

Abstract
The recombination signals (RS) that guide V(D)J recombination are phylogenetically conserved but retain a surprising degree of sequence variability, especially in the nonamer and spacer. To characterize RS variability, we computed the position-wise information, a measure correlated with sequence conservation, for each nucleotide position in an RS alignment and demonstrate that most position-wise information is present in the RS heptamers and nonamers. We have previously demonstrated significant correlations between RS positions and here show that statistical models of the correlation structure that underlies RS variability efficiently identify physiologic and cryptic RS and accurately predict the recombination efficiencies of natural and synthetic RS. In scans of mouse and human genomes, these models identify a highly conserved family of repetitive DNA as an unexpected source of frequent, cryptic RS that rearrange both in extrachromosomal substrates and in their genomic context.
Keywords: recombination signal sequence, cryptic recombination signal, recombination efficiency, recombination signal models, illegitimate V(D)J recombination
Introduction
The rearrangements of V, D, and J gene segments are mediated by RAG1 and RAG2, products of the recombination activating genes, Rag-1 and Rag-2 (for a review, see reference 1). RAG1 and RAG2 function as a DNA recombinase (2, 3) that recognizes recombination signals (RS)* consisting of conserved nucleotide heptamers and nonamers separated by less conserved strings of 12 ± 1 or 23 ± 1 nucleotides (4, 5). Efficient physiologic V(D)J recombination occurs only between 12- and 23-bp spacer signals, defining the 12/23 rule (6).
DNA recombination is phylogenetically ancient and widespread. RAG1 has substantial homologies with Hin that mediates DNA inversions in Salmonella (7, 8), and the inversion signals recognized by Hin are similar to the consensus RS with a 23-bp spacer (23-RS; reference 9). The V(D)J recombinase has also been shown to have latent transposase activity (10–12). These findings support the idea that RAG1 and RAG2 originated from a transposable element that was captured and enslaved by the vertebrate immune system; physiologic RS associated with Ig and Tcr gene segments are thought to be relics of this process. Other functional signals have been located at genomic sites that are not adjacent to a V, D, or J gene segment. These cryptic RS (cRS) may be responsible for some chromosomal translocations (for a review, see reference 13) and receptor editing (for a review, see reference 14). Undoubtedly, some of these cRS arise by chance; others, however, may be traced to the evolutionary origins of V(D)J recombination.
Physiologic RS are highly variable and the import of this genetic diversity is not understood (for a review, see reference 15). Studies using extrachromosomal recombination assays have shown that, while mutating the CAC trinucleotide in the first three positions of the RS heptamer dramatically reduced recombination efficiency, at least some mutations were tolerated at every other RS position and, at many positions, mutations had almost no affect on recombination (16–18). Consistent with the observation that nonamer positions are more tolerant to changes than heptamer positions, known cRS often contain recognizable heptamers but lack identifiable nonamers (14). Because of this variability, cRS can only be identified empirically, by observing their participation in illegitimate rearrangements.
We demonstrated previously that strong pair-wise correlations exist between RS positions, especially among positions in 23-RS (19). To understand the significance of these correlations, we developed statistical models of the correlation structure underlying RS variability; these models indicate that higher order correlations, between three or more positions, are also present (19). While most positions in the RS are correlated with at least one other position, the correlations can be ranked by their relative strength. Strong correlations substantially overlap sites of DNA ethylation/methylation interference present in RS complexed with RAG1/RAG2 (19, 20), suggesting that the correlations may be relevant to recombinase/RS interaction.
Our models compute a recombination signal information content (RIC) score to rate the potential function of any RS-length sequence and also to serve as a search procedure for RS. Retrospective analyses indicated that RIC scores are strongly correlated with RS efficiency and could locate known physiologic- and cRS in their genomic contexts (19). Here, we show that our models of RS structure accurately predict the activity of physiologic RS and identify new, functional cRS in the mammalian genome. In addition, we demonstrate Ig- and Tcr-associated patterns of RS variability that could influence receptor rearrangement. For the first time, the identity and efficacy of RS can be predicted from DNA sequence by a precise and rigorous algorithm. The ability to predict RS function and efficiency from these models opens the possibility of directed mutational analyses of RS structure and suggests that recombinase/RS interaction depends upon the cooperative influence of widely dispersed nucleotides in the RS.
Materials and Methods
RS Sequence Set.
We analyzed 356 physiologic mouse RS from all Tcr and Ig loci (available at http://www.duke.edu/~lgcowell). A detailed description of the data can be found in reference 19. When 12- and 23-RS were analyzed as a pooled set, positions 1 through 13 of 12- and 23-RS were aligned, and positions 14 through 28 of 12-RS were aligned to positions 25 through 39 of 23-RS.
Genomic Sequence Set.
The following mouse DNA sequences were analyzed in this study: 212,133 bp of chromosome 8 sequence (NCBI accession no. AC084823), 199,101 bp of sequence from the Tcr β locus (accession no. AE000665), and 3,926 bp from the DH locus (accession no. AF018146).
Calculation of Position-wise Information.
Information (I) is calculated from the Shannon entropy (21). The Shannon entropy at the i th position in an alignment is given by
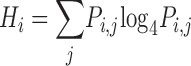 |
(1) |
where P i, j is the probability of nucleotide j at position i. The genomic entropy is
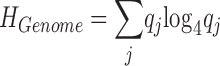 |
(2) |
where q j is the probability of nucleotide j in the genome. The position-wise information content (22) is computed I i = HGenome − H i; the unit is 0.5 bits. For a DNA sequence alignment, I is correlated with sequence conservation: maximum I is 1 and indicates strict conservation; minimum I is 0 and indicates that no nucleotide is more frequent than any other.
Statistical Models of RS Structure.
We developed statistical models of RS correlation structure for 12-RS and 23-RS (19). Briefly, each model computes a score for any sequence of appropriate length (i.e. 28-bp sequences for the 12-RS model and 39-bp sequences for the 23-RS model) by taking the natural logarithm of the probability of observing the sequence as estimated by the nucleotide composition of the RS sequence set. The smallest model assumes that all nucleotide positions in RS are independent and is based on the set of probability distributions for the four nucleotides at each RS position, i.e. the probability of observing nucleotide X at RS position i for all positions i. The models were enlarged by the step-wise incorporation of correlation between one RS position and at least one other RS position. Correlations are included in the models by forming joint probability distributions for the correlated positions, e.g. the probability of observing nucleotide X at position i and nucleotide Y at position k, or the probability of observing nucleotides X, Y, and Z at positions i, k, and l, respectively. Joint probability distributions are formed when they increase the average probability of observing the set of physiologic RS. The final RS models define the set of probability distributions that assign the highest average probability to physiologic 12- and 23-RS.
The score (log P) for a sequence is a value between -∞ and 0. If RS were strictly conserved, sequences identical to the RS would have log P = 0 and all other sequences would have log P = -∞. RS are not strictly conserved, however, but the models were selected such that RS have higher log P on average than non-RS. We define the log P of a sequence as its RIC. RIC is computed as follows: RIC 12 = ln[P 1 P 2 P 3,15,25 P 4,5 P 6,28 P 7,8,19 P 9,26 P 10,12 P 11,27 P 13,14,23 P 16,17,18 P 20,21,22 P 24] for 12-RS and RIC 23 = ln[P 1 P 2 P 3 P 4,14 P 5,39 P 6 P 7,24,25 P 8,9,21 P 10,16 P 11,12 P 13,22 P 15,23 P 17,18 P 19,27,30,31,32,33,37 P 20,26 P 28,29 P 34,38 P 35,36] for 23-RS. P 1 is the marginal probability distribution for the four nucleotides at position 1, and P 3,15,25 is the joint probability distribution for the 64 triplets at positions 3, 15, and 25. The presence of the joint probability function indicates that these three positions are correlated in the RS alignment. Correlation between positions may be observed because the positions act cooperatively to influence recombination or because RS share a common ancestry.
Only very low levels of extrachromosomal recombination are observed for RS not beginning with CAC (16, 17), so it is often assumed that CAC at positions 1–3 of the heptamer is required for recombination. The set of functional, physiologic RS reported by Ramsden et al. (18) includes an RS with heptamer CAGAGTG, however. Therefore, our models assign a probability of 0, and therefore RIC = -∞, to any sequence not beginning with CA.
Correlation Between RIC and Recombination Efficiency.
Spearman's rank correlation coefficient (r S) was used to detect correlation between RIC and measured recombination efficiencies.
Recombinationally Active Cell Lines.
The 103/BCL2 cell line was obtained by the transformation of mouse pre-B cells with a temperature-sensitive Abelson murine leukemia virus and the subsequent transfection with human Bcl-2 (23). 103/BCL2 proliferates at 34°C and expresses low levels of recombinase mRNA, protein, and activity (D. Ramsden, personal communication, and unpublished data). At 34°C, 103/BCL2 can support the rearrangement of efficient extrachromosomal recombination substrates (e.g., pJH290) but not rearrangement at the endogenous Igκ locus as detected by Southern blotting (23; and unpublished data). After as little as 12 h at 39°C, 103/BCL2 upregulates recombinase activity and rearranges the endogenous Igκ locus (23). 103/BCL2 cells were maintained at 34°C in RPMI 1640 supplemented with 10% FCS, 100 U/ml penicillin and streptomycin, 0.5 mg/ml Geneticin, and 0.55 μM 2-mercaptoethanol.
5B3 cells are M12 cells stably transfected with tetracycline-sensitive Rag1 and Rag2 (Tet-R1 and Tet-R2) vectors (24) modified to encode a RAG2-GFP fusion protein capable of supporting V(D)J recombination (25). Upon culture in the absence of tetracycline, 5B3 cells become GFP-positive and exhibit rearrangements of the endogenous Vλ and Jλ loci (25). 5B3 cells were cultured at 37°C in supplemented RPMI1640 (10% FCS, 100 U/ml penicillin and streptomycin, 3 mM histidinol, 10% glutamine, and 0.55 μM 2-mercaptoethanol) with or without tetracycline (0.5 μg/ml).
Extrachromosomal Recombination Assay.
We measured the efficiency of 18 physiologic and 9 synthetic RS by standard methods using extrachromosomal recombination templates. Briefly, recombination efficiencies of 12- and 23-RS were determined in pJH290 or a variant, p290T (see below), by the method of Hesse et al. (26). Both plasmids are coding joint substrates. In pJH290, a prokaryotic terminator of transcription is flanked by a 12- and a 23-RS; when pJH290 is transfected into recombination-competent 103/BCL2 (23), V(D)J recombination deletes a 300-bp fragment containing the RS and intervening sequence; free coding ends are recombined to form a coding joint in place of the deleted fragment. After alkaline lysis extraction of the plasmid, DH10B bacteria are transformed and the rearrangement status of plasmids in single bacterial colonies is assessed by PCR; 900-bp products represent intact pJH290, and 600-bp products indicate deletional rearrangements.
RS variants were introduced into plasmids by ligating representative 12- or 23-RS oligomers (Integrated DNA Technologies) into pJH290 digested with SalI or BamHI (NEB), respectively. SalI and BamHI restriction sites flank the 12-RS and 23-RS, respectively. Two additional modifications are present in p290T 23-RS variants, both created by the insertion of the 23-RS oligomer into the pJH290 backbone. All 23-RS oligomers carried a 4-bp deletion between the BamHI adhesive end and the nonamer, and both BamHI adhesive ends of 23-RS inserts were modified (to GGATCT). This modification produces a T substitution at the coding and signal flank of the 23-RS and renders the p290T plasmid resistant to BamHI digestion. These modifications, most likely the T substitution at the coding flank (27, 28), result in a 2-fold decrease in recombination efficiency compared to pJH290 (unpublished data). All pJH290 and p290T RS variants were confirmed by DNA sequencing.
10 μg of pJH290, p290T, or their RS variants were electroporated into 5 × 106 103/BCL2 cells. 103/BCL2 cells were washed with RPMI 1640 supplemented with 25 mM HEPES (Invitrogen) and resuspended to 1 × 107 cells/ml. 0.5 ml of this suspension was transferred into electroporation cuvettes (0.4 cm; Bio-Rad Laboratories) and incubated with 10 μg of recombination substrate for 5 min at room temperature and 10 min on ice. Samples were electroporated (250 V, 960 μF, 0 Ω), and transfectant cells were immediately chilled on ice (10 min), diluted to 5.5 ml with supplemented RPMI 1640, and incubated at 34°C overnight. Transfectant cultures were transferred to flasks containing 25 ml of supplemented RPMI for 24 h at 34°C and then incubated at 39°C for 48 h. Plasmid DNA was recovered by alkaline lysis extraction (29) and digested with DpnI (NEB) in a total volume of 100 μl. Digested plasmid DNA was purified by phenol:chloroform extraction (29) into a 10 μl volume of water. DH10B bacteria (Invitrogen) were transformed with 1 μl of digested, purified plasmid. Bacterial transformants were streaked on ampicillin (50 μg/ml) Luria broth (LB) agar plates.
Determination of Recombination Frequencies.
We measured recombination efficiency (R) by two similar methods. Analysis of low (R < 1%) efficiency cRS was based on bacterial colonies carrying plasmid substrates that impart constitutive ampicillin resistance (ampr) and conditional chloramphenicol (camr) resistance (26). R was estimated by the ratio of bacterial transformants exhibiting conditional (amprcamr) and constitutive (ampr) drug resistance (26); final values for R were averaged from ≥5 independent electroporations.
For high efficiency (R > 1%) physiologic and synthetic RS, we screened recombination templates in ampr bacterial colonies directly by PCR. This approach reduces the assay's sensitivity (frequencies < 0.3%) but precludes selection for spurious double-resistance. For the estimation of R by PCR, ampr colonies were randomly picked and expanded overnight at 37°C in 150 μl of LB containing 50 μg/ml ampicillin. The region of pJH290 and p290T flanked by the 12- and 23-RS was amplified by common PCR primers (290For, 5′-ATTAATGCAGCTGGCACG-3′, and 290Rev, 5′-CACTATCCCATATCACCA-3′) using Taq polymerase (Invitrogen). Amplifications were performed on 5 μl of template in a 50 μl reaction. Cycling parameters were: 94°C, 5 min; 28 cycles of 94° C, 1 min, 55° C, 1 min, and 72° C, 1 min. 10 min at 72°C ended the PCR program. PCR products were electrophoresed over 1% agarose gels to identify unmodified (900-bp product) and rearranged (600-bp product) plasmids. Final values for R (nos. rearranged plasmids ÷ [nos. rearranged plasmids + nos. unmodified plasmids]) were averaged from five independent electroporations. All 600-bp products from rearranged plasmids were sequenced and possessed typical coding joints (unpublished data).
Ligation-mediated PCR.
To assay for RAG-induced, double-strand DNA breaks (dsb) in 5B3 cells, various ligation-mediated PCR (LM-PCR) reactions were performed (30-33). Briefly, genomic DNA was isolated from 5B3 cells cultured for 48 h in the presence (Teton) or absence (Tetoff) of tetracycline; the recovered DNA was subsequently ligated to the BW-LC linker (BW-LC1, 5′-AGCAACTGACGTGGAATCGCCAGAC-3′; BW-LC2, 5′-GTCTGGCGATTCC-3′; references 30 and 31). Ligated and unligated controls were amplified using locus-specific primer sets and Thermalase polymerase (Invitrogen). The locus-specific primers were γ-satellite (BW-LCHlong and γ1long: 5′-ACTGACGTGGAATCGCCAGACCAC-3′ and 5′-TTCCGTGATTTTCAGTTTTCTCGCC-3′, respectively), Vλ (25, 32), and Dβ (33). The PCR program for the amplification of γ-satellite DNA was: 98°C, 2 min, 28 cycles of 98°C, 30 s, 66°C, 30 s, and 72°C, 30 s and termination by 72°C for 10 min.
PCR products were electrophoresed over 0.8% agarose gels and transferred to nylon membranes (PerkinElmer; reference 29). LM-PCR products containing γ-satellite DNA were detected by hybridization (30, 31) with a 32P-labeled probe (BW-LCγ: 5′-GGAATCGCCAGACCACTGTAGGACCTGGAA-3′) that overlaps the BW-LC linker and a portion of the 234-bp γ-satellite repeat (34); hybridization was quantitated in a Storm phosphoimager (Amersham Biosciences). Hybridizations specific for Vλ and Dβ PCR products were carried out as described (25, 32, 33).
LM-PCR products were gel purified (QIAGEN) and ligated into the pCR2.1TOPO vector (Invitrogen) following the manufacturer's directions. TOP10 bacteria (Invitrogen) were transformed with the pCR2.1TOPO plasmid carrying LM-PCR inserts and streaked onto LB-agar plates supplemented with ampicillin (50 μg/ml) for blue/white colony selection as directed by the manufacturer. Single white colonies were picked and expanded overnight at 37°C in 3 ml LB supplemented with ampicillin (50 μg/ml). Cloned plasmid inserts were then purified by alkaline lysis extraction (QIAGEN) and sequenced with the M13 reverse primer by the Duke University DNA Sequencing Facility.
Results
Patterned Genetic Variability in Mouse RS.
Alignment of 356 physiologic RS from all mouse Ig and Tcr loci reveals extensive sequence variability (19). To characterize this variability, we computed I
i for each nucleotide position in the RS alignment. I
i is proportional to sequence conservation; maximally informative positions are invariant whereas at minimally informative positions, nucleotides are present at frequencies equal to their genomic usage. The distribution of I
i along the RS alignment is shown in Fig. 1 . I
i averaged over the length of the RS is 0.34. The heptamer has a higher mean position-wise information than the nonamer (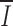 H = 0.78;
H = 0.78; 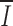 N = 0.53), and relatively little position-wise information (
N = 0.53), and relatively little position-wise information (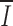 S = 0.15) is contained in the spacer (Fig. 1). Different alignments of 12- and 23-bp spacers did not increase I
S (unpublished data), and we did not find that 12-bp spacers are most similar to the first 12 nucleotides of 23-bp spacers (18). Separate alignments of 12- and 23-RS reveal greater conservation in 12-RS (
S = 0.15) is contained in the spacer (Fig. 1). Different alignments of 12- and 23-bp spacers did not increase I
S (unpublished data), and we did not find that 12-bp spacers are most similar to the first 12 nucleotides of 23-bp spacers (18). Separate alignments of 12- and 23-RS reveal greater conservation in 12-RS (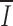 12 = 0.49;
12 = 0.49; 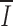 23 = 0.33), and separate alignments of Ig and Tcr RS reveal greater conservation in Ig RS (
23 = 0.33), and separate alignments of Ig and Tcr RS reveal greater conservation in Ig RS (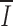 Ig = 0.46;
Ig = 0.46; 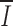 TCR = 0.30).
TCR = 0.30).
Figure 1.

Information (I i) at each position in an alignment of physiologic 12- and 23-RS. Position in the alignment is shown on the x axis, and I i at that position is given by the height of the bar. The most frequent nucleotide at highly conserved positions is indicated on the bar. The average amount of information contained in the heptamer, spacer, and nonamer regions is noted.
We computed I
i for each position in the alignment under two additional nucleotide classifications: the strength of hydrogen bonding, weak (T:A) vs. strong (G:C), and purine/pyrimidine. Higher position-wise information under one of these schemes would result if selection maintained nucleotide properties rather than particular nucleotides. 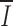 was 0.28 under the weak/strong classification and 0.31 under purine/pyrimidine classification, indicating conservation for specific nucleotides. The distribution of I
i along the RS under both classifications was indistinguishable from that shown in Fig. 1 (unpublished data).
was 0.28 under the weak/strong classification and 0.31 under purine/pyrimidine classification, indicating conservation for specific nucleotides. The distribution of I
i along the RS under both classifications was indistinguishable from that shown in Fig. 1 (unpublished data).
For any 28- or 39-bp sequence, the corresponding RS model computes a score, RIC
12 or RIC
23, respectively (19). For mouse 12-RS, the mean RIC
12 score (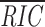 12) is –18.47; the highest RIC
12 is associated with Vκ4-86 (−8.02) and the lowest with Jβ1-2 (−48.16). RIC
12 scores are ranked in Fig. 2 A. The 100 highest RIC
12 scores are similar, but the remaining 101 decrease more rapidly. When
12) is –18.47; the highest RIC
12 is associated with Vκ4-86 (−8.02) and the lowest with Jβ1-2 (−48.16). RIC
12 scores are ranked in Fig. 2 A. The 100 highest RIC
12 scores are similar, but the remaining 101 decrease more rapidly. When 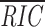 12 and the mean rank for each locus containing 12-RS are plotted (Fig. 2 A), there is substantial overlap between RIC
12 scores and between ranks across loci (Fig. 2, A and B). Nonetheless, RIC scores for Ig and Tcr RS are clearly separated. Ig 12-RS have higher RIC
12 on average than Tcr 12-RS (−13.72 and −27.98, respectively; Fig. 2 B); higher scores for Ig RS is consistent with their lower variability.
12 and the mean rank for each locus containing 12-RS are plotted (Fig. 2 A), there is substantial overlap between RIC
12 scores and between ranks across loci (Fig. 2, A and B). Nonetheless, RIC scores for Ig and Tcr RS are clearly separated. Ig 12-RS have higher RIC
12 on average than Tcr 12-RS (−13.72 and −27.98, respectively; Fig. 2 B); higher scores for Ig RS is consistent with their lower variability.
Figure 2.

(A) Rank ordered RIC 12 scores. The rank of each RIC 12 score is shown on the x axis, and the RIC 12 value is shown on the y axis. Each open circle corresponds to one of the physiologic 12-RS in our data set. Each filled circle corresponds to the mean RIC 12 and mean rank computed over the RS within one locus. The error bars show the range of one standard deviation. (B) Histogram of RIC 12 computed for physiologic Ig and Tcr 12-RS and for 28-bp segments taken from chromosome 8 DNA (AC084823). RIC 12 is shown on the x axis. The height of the bar indicates the frequency of physiologic RS or of 28-bp sequences in AC084823 receiving a finite score with RIC at that level.
RIC is based on the product of probabilities; the longer 23-RS therefore have lower RIC values than 12-RS. 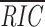 23 for the mouse RS studied is –32.39. The RS associated with Dδ1 receives the lowest RIC
23 (−69.68), and that of VH1S60 receives the highest (−15.83). The range of RIC
23 scores is broader than for 12-RS (54 vs. 40 RIC units), consistent with higher sequence variability of 23-RS. RIC
23 scores within loci are also more variable than observed for 12-RS, and Ig 23-RS tend to have higher RIC
23 values than Tcr 23-RS (unpublished data).
23 for the mouse RS studied is –32.39. The RS associated with Dδ1 receives the lowest RIC
23 (−69.68), and that of VH1S60 receives the highest (−15.83). The range of RIC
23 scores is broader than for 12-RS (54 vs. 40 RIC units), consistent with higher sequence variability of 23-RS. RIC
23 scores within loci are also more variable than observed for 12-RS, and Ig 23-RS tend to have higher RIC
23 values than Tcr 23-RS (unpublished data).
Resolution of RS from Surrounding DNA.
To resolve RS, we characterized the RIC 12 and RIC 23 distributions of non-RS DNA (19). From these background RIC distributions, we set threshold RIC scores, −40 for RIC 12 and −60 for RIC 23, that balance the numbers of physiologic RS with subthreshold scores and non-RS with scores above threshold (19). Putative RS are resolved from the genomic background by RIC ≥ threshold. Only five of the 356 (1.4 × 10−2) physiologic RS score below threshold, and the frequency of non-RS DNA sequences having RIC above threshold is 5 × 10−4 (19). This is not a false positive rate, however, as these high scoring sequences may function as RS.
To identify known, functional RS, we searched >450 kb of genomic DNA containing 39 physiologic RS to demonstrate whether RIC scores could resolve 12- and 23-RS (19). Fig. 3 shows RIC 12 values for a region of sequence AE000665 containing Jβ1-1 (−29.77) and Jβ1-2 (−38.81) and a region of sequence AF018146 containing DHFL16.1 (5′: −13.63, 3′: −15.53). Jβ1-2 is the lowest scoring physiologic 12-RS. Thus, RIC scores efficiently resolve physiologic 12-RS from the genomic background; results for 23-RS are similar (unpublished data). The only RS scoring below threshold are those associated with pseudogenes (19). RS flanking pseudogenes can not be selected, and we expect their RIC to be below threshold but above background.
Figure 3.

RIC 12 values for 28-bp segments from genomic regions containing DHFL16.1 or Jβ1-1 and Jβ1-2. RIC 12 are given on the y axis, and position in the sequence is shown on the x axis. RIC 12 values from the DHFL16.1 region are shown in the left panel, and those from the Jβ region are shown in the right panel. Each point represents the RIC 12 for the 28-bp segment beginning at that position.
Prediction of RS Efficiencies.
Previously, we computed Spearman's rank correlation between RIC and published recombination frequencies (17); RIC 12 and RIC 23 scores correlated well with extrachromosomal measurements of R (19). These published frequencies, however, were determined for a single 12- and 23-RS pair; most other RS tested differed from these RS by only 1-2 point mutations (17).
To determine whether RIC scores predict the functional efficiency of highly variable physiologic RS, we calculated RIC for 28 physiologic and synthetic RS and determined R for each in a standard extrachromosomal assay (Fig. 4 , and Table I). RIC 12 scores for 10 physiologic 12-RS correlated strongly with recombination (r S = 0.81) explaining 66% of the observed variation. RIC 23 scores for 18 physiologic and synthetic 23-RS also correlated well (r S = 0.76), explaining 58% of recombination variability.
Figure 4.

Correlation between recombination efficiency (x axis) and RIC (y axis). The recombination efficiency of the 23-RS (synthetic [gray triangle] and physiologic [black triangle]) is given by the top x axis and that for the 12-RS (open circle) is given by the bottom x axis. Spearman's rank correlation coefficients for the 12- and 23-RS are 0.81 and 0.76, respectively. Rank correlations computed for the physiologic and synthetic 23-RS separately are r S = 0.55 and r S = 0.93, respectively.
Table I.
Predicted and Observed Recombination Efficiencies for Physiologic and Synthetic RS
| Test vector | RIC | R (%) | Assoc. gene segment |
RS |
|---|---|---|---|---|
| p290T-2310 | −22.5 | 3.5 ± 1.2 | VH7S3*01 | GATCT CACAGTG AGGGTACTTCAGTGTGAGCCTAG ACACAAACC AGATCC |
| p290Tspa3 | −27.7 | 6.1 ± 1.6 | Synthetic | GATCT CACAGTG ACGGAGATAAAGGAGGAAGCAGG ACAAAAACC AGATCC |
| p290Ta | −28.8 | 7.6 ± 2.1 | Jk1*1 | GATCT CACAGTG GTAGTACTCCACTGTCTGGCTGT ACAAAAACC AGATCC |
| p290Thep3 | −28.7 | 3.7 ± 2.4 | Synthetic | GATCT CACAGTA GTAGTACTCCACTGTCTGGCTGT ACAAAAACC AGATCC |
| p290Thepγ | −30.2 | 5.5 ± 1.4 | Synthetic | GATCT CACACTG GTAGTACTCCACTGTCTGGCTGT ACAAAAACC AGATCC |
| p290T-2304 | −31.1 | 2.4 ± 1.1 | Vα7-4*01 | GATCT CACAGTG CTCTCCAGGCACCTGCGGGCTGC ACCCAAACC AGATCC |
| p290Tnon3 | −31.4 | 5.6 ± 2.8 | Synthetic | GATCT CACAGTG GTAGTACTCCACTGTCTGGCTGT ACAGAAACT AGATCC |
| p290T-2305 | −32.7 | <0.4 | VH7S4*02 | GATCT CACAGTG AGGGTACTTCAGTGTGAGCCTAG AAAAAAACC AGATCC |
| p290Thepnon3 | −32.8 | 2.9 ± 1.5 | Synthetic | GATCT CACAGTA GTAGTACTCCACTGTCTGGCTGT ACAGAAACT AGATCC |
| p290Tnonγ | −33.8 | 1.9 ± 1.3 | Synthetic | GATCT CACAGTG GTAGTACTCCACTGTCTGGCTGT ACTGAAAAT AGATCC |
| p290Thepnonγ | −35.7 | 1.0 ± 0.4 | Synthetic | GATCT CACACTG GTAGTACTCCACTGTCTGGCTGT ACTGAAAAT AGATCC |
| p290T-2301 | −35.8 | 0.6 ± 0.6 | Vβ13-2*1 | GATCT CACAGTG ATGTGGGGTTTCCTCCCCTCTGC ACAGAAAGG AGATCC |
| p290T3 | −36.8 | 2.5 ± 1.3 | Vλ3 | GATCT CACAGTA ACGGAGATAAAGGAGGAAGCAGG ACAGAAACT AGATCC |
| p290T-2306 | −40.0 | 2.4 ± 0.8 | Vδ1*01 | GATCT CACAGTG GTCTACAGTCAGCCACAGGCTGT CTCCAAACC AGATCC |
| p290T-2302 | −40.1 | <0.4 | JH1*1 | GATCT CACAGTC TCTGTTCTGCCTCTGTTCCTATA CTAAAACTC AGATCC |
| p290T-2303 | −40.6 | 1.2 ± 0.5 | Vα4-4/Vδ10*01 | GATCT CACAGTG CTCCCAGCAAGGCTGGAGCCTGG CCCCAAACT AGATCC |
| p290Tspaγ | −49.8 | 0.9 ± 0.6 | Synthetic | GATCT CACAGTG AAGGACCTGGAATAGGCAAGAAA ACAAAAACC AGATCC |
| p290TMDLCγ | −57.1 | <0.3 | Putative cRS | GATCT CACACTG AAGGACCTGGAATAGGCAAGAAA ACTGAAAAT AGATCC |
| pJH290 12-RSb | Synthetic | TCGAC CACAGTG CTACAGACTGGA ACAAAAACC CTGCAG | ||
| p290-1201 | −8.0 | 30.9 ± 8.9 | Vκ4-86 | TCGAC CACAGTG ATACAGACTGGA ACAAAAACC CTGCAG |
| p290-1202 | −12.4 | 36.7 ± 3.8 | Vκ4-79 | TCGAC CACAGTG AAACAGACTAGA ACAAAAACC CTGCAG |
| p290-1204 | −13.7 | 11.4 ± 4.1 | Vκ10-94 | TCGAC CACAATG ATATAAGTCATA ACATAAACC CTGCAG |
| p290-1203 | −14.2 | 18.0 ± 5.9 | Vκ8-27 | TCGAC CACAATG CTTCAGCCTCCT ACAAAAACC CTGCAG |
| p290-1205 | −18.7 | 3.3 ± 1.0 | Vκ17-121 | TCGAC CACAGTG CTATGTCCTCTT ACAGAAACC CTGCAG |
| p290-1206 | −24.7 | 15.9 ± 5.0 | Jα34 | TCGAC CACAGTG ATATCATGTTCT ACAAAAACC CTGCAG |
| p290-1207 | −28.4 | 16.3 ± 2.8 | Jα12 | TCGAC CACAGTG TTTCTTAGTCAG TCAAAAACA CTGCAG |
| p290-1208 | −32.3 | 6.6 ± 1.4 | Jα4 | TCGAC CACAGTA GAAAGGTGCTTT ACAAGAATT CTGCAG |
| p290-1209 | −37.8 | 1.1 ± 0.7 | Jβ2-2 | TCGAC CACAGTC GTCGAAATGCTG GCACAAACC CTGCAG |
| p290-1210 | −48.2 | <0.3 | Jb1-2 | TCGAC CACAGTC GTCGAAATGCTG GCACAAACC CTGCAG |
Recombination efficiency of physiologic and synthetic RS. The recombination efficiency, R, is measured by PCR as described in Materials and Methods. The 12- and 23-RS are listed with coding- and signal-flanks in italics, heptamer and nonamer in bold, and the spacer in plain text.
12-RS variants were tested for rearrangement to this 23-RS.
23-RS variants were tested for rearrangement to this 12-RS. This RS is synthetic; its R and RIC are therefore not included in Fig. 4.
To determine if RIC predicts function in RS not used for model development, we computed r S for physiologic and synthetic 23-RS separately. Recombination efficiencies of synthetic RS were predicted with very high accuracy, r S = 0.93, explaining 86% of observed variability. Thus, RIC scores are effective predictors of RS function, even when diverse or synthetic signals are analyzed.
Nucleotide differences in heptamers, nonamers, or spacers can profoundly and synergistically affect recombination (35). Nevertheless, RIC accurately predicts dissimilar recombination efficiencies in similar RS and similar efficiencies in RS that differ substantially. For example, 290Tspaγ and 290Tspa3 (Table I) share consensus heptamers and nonamers but differ in their spacers. RIC 23 scores correctly predict that 290Tspa3 (RIC 23 = −27.7; R = 6.1%) will rearrange with higher efficiency than 290Tspaγ (RIC 23 = −49.8; R = 0.9%). The physiologic 23-RS 2305 and 2310 (Table I) differ from each other by only two nucleotides in a non-consensus nonamer; the large difference in their RIC 23 scores (10 RIC 23 units) is consistent with their very different recombination efficiencies (<0.004 and 3.5%, respectively). Reciprocally, the 12-RS 1206 and 1207 (Table I) have consensus heptamers but differ at nine positions in their spacers and two in their nonamers; whereas the 1206 nonamer is consensus, the 1207 nonamer is not. Despite these differences, RIC 12 for these RS differ by <4 RIC 12 units, and their recombination efficiencies are similar (Table I).
Recognition of Known cRS.
The ability to predict R for RS that are similar or dissimilar implies that our statistical models capture some fundamental quality(ies) of RS structure. To test the limits of our models, we used RIC scores to identify cRS and to predict cRS activity. cRS were not used for model development and are likely under less stringent selection than physiologic RS.
Lewis et al. (36) reported 14 cRS (one 23-RS and 13 12-RS) that mediated illegitimate V(D)J recombination in plasmids transfected into RAG-expressing cells. To determine retrospectively whether these cRS could be resolved from the plasmid backbone, we computed RIC scores for each cRS. The 23-cRS scored −53.84, well above the physiologic threshold, indicating strong RS function. The average RIC 12 score for the 12-cRS was −50.3, below the physiologic threshold but well above the mean of −60.07 for non-RS DNA (19). These RIC scores could not be compared to the activity of the plasmid cRS because those data have not been reported (36). Nonetheless, the results show that the (fortuitous) cRS present in the pJH288 plasmid could be identified by RIC and that our models might be used to search prospectively for cRS.
To extend our analyses of cRS, we searched 234 mouse and 229 human VH gene segments (37) for cRS in 3′→5′ orientation (Table II); 12-cRS near the 3′ end of VH gene segments can mediate receptor editing (38, 39). Our search located 51 (out of 111,990 possible signals) potential cRS with RIC 12 > −40.0 (Table II). Virtually all (50/51) of these were from mouse VH gene segments, a bias that may reflect the mouse data set used for model-building. Half (26/51) of these 12-cRS lie within 12 bp of the VH segment's 3′ end where receptor editing is observed. The cRS with the highest RIC 12 (−29.28) is located 6 bp from the 3′ end of mouse VH2S5 (Fig. 5) .
Table II.
Cryptic RS Identified in VH Gene Segments by RIC12 Scores
| Cryptic RS, RIC
12 >−45
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. VH genes |
Total
|
<12 bp from 3′ end of gene segment |
Frequency of VH with cRS <12 bp from 3′ endc |
||||||
| Gene family (mouse nomenclature) |
Ma | Hb | M | H | M | H | M | H | |
| VH1 | (J558) | 144 | 32 | 101 | 27 | 41 | 26 | 0.3 | 0.8 |
| VH2 | (Q52) | 5 | 23 | 5 | 29 | 5 | 14 | 1.0 | 0.6 |
| VH3 | (36-60) | 8 | 88 | 3 | 41 | 3 | 25 | 0.4 | 0.3 |
| VH4 | (X-24) | 2 | 72 | 3 | 16 | 2 | 14 | 1.0 | 0.2 |
| VH5 | (7183) | 32 | 8 | 31 | 5 | 28 | 0 | 0.9 | 0.0 |
| VH6 | (J606) | 4 | 2 | 0 | 0 | – | – | – | – |
| VH7 | (S107) | 8 | 4 | 6 | 6 | 6 | 3 | 0.8 | 0.8 |
| VH8 | (3609) | 8 | – | 2 | – | 0 | – | 0.0 | – |
| VH9 | (VGAM3-8) | 9 | – | 5 | – | 0 | – | 0.0 | – |
| VH10 | (VH10) | 6 | – | 6 | – | 6 | – | 1.0 | – |
| VH11 | (CP3) | 1 | – | 1 | – | 0 | – | 0.0 | – |
| VH12 | (CH27) | 1 | – | 0 | – | – | – | – | – |
| VH13 | (3609N) | 1 | – | 1 | – | 0 | – | 0.0 | – |
| VH14 | (SM7) | 4 | – | 3 | – | 2 | – | 0.5 | – |
| VH15 | NA | 1 | – | 0 | – | – | – | – | – |
RIC 12 scores were computed for all 28-bp segments in mouse and human VH gene segments. The RIC 12 scores were used to identify cRS embedded in the 3′ end of the gene segments. Some sequences contain multiple cRS.
Mouse VH reference directory set (37) includes allelic variants.
Human VH reference directory set (37) includes allelic variants.
Frequency equals nos. cRS < 12 bp from 3′ end divided by no. VH genes.
Figure 5.

RIC 12 values for 28-bp segments from the mouse VH2S5 gene segment. Other details as in the legend to Fig. 3.
The number of cRS in VH gene segments with RIC 12 > −40 probably underestimates the prevalence of functional signals; we expect cRS to have lower scores than physiologic RS. For example, a 12-cRS in the 3H9 transgene has RIC 12 = −45.32 but is known to mediate VH replacement (39). RIC 12 scores > −45 identify 290 cRS in human (123) and mouse (167) VH segments and indicate that >50% of human and mouse VH gene families contain gene segments with potentially functional cRS (Table II). In VH families where cRS can be identified, fully 30–100% of gene segments (including allelic forms) carry potential cRS, a finding consistent with their conservation for H-chain editing (39, 40).
To determine if RIC 12 scores could predict function in VH cRS, five potential 12-cRS from mouse (VH2S2, VH2S5, VH5S1) or human (VH3-64, VH7-81) VH gene segments were tested in an extrachromosomal recombination assay (Table III). The 12-cRS tested had RIC 12 scores ranging from −29.3 to −40.2 and were located near the 3′ end of a VH gene segment. The Jβ2-2 12-RS (RIC 12 = −37.8) and the cRS present in VH 3H9 (RIC 12 = −45.3; reference 39) were also tested.
Table III.
Recombination Efficiencies of Various cRS
| Test vector | RIC | R (%) | Embedded in | cRS |
|---|---|---|---|---|
| p290-3H9 | −45.32 | <0.01 | 3H9 transgene | TCGAC CACAGAA GTAGACCGCAGA GTCCTCAGA CTGCAG |
| p290-m2S2 | −31.18 | 0.03 ± 0.03 | Mouse VH2S2*01 | TCGAC CACAGTA ATATATGGCTGT GTCATTAGA CTGCAG |
| p290-m2S5 | −29.26 | 0.4 ± 0.2 | Mouse VH2S5*01 | TCGAC CACAGTA ATATATGGCTGT GTCATTAGC CTGCAG |
| p290-m5S1 | −37.62 | 0.6 ± 0.4 | Mouse VH5S1*01 | TCGAC CACAGTA ATACAAGGCTGT GTCCTCAGA CTGCAG |
| p290-h364 | −40.23 | 0.5 ± 0.3 | Human VH3-64 | TCGAC CACAGTA ATACACAGCCAT GTCCTCAGC CTGCAG |
| p290-h781 | −40.23 | 0.4 ± 0.3 | Human VH7-81 | TCGAC CACAGTA ATACATGGCCAT GTCCTCAGC CTGCAG |
| p290-1209a | −37.8 | 0.7 ± 0.1 | Mouse Jβ2-2 | TCGAC CACAGTC GTCGAAATGCTG GCACAAACC CTGCAG |
| p290γMD | −56.7 | 0.02 ± 0.02 | γ-satellite | GATCC CACACTG AAGGACCTGGAATAGGCAAGAAA ACTGAAAAT CTCGG |
| p290γ01b | −53.2 | 0.6 ± 0.4 | γ-satellite | GATCC CACTGTA GGACATGGAATATGGCAAGACAA CTGAAAATC CTCGG |
| p290γ12 | −53.8 | <0.01 | γ-satellite | GATCC CACTCTA GCACATGGAATACGGCAGGAAAC TGAAAATCA CTCGG |
Recombination efficiency of cRS. Recombination efficiency, R, is measured as the ratio of ampr and camr double-resistant colonies to ampr colonies from a given transformation reaction (see Materials and Methods). When different volumes of the transformation reaction were streaked on plates, colony numbers were normalized per mL before the ratio was calculated.
This RS is the physiologic RS adjacent to Jβ2-2, not a cRS.
R of this cRS is measured by methods as in Table I.
All the putative cRS, except that present in 3H9 (i.e., RIC 12 ≥ −40.3), rearranged in pJH290 (Table III). Both human VH cRS and the mouse VH2S5 and VH5S1 cRS supported deletional rearrangements with efficiencies (0.4–0.6%) equivalent to the Jβ2-2 12-RS (0.7%). Rearrangements of the third mouse cRS (VH2S2) were observed at 10-fold lower frequencies (0.03%); all rearranged plasmids were sequenced and confirmed to contain bona fide coding joints (unpublished data). Thus, RIC scores identified functional VH cRS even though these cryptic sequences were not used to generate our RS models. The absence (<0.01%) of detectable rearrangements to the known cRS of 3H9 (RIC 12 = −45.3) suggests that the number of cRS capable of supporting VH replacement in vivo may well exceed our estimate of 290 functional signals (Table II).
Prospective Identification of Novel cRS.
The ability of RIC scores to identify functional cRS in VH gene segments indicated that our models might locate unknown cRS. We therefore searched >10.5 Mb of mouse and human cDNA and genomic DNA for potential 12- and 23-cRS. Using RIC scores that indicate physiologic thresholds of activity (RIC 12 ≥ −40 and RIC 23 ≥ −60; reference 19), we identified 4,746 12-cRS and 16,439 23-cRS, yielding a frequency of 5 × 10−4 cRS/bp. This value is lower than that estimated by Lewis et al., 1.7 × 10−3 (36), from illegitimate rearrangements in plasmids but indicates that some 0.5–1 × 106 cRS capable of efficient rearrangement are present in the mammalian genome.
Some of the potential cRS identified by this search are embedded in the 234-bp repeat of mouse γ-satellite DNA (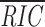 23 = –64.2 ± 5.1; reference 34) and in a highly similar repeat present in the human genome (41). Some of these potential cRS, e.g. γ01, γ12, and γMD (Table III), have RIC
23 scores (−59.9 to −53.2) indicative of efficient recombination; we cloned three of these cRS into pJH290 (p290γ01, p290γ12, p290γMD) to determine their recombination efficiencies (Table III).
23 = –64.2 ± 5.1; reference 34) and in a highly similar repeat present in the human genome (41). Some of these potential cRS, e.g. γ01, γ12, and γMD (Table III), have RIC
23 scores (−59.9 to −53.2) indicative of efficient recombination; we cloned three of these cRS into pJH290 (p290γ01, p290γ12, p290γMD) to determine their recombination efficiencies (Table III).
Two of the three γ-satellite cRS mediated detectable levels of V(D)J recombination (Table III). p290γ01 had a recombination efficiency (0.6%) equivalent to the mouse Jβ2-2 RS (Table III), and p290-γMD also rearranged, albeit 30-fold less efficiently (0.02%). We were unable to detect rearrangement (<0.01%) in p290-γ12. Sequencing confirmed that all rearrangements were to the heptamer-like motif of the γ-satellite cRS rather than spurious rearrangements to cryptic signals in the plasmid backbone (unpublished data).
To determine if endogenous γ-satellite DNA could rearrange in vivo, we performed a LM-PCR (31) specific for signal end cleavage in the γ-satellite repeat using the recombinase-inducible cell line, 5B3 (25). After 48 h of culture in the absence of tetracycline (Tetoff), ∼70% of 5B3 cells become GFP+, indicating the production of transgenic RAG1 and RAG2:GFP (Fig. 6 A). Ligase-dependent PCR products consistent with blunt-ended dsb at γ-satellite cRS heptamers are present at low levels even in 5B3 cells cultured in medium containing tetracycline (Teton), but analogous dsb in the endogenous Igλ and Tcrβ loci are undetectable (Fig. 6 B). Under Tetoff conditions, γ-satellite dsb increase 8- to 16-fold and dsb in the Igλ locus become abundant. In our hands, Tetoff 5B3 cells also exhibit low levels of dsb in the endogenous Dβ loci (Fig. 6 B). Thus, dsb consistent with cleavage at the γ-satellite heptamer-like element are induced in 5B3 cells under conditions that promote rearrangement intermediates in the endogenous Igλ and Tcrβ loci. Sequence analysis confirmed that LM-PCR products produced from Vλ signal end (SE) and Dβ5'SE-specific primers represented authentic recombination intermediates (unpublished data). We interpret the presence of γ-satellite dsb under Teton conditions as the result of imperfect silencing of the RAG transgenes and the extraordinary abundance of γ-satellite DNA in the mouse genome (34) but can not exclude the possibility that γ-satellite DNA is exceptionally fragile.
Figure 6.

LM-PCR detects in vivo RAG-mediated double strand breaks in the γ-satellite repeat. For detailed protocol, see Materials and Methods. 5B3 cells were incubated for 48 h with or without tetracycline and subsequently analyzed by flow cytometry (A) for expression of the RAG2-GFP fusion protein. (B) Genomic DNA was isolated, ligated (+ Ligase) to the BW-LC linker, and LM-PCR of genomic DNA was performed on serial 2-fold diluted samples. The initial amount of template used for the CD14 PCR and γ-satellite LM-PCR was ∼5 ng. The CD14 PCR is included to show normalization of template. The Dβ and Vλ LM-PCR use an initial template of 40 ng. No ligase controls are indicated as (−Ligase), while Teton or Tetoff refers to the presence or absence of tetracycline in the media, respectively. γ-sat, Dβ, and Vλ LM-PCR products are detected by radioactive-oligonucleotide hybridization.
If γ-satellite DNA were exceptionally prone to mechanical shearing or to cleavage by mechanisms unrelated to V(D)J recombination, we should observe frequent dsb at sites other than the γ-satellite heptamer. We therefore cloned and sequenced equal numbers (n = 17) of LM-PCR products recovered from 5B3 cells grown under Teton or Tetoff conditions and compared them to the γ-satellite consensus generated in Vector NTI (Informax) from the 31 published γ-satellite repeat elements (34; Fig. 7 A). Comparison to the consensus sequence permitted our detection of artifactual CAC-bearing heptamers introduced during PCR amplification (42). Our sequence analysis demonstrated that virtually all (32/34) of the sequenced LM-PCR products represented the BW-LC linker fused to a γ-satellite cRS heptamer-like element (Fig. 7 A). Half (9/17) of the γ-satellite sequences recovered from 5B3 cells under Teton conditions are repeats; five repeats were recovered under Tetoff conditions (Fig. 7 A). Identical γ-satellite motifs were amplified under Teton and Tetoff conditions, even though LM-PCR product was increased ∼10-fold in Tetoff cells (Fig. 6). Recovery of identical LM-PCR products under Teton and Tetoff conditions is consistent with low levels of constitutive recombinase activity in 5B3. Alignment of Teton and Tetoff γ-satellite LM-PCR products illustrates the lower diversity of the Teton group (Fig. 7 A) and suggests that these sequences might represent the more efficient group, even though average RIC 23 scores for the Teton and Tetoff γ-satellite signals do not significantly (P > 0.5) differ (−74.4 versus −72.9, respectively).
Figure 7.



(A) Table of γ-satellite cRS within cloned LM-PCR products. BW indicates the BW-linker sequence. * identifies groups of identical cRS. Sequence logos (reference 72) depict the information content for each position in an alignment of the Teton and Tetoff groups of cloned cRS. The sequence logos were generated at http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi. (B) cRS in γ-satellite DNA. The consensus γ-satellite sequence was generated in Vector NTI from the 31 published γ-satellite repeat elements (reference 34). Nucleotide position in the consensus is shown on the x axis. The squares indicate the location of CA dinucleotides in the consensus and the RIC score (y axis, right) for the 39-bp sequence beginning with that CA. The location of CAC trinucleotides is indicated by the horizontal black lines. The height of the bars (gray, Tetoff; black, Teton) indicates the number of LM-PCR products (y axis, left) that align to that location in the γ-satellite consensus.
Comparisons of each γ-satellite LM-PCR product (Fig. 7 B) to the consensus γ-satellite repeat demonstrate favored sites for linker ligation; with a single exception all of these are compatible with recombinase-mediated DNA cleavage (Fig. 7 B). The 234-bp γ-satellite repeat contains nine CA dinucleotides that represent potential cRS and are evaluated by the 23-RS model (Fig. 7 B); the canonical CAC trinucleotide of the RS heptamer is found at six of these (Fig. 7 B). Two Tetoff LM-PCR products indicated ligations to sites other than a CAC trinucleotide. The first, at position 96, indicated linker ligation to a consensus AAC and the second (position 98) to CAT (Fig. 7 B). These LM-PCR products are atypical for recombination intermediates and may represent dsb unassociated with recombinase activity. Of the six potential γ-satellite cRS beginning with CAC, three – at positions 44, 102, and 158 (Fig. 7 B) – account for most (81%, 26/32) dsb events. The 23-cRS at positions 44 and 158 of the consensus repeat exhibit near-physiologic RIC 23 scores and were predicted by our model. The model predicts only a low recombination frequency for the major cRS at position 102 of the γ-satellite consensus (Fig. 7 B). We also scored the 31 published γ-satellite repeat elements (34) used to generate the consensus. 26 of the 31 have a CA, and thus a potential cRS, at position 102. The average score for the 26 cRS is –75.86, and the model predicts a higher recombination efficiency for 15 of the 26 than for the consensus position 102 cRS. Thus, to our knowledge, this is the first prospective identification of a cRS based solely on primary sequence analysis.
Discussion
The information I present in individual positions of the RS alignment is predominately in the RS heptamer and nonamer (43; Fig. 1). Genetic variability elsewhere reduces the average I
i to 34% of the 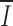 if all RS were identical. We find this level of
if all RS were identical. We find this level of 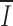 to be surprisingly low for a signal mediating the introduction of dsb in DNA; promiscuous recombination driven by poorly regulated DNA cleavage would cause significant damage to the cell. We have previously shown that RS positions are correlated (19); these correlations could increase the specificity of the signal contained in the RS and reduce promiscuous binding. We introduced a model of RS correlation structure that computes a score, RIC, for any RS-length sequence (19). RIC efficiently identifies physiologic RS and known cRS (Figs. 2, 3, and 5; Tables I and III) and is strongly predictive of recombination efficiency (Fig. 4). Together these results suggest that RIC captures biologically important RS characteristics. In fact, the strongest correlations in the models overlap regions of RS/recombinase contact (19, 20).
to be surprisingly low for a signal mediating the introduction of dsb in DNA; promiscuous recombination driven by poorly regulated DNA cleavage would cause significant damage to the cell. We have previously shown that RS positions are correlated (19); these correlations could increase the specificity of the signal contained in the RS and reduce promiscuous binding. We introduced a model of RS correlation structure that computes a score, RIC, for any RS-length sequence (19). RIC efficiently identifies physiologic RS and known cRS (Figs. 2, 3, and 5; Tables I and III) and is strongly predictive of recombination efficiency (Fig. 4). Together these results suggest that RIC captures biologically important RS characteristics. In fact, the strongest correlations in the models overlap regions of RS/recombinase contact (19, 20).
On average, RIC scores for Ig RS are higher than for Tcr RS, possibly due to the slight overrepresentation (56%) of Ig RS in our data set. We doubt, however, that this small surplus alone could be the cause. Genetic variability in Tcr RS is > in Ig RS. Why? It is unlikely that recombinase-RS interaction differs in B and T cells, but if it did, discrete patterns of mutual information (MI) in Tcr and Ig RS should exist. Instead, I and MI are patterned similarly for all RS groups in our data set, and we observe lower levels of sequence conservation in Tcr RS. It may be that genetic variability in Tcr RS is expanded to influence the TCR repertoire (44). While Ig RS could also bias the Ig repertoire (15, 45, 46), the MHC-restriction of TCR may have favored biased associations of Tcr V, D, and J gene segments (47-49). Increased variability in Tcr RS could serve to increase favored Tcr rearrangements by preferentially guiding rearrangement partners.
We also find greater variability among 23-RS than among 12-RS. The recombinase may interact differently with 12-RS than with 23-RS, due to their different lengths and/or to enforce the 12/23 rule, resulting in more stringent sequence constraints for 12-RS. For example, Swanson and Desiderio (20) observed ethylation/methylation interference at 11/12 spacer positions in 12-RS but at only 3 positions in 23-bp spacers. RS spacers may be bent when bound to the recombinase (50); this bending, or other structural constraints such as rotational phasing, may constrain the shorter spacers more severely. HMG1 and HMG2 have a more pronounced effect on the binding and bending of 23-RS than of 12-RS (51), and the RS positions contacted by HMG1 differ between the two types of RS (52).
It is also possible that the increased variability among 23-RS results from a unique role in the regulation of ordered assembly and/or allelic exclusion at the H, β, and δ loci. There is strong evidence that 12-RS regulate the precise targeting of Dβ gene segments to Jβ gene segments and Vβ gene segments to Dβ gene segments (47, 48). These results do not rule out a role for 23-RS, however, and they demonstrate that RS can play a significant role in regulating ordered assembly at the β locus. The high level of variability among 23-RS can be explained by hypothesizing that there are two groups of 23-RS, those that participate in the first stage of rearrangement (JH, Dβ, and Vδ) and those that participate in the second stage of rearrangement (VH, Vβ, and Dδ). The specificity of the signal in the two sets of RS may differ, or the V-to-DJ type rearrangers may simply be less efficient. Indeed, Liang et al. (53) have recently demonstrated that recombinase activity mediated by core or full length RAG2 distinguishes between D→J and V→DJ 23-RS groups. This finding supports the notion that patterned variability among RS could provide a mechanism for regulating receptor assembly and allelic exclusion at the β, H, and δ loci (53).
RIC scores for physiologic RS lie well outside background distributions (Fig. 2 B), allowing us to define thresholds that discriminate between RS and non-RS. When correlations between positions in RS are ignored, as in consensus models, scores for non-RS increase and resolution of RS becomes problematic (19). Not only do higher RIC values identify physiologic RS located in the Tcr and Ig loci (19; Fig. 3), but RIC scores are also highly correlated with recombination efficiency (19; Fig. 4). Determinations of 12- and 23-RS efficiencies in a standard extrachromosomal recombination assay (26) revealed very high correlations between measured and predicted recombination efficiencies (r S = 0.81 and 0.76 for 12- and 23-RS, respectively) even for synthetic signals not present in nature (Fig. 4). Analogous models that ignore associations between nucleotide positions in RS never predict recombination better and sometimes much less well than RIC (19).
Of particular interest is the ability of RIC to predict dissimilar recombination efficiencies in similar RS. The physiologic RS p290T-2305 and p290T-2310 differ at only two nonamer positions (Table I). These signals are respectively associated with the VH7S4 and VH7S3 gene segments (54, 55); in the unselected B cell repertoire, VH7S3 is ∼8-fold more frequent than VH7S4 (54, 55). Even though p290T-2305 and p290T-2310 RS share 95% sequence identity, their RIC 23 scores are very different: −32.7 and −22.5, respectively (Table I). This difference correlates with their relative activities in extrachromosomal substrates (Table I and Fig. 4) and with their usage in vivo (54, 55).
Given the ability of our models to identify physiologic RS and accurately predict their efficiencies, we searched mouse and human VH gene segments to determine if the models could also identify embedded 12-cRS (14). RIC 12 scores located known and novel VH cRS and predicted that efficient 3′ cRS, located where receptor editing could result in a functional H chain, are common (Fig. 5 and Table II). Our genomic scans indicate that >50% of gene segments comprising six mouse and three human VH gene families contain putative cRS in this location (Table II), a result consistent with other analyses based on heptamer-like motifs conserved in Ig loci (for a review, see reference 14).
In contrast to searches for cRS “heptamers”, our models predict recombination efficiencies based on the entire RS sequence, allowing for the identification of cRS in VH gene segments that are likely to function. All five of the potential VH-associated cRS that we selected for testing in an extrachromosomal recombination assay exhibited detectable activity; four at levels similar to physiologic RS (Table II). To our knowledge, this is the first evidence that VH cRS can support V(D)J recombination efficiently, at levels near that of some physiologic RS. Our findings are consistent with models of B cell development where VH replacement contributes significantly to the BCR repertoire (14).
Previously, a single VH-associated cRS was predicted by Feeney and colleagues (56) based on the presence of a heptamer-like motif (CACAGTA) and its 3′ location in a VH gene segment. No recombination events mediated by this cRS were detected (56), but the identical cRS was functional in our hands (p290-m5S1, Table II). We speculate that the detection method used by Nadel et al. (56) was less sensitive than our own to infrequent recombination events.
The prevalence of cRS at the 3′ end of VH gene segments has led to speculation that these signals are conserved for VH gene replacement (14). Studies of IgH knock-in mice (39, 57, 58) have clearly demonstrated the possibility of VH gene replacement in vivo; however, the strong selective forces acting on B cells in these animals may emphasize rare or antigen-independent replacements (39, 56-58). For example, it is not clear if the VH replacements observed in IgH knock-in mice occur at a stage of B cell development consistent with (self) antigen-driven selection (39, 56-58).
We also identified cRS within the 234 bp-repeat of γ-satellite DNA. γ-satellite DNA is a highly repetitive, tandemly arrayed element that comprises ∼6% of the mouse genome (34). A highly similar (∼95%) repeat is also present in human DNA (reference 41 and unpublished data) suggesting this repeat is phylogenetically conserved. The abundance and conservation of this complex DNA motif suggest that the γ-satellite repeat may represent a link between physiologic RS and the transposon ancestor of RAG1/2 (11, 12). cRS in γ-satellite DNA rearrange with variable efficiencies in extrachromosomal substrates (Table III), but at least one γ-satellite cRS, p290γ01, rearranges as efficiently as the Jβ2-2 RS (Table III). γ-satellite cRS can function in vivo; LM-PCR products consistent with γ-satellite rearrangement intermediates are substantially increased in 5B3 cells under the Tetoff culture conditions that up-regulate expression of RAG1 and RAG2:GFP and initiate V(D)J rearrangement in the endogenous Igλ and Tcrβ loci (Figs. 6 and 7). At least some γ-satellite DNA is accessible to enzymatic machinery, as demonstrated by abundant γ-satellite RNA transcripts and recurrent integration of active transgenes into γ-satellite DNA (59–61). Functional cRS are also present in CA dinucleotide repeats (36). In contrast to CA repeats, however, γ-satellite cRS are complex, closely resemble physiologic RS, are more abundant than CA repeats (34, 62), and rearrange more efficiently (Table III and reference 36). The abundance of γ-satellite cRS may make them a common substrate for illegitimate V(D)J rearrangement and a potential site for RAG-mediated genomic remodeling (63), or a frequent and safe site where RAG-induced dsb can harmlessly rearrange.
RIC 23 scores identified two of the three efficient cRS in the γ-satellite consensus. The model's prediction of only low recombination efficiency for the major cRS at position 102 of the consensus indicates that further additional work is necessary to model and understand RAG/RS interaction. Nonetheless, even in their current iteration, our statistical models are capable of identifying functional RS in the genome and offer the basis for rational analyses of RS structure by mutagenesis.
RS variability is sufficient to preclude exhaustive measurements of recombination efficiencies and effective searches for cRS. Except for the 12/23 rule (6) and the requirement for a CAC heptamer (16, 17), RS function can not be predicted (64, 65). Surprisingly, methods for the characterization of variable DNA sequence motifs have been available for 20 years (66–71), but until now, RS have only been described using consensus methods (18). RIC and these older probabalistic methods (66–71) will always outperform consensus methods for representing variable DNA motifs because they do not censor the information present in genetic diversity. Additionally, RIC incorporates correlation structures ignored by previous methods, increasing its ability to resolve and evaluate DNA motifs (19). RIC accurately identifies RS and predicts recombination efficiencies for physiologic, synthetic, and cRS (Figs. 4 and 6; Tables I and III). In addition, the statistical models that generate RIC can scan genomes for cRS. Our frequency estimates for fortuitous 12- and 23-RS (1–4 × 10−4) are 10-fold below earlier, empirical estimates (36). This higher level of discrimination is important when searching for cRS that may participate in illegitimate rearrangements, e.g. potential cRS in VH gene segments and at breakpoints of chromosomal translocations (13). Statistical models of RS structure are designed to aid empirical studies by focusing experiments on the most promising candidate structures; RIC's place in the study of V(D)J recombination is to identify and prospectively evaluate RS, ending roundups of the usual suspects.
Acknowledgments
We are grateful to Dr. D. Ramsden (University of North Carolina, Chapel Hill) for expert advice and the pJH290 substrate, and to Dr. E. Oltz (Vanderbilt University) who provided the 5B3 cell line. We are also grateful to Dr. N. Rosenberg (Tufts University) for the 103/BCL2 cell line. We thank Drs. D. Ramsden and M. Schlissel (University of California, Berkeley) for their comments on the manuscript.
L.G. Cowell received a Bioinformatics and Genome Technology postdoctoral fellowship from Duke University and support from National Institutes of Health training grant T32 AI52077. This work was supported in part by U.S. Public Health Service grants AI24335 and AI49326 (to G. Kelsoe).
L.G. Cowell and M. Davila contributed equally to this work.
Footnotes
Abbreviations used in this paper: ampr, ampicillin resistant; camr, chloramphenicol resistant; cRS, cryptic RS; dsb, double-strand breaks; I, position-wise information; MI, mutual information; LM-PCR, ligation-mediated PCR; RIC, RS information content; RS, recombination signal; r S, Spearman's rank correlation coefficient; 12-RS, 12-bp spacer RS; 23-RS, 23-bp spacer RS.
References
- 1.Fugmann, S.D., A.I. Lee, P.E. Shockett, I.J. Villey, and D.G. Schatz. 2000. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 18:495–527. [DOI] [PubMed] [Google Scholar]
- 2.Oettinger, M.A., D.G. Schatz, C. Gorka, and D. Baltimore. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 248:1517–1523. [DOI] [PubMed] [Google Scholar]
- 3.Schatz, D.G., M.A. Oettinger, and D. Baltimore. 1989. The V(D)J recombination activating gene, RAG-1. Cell. 59:1035–1048. [DOI] [PubMed] [Google Scholar]
- 4.Sakano, H., K. Huppi, G. Heinrich, and S. Tonegawa. 1979. Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature. 280:288–294. [DOI] [PubMed] [Google Scholar]
- 5.Akira, S., K. Okazaki, and H. Sakano. 1987. Two pairs of recombination signals are sufficient to cause immunoglobulin V-(D)-J joining. Science. 238:1134–1138. [DOI] [PubMed] [Google Scholar]
- 6.Tonegawa, S. 1983. Somatic generation of antibody diversity. Nature. 302:575–581. [DOI] [PubMed] [Google Scholar]
- 7.Difilippantonio, M.J., C.J. McMahan, Q.M. Eastman, E. Spanopoulou, and D.G. Schatz. 1996. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell. 87:253–262. [DOI] [PubMed] [Google Scholar]
- 8.Spanopoulou, E., F. Zaitseva, F.H. Wang, S. Santagata, D. Baltimore, and G. Panayotou. 1996. The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. 87:263–276. [DOI] [PubMed] [Google Scholar]
- 9.Simon, M., J. Zieg, M. Silverman, G. Mandel, and R. Doolittle. 1980. Phase variation: evolution of a controlling element. Science. 209:1370–1374. [DOI] [PubMed] [Google Scholar]
- 10.Lee, G.S., M.B. Neiditch, R.R. Sinden, and D.B. Roth. 2002. Targeted transposition by the V(D)J recombinase. Mol. Cell. Biol. 22:2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal, A., Q.M. Eastman, and D.G. Schatz. 1998. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 394:744–751. [DOI] [PubMed] [Google Scholar]
- 12.Hiom, K., M. Melek, and M. Gellert. 1998. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 94:463–470. [DOI] [PubMed] [Google Scholar]
- 13.Davila, M., S. Foster, G. Kelsoe, and K. Yang. 2001. A role for secondary V(D)J recombination in oncogenic chromosomal translocations? Adv. Cancer Res. 81:61–92. [DOI] [PubMed] [Google Scholar]
- 14.Fanning, L., F.E. Bertrand, C. Steinberg, and G.E. Wu. 1998. Molecular mechanisms involved in receptor editing at the Ig heavy chain locus. Int. Immunol. 10:241–246. [DOI] [PubMed] [Google Scholar]
- 15.Feeney, A.J., A. Tang, and K.M. Ogwaro. 2000. B-cell repertoire formation: role of the recombination signal sequence in non-random V segment utilization. Immunol. Rev. 175:59–69. [PubMed] [Google Scholar]
- 16.Akamatsu, Y., N. Tsurushita, F. Nagawa, M. Matsuoka, K. Okazaki, M. Imai, and H. Sakano. 1994. Essential residues in V(D)J recombination signals. J. Immunol. 153:4520–4529. [PubMed] [Google Scholar]
- 17.Hesse, J.E., M.R. Lieber, K. Mizuuchi, and M. Gellert. 1989. V(D)J recombination: a functional definition of the joining signals. Genes Dev. 3:1053–1061. [DOI] [PubMed] [Google Scholar]
- 18.Ramsden, D.A., K. Baetz, and G.E. Wu. 1994. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res. 22:1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowell, L.G., M. Davila, T.B. Kepler, and G. Kelsoe. 2002. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biology. 3:research0072.1–research0072.20. [DOI] [PMC free article] [PubMed]
- 20.Swanson, P.C., and S. Desiderio. 1998. V(D)J recombination signal recognition: distinct, overlapping DNA-protein contacts in complexes containing RAG1 with and without RAG2. Immunity. 9:115–125. [DOI] [PubMed] [Google Scholar]
- 21.Shannon, C.E., and W. Weaver. 1949. The Mathematical Theory of Communication. University of Illinois Press, Urbana. v (i.e. vii), 117 pp.
- 22.Schneider, T.D., G.D. Stormo, L. Gold, and A. Ehrenfeucht. 1986. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 188:415–431. [DOI] [PubMed] [Google Scholar]
- 23.Chen, Y.Y., L.C. Wang, M.S. Huang, and N. Rosenberg. 1994. An active v-abl protein tyrosine kinase blocks immunoglobulin light-chain gene rearrangement. Genes Dev. 8:688–697. [DOI] [PubMed] [Google Scholar]
- 24.Shockett, P., M. Difilippantonio, N. Hellman, and D.G. Schatz. 1995. A modified tetracycline-regulated system provides autoregulatory, inducible gene expression in cultured cells and transgenic mice. Proc. Natl. Acad. Sci. USA. 92:6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sikes, M.L., A. Meade, R. Tripathi, M.S. Krangel, and E.M. Oltz. 2002. Regulation of V(D)J recombination: A dominant role for promoter positioning in gene segment accessibility. Proc. Natl. Acad. Sci. USA. 99:12309–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hesse, J.E., M.R. Lieber, M. Gellert, and K. Mizuuchi. 1987. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell. 49:775–783. [DOI] [PubMed] [Google Scholar]
- 27.Gerstein, R.M., and M.R. Lieber. 1993. Coding end sequence can markedly affect the initiation of V(D)J recombination. Genes Dev. 7:1459–1469. [DOI] [PubMed] [Google Scholar]
- 28.Yu, K., and M.R. Lieber. 1999. Mechanistic basis for coding end sequence effects in the initiation of V(D)J recombination. Mol. Cell. Biol. 19:8094–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 545 pp.
- 30.Han, S., S.R. Dillon, B. Zheng, M. Shimoda, M.S. Schlissel, and G. Kelsoe. 1997. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science. 278:301–305. [DOI] [PubMed] [Google Scholar]
- 31.Schlissel, M.S., and D. Baltimore. 1989. Activation of immunoglobulin kappa gene rearrangement correlates with induction of germline kappa gene transcription. Cell. 58:1001–1007. [DOI] [PubMed] [Google Scholar]
- 32.Bendall, H.H., M.L. Sikes, and E.M. Oltz. 2001. Transcription factor NF-kappa B regulates Ig lambda light chain gene rearrangement. J. Immunol. 167:264–269. [DOI] [PubMed] [Google Scholar]
- 33.McMahan, C.J., and P.J. Fink. 1998. RAG reexpression and DNA recombination at T cell receptor loci in peripheral CD4+ T cells. Immunity. 9:637–647. [DOI] [PubMed] [Google Scholar]
- 34.Vissel, B., and K.H. Choo. 1989. Mouse major (gamma) satellite DNA is highly conserved and organized into extremely long tandem arrays: implications for recombination between nonhomologous chromosomes. Genomics. 5:407–414. [DOI] [PubMed] [Google Scholar]
- 35.Nadel, B., A. Tang, G. Escuro, G. Lugo, and A.J. Feeney. 1998. Sequence of the spacer in the recombination signal sequence affects V(D)J rearrangement frequency and correlates with nonrandom Vkappa usage in vivo. J. Exp. Med. 187:1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis, S.M., E. Agard, S. Suh, and L. Czyzyk. 1997. Cryptic signals and the fidelity of V(D)J joining. Mol. Cell. Biol. 17:3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefranc, M.-P. IMGT, the international ImMunoGeneTics database http://imgt.cines.fr (Initiator and coordinator: Marie-Paule Lefranc, Montpellier, France).
- 38.Kleinfield, R., R.R. Hardy, D. Tarlinton, J. Dangl, L.A. Herzenberg, and M. Weigert. 1986. Recombination between an expressed immunoglobulin heavy-chain gene and a germline variable gene segment in a Ly 1+ B-cell lymphoma. Nature. 322:843–846. [DOI] [PubMed] [Google Scholar]
- 39.Chen, C., Z. Nagy, E.L. Prak, and M. Weigert. 1995. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 3:747–755. [DOI] [PubMed] [Google Scholar]
- 40.Chen, C., M.Z. Radic, J. Erikson, S.A. Camper, S. Litwin, R.R. Hardy, and M. Weigert. 1994. Deletion and editing of B cells that express antibodies to DNA. J. Immunol. 152:1970–1982. [PubMed] [Google Scholar]
- 41.Krizman, D.B., R.F. Chuaqui, P.S. Meltzer, J.M. Trent, P.H. Duray, W.M. Linehan, L.A. Liotta, and M.R. Emmert-Buck. 1996. Construction of a representative cDNA library from prostatic intraepithelial neoplasia. Cancer Res. 56:5380–5383. [PubMed] [Google Scholar]
- 42.Schlissel, M., A. Constantinescu, T. Morrow, M. Baxter, and A. Peng. 1993. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5'-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 7:2520–2532. [DOI] [PubMed] [Google Scholar]
- 43.Glusman, G., L. Rowen, I. Lee, C. Boysen, J.C. Roach, A.F. Smit, K. Wang, B.F. Koop, and L. Hood. 2001. Comparative genomics of the human and mouse T cell receptor loci. Immunity. 15:337–349. [DOI] [PubMed] [Google Scholar]
- 44.Livak, F., D.B. Burtrum, L. Rowen, D.G. Schatz, and H.T. Petrie. 2000. Genetic modulation of T cell receptor gene segment usage during somatic recombination. J. Exp. Med. 192:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramsden, D.A., and G.E. Wu. 1991. Mouse kappa light-chain recombination signal sequences mediate recombination more frequently than do those of lambda light chain. Proc. Natl. Acad. Sci. USA. 88:10721–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Connor, A.M., L.J. Fanning, J.W. Celler, L.K. Hicks, D.A. Ramsden, and G.E. Wu. 1995. Mouse VH7183 recombination signal sequences mediate recombination more frequently than those of VHJ558. J. Immunol. 155:5268–5272. [PubMed] [Google Scholar]
- 47.Bassing, C.H., F.W. Alt, M.M. Hughes, M. D'Auteuil, T.D. Wehrly, B.B. Woodman, F. Gartner, J.M. White, L. Davidson, and B.P. Sleckman. 2000. Recombination signal sequences restrict chromosomal V(D)J recombination beyond the 12/23 rule. Nature. 405:583–586. [DOI] [PubMed] [Google Scholar]
- 48.Sleckman, B.P., C.H. Bassing, M.M. Hughes, A. Okada, M. D'Auteuil, T.D. Wehrly, B.B. Woodman, L. Davidson, J. Chen, and F.W. Alt. 2000. Mechanisms that direct ordered assembly of T cell receptor beta locus V, D, and J gene segments. Proc. Natl. Acad. Sci. USA. 97:7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrier, P., L.R. Covey, H. Suh, A. Winoto, L. Hood, and F.W. Alt. 1989. T cell receptor DJ but not VDJ rearrangement within a recombination substrate introduced into a pre-B cell line. Int. Immunol. 1:66–74. [DOI] [PubMed] [Google Scholar]
- 50.Sadofsky, M.J. 2001. The RAG proteins in V(D)J recombination: more than just a nuclease. Nucleic Acids Res. 29:1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aidinis, V., T. Bonaldi, M. Beltrame, S. Santagata, M.E. Bianchi, and E. Spanopoulou. 1999. The RAG1 homeodomain recruits HMG1 and HMG2 to facilitate recombination signal sequence binding and to enhance the intrinsic DNA-bending activity of RAG1-RAG2. Mol. Cell. Biol. 19:6532–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mo, X., T. Bailin, S. Noggle, and M.J. Sadofsky. 2000. A highly ordered structure in V(D)J recombination cleavage complexes is facilitated by HMG1. Nucleic Acids Res. 28:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang, H.E., L.Y. Hsu, D. Cado, L.G. Cowell, G. Kelsoe, and M.S. Schlissel. 2002. The “dispensable” portion of RAG-2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 17:639–651. [DOI] [PubMed] [Google Scholar]
- 54.Siu, G., E.A. Springer, H.V. Huang, L.E. Hood, and S.T. Crews. 1987. Structure of the T15 VH gene subfamily: identification of immunoglobulin gene promotor homologies. J. Immunol. 138:4466–4471. [PubMed] [Google Scholar]
- 55.Love, V.A., G. Lugo, D. Merz, and A.J. Feeney. 2000. Individual V(H) promoters vary in strength, but the frequency of rearrangement of those V(H) genes does not correlate with promoter strength nor enhancer-independence. Mol. Immunol. 37:29–39. [DOI] [PubMed] [Google Scholar]
- 56.Nadel, B., A. Tang, and A.J. Feeney. 1998. V(H) replacement is unlikely to contribute significantly to receptor editing due to an ineffectual embedded recombination signal sequence. Mol. Immunol. 35:227–232. [DOI] [PubMed] [Google Scholar]
- 57.Taki, S., F. Schwenk, and K. Rajewsky. 1995. Rearrangement of upstream DH and VH genes to a rearranged immunoglobulin variable region gene inserted into the DQ52-JH region of the immunoglobulin heavy chain locus. Eur. J. Immunol. 25:1888–1896. [DOI] [PubMed] [Google Scholar]
- 58.Bertrand, F.E., R. Golub, and G.E. Wu. 1998. V(H) gene replacement occurs in the spleen and bone marrow of non-autoimmune quasi-monoclonal mice. Eur. J. Immunol. 28:3362–3370. [DOI] [PubMed] [Google Scholar]
- 59.Rudert, F., S. Bronner, J.M. Garnier, and P. Dolle. 1995. Transcripts from opposite strands of gamma satellite DNA are differentially expressed during mouse development. Mamm. Genome. 6:76–83. [DOI] [PubMed] [Google Scholar]
- 60.Allen, M.J., A.J. Jeffreys, M.A. Surani, S. Barton, M.L. Norris, and A. Collick. 1994. Tandemly repeated transgenes of the human minisatellite MS32 (D1S8), with novel mouse gamma satellite integration. Nucleic Acids Res. 22:2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sabbattini, P., A. Georgiou, C. Sinclair, and N. Dillon. 1999. Analysis of mice with single and multiple copies of transgenes reveals a novel arrangement for the lambda5-VpreB1 locus control region. Mol. Cell. Biol. 19:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stallings, R.L., A.F. Ford, D. Nelson, D.C. Torney, C.E. Hildebrand, and R.K. Moyzis. 1991. Evolution and distribution of (GT)n repetitive sequences in mammalian genomes. Genomics. 10:807–815. [DOI] [PubMed] [Google Scholar]
- 63.Lee, S.S., D. Fitch, M.F. Flajnik, and E. Hsu. 2000. Rearrangement of immunoglobulin genes in shark germ cells. J. Exp. Med. 191:1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marculescu, R., T. Le, P. Simon, U. Jaeger, and B. Nadel. 2002. V(D)J-mediated translocations in lymphoid neoplasms: a functional assessment of genomic instability by cryptic sites. J. Exp. Med. 195:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu, K., A. Taghva, and M.R. Lieber. 2001. The cleavage efficiency of the human immunoglobulin heavy chain VH elements by the RAG complex: Implications for the immune repertoire. J. Biol. Chem. 277:5040–5046. [DOI] [PubMed] [Google Scholar]
- 66.Stormo, G.D., T.D. Schneider, L. Gold, and A. Ehrenfeucht. 1982. Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 10:2997–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stormo, G.D. 1990. Consensus patterns in DNA. Methods Enzymol. 183:211–221. [DOI] [PubMed] [Google Scholar]
- 68.Vanet, A., L. Marsan, and M.F. Sagot. 1999. Promoter sequences and algorithmical methods for identifying them. Res. Microbiol. 150:779–799. [DOI] [PubMed] [Google Scholar]
- 69.Roulet, E., P. Bucher, R. Schneider, E. Wingender, Y. Dusserre, T. Werner, and N. Mermod. 2000. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 297:833–848. [DOI] [PubMed] [Google Scholar]
- 70.Berg, O.G., and P.H. von Hippel. 1987. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 193:723–750. [DOI] [PubMed] [Google Scholar]
- 71.Crowley, E.M., K. Roeder, and M. Bina. 1997. A statistical model for locating regulatory regions in genomic DNA. J. Mol. Biol. 268:8–14. [DOI] [PubMed] [Google Scholar]
- 72.Stephens, R.M., and T.D. Schneider. 1992. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J. Mol. Biol. 228:1124–1136. [DOI] [PubMed] [Google Scholar]