

Abstract
T lymphocytes expressing the T cell receptor (TCR)-γδ recognize unknown antigens on tumor cells. Here we identify metabolites of the mevalonate pathway as the tumor ligands that activate TCR-γδ cells. In tumor cells, blockade of hydroxy-methylglutaryl-CoA reductase (HMGR), the rate limiting enzyme of the mevalonate pathway, prevents both accumulation of mevalonate metabolites and recognition by TCR-γδ cells. When metabolite accumulation is induced by overexpressing HMGR or by treatment with nitrogen-containing bisphosphonate drugs, tumor cells derived from many tissues acquire the capacity to stimulate the same TCR-γδ population. Accumulation of mevalonate metabolites in tumor cells is a powerful danger signal that activates the immune response and may represent a novel target of tumor immunotherapy.
Keywords: antigen recognition, tumor antigen, HMGR, IPP, bisphosphonate drugs
Introduction
Human T cells expressing the TCR-γδ represent a unique lymphocyte population with an unusual tissue distribution. These cells are present in both organized lymphoid tissues as well as in the skin- and gut-associated lymphoid systems without any special tropism for epithelia (1). Information has been accumulating regarding the microbial antigens they recognize. A series of small phosphorylated nonpeptidic metabolites which stimulate the Vγ9/Vδ2 population have been identified. Amongst these isopentenylpyrophosphate (IPP)* is a prototype ligand (2, 3). Some of these ligands were purified from microbial cells and are intermediate metabolites of farnesylpyrophosphate (FPP) synthesis (4, 5). These products are essential for cell survival and are ubiquitous. Therefore, this unique type of antigen specificity was suggested to be best suited for activation of “sentinel” cells independently of any unique antigens derived from individual microbes (6).
An important aspect of antigen specificity of human TCR-γδ cells is their capacity to recognize and kill tumor targets. T cells expressing the Vγ9/Vδ2 heterodimer, the same TCR stimulated by bacterial phosphorylated metabolites, recognize bone marrow–derived tumor cells such as the non-Hodgkin B cell lymphoma line Daudi both in vitro (7) and in a SCID animal model in vivo (8). TCR Vγ9/Vδ2 also recognize and kill the B cell lymphoma RPMI-8233 (9), the T cell lymphoma MOLT-4 (10) and the erythroleukemia line K562 (11). In no case has the activating ligand been identified.
To identify the tumor antigens which activate Vγ9/Vδ2 cells, we tested the hypothesis that in tumor cells TCR-γδ cells recognize phosphorylated nonpeptidic ligands resembling those produced by microbial cells. This hypothesis was prompted by the reported increased expression and function of HMGR, the rate limiting enzyme in the mevalonate pathway (Fig. 1) in haematological malignancies (12) and mammary carcinoma cells (13). Mevalonate pathway dysregulation in tumor cells might lead to accumulation of phosphorylated mevalonate metabolites and activation of Vγ9/Vδ2 cells. Most of the studies herein were performed using Daudi cells. We also found that a breast carcinoma line (YMB-1) activates the Vγ9/Vδ2 population using the same mechanism as Daudi cells.
Figure 1.

Mevalonate metabolic pathway. The compounds used in this study and the affected enzymes are indicated in frames.
Materials and Methods
Chemicals.
IPP, mevastatin, 7-dehydrocholesterol (7-DHC), farnesol, and monensin (Mon) were purchased from Sigma-Aldrich. Zoledronate (ZOL), C14-labeled ZOL, pamidronate (PAM), and etidronate were supplied as hydrated disodium salts by Novartis. PHA was purchased from Murex Biotech, 3H-IPP from ARC.
Cells.
The following cell lines were used: A-375 cells, a malignant melanoma; A-431, an epidermoid cancer; Daudi, a Burkitt's lymphoma; THP-1, a monocytic leukemia; CEM 1.3, a T cell lymphoma; Colo 201, a coloncarcinoma line; HEP-G2 a hepatocarcinoma and K562 an erythroleukemia line all obtained from American Type Culture Collection. YMB-1, HMC-1–8, and MRK-nu-1 mammary carcinomas were obtained from Health Science Research Resources Bank, Osaka, Japan. Primary human lung fibroblasts were obtained from M. Bihl; hepatoblastoma cell line HuH6 (E. Köhler); glioblastomas U118 and BS125.3.2 (A. Merlo; all from University Hospital, Basel, Switzerland); astrocytoma A-243 was established from an anaplastic astrocytoma (WHO grade III). TCR Vγ9/Vδ2 T cell clones were obtained, cultured, and restimulated as described (10). Tumor and primary cells were cultured as described (10). Most of the experiments were performed with more than one TCR Vγ9/Vδ2 T cell clones, always showing similar results and were done at least three times. Activation assays with ZOL were performed using a large panel of previously described TCR-γδ clones (10), (online supplemental Table S1). These experiments confirmed that a TCR composed of Vγ9-Cγ1 and Vδ2 chains is required for recognition of Daudi cells or for ZOL-pulsed APCs.
T Cell Stimulation Assays.
Cells used as stimulatory APCs were incubated with different doses of bisphosphonates for 3 h at 37°C or at 4°C followed by three washes before plating (5 × 104/well) and subsequent addition of T cells (5 × 104/well). In some experiments mevastatin (25 μM) was added 2 h before bisphosphonates and again after removal of bisphosphonates to maintain a constant concentration during incubation with T cells. Farnesol and 7-DHC were pulsed on APC for 12 h and rinsed out before addition of T cells. Stimulation with IPP (10 μM) and PHA (1 μg/ml) served as positive controls to exclude unspecific or toxic effects of the tested drugs. To study the kinetics of activation induced by nitrogen-containing bisphosphonate drugs (nBP), Daudi cells were pulsed with 50 μM PAM or ZOL for selected periods (1–18 h), washed three times, and incubated with TCR-γδ T cell clone G2B9 for 12 h.
TNFα Release Assay.
APC (5 × 104/well) and T cells (5 × 104/well) were incubated in 96-well plates (BD Biosciences) in triplicate for 12 h. Ligand and drug concentrations used are indicated in the figure legends. TNFα released into the supernatant was measured by ELISA (Immunokontakt) and is expressed as mean pg/ml or ng/ml ± SD.
14C-ZOL Uptake.
Daudi cells were pulsed with 14C-ZOL (25 μg/ml, corresponding to 700 nCi/ml) for 3 h at 37°C or at 4°C, and washed thrice. Some experiments included monensin (20 μM) and NaN3 (0.05%) during the ZOL pulse. Radioactivity incorporated into 106 cells was counted using scintillation liquid (IRGA-SAFE PLUS, Packard Bioscience BV) and a β-counter (TR 1900, Canberra Packard). Triplicate samples were evaluated.
Immunoprecipitation and Western Blotting.
Daudi cells (2 × 107/group) either untreated or treated with 7-DHC (100 μg/ml) or farnesol (50 μM) were lysed and immunoprecipitated with anti-HMGR rabbit polyclonal antiserum (gift of R. Simoni, Stanford University, Palo Alto, CA) using protein G-Sepharose (Amersham Biosciences). Western blotting, after SDS-separation was performed according to standard protocols. Blots were sequentially incubated with anti-HMGR A9 mAb (American Type Culture Collection) and goat anti–mouse horseradish peroxidase-conjugated Ig (Southern Biotechnology Associates, Inc.) and HMGR detected using chemiluminescence substrate (Supersignal; Pierce Chemical Co.).
Mevalonate Pathway Assay.
Daudi cells (5 × 108) were lysed according to reference 14. Cell lysates were concentrated four times using Biomax-10K filters (Millipore) and 125 μl aliquots were incubated for 60 min at 37°C under conditions described (14) in a total volume of 500 μl. The reaction was stopped by addition of an equal volume of methanol and cooling on ice. The sample was clarified by centrifugation at 9,000 rpm and stored at −20°C until HPLC separation or phosphatase treatment. HPLC separation was performed as reported (14) with some modifications. Briefly, a Spherisorb SAX 5μ HPLC column (4.6 mm × 25 cm; VDS Optilab) was used together with the buffers described in reference 14. The gradient was 0–2 min 0% B, 2–13.5 min 57% B, 13.5–14.5 min 99% B, 14.5–35 min 99% B. Radioactivity was detected online with a β scintillation detector (Packard Instrument Co.) using Ultima Flo M (Packard Instrument Co.) as scintillant. UV monitoring (254 nm) was also applied. Separate HPLC runs under identical conditions were performed using material obtained by incubation with nonradioactive mevalonate. Fractions were collected every 15 s and examined in T cell activation assays. Active fractions were further analyzed by mass spectrometry. Fractions were lyophilized and tested at a final dilution of 1:12 using 5 × 104/well THP-1 cells as APCs and 5 × 104/well G2B9 TCR-γδ T cells. Released TNFα was measured by ELISA. In some experiments Daudi cell extract was treated with alkaline phosphatase as described (15).
Generation of HMGR Stable Transfectants.
Hamster HMGR cDNA (pRed-227; American Type Culture Collection) was subcloned into the XhoI/NotI sites of the BCMGSNeo expression vector. Daudi cells were transfected with 10 μg DNA by electroporation, and selected using G418 (Calbiochem).
Intracellular HMGR Staining of Transfected Cells.
Daudi cells fixed with 2% paraformaldehyde were incubated for 30 min with biotinylated mouse anti-HMGR A9 mAb or isotype-matched irrelevant mAb in PBS containing 0.1% saponin, revealed with phycoerythrin-coupled Streptavidin (Southern Biotechnology Associates, Inc.), and analyzed using a FACScan™ and the CELLQuest program (BD Biosciences).
Mass Spectrometry Analysis.
After desalting, samples were dissolved in 10 μl of 50% methanol. A few microliters were loaded into a nanospray tip (Protana Engineering A/S). The spray was initiated at a voltage of 1,100 V. Spectra were recorded on a Finnigan TSQ7000 instrument (Finnigan) in negative ion mode set to 1 D resolution.
Online Supplemental Material.
Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20021500/DC1.
Results and Discussion
We found that YMB-1 cells, a solid breast carcinoma, activate the same Vγ9/Vδ2 population as that stimulated by Daudi Burkitt's lymphoma cells. To test the role of HMGR in generating TCR-γδ stimulatory ligands, both Daudi and YMB-1 cells were treated with mevastatin, an inhibitor of the HMGR catalytic site (16). Mevastatin treatment of both cell lines efficiently inhibited activation of TCR-γδ cells expressing the TCR Vγ9/Vδ2 (Fig. 2 A). In control experiments, the same T cell population was not inhibited when stimulated with exogenous IPP or with PHA in the presence of mevastatin, thus excluding a toxic effect of the drug (Fig. 2 A). Involvement of HMGR was further investigated by treating Daudi cells with 7-DHC or farnesol, two endogenous metabolites that facilitate degradation of HMGR by a negative feedback mechanism (17, 18). Both compounds reduced intracellular levels of HMGR protein as shown by immunoprecipitation and Western blotting (Fig. 2 B), and significantly inhibited the capacity of Daudi cells to activate TCR-γδ cells (Fig. 2 C). In control experiments neither compound modified TCR-γδ response to IPP, thus excluding nonspecific toxic effects (Fig. 2 C). Taken together these experiments show that HMGR is a key player in the stimulation of TCR-γδ lymphocytes by Daudi and YMB-1 cells, and suggest that phosphorylated metabolites generated in the mevalonate pathway are the stimulatory antigens in these cells.
Figure 2.

Activation of TCR-γδ cells by tumor cells is dependent upon the intracellular levels and activity of HMGR. (A) Mevastatin prevents response of the TCR-γδ T cell clone G2B9 stimulated by Daudi and YMB-1 ligands, but has no effect on stimulation with IPP or PHA. Results show TNFα mean release ± SD. (B) HMGR protein levels are down-regulated by farnesol or 7-DHC as detected by immunoprecipitation and Western blot. (C) Treatment of Daudi cells with farnesol or 7-DHC prevents stimulation of the G2B9 cells. In control experiments the same treatments of Daudi cells did not affect stimulation with exogenous IPP.
To confirm this hypothesis two series of experiments were performed. First, the HMGR gene was transfected into Daudi cells. Transfected cells (Daudi-HMGR) expressed high levels of HMGR as shown by intracellular staining with a specific mAb (Fig. 3 A) and acquired a stronger capacity to stimulate TCR-γδ cells (Fig. 3 B). Treatment of Daudi-HMGR with mevastatin prevented TCR-γδ cell activation (Fig. 3 B). Importantly, when used at suboptimal doses, mevastatin completely inhibited stimulation by Daudi-HMGR only when added at least 12 h before the assay, probably due to increased intracellular HMGR levels.
Figure 3.
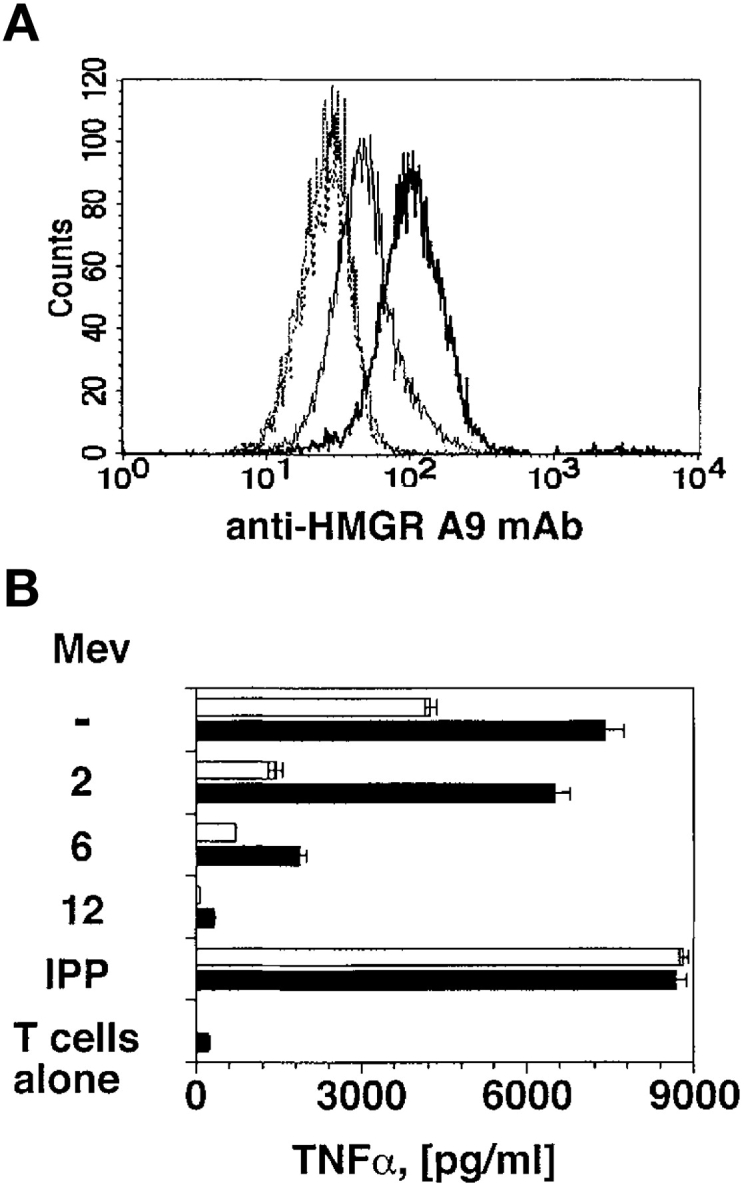
HMGR-Daudi transfectants are potent stimulators. (A) Daudi cells transfected with the HMGR gene express higher levels of HMGR protein (bold line) than nontransfected Daudi cells (thin line). No difference between the two cell types was detected using an irrelevant mAb (dotted lines). (B) Daudi-HMGR cells (black bars) show increased capacity to stimulate Vγ9/Vδ2 cells as compared with nontransfected Daudi cells (white bars). Mevastatin was added at suboptimal doses (10 μM) 2, 6, or 12 h before incubation with T cells. Stimulation with IPP was used as positive control.
In the second experimental series, Daudi cells were treated with nBP, which inhibit FPP synthase and induce accumulation of IPP, dimethylallylpyrophosphate, and geranylpyrophosphate (14). These metabolites are substrates of FPP synthase and are capable of activating TCR-γδ cells when added exogenously to APCs (2, 3, 19). Two structurally different nBP, PAM and ZOL, were tested at equimolar concentrations and compared with etidronate, a non-nBP that does not inhibit FPP synthase (14, 20). ZOL- and PAM-treated APCs, but not etidronate-treated APCs, stimulated TCR-γδ cells (online supplemental Fig. S1). These results confirm previous studies performed with other nBP (21, 22) and strongly suggest that accumulation of phosphorylated metabolites within APCs is an important event preceding stimulation of TCR-γδ cells.
A striking feature of the TCR-γδ cell stimulation with exogenously added phosphorylated metabolites is a requirement for the permanent presence of phosphorylated ligands during the assay. Indeed, if bacterial ligands or IPP are added to APCs at normal stimulatory doses and then the cells are washed, the stimulatory capacity of APCs is immediately lost, presumably because the activatory compounds do not remain associated to the APC surface in a stable and active form (23). To examine whether stimulation with nBP follows the same rules as IPP, THP-1 cells were pulsed for 2 h with ZOL, PAM, or IPP, and then extensively washed before being used to stimulate TCR-γδ cells. After pulsing with ZOL and PAM, but not with IPP, THP-1 cells retained a strong stimulatory capacity (Fig. 4 A). These data confirm the recent observation that the nBP risedronate and PAM can be pulsed on primary monocytes and on different tumor cell lines (22, 24). We also examined the kinetics of activation by pulsing Daudi cells with ZOL or PAM at equimolar concentrations for various time periods. ZOL induced its effects more rapidly than PAM with 80% of maximal stimulation already observed after a 2-h pulse. This differential likely reflects the greater potency of ZOL in inhibiting FPP synthase (20). However, both nBP showed equal activity after a prolonged pulsing time (Fig. 4 B), suggesting that they do not differ in their mechanism of action.
Figure 4.

ZOL and PAM activate TCR-γδ cells after internalization in the APCs and accumulation of mevalonate metabolites. (A) THP-1 cells stimulate the T cell clone G2D7 after 2 h pulse (black bars) with ZOL or PAM, but not with IPP. Overnight incubation (white bars) with PAM and ZOL was used as positive control. Similar results were obtained by measuring IFN-γ release and using a second TCR Vγ9/Vδ2 clone (data not depicted). (B) ZOL (•) is faster than PAM (○) in inducing TCR-γδ cell stimulatory ligands in Daudi cells. (C) ZOL requires cellular internalization to become stimulatory. Daudi cells were pulsed at 4 or at 37°C. In one control group ZOL was added back to exclude inhibitory effects of incubation at 4°C. (D) Daudi cells have reduced stimulatory capacities when incubated with monensin (Mon) and ZOL. IPP stimulation was used as positive control. (E) Intracellular uptake by Daudi cells of 14C-ZOL. To exclude surface binding, pulse with 14C-ZOL was also performed at 4°C or in the presence of monensin. (F) Mevastatin completely inhibits activation only when added simultaneously with ZOL. Mevastatin was added to Daudi cells together with ZOL, 1 or 3 h later. 3 h after ZOL, Daudi cells were washed and T cells were added. Mevastatin was added back to keep its concentration constant during the entire culture period. Supernatants were harvested 12 h later.
As FPP synthase, the target of nBP, is present within cells, it is likely that nBP must be internalized before becoming active. The fact that nBP are active after pulsing and washing of APCs supports this possibility. To address whether nBP internalization into the APCs is mandatory for activation of Vγ9/Vδ2 T cells, pulsing was conducted under conditions which inhibit pinocytosis and receptor-mediated endocytosis. Daudi cells were pulsed with ZOL either at 4°C (Fig. 4 C) or in the presence of monensin (Fig. 4 D). Both conditions strongly inhibited activation of Vγ9/Vδ2 T cells by ZOL-pulsed APCs, while activation by IPP was minimally affected (Fig. 4 D). In control experiments ZOL vigorously stimulated TCR-γδ cells when included after incubation at 4°C and for the entire period of the assay (Fig. 4 C), demonstrating that the 4°C incubation did not render the APCs poorly stimulatory. To obtain further proof of internalization, Daudi cells were pulsed with 14C-labeled ZOL under the same experimental conditions. 14C-ZOL was taken up by cells during pulsing at 37°C, but not at 4°C or in the presence of monensin (Fig. 4 E). Together, these data show that internalization of ZOL is prerequisite for its activity, and establish its distinction from IPP, which is active without being internalized (23).
These findings suggest that nBP activate TCR-γδ by inducing intracellular accumulation of stimulatory metabolites, although it cannot be formally excluded that mimicry of IPP is also applying, at least in some cases. The hypothesis based on the effects exerted by accumulating metabolites was examined by incubating Daudi cells with ZOL and mevastatin together. As shown in Fig. 4 F, mevastatin completely abolished activation of Vγ9/Vδ2 T cells when added simultaneously with ZOL, but was only 50% inhibitory when added 1 h later. Mevastatin was not inhibitory when added 3 h after ZOL, suggesting that accumulation of the stimulatory ligands had already taken place. These data accord with those showing that a pulse with ZOL for 3 h is sufficient to induce maximal stimulatory activity in APCs (Fig. 4 B). The latter results exclude that mevastatin is toxic, and further support the conclusion that the inhibitory action of mevastatin depends upon its prevention of accumulation of mevalonate metabolites during a ZOL pulse.
To identify the metabolites important in tumor cell recognition, we undertook their biochemical purification and examined their TCR-γδ stimulatory capacities. Daudi cell extracts were incubated with 3H-labeled mevalonate, the first metabolite in the mevalonate cascade, and the radioactive downstream metabolic products were detected in HPLC fractions with the same retention time as reference 3H-labeled IPP (Fig. 5, A–C) . Importantly, the same fractions (which elute at 15–16 min) were also capable of activating Vγ9/Vδ2 T cells (Fig. 5 E). Furthermore, the TCR-γδ stimulatory capacity was sensitive to alkaline phosphatase treatment (Fig. 5 D), as previously shown for mycobacterial products activating Vγ9/Vδ2 T cells (15). Mass spectrometry analysis of the HPLC purified and TCR-γδ stimulatory fraction eluting at 15.5 min (Fig. 5 F) confirmed the presence of a compound with the same mass as IPP (Fig. 5 F, insert).
Figure 5.

The intracellular Daudi compounds activating TCR-γδ cells are metabolites of the mevalonate pathway. (A–C) The stimulatory Daudi ligands have the same retention time as IPP. Panel A shows the radioactive compounds generated with Daudi cell extracts after addition of 3H-mevalonic acid, panel B the reference 3H-IPP, panel C the reference 3H-mevalonic acid (3H-Mev). (D) The stimulatory ligands present in Daudi cell extracts are phosphatase sensitive. The semipurified material was treated with alkaline phosphatase (AP) and then tested in TCR-γδ cell stimulation assays. IPP treated identically was used as positive control for AP activity. (E) Bioactivity of TCR-γδ stimulatory ligands from Daudi cells. HPLC fractions were tested at a final dilution of 1:12 using THP-1 cells as APCs and the T cell clone G2B9. (F) Ion-spray Mass spectrometry of the most active fraction from panel E. The arrow indicates the mass of IPP. The insert shows the mass of reference IPP.
As the mevalonate pathway is fundamental for cell survival and growth (25), all cells generate phosphorylated mevalonate metabolites. Therefore, given the presence of the appropriate adhesion and presentation molecules, it is possible that all cells treated with nBP can stimulate TCR-γδ cells. This hypothesis was tested by pulsing a variety of tumor cell lines derived from different tissues with ZOL and assaying their subsequent capacity to stimulate Vγ9/Vδ2 T cells. Tumor cell lines established from distinct tissues, as well as primary human lung fibroblasts, were used as APCs either in the presence of IPP or after ZOL pulsing. All tested lines, although with clear differences, were capable of activating TCR-γδ cells under both stimulatory conditions (online supplemental Table S2). Thus, cells from many different tissues have the capacity to present accumulated phosphorylated ligands in a stimulatory manner to TCR-γδ cells. These findings indicate that presentation of endogenous and exogenous ligands involves nonpolymorphic molecules with a wide tissue distribution.
Recognition of cells which overproduce nonpeptidic phosphorylated metabolites generated by the mevalonate pathway may allow the immune system to target cells with significant metabolic abnormalities. This strategy has the following important advantages. First, mevalonate intermediates are necessary for sterol synthesis, cell growth, and membrane integrity, and thus tumor cells which up-regulate this pathway become more visible to the immune system. Second, an alternative pathway for production of FPP does not exist in mammalian cells (26) and this prevents selection of negative mutants escaping immunosurveillance. Third, the intracellular concentration of isoprenoids is finely controlled by HMGR levels, which is one of the most tightly regulated enzymes in mammalian cells (25). Several signals such as cholesterol levels (27), insulin and thyroid hormones (28), retinoids (29), and cell stress (30) contribute toward increasing HMGR levels and the generation of mevalonate pathway products. Accumulation of these metabolites in excess of physiological levels represents the alarm signal for activating TCR-γδ lymphocytes. Importantly, this accumulation can be induced in vivo by using bisphosphonate drugs, such as ZOL, which are already in clinical use. Tumor targeting of these drugs may provide a novel approach to tumor immunotherapy that exploits the killing capacity and the ligand specificity of TCR-γδ cells.
What is the immunological value of this unique type of antigen recognition? It offers a general mechanism whereby one T cell population could survey all tissues for both normal and transformed cells that are metabolically altered in the mevalonate pathway. Furthermore, the same strategy also carries the advantage of functioning as a surveillance system for bacterial infection (6). It remains to be investigated which type of evolutionary benefit this property has endowed primates, the only order of mammals using this unique immunological surveillance mechanism.
Acknowledgments
We thank J. Green, R. Landmann, E. Palmer, T. Resink, G. Spagnoli, and colleagues in our laboratory for discussions and critical reading of the manuscript. We also thank G. Spagnoli, A. Wodnar-Filipowicz, M. Bihl, A. Merlo, E. Koehler, and R. Simoni for providing cell lines and reagents and V. Jäggin for HPLC analysis.
This work was supported by the Swiss National Fund, European Community and Novartis Pharma AG, Basel.
The online version of this paper contains supplemental material.
Footnotes
Abbreviations used in this paper: FPP, farnesylpyrophosphate; IPP, isopentenylpyrophosphate; PAM, pamidronate; ZOL, zoledronate.
References
- 1.Porcelli, S., M.B. Brenner, and H. Band. 1991. Biology of the human γδ T-cell receptor. Immunol. Rev. 120:137–183. [DOI] [PubMed] [Google Scholar]
- 2.Burk, M.R., L. Mori, and G. De Libero. 1995. Human V gamma 9-V delta 2 cells are stimulated in a cross-reactive fashion by a variety of phosphorylated metabolites. Eur. J. Immunol. 25:2052–2058. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka, Y., C.T. Morita, Y. Tanaka, E. Nieves, M.B. Brenner, and B.R. Bloom. 1995. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature. 375:155–158. [DOI] [PubMed] [Google Scholar]
- 4.Belmant, C., E. Espinosa, R. Poupot, M.A. Peyrat, M. Guiraud, Y. Poquet, M. Bonneville, and J.J. Fournie. 1999. 3-Formyl-1-butyl pyrophosphate A novel mycobacterial metabolite-activating human gammadelta T cells. J. Biol. Chem. 274:32079–32084. [DOI] [PubMed] [Google Scholar]
- 5.Feurle, J., E. Espinosa, S. Eckstein, F. Pont, V. Kunzmann, J.J. Fournie, M. Herderich, and M. Wilhelm. 2002. Escherichia coli produces phosphoantigens activating human gamma delta T cells. J. Biol. Chem. 277:148–154. [DOI] [PubMed] [Google Scholar]
- 6.De Libero, G. 1997. Sentinel function of broadly reactive human gamma delta T cells. Immunol. Today. 18:22–26. [DOI] [PubMed] [Google Scholar]
- 7.Fisch, P., M. Malkovsky, S. Kovats, E. Sturm, E. Braakman, B.S. Klein, S.D. Voss, L.W. Morrissey, R. DeMars, W.J. Welch, et al. 1990. Recognition by human V gamma 9/V delta 2 T cells of a GroEL homolog on Daudi Burkitt's lymphoma cells. Science. 250:1269–1273. [DOI] [PubMed] [Google Scholar]
- 8.Malkovska, V., F.K. Cigel, N. Armstrong, B.E. Storer, and R. Hong. 1992. Antilymphoma activity of human gamma delta T-cells in mice with severe combined immune deficiency. Cancer Res. 52:5610–5616. [PubMed] [Google Scholar]
- 9.Selin, L.K., S. Stewart, C. Shen, H.Q. Mao, and J.A. Wilkins. 1992. Reactivity of gamma delta T cells induced by the tumour cell line RPMI 8226: functional heterogeneity of clonal populations and role of GroEL heat shock proteins. Scand. J. Immunol. 36:107–117. [DOI] [PubMed] [Google Scholar]
- 10.De Libero, G., G. Casorati, C. Giachino, C. Carbonara, N. Migone, P. Matzinger, and A. Lanzavecchia. 1991. Selection by two powerful antigens may account for the presence of the major population of human peripheral gamma/delta T cells. J. Exp. Med. 173:1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Fabrizio, L., Y. Kimura, R. Ware, L. Rogozinski, and L. Chess. 1991. Specific triggering of gamma, delta T cells by K562 activates the gamma, delta T cell receptor and may regulate natural killer-like function. J. Immunol. 146:2495–2503. [PubMed] [Google Scholar]
- 12.Harwood, H.J., Jr., I.M. Alvarez, W.D. Noyes, and P.W. Stacpoole. 1991. In vivo regulation of human leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase: increased enzyme protein concentration and catalytic efficiency in human leukemia and lymphoma. J. Lipid Res. 32:1237–1252. [PubMed] [Google Scholar]
- 13.Asslan, R., A. Pradines, C. Pratx, C. Allal, G. Favre, and F. Le Gaillard. 1999. Epidermal growth factor stimulates 3-hydroxy-3-methylglutaryl-coenzyme A reductase expression via the ErbB-2 pathway in human breast adenocarcinoma cells. Biochem. Biophys. Res. Commun. 260:699–706. [DOI] [PubMed] [Google Scholar]
- 14.Bergstrom, J.D., R.G. Bostedor, P.J. Masarachia, A.A. Reszka, and G. Rodan. 2000. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch. Biochem. Biophys. 373:231–241. [DOI] [PubMed] [Google Scholar]
- 15.Constant, P., F. Davodeau, M.A. Peyrat, Y. Poquet, G. Puzo, M. Bonneville, and J.J. Fournie. 1994. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science. 264:267–270. [DOI] [PubMed] [Google Scholar]
- 16.Istvan, E.S., and J. Deisenhofer. 2001. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 292:1160–1164. [DOI] [PubMed] [Google Scholar]
- 17.Correll, C.C., L. Ng, and P.A. Edwards. 1994. Identification of farnesol as the non-sterol derivative of mevalonic acid required for the accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 269:17390–17393. [PubMed] [Google Scholar]
- 18.Honda, M., G.S. Tint, A. Honda, L.B. Nguyen, T.S. Chen, and S. Shefer. 1998. 7-Dehydrocholesterol down-regulates cholesterol biosynthesis in cultured Smith-Lemli-Opitz syndrome skin fibroblasts. J. Lipid Res. 39:647–657. [PubMed] [Google Scholar]
- 19.Morita, C.T., R.A. Mariuzza, and M.B. Brenner. 2000. Antigen recognition by human gamma delta T cells: pattern recognition by the adaptive immune system. Springer Semin. Immunopathol. 22:191–217. [DOI] [PubMed] [Google Scholar]
- 20.Dunford, J.E., K. Thompson, F.P. Coxon, S.P. Luckman, F.M. Hahn, C.D. Poulter, F.H. Ebetino, and M.J. Rogers. 2001. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 296:235–242. [PubMed] [Google Scholar]
- 21.Kunzmann, V., E. Bauer, J. Feurle, F. Weissinger, H.P. Tony, and M. Wilhelm. 2000. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 96:384–392. [PubMed] [Google Scholar]
- 22.Das, H., L. Wang, A. Kamath, and J.F. Bukowski. 2001. Vgamma2Vdelta2 T-cell receptor-mediated recognition of aminobisphosphonates. Blood. 98:1616–1618. [DOI] [PubMed] [Google Scholar]
- 23.Morita, C.T., E.M. Beckman, J.F. Bukowski, Y. Tanaka, H. Band, B.R. Bloom, D.E. Golan, and M.B. Brenner. 1995. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity. 3:495–507. [DOI] [PubMed] [Google Scholar]
- 24.Kato, Y., Y. Tanaka, F. Miyagawa, S. Yamashita, and N. Minato. 2001. Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J. Immunol. 167:5092–5098. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein, J.L., and M.S. Brown. 1990. Regulation of the mevalonate pathway. Nature. 343:425–430. [DOI] [PubMed] [Google Scholar]
- 26.Edwards, P.A., and J. Ericsson. 1999. Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annu. Rev. Biochem. 68:157–185. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi, M., J.L. Goldstein, and M.S. Brown. 1988. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem. 263:8929–8937. [PubMed] [Google Scholar]
- 28.Ness, G.C., and C.M. Chambers. 2000. Feedback and hormonal regulation of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase: the concept of cholesterol buffering capacity. Proc. Soc. Exp. Biol. Med. 224:8–19. [DOI] [PubMed] [Google Scholar]
- 29.Dimitroulakos, J., and H. Yeger. 1996. HMG-CoA reductase mediates the biological effects of retinoic acid on human neuroblastoma cells: lovastatin specifically targets P-glycoprotein-expressing cells. Nat. Med. 2:326–333. [DOI] [PubMed] [Google Scholar]
- 30.Shack, S., M. Gorospe, T.W. Fawcett, W.R. Hudgins, and N.J. Holbrook. 1999. Activation of the cholesterol pathway and Ras maturation in response to stress. Oncogene. 18:6021–6028. [DOI] [PubMed] [Google Scholar]