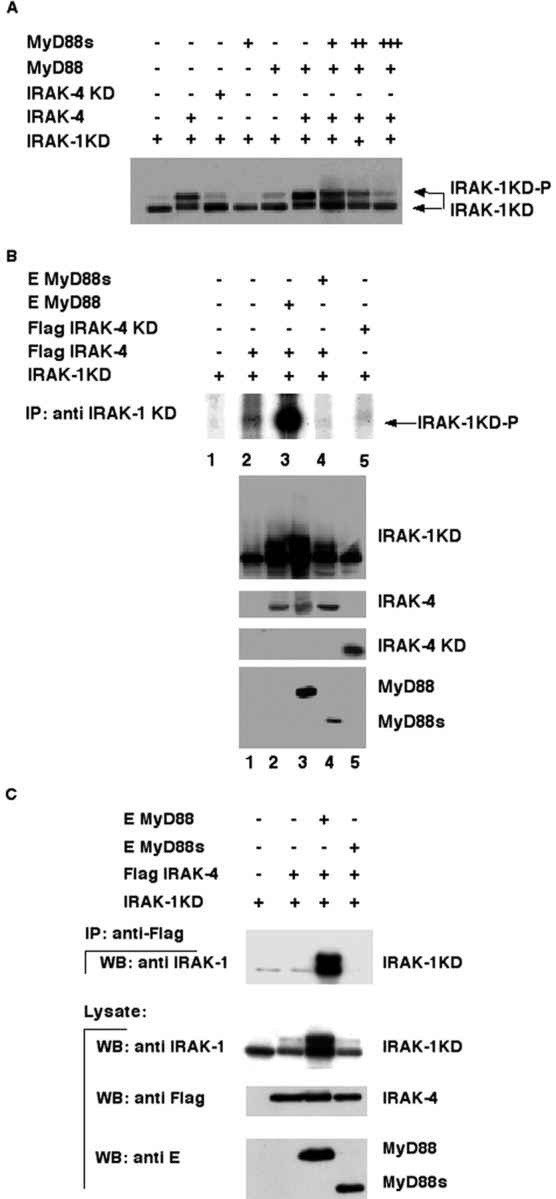
IRAK-4–induced IRAK-1 phosphorylation is regulated by MyD88/MyD88s. (A) IRAK-1KD (IRAK-1D340N) was cotransfected in 293T cells with the indicated expression vectors. Total cell lysates were prepared, separated by 10% SDS-PAGE, and analyzed by Western blotting with anti–IRAK-1 antibodies. Expression of IRAK-4, MyD88, and/or MyD88s was confirmed by Western analysis with the appropriate antibodies. (B) IRAK-1KD was immunoprecipitated from cell extracts coexpressing the indicated proteins and then subjected to an in vitro kinase assay. Phosphorylation of IRAK-1KD was then detected after SDS-PAGE and autoradiography (top). The expression levels of the indicated proteins (bottom panels), transfected as in the top panel lanes 1–5, were confirmed by Western blotting with the appropriate antibodies. (C) IRAK-4 was coimmunoprecipitated using an anti-Flag antibody from lysates of 293T cells cotransfected with the indicated combinations of expression plasmids and immunoblotted with an anti–IRAK-1 antibody (top). On the middle and bottom, Western blotting of cell lysates using anti–IRAK-1, anti-Flag, and anti-E antibodies to detect IRAK-1KD, IRAK-4, and MyD88/MyD88s, respectively, is shown. IP, immunoprecipitate; WB, Western blot.
As MyD88 binds to IRAK-1KD (MyD88 does not bind the hyperphosphorylated form of IRAK-1 induced by its overexpression; references 19 and 22) and was recently reported to bind IRAK-4 (20), the simplest explanation for the observed finding was that MyD88 modulates contact of IRAK-1 and IRAK–4. To test this, IRAK-1KD and IRAK-4 interactions were analyzed in the presence or absence of MyD88 and/or MyD88s (Fig. 2 C). As previously reported, IRAK-4 and IRAK-1KD do not directly associate (20). However, the addition of MyD88 but not MyD88s (discussed below) permitted assembly of a complex containing both IRAKs. MyD88 thereby appears to act like a hinge inducing the proximity of IRAK-1 and IRAK-4. Interestingly, phosphorylated IRAK-1 is stably detected together with MyD88, IRAK-1, and IRAK-4, suggesting that MyD88–IRAK-1 interactions are destabilized only after multiple sites are phosphorylated on IRAK-1 (Fig. 2 C).
MyD88s Blocks IRAK-4–induced IRAK-1 Phosphorylation.
Unlike MyD88, MyD88s does not stimulate IRAK-4–induced IRAK-1KD phosphorylation (Fig. 2, A and B). In fact, in vitro phosphorylation of IRAK-1KD induced by IRAK-4 coexpression was completely inhibited when MyD88s was coexpressed (Fig. 2 B, compare lanes 2 and 4). In addition, MyD88s inhibited MyD88's stimulatory effect on IRAK-4–induced IRAK-1KD phosphorylation in a dose-dependent manner (Fig. 2 A).
MyD88s Does not Bind to IRAK-4 and Blocks Recruitment of IRAK-4 to the IL-1Rs.
To characterize the underlying mechanism by which MyD88s blocks IRAK-1 phosphorylation, we initially analyzed if MyD88 and IRAK-4 associate. We did not expect the contrary, considering that MyD88 binds IRAK-1 primarily through DD–DD interactions (19, 22, and unpublished data). Surprisingly, MyD88s–IRAK-4 complexes were not detected (Fig. 3 A, left), despite the strong association of MyD88s and IRAK-1KD under similar conditions of coimmunoprecipitation (Fig. 3 A, right). Therefore, this suggested that the ID of MyD88 is required for this association with IRAK-4. To confirm this, the precise region of MyD88 mediating its interaction with IRAK-4 was mapped by yeast 2 hybrid and coimmunoprecipitation binding assays (Fig. 3 B and unpublished data). These assays confirm that MyD88 does, and MyD88s does not, interact with IRAK4. However, the ID in itself is insufficient, suggesting that MyD88 interacts with IRAK-4 via a peptide spanning both the ID and adjacent amino acids in the DD (we cannot exclude that the first 17 amino acids of the TIR are important for binding) or that the ID induces a conformation of MyD88, exposing residues in the DD that are critical for interactions between the two proteins.
Taken together, the results described above suggested that MyD88s acts as a negative regulator by its incapacity to bind to IRAK-4 and thus prevents IRAK-4–induced IRAK-1 phosphorylation. IRAK-4 is presumably recruited to the activated IL-1R complex raising the question of whether MyD88s also blocks IRAK-4 recruitment to the activated receptors. To address this question, coimmunoprecipitation of the IL-1Rs with associating proteins (MyD88, IRAK-1KD, and IRAK-4-KD) was performed in the presence or absence of MyD88s and/or Tollip, which we have previously shown to be important for IRAK-1 recruitment to the IL-1Rs (12). More IRAK-4 was found to associate with the IL-1Rs in the presence of Tollip, suggesting that Tollip may also be involved in IRAK-4 recruitment (Fig. 3 C). However, despite Tollip, the presence of MyD88s dramatically inhibited IRAK-4 but not IRAK-1 association with the IL-1Rs (Fig. 3 C).
In summary, we have demonstrated why the ID of MyD88 is critical for the activation of NF-κB. Via this domain, MyD88 plays an active role in the phosphorylation and activation of IRAK-1. Our results are compatible with the following sequence of events (Fig. 4) . After their recruitment to the IL-1R–TLR complexes, MyD88, most probably as a dimer (13), binds IRAK-1 and IRAK-4, inducing the close proximity of the IRAK-1 and IRAK-4 kinase domains. This in turn allows IRAK-4 to phosphorylate critical residue(s) in the kinase activation loop of IRAK-1, triggering IRAK-1's own kinase activity. Threonine-387 and Serine-376 are potential phosphorylation sites in the activation loop of IRAK-1 based on the reduced ability of IRAK-4 to phosphorylate short peptides with mutations at these residues (20). Once activated, IRAK-1 likely autophosphorylates residues in its NH2 terminus resulting in the hyperphosphorylated form detected after stimulation (5, 9). It is this multiphosphorylated form of IRAK-1 that is likely targeted for degradation (10). Under chronic conditions of inflammation or after prolonged exposure to LPS MyD88s is expressed (11). As a result, the above sequence of events is halted. IRAK-4 is not recruited to the IL-1Rs/TLRs and therefore IRAK-1 is not phosphorylated/activated and as a consequence signal transmission is interrupted. In conclusion, MyD88s acts as a negative regulator of IL-1β/LPS-induced NF-κB activation by preventing IRAK-4's access to its substrate.
Figure 4.

Model for MyD88/MyD88s regulation of IRAK-4–induced IRAK-1 phosphorylation. On the left, MyD88 links IRAK-1 with IRAK-4–activating NF-κB. IL-1 stimulation of the IL-1Rs induces assembly of a signaling complex containing MyD88 and IRAK-1 and IRAK-4. Dimers of MyD88 bind IRAK-1 and IRAK-4 bringing the respective kinase domains in close association (step 1). This results in the phosphorylation of IRAK-1 by IRAK-4, which in turn likely induces the kinase activity of IRAK-1 leading to its autophosphorylation (step 2). IRAK-4 is depicted activated at the IL-1Rs but this has not been formally demonstrated. Phosphorylated IRAK-1 first binds TNFR-associated factor 6 (not depicted) activating NF-κB and then is ubiquitinated (multiple arrows attached to IRAK-1) and degraded. For simplicity, other adaptor molecules such as Tollip have been omitted (12). On the right, inhibition of IL-1 signaling by MyD88s is shown. MyD88s (depicted as a shorter molecule missing the ID) does not interact with IRAK-4. IRAK-4 is not recruited to the IL-1Rs, thereby preventing the association of IRAK-1 and IRAK-4 and thus the phosphorylation of IRAK-1. As a result, there is no NF-κB activation.
Septic shock is characterized by tissue and organ damage resulting from hyperproduction of cytokines. Septic shock survivors have an increased incidence of bacterial infections and suppressed monocyte responses to LPS. This “endotoxin tolerance” is a transient state of LPS refractoriness after the initial, nonlethal exposure to LPS. An understanding of the mechanisms that elicit endotoxin tolerance is critical for unraveling the molecular basis of the septic shock syndrome, yet despite numerous studies these mechanisms remain largely unknown. Recently, it has been suggested that IRAK-3 (IRAK-M) is a key component of this important control system (23). IRAK-3 lacks kinase activity, is only expressed upon prior endotoxin treatment, and blocks TLR-induced signaling. IRAK-3–deficient cells, however, still retain some capacity to develop LPS tolerance, indicating the presence of additional control mechanisms. Aside from the proposed down-regulation of TLR4 (24), MyD88s also appears to be a likely candidate for tolerance induction as the expression of MyD88s is inducible by LPS and TNF (11). Thus, MyD88s and IRAK-3 might both contribute to endotoxin tolerance.
Acknowledgments
We would like to give special thanks to Dr. S. Akira for MyD88-deficient mice. Also thanks to Filippo Volpe and Barbara Maschera for IRAK-1/KD and Fabio Martinon for providing the EST encoding IRAK-4.
This work was supported by a grant of the Swiss National Science Foundation.
K. Burns and S. Janssens contributed equally to this work.
R. Beyaert and J. Tschopp contributed equally to this work.
References
- 1.Croston, G.E., Z. Cao, and D.V. Goeddel. 1995. NF-kappa B activation by interleukin-1 (IL-1) requires an IL-1 receptor-associated protein kinase activity. J. Biol. Chem. 270:16514–16517. [DOI] [PubMed] [Google Scholar]
- 2.Cao, Z., W.J. Henzel, and X. Gao. 1996. IRAK: a kinase associated with the interleukin-1 receptor. Science. 271:1128–1131. [DOI] [PubMed] [Google Scholar]
- 3.Kanakaraj, P., P.H. Schafer, D.E. Cavender, Y. Wu, K. Ngo, P.F. Grealish, S.A. Wadsworth, P.A. Peterson, J.J. Siekierka, C.A. Harris, et al. 1998. Interleukin (IL)-1 receptor–associated kinase (IRAK) requirement for optimal induction of multiple IL-1 signaling pathways and IL-6 production. J. Exp. Med. 187:2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swantek, J.L., M.F. Tsen, M.H. Cobb, and J.A. Thomas. 2000. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164:4301–4306. [DOI] [PubMed] [Google Scholar]
- 5.Li, X., M. Commane, Z. Jiang, and G.R. Stark. 2001. IL-1-induced NFkappa B and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK). Proc. Natl. Acad. Sci. USA. 98:4461–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian, Y., M. Commane, J. Ninomiya-Tsuji, K. Matsumoto, and X. Li. 2001. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFkappa B. J. Biol. Chem. 276:41661–41667. [DOI] [PubMed] [Google Scholar]
- 7.Takaesu, G., J. Ninomiya-Tsuji, S. Kishida, X. Li, G.R. Stark, and K. Matsumoto. 2001. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol. Cell. Biol. 21:2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao, Z., J. Xiong, M. Takeuchi, T. Kurama, and D.V. Goeddel. 1996. TRAF6 is a signal transducer for interleukin-1. Nature. 383:443–446. [DOI] [PubMed] [Google Scholar]
- 9.Hu, J., R. Jacinto, C. McCall, and L. Li. 2002. Regulation of IL-1 receptor-associated kinases by lipopolysaccharide. J. Immunol. 168:3910–3914. [DOI] [PubMed] [Google Scholar]
- 10.Yamin, T.T., and D.K. Miller. 1997. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J. Biol. Chem. 272:21540–21547. [DOI] [PubMed] [Google Scholar]
- 11.Janssens, S., K. Burns, J. Tschopp, and R. Beyaert. 2002. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr. Biol. 12:467–471. [DOI] [PubMed] [Google Scholar]
- 12.Burns, K., J. Clatworthy, L. Martin, F. Martinon, C. Plumpton, B. Maschera, A. Lewis, K. Ray, J. Tschopp, and F. Volpe. 2000. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol. 2:346–351. [DOI] [PubMed] [Google Scholar]
- 13.Burns, K., F. Martinon, C. Esslinger, H. Pahl, P. Schneider, J.L. Bodmer, F. Di Marco, L. French, and J. Tschopp. 1998. MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 273:12203–12209. [DOI] [PubMed] [Google Scholar]
- 14.De Valck, D., K. Heyninck, W. Van Criekinge, R. Contreras, R. Beyaert, and W. Fiers. 1996. A20, an inhibitor of cell death, self-associates by its zinc finger domain. FEBS Lett. 384:61–64. [DOI] [PubMed] [Google Scholar]
- 15.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 9:143–150. [DOI] [PubMed] [Google Scholar]
- 16.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 17.Li, X., M. Commane, C. Burns, K. Vithalani, Z. Cao, and G.R. Stark. 1999. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol. Cell. Biol. 19:4643–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wesche, H., X. Gao, X. Li, C.J. Kirschning, G.R. Stark, and Z. Cao. 1999. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 274:19403–19410. [DOI] [PubMed] [Google Scholar]
- 19.Muzio, M., J. Ni, P. Feng, and V.M. Dixit. 1997. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 278:1612–1615. [DOI] [PubMed] [Google Scholar]
- 20.Li, S., A. Strelow, E.J. Fontana, and H. Wesche. 2002. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA. 99:5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki, N., S. Suzuki, G.S. Duncan, D.G. Millar, T. Wada, C. Mirtsos, H. Takada, A. Wakeham, A. Itie, S. Li, et al. 2002. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 416:750–756. [DOI] [PubMed] [Google Scholar]
- 22.Wesche, H., W.J. Henzel, W. Shillinglaw, S. Li, and Z. Cao. 1997. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 7:837–847. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi, K., L.D. Hernandez, J.E. Galan, C.A. Janeway, Jr., R. Medzhitov, and R.A. Flavell. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 110:191–202. [DOI] [PubMed] [Google Scholar]
- 24.Sato, S., F. Nomura, T. Kawai, O. Takeuchi, P.F. Muhlradt, K. Takeda, and S. Akira. 2000. Synergy and cross-tolerance between toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J. Immunol. 165:7096–7101. [DOI] [PubMed] [Google Scholar]