

Abstract
Dendritic cells (DCs)* fulfill an important regulatory function at the interface of the innate and adaptive immune system. The thymus and activation-regulated chemokine (TARC/CCL17) is produced by DCs and facilitates the attraction of activated T cells. Using a fluorescence-based in vivo reporter system, we show that CCL17 expression in mice is found in activated Langerhans cells and mature DCs located in various lymphoid and nonlymphoid organs, and is up-regulated after stimulation with Toll-like receptor ligands. DCs expressing CCL17 belong to the CD11b+CD8−Dec205+ DC subset, including the myeloid-related DCs located in the subepithelial dome of Peyer's patches. CCL17-deficient mice mount diminished T cell–dependent contact hypersensitivity responses and display a deficiency in rejection of allogeneic organ transplants. In contrast to lymphoid organs located at external barriers of the skin and mucosa, CCL17 is not expressed in the spleen, even after systemic microbial challenge or after in vitro stimulation. These findings indicate that CCL17 production is a hallmark of local DC stimulation in peripheral organs but is absent from the spleen as a filter of blood-borne antigens.
Keywords: CCL17/TARC, dendritic cells, transplant rejection, contact hypersensitivity, Toll-like receptors
Introduction
Efficient activation of T lymphocytes requires the recognition of peptide/MHC complexes and costimulatory molecules on the surface of professional APCs in the environment of organized secondary lymphoid tissues. Dendritic cells (DCs)* represent professional APCs, able to transport antigen from sites of infection to the local draining LNs and to initiate primary immune responses (1, 2). In addition to membrane-bound receptor/ligand interactions, the secretion of chemokines and immunostimulatory cytokines by DCs is required for optimal attraction and differentiation of antigen-specific T cells. Several subsets of DCs have been classified on the basis of surface marker expression, such as the CD11b+CD8− myeloid-related and the CD11b− CD8+ lymphoid-related subsets (3) as well as the B220+ plasmacytoid DC population (4). All of these subsets can be found in the spleen, where they are either generated locally from hemopoietic precursors or recruited from the blood (2, 5). Prototype lymphatic migratory DCs are the Langerhans cells (LCs) which efficiently phagocytose and process antigens in the skin and subsequently locate to the local draining LNs (6, 7). In addition, a subset of intestinal DCs was shown to migrate from the lamina propria (LP) and Peyer's Patches (PPs) to the mesenteric LN (mLN) through the afferent lymph (8).
Chemotactic attraction of T lymphocytes by DCs is mediated through various constitutive and inducible chemokines. DC-derived CC chemokine 1 (DC-CK1) (CCL18) and EBV-ligand chemokine (ELC) (CCL19) attract naive T cells into the T cell zone, while the inflammatory chemokines monocyte chemoattractant protein (MCP)-1 (CCL2), macrophage inflammatory protein (MIP)-1α (CCL3), thymus and activation-regulated chemokine (TARC/CCL17), and macrophage-derived chemokine (MDC) (CCL22) mainly recruit activated and memory T cells (9–13). Although differential effects of the inducible, DC-derived chemokines on polarization of Th1- versus Th2-dominated T cell responses have been described, this issue is still controversial (13–17). CCL17 and CCL22 bind to the chemokine receptor CCR4, which is highly expressed on Th2 cells and the CLA+ subset of skin-homing memory T cells (14, 18, 19). Additionally, it has also been identified on Langerhans cells, monocytes, NK cells, and platelets. A functional role of these CCR4 ligands has been implied in the pathogenesis of allergic airway inflammation, atopic dermatitis, and septic shock syndrome (20, 21).
To obtain better insight into the regulation of CCL17 expression and the participation of CCL17-expressing cells in immune responses, we employed a reporter mouse model which allows tracking of CCL17-expressing cells in vivo by insertion of an enhanced green fluorescent protein (EGFP) cassette into the endogenous murine CCL17 locus. Using this approach, we demonstrate that CCL17 is expressed in a population of mature myeloid-related DCs located in most peripheral lymphoid and nonlymphoid organs. Intriguingly, however, CCL17 expression cannot be induced in the spleen, even in the presence of systemic bacterial infection, indicating that CCL17-mediated attraction of activated/effector T cells is favored at sites of prevalent environmental antigenic stimulation, like LNs and the mucosal immune system. Using CCL17-knockout mice we further show that absence of this chemokine leads to reduced contact hypersensitivity responses and delayed allograft rejection.
Materials and Methods
Targeting the Murine ccl17 Locus by Homologous Recombination.
A P1 phage library generated from murine genomic DNA of the strain 129/ola (GenomeSystems) was screened by PCR using the following ccl17-specific primers: pCCL17-US 5′-CAT GTG AAG AAG GCC ATC AGA TTG GTG-3′ and pCCL17-DS 5′-GAG GGA GGA AGG CTT TAT TCC GTT GC-3′. The resulting P1 clone was mapped by Southern blot hybridization and fragments containing the coding sequence and parts of the promoter were subcloned into pBluescript (Stratagene). The targeting vector was constructed in pGEM-4Z in a way that an eGFP cDNA together with a polyA signal and followed by a neomycin resistance cassette were inserted in the second exon of the ccl17 gene. Potential endogenous initiation codons in the first exon were mutated to ATC leaving the start codon of the inserted EGFP as the only possible start for translation. An additional heterologous polyadenylation signal was inserted following the EGFP gene to ensure transcript stability. A herpes simplex virus thymidine kinase (HSV-TK) cassette was inserted upstream of the targeted sequence (Fig. 1 A). E14.1 ES cells were electroporated with the linearized targeting vector and G418- and gancyclovir-resistant ES cell clones were picked. The neomycin resistance cassette was flanked by FRT recombination sites which allowed its removal from the ccl17 locus by FLP recombinase expression in vitro in the targeted ES cell lines. Homologous recombination was detected by genomic Southern blot hybridization. Correctly targeted ES cell clones were injected into pseudopregnant foster mothers. Resulting chimeric mice were backcrossed to C57BL/6 mice, and germline transmission of the targeted allele was confirmed by Southern blot analysis with a 5′ flanking probe (see Fig. 1 A) of genomic tail DNA digested with XbaI (Fig. 1 B).
Figure 1.

Generation of CCL17 reporter and CCL17-deficient mice. (A) Targeting strategy for insertion of the EGFP cDNA into the murine CCL17 locus. The murine genomic CCL17 locus with partial restriction map (top), the targeting construct (middle), and the targeted allele before and after neomycin deletion (bottom) are shown. The 3 exons of the CCL17 gene are indicated as gray boxes and the hybridization probe used for Southern blot analysis as a black bar. The EGFP reporter gene and the neomycin resistance cassette are indicated as open boxes, FRT-sites as black arrows. Restriction sites: B, BamHI; Xa, XbaI; Xo, XhoI. (B) Representative Southern blot analysis of XbaI digested genomic DNA from targeted ES cell clone and mouse tail biopsies from WT (CCL17+/+) and heterozygous CCL17 mutant (CCL17E/+) littermates. The WT and targeted allele give signals at 6 and 4 kb, respectively, after hybridization with the 5′ flanking probe. (C) CCL17 production by BM-derived DCs from CCL17E/+, CCL17+/+, and homozygous CCL17-deficient (CCL17E/E) mice. BM-derived DCs from CCL17E/+ and CCL17E/E mice were sorted into EGFP+CD11c+ (black bars) and EGFP− CD11c+ DCs (white bars) and those from CCL17+/+ mice as CD11c+ cells. All cells were cultured in vitro for 15 h. CCL17 production was measured by ELISA. * CCL17 protein was not detected in the supernatants of BM-derived DCs from CCL17E/E mice. Shown are data from two independent sorts from each group. (D) CCL17 production by sorted EGFP+CD11c+ (black bars) and EGFP-CD11c+ DC (white bars) from CCL17E/+ mice. BM-derived DCs from CCL17E/+ mice were stimulated with LPS for 15 h in vitro before cell sorting. Sorted EGFP+CD11c+ (black bars) and sorted EGFP-CD11c+ DCs (white bars) were cultured in vitro for 15 h. CCL17 production was measured by ELISA.
Mouse Breeding.
Genotyping for the reporter allele was performed by PCR on DNA from tail biopsies with the following primers: pCCL17–2M 5′-ACT CTC AGG ACA CCT GCT TCC-3′, pCCL17-bgHpA3 5′-GGG GCA AAC AAC AGA TGG C-3′ and pCCL17-P4 5′-GAG ACC CTT GAG CCT GAG AG-3′. CCL17/EGFP reporter mice were on a mixed C57BL/6/129ola genetic background and were used as heterozygous CCL17E/+ or homozygous CCL17E/E mutants in all experiments. CCL17+/+ littermates or C57BL/6 mice were used as controls.
Mice were bred in the SPF animal facility of the Institute for Medical Microbiology, Immunology and Hygiene of the Technical University of Munich according to German guidelines for animal care.
DC Preparation.
Bone marrow (BM)-derived DCs were generated as described previously (9). Briefly, BM cells were removed from femurs and tibias of reporter mice and control mice, filtered through a nylon mesh, and 5 × 105 cells/ml were cultured in RPMI 1640 supplemented with 10% vol/vol heat-inactivated FCS, l-glutamine, penicillin-streptomycin, HEPES, 2-ME (all from GIBCO BRL), and 10% supernatant of GM-CSF transfected X63Ag8–653-cells (22). On day 3, 75% of the culture supernatants were aspirated and replaced with complete medium. On day 6, the nonadherent cells were harvested and enriched for DCs as described below. To enrich DCs from organs of neonatal or 6–12-wk-old mice, thymus, LN, spleen, lung, colon, or PPs were digested with collagenase (collagenase D; Roche) and DNase (DNase I; Boehringer) before preparation of a single-cell suspension. Anti-CD11c (N418) magnetic microbeads were used to positively enrich DCs from BM cultures or total organ cell suspensions (Miltenyi Biotec). In some experiments DC subsets from CCL17E/+ mice were sorted by flow cytometry (MOFLO) on the basis of the expression of CD11c and EGFP. Purity was 98%.
Skin-derived DCs were prepared by splitting ear skin into dorsal and ventral portions and placing them dermal side down on a 1% trypsin-PBS solution, before peeling epidermal sheets from the underlying dermis and mechanically agitating them over a stainless steel mesh. Resulting cell suspensions were cultured under the same conditions as BM-derived DCs for 24 h.
Histology and In Vitro Phagocytosis Analysis Using Confocal Microscopy.
Tissue samples were fixed for 30 min up to 2 h in 4% paraformaldehyde (PFA) at 4°C, washed twice in PBS, and subsequently saturated in 20% sucrose for 8 h before being embedded in tissue-freezing medium (Leica) and snap-frozen in 2-methylbutane (Merck) prechilled with liquid nitrogen. Cryostat sections (7 μm) were fixed in acetone (Merck). Frozen sections were thawed and mounted with Fluoromount-G (Southern Biotechnology Associates, Inc.). For measurement of solid particle phagocytosis in vitro, purified BM-derived DCs were incubated on cover slides for 2 h at 37°C with carboxy-modified microspheres that fluorescinate in the red (488 nm/605 nm; size: 1 μm diameter; Molecular Probes), the uptake was stopped with ice-cold 0.1% sodium azide-1% FCS-PBS and fixation with 4% PFA. Analysis was performed using a laser scanning microscope (Carl Zeiss MicroImaging, Inc. LSM 510). For paraffin sections specimens were fixed in 4% buffered formalin for 14 to 24 h and routinely embedded in paraffin. Tissue sections were obtained and dried over night at 56°C, dewaxed, rehydrated, and rinsed in buffer. Pressure cooking in 0.01 M citrate buffer (pH 6.0) was used for antigen retrieval. Sections were subsequently stained with antibodies against CD45R (Linaris) and detected using a biotinylated goat anti rat secondary antibody (Dako) with Streptavidin PE (BD Biosciences).
In Vitro and In Vivo Stimulation of BM-, Organ-, or Skin-derived DCs and B Cells.
Mice were injected intraperitoneally or intravenously with a sublethal dose of 300 μg LPS (Sigma-Aldrich), or 20 nmol CpG-oligodeoxynucleotide (ODN) 1668 (Tib Molbiol) and Listeria monocytogenes (50,000 listeria/mouse), and organs or organ-derived DCs were analyzed 15 h (LPS and CpG-ODN) or 4 d (listeria) later. Purified BM or organ-derived DCs were stimulated in vitro for 24 h with 100 ng/ml PAM3Cys (EMC microcollections), anti-CD40 (10 μg/ml; BD Biosciences) 10 ng/ml TNF-α (R&D Systems), 100 ng/ml Escherichia coli LPS Serotype 0127:B8 (Sigma-Aldrich), CPG-ODN 1668 (1 μM), 10 ng/ml IL-1-α (R&D Systems), or poly(I:C) (50 μg/ml; Sigma-Aldrich). B cells were purified from spleen and LNs of CCL17E/+ mice using FACS sorting (MOFLO) on the basis of the expression of B220 followed by incubation with LPS (10 μg/ml), anti-IgM (10 μg/ml), or anti-CD40 (10 μg/ml; BD Biosciences) for 48 h.
Flow Cytometry.
Fluorescence staining was performed using the following antibodies purchased from BD Biosciences: anti-CD11c (PE labeled or biotinylated), anti–I-Ab (PE labeled or biotinylated), anti-CD40 (PE labeled), anti-CD86 (PE labeled), anti-CD11b (PE labeled), and anti-CD8 (PE labeled). Intracellular cytokine production was measured using anti–IL-12p40/p70 antibodies according to the manufacturers instructions (BD Biosciences). Fluorescence was analyzed using a FACSCalibur™ flow cytometer and CELLQuest™ software (both Becton Dickinson).
Measurement of Proliferative Responses and Cytokines.
For Ag-specific activation of TCR-transgenic T cells titrated numbers of BM- or LN-derived DCs were pulsed with the peptide for the dominant epitope of the human C-reactive protein (CRP; 1 μg/ml) or 5 × 103 BM- or LN-derived DCs were pulsed with titrated amount of CRP-peptide and cocultered with magnetic-activated cell sorting (MACS) purified LNs and splenic CD4 cells from DEP-tg mice (23) for 72 h. Proliferation was measured after addition of 1 μCi/well 3[H]-thymidine (Amersham Biosciences) during the last 17 h of the culture.
Culture supernatants were harvested at the indicated time points and cytokine concentration was assessed by sandwich ELISA for CCL17, IFN-γ, and IL-10 according to the manufacturer's instructions (R&D Systems). Detection limits were 5 pg/ml for CCL17, 2 pg/ml for IFN-γ, and 4 pg/ml for IL-10.
In Vivo Skin Sensitization.
Tetramethylrhodamine-5-(and-6)-isothiocyanate (TRITC) was obtained from Molecular Probes. A stock solution of 10% TRITC in DMSO was diluted to 1% in a solvent 50:50 (vol/vol) of acetone and dibutylphtalate just before application. Mice were painted at the shaved abdomen with 0.4 ml of TRITC solution. Inguinal LNs were isolated as the draining LNs. Contact hypersensitivity against dinitrofluorobenzene (DNFB; Sigma-Aldrich) was induced and analyzed as described (24). Briefly, 17 μl of 0.4% DNFB solution in acetone:olive oil (4:1) was applied to the shaved abdomen on two consecutive days. 3 d later mice were challenged with 8.5 μl on both sides of one ear. The contralateral ear was treated with vehicle only. Ear thickness of both ears was measured using an engineer's micrometer (Mitutoyo) to calculate ear swelling. Alternatively, 0.5% FITC (Sigma-Aldrich) in acetone:dibutylphthalate (1:1) was used following the same protocol with the exception that 100 μl solution was applied for sensitization and 10 μl on both sides of the ear for elicitation.
Mouse Heart Transplantation.
Vascularized heterotopic heart transplantation was performed as described previously (25). In brief, donor hearts were harvested from anaesthesized BALB/c (H-2d, allogeneic) mice and placed in Brettschneider's solution (HTK; Köhler Chemie) until grafting. The donor aorta and pulmonary artery were anastomosed to the abdominal aorta and vena cava of the H-2b (CCL17E/+ or CCL17E/E) recipient mice, respectively. The graft was monitored daily for the presence of palpable contractions, and was considered rejected, when contractions ceased as confirmed by laparotomy. Gallium nitrate, GN [Ga(NO3)] (Sigma-Aldrich) was dissolved in 5% sodium citrate dihydrate buffer and administered subcutaneously. Transplant recipients were treated with 30 mg/kg of GN on the day of transplantation, day +1, +2, +3 and subsequently every other day until day 30. No additional immunosuppression was given to the mice.
Results
Generation of CCL17-EGFP Reporter Mice.
An EGFP cDNA was inserted into the murine ccl17 locus by homologous recombination in embryonic stem cells (Fig. 1 A). After FLP-mediated deletion of the neomycin resistance cassette two independent targeted ES cell lines were used to generate CCL17-EGFP knockin mice. Mice heterozygous for the mutated allele (hereafter referred to as CCL17E/+) were identified by Southern blot analysis of genomic tail DNA (Fig. 1 B). To verify that EGFP-expressing cells indeed represent CCL17-producing cells BM-derived DCs from CCL17E/+ mice were sorted into EGFP+ and EGFP− subpopulations and cultured for 15 h. CCL17 protein was detected by ELISA in supernatants of EGFP+CD11c+ DCs from CCL17E/+ reporter mice in equivalent amounts to those of total CD11c+ DCs from CCL17+/+ WT mice (Fig. 1 C). Residual CCL17 production in the sorted EGFP−CD11c+ DC can be explained by the fact that approximately half of the cells up-regulated CCL17/EGFP expression subsequent to cell sorting (unpublished data). LPS stimulation before cell sorting reduced the proportion of contaminating EGFP+CD11c+ DCs in the EGFP−CD11c+ DC fraction to 15%. In these cultures CCL17 production was 14-fold reduced compared with those of EGFP+ CD11c+ DCs (Fig. 1 D). Thus, expression of EGFP correlates with CCL17 production in the CCL17E/+ mice, excluding the possibility that the ccl17 locus is allelicly excluded. Therefore, EGFP+ cells from CCL17E/+ mice are hereafter referred to as CCL17+ cells. CCL17 protein was not detectable in the supernatants of cultured BM-derived DCs from homozygous CCL17-deficient mice (hereafter referred to as CCL17E/E mice; Fig. 1 C).
CCL17-expressing Cells Reside in Primary and Secondary Lymphoid Organs but Not in the Spleen.
We determined the localization of CCL17+ cells in tissue sections by confocal microscopy. CCL17+ cells were clearly identifiable in the thymus, LN, lung, and the intestine of CCL17E/+ mice (Fig. 2) . In LNs, CCL17+ cells were distributed in the paracortical T cell zones accumulating in the perifollicular region and only few cells were located in the follicles (Fig. 3 A). In the cutaneous LN (cLN), they are additionally found in the subcapsular sinus, the site of entry of skin-derived DCs (Fig. 2). In the lung, CCL17+ cells accumulated in the peribronchial regions beneath the airway epithelium. CCL17+ cells were also found in the PPs, forming a cell layer in the subepithelial dome, and scattered in the interfollicular region. In the LP of the colon, CCL17+ cells are located in the basal crypts underneath the epithelium. Surprisingly, we could not detect CCL17+ cells in the spleen (Fig. 2), in contrast to all other lymphoid and nonlymphoid organs investigated.
Figure 2.

Localization of green fluorescent cells in CCL17E/+ mice. Organs were isolated from CCL17E/+ mice, fixed with 4% PFA followed by incubation in 20% sucrose and cryostat sections were analyzed by confocal microscopy. Paraffin sections were used for analysis of the colon.
Figure 3.
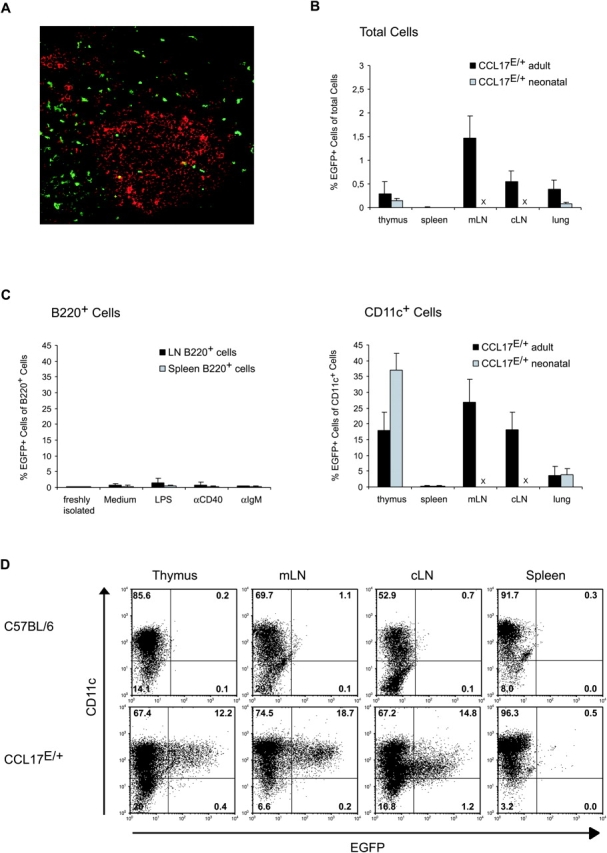
CCL17 is expressed by a subpopulation of DCs. (A) Paraffin sections of cLNs from CCL17E/+ mice were stained with anti-CD45R Abs and a biotinylated goat anti rat secondary antibody followed by Streptavidin PE (red) for detection of B220+ cells. (B) Frequency of CCL17+ cells among total cells and among MACS-enriched CD11c+ cells (C, right) from collagenase-digested organs of adult (black bars) or neonatal (gray bars) reporter mice. B cells were sorted as B220+ cells from spleen or LNs of adult CCL17E/+ mice and stimulated with the indicated substances for 48 h (C, left). × LNs were not investigated in neonatal reporter mice. (D) MACS enriched DCs isolated from collagenase digested thymus, mesenteric LN (mLN), cutaneous LN (cLN), and spleen from control C57BL/6 and CCL17E/+ mice were stained for CD11c and analyzed by flow cytometry. The percentages of cells are indicated within each quadrant.
Using flow cytometry, the frequency of CCL17+ cells was shown to be in the range of 0.25–2% of total cells in the thymus, LN, and lung of neonatal as well as adult mice (Fig. 3 B). These cells did not express markers specific for T cells, NK cells, or macrophages identified as CD11b+ CD11c− cells (unpublished data). Furthermore, CCL17 was rarely expressed in B220+ B cells, even after stimulation with LPS, anti-CD40, or anti-IgM for 48 h (Fig. 3, A and C, left). In contrast, CCL17+ cells accounted for 18% of CD11c+ DCs in the thymus, 27 or 17% in the mLN or cLN, respectively, and ∼2% of CD11c+ cells in the lung (Fig. 3 C, right). In line with the immunohistological data and our previous findings that CCL17 mRNA is not expressed in splenic DCs (9), CCL17+ cells could not be detected among CD11c+ MACS-enriched splenic DCs (<0.6%; Fig. 3, B and C). CCL17+ DCs were also absent from the spleen of neonatal reporter mice, whereas the frequency of CCL17+ DCs in the neonatal thymus appeared higher as compared with adult mice (Fig. 3 C). These findings demonstrate, that CCL17 is specifically produced by a subset of DCs which is widely distributed in most lymphoid and nonlymphoid tissues of the mouse with the marked exception of the spleen.
CCL17+ DCs Display Phenotypic and Functional Features of Mature DCs.
CCL17+ DCs express high levels of the CD11c leukocyte integrin in the thymus and mLNs, whereas those in cLNs predominantly possessed intermediate levels of CD11c (Fig. 3 D). Surface MHC class II and costimulatory molecules such as CD40 and CD86 were significantly enhanced in CCL17+ DCs as compared with CCL17− DCs in thymus and LNs (Fig. 4 A). Additionally, CCL17+ DCs in the LNs expressed the DEC-205 multilectin receptor (26, 27), displayed intermediate/high levels of CD11b, and were CD8− (Fig. 4 A). This was also true for CCL17+ DCs in the thymus and PPs (Fig. 4 A, and unpublished data). Notably, CCL17+ DCs did not express the markers B220 and Gr-1, which in mice identify plasmacytoid DC (reference 4; unpublished data). Next, we investigated the capacity of CCL17+ DCs to prime antigen-specific T cells in vitro. Purified murine CD4 T cells expressing a transgenic TCR specific for the human CRP in the context of I-Ab (23) were cultured with sorted CCL17+ or CCL17− DCs in the presence of hCRP-peptide (89–101). CCL17+ BM-derived DCs induced vigorous T cell proliferation in contrast to CCL17− DCs (Fig. 4 B). LN-derived CCL17+ DCs also displayed an enhanced stimulatory ability, even though the difference was less pronounced in this case (Fig. 4 B, middle and bottom). In addition, BM-derived CCL17+ DCs were highly efficient in stimulating allogeneic T cells in a mixed lymphocyte response (unpublished data). While CCL17+ DCs induced high IFN-γ production by transgenic CD4 T cells, CCL17− BM- or LN-derived DCs were unable to stimulate IFN-γ production (Fig. 4 C). In contrast, splenic DCs which we show here to be consistently CCL17-negative were able to induce T cell responses to similar levels as CCL17+ LN DCs in vitro (unpublished data). We also detected IL-10 at a level of 60 pg/ml in cultures stimulated with CCL17+ BM-derived DCs but less than 4 pg/ml in those of CCL17− DCs. Hence, in agreement with the enhanced stimulatory capacity of CCL17+ DCs, this DC subset can induce IFN-γ as well as IL-10 production in antigen-specific T cells in vitro.
Figure 4.

CCL17+ DCs exhibit a myeloid-related phenotype and possess enhanced stimulatory but diminished phagocytotic capacity. (A) Cytofluorimetric analysis of DCs isolated by MACS from collagenase digested cLNs and thymus of CCL17E/+ mice. The histograms show expression of surface MHC class II (I-Ab), CD86, and CD40 (top histograms) and lineage-related markers DEC205, CD8, and CD11b (bottom histograms) on gated CCL17+ DCs (shaded histograms) and CCL17− DCs (solid lines). The figure shows one of four representative experiments with similar results. (B) DCs were generated from the BM (top) or isolated from LNs (middle and bottom) of CCL17E/+ mice and sorted into CCL17+ DCs (filled diamonds) and CCL17− DCs (open diamonds). DC subpopulations were pulsed with 1 μg/ml CRP peptide (89–101) and cultured with 2 × 104 MACS purified CD4+ T cells from Dep TCR transgenic mice (top and middle). 5 × 103 DCs were pulsed with titrated amounts of CRP peptide (89–101) and incubated with purified CD4+ T cells from Dep TCR transgenic mice (bottom). T cell proliferation was determined by (3H)-thymidine incorporation during the final 17 h of a 72 h culture. Data represent the means ± SD of triplicate determinations from one of two similar experiments. (C) IFN-γ production by CD4+ Dep transgenic T cells after stimulation with CCL17+ DCs (black bars) or CCL17− DCs (white bars). Supernatants were collected from the cultures described in (B) 72 h after stimulation with BM DCs or LN DCs, respectively, and IFN-γ secretion was assessed by ELISA. Data are the means plus SD of duplicate determinations. Shown is one of two similar experiments. (D) Diminished phagocytosis of BM-derived CCL17+ DCs compared with CCL17− DCs. Purified BM-derived DCs from CCL17E/+ mice were incubated for 2 h with fluorescent microspheres before fixation with 4% PFA on coverslides. A representative field of cells analyzed by confocal microscopy is shown.
The phagocytotic capacity of the CCL17+ DC subset was examined by culturing purified BM-derived DCs in the presence of fluorescent particles. Visualization of phagocytosis by confocal microscopy revealed that both CCL17+ and CCL17− DC subsets engulfed the particles (Fig. 4 D). However, the number of fluorescent particles per cell was much lower in the CCL17+ DC population, indicating that they were less efficient in bead uptake. Only 21 ± 2% of CCL17+ DCs phagocytosed more than 4 beads compared with 60 ± 9% of CCL17− DCs as verified by FACS® analysis (unpublished data). In summary, CCL17-producing DCs represent a mature subset of DCs with diminished ability of phagocytosis but high capacity of inducing T cell–mediated immune responses.
Epidermal LCs Express CCL17 after Maturation and upon Entry in Cutaneous LN.
EGFP fluorescence could not be detected in skin sections of naive CCL17E/+ reporter mice, indicating that CCL17 was not constitutively produced by epidermal LC (Fig. 5 A). Next, we prepared epidermal cell suspensions from ear sheets and analyzed CCL17 expression after cytokine exposure in vitro. Whereas no CCL17 expression was found in freshly isolated LCs, 11 or 13% of LCs produced CCL17 after treatment with IL-1α or TNF-α, respectively (Fig. 5 B). The phenotype of CCL17+ DCs in cLNs resembled the phenotype described for skin-derived DCs, i.e., high expression of MHC class II and CD40, and intermediate to high levels of CD11c (Fig. 3 D; reference 28). To investigate whether CCL17+ DCs in cLNs were descendants from LCs, we painted the skin with TRITC. 24 h after application of TRITC we assessed the presence of red fluorescent cells in the draining LNs (Fig. 5 C). All TRITC-positive cells that emigrated from the skin to the draining LNs coexpressed CCL17, demonstrating that LCs up-regulate CCL17 in the LNs after antigen uptake and maturation, in agreement with results presented by Merad et al. (29).
Figure 5.
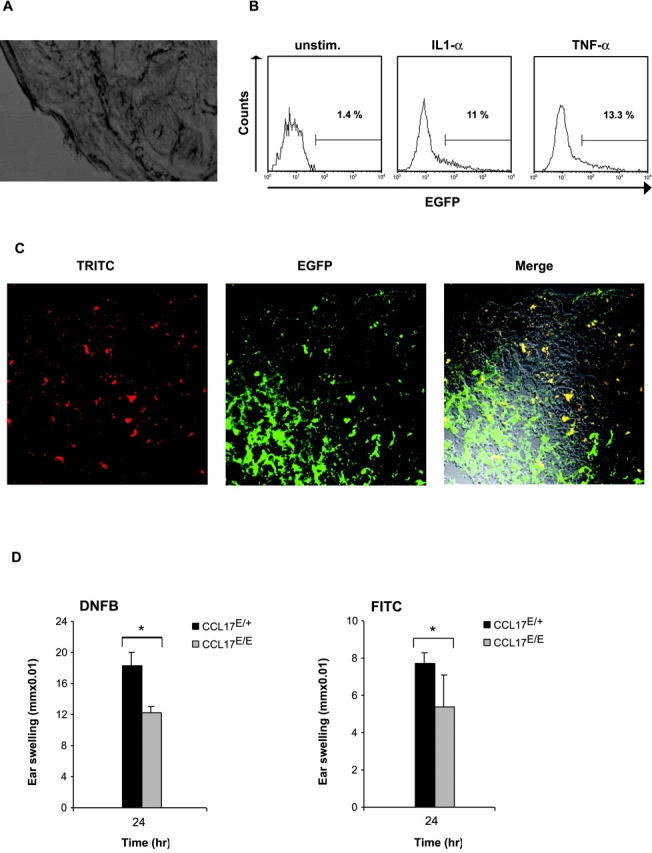
Expression of CCL17 in epidermal LCs upon maturation and impairment of contact hypersensitivity responses in the absence of CCL17. (A) Absence of CCL17-expression in the skin of CCL17E/+ mice. Cryostat sections of tail skin of reporter mice fixed with 4% PFA were analyzed by confocal analysis. (B) Cell suspensions were prepared by trypsinization of epidermal ear sheets of reporter mice and the phenotype of LCs was determined by flow cytometry. LCs were gated based on expression of MHC class II I-Ab and analyzed for CCL17/EGFP expression before or 15 h after culture in cytokine-supplemented medium (GMCSF + IL-1α [10 ng/ml] or GMCSF + TNF-α [10 ng/ml]). (C) Up-regulation of CCL17 by epidermal LCs after contact sensitization and migration to the draining LN. Mice were painted with TRITC dissolved in butylphtalate and acetone (1:1) 24 h before cryostat sections of draining LNs were analyzed by confocal microscopy. Skin-derived LCs in the LN can be identified by TRITC uptake and appear as red-fluorescent cells (first from left). CCL17-expression (green) and merged confocal analysis of green and red fluorescence are shown (second and third from left). TRITC+ cells coexpressing CCL17 can be identified by yellow fluorescence. (D) CCL17-deficient mice display reduced contact hypersensitivity responses to DNFB and FITC. CHS was induced in CCL17-deficient (CCL17E/E) and CCL17E/+ control mice by sensitization with 0.4% DNFB or 0.5% FITC on day 0 and day 1. On day 4, sensitized mice were ear challenged with 0.4% DNFB or 0.5% FITC. Ear thickness was measured before and 24 h after challenge. Data represent mean swelling values ± SD obtained with 11 (DNFB) and 10 (FITC) mice in two independent experiments. The differences between mean values derived from CCL17E/E and CCL17E/+ are statistically significant (P < 0.01).
Impaired Contact Hypersensitivity Response in the Absence of CCL17.
To evaluate the functional importance of CCL17 production by activated Langerhans cells, homozygous CCL17E/E mice carrying a genetic CCL17 deficiency and heterozygous CCL17E/+ mice were sensitized by abdominal painting with DNFB or FITC on two consecutive days. 3 d later mice were challenged with the respective haptens. The contact hypersensitivity (CHS) response to DNFB and FITC measured at 24 h after challenge was significantly diminished in CCL17E/E mice compared with CCL17E/+ control mice as demonstrated by a reduced ear thickness (P < 0.01; Fig. 5 D). Thus, CCL17 plays a crucial role in the development of CHS responses.
Prolonged Survival of Cardiac Allografts in the Absence of CCL17.
We studied the impact of CCL17 in allograft rejection in a model of heterotopic heart transplantation. To establish a model of chronic transplant rejection recipient mice were treated with gallium nitrate known to promote long-term allograft survival due to immunosuppression (30). We observed a significantly enhanced acceptance of allogeneic H-2d heart grafts when transplanted into CCL17-deficient H-2b recipient mice and also a prolonged allograft survival time of more than 35 d as compared with CCL17E/+ H-2b recipients (P < 0.0001; Mann-Whitney test; Fig. 6) . 85% of CCL17E/E mice still accepted the allograft after 60 d, while it was already rejected in 40% of CCL17E/+ recipients. Even 100 d after transplantation allografts were still accepted by 66% of CCL17E/E mice, but only by 30% of CCL17E/+ mice. Indefinite survival of cardiac grafts can be seen in a syngeneic donor/recipient strain combination serving as a control. The markedly enhanced survival of cardiac allografts and the deficiency in allograft rejection in CCL17E/E mice suggests that absence of CCL17 expression in recipient DCs may be responsible for delayed graft rejection.
Figure 6.

Enhanced survival of cardiac allografts in the absence of CCL17. Heterotopic mouse heart transplantation using BALB/c donor hearts and either CCL17E/E, CCL17E/+, or BALB/c mice as recipients was performed as described in Materials and Methods. Recipient mice were injected intraperitoneally with 30 mg/kg of Gallium nitrate on the day of transplantation, day +1, +2, +3, and subsequently every other day until day 30. Cardiac allografts were monitored daily for survival. Results are expressed as percentage of mice with a viable allograft.
Stimulation with Microbial Compounds or Inflammatory Cytokines Augments CCL17 Expression.
To explore the impact of bacterial products or inflammatory mediators on CCL17 expression, BM-derived DCs were stimulated with various known Toll-like receptor (TLR) ligands and TNF-α. The gram-negative bacterial compound LPS as well as the synthetic lipopeptide PAM3Cys induced a twofold increase in the percentage of CCL17-expressing DCs after 15 h of culture and also up-regulated the level of CCL17 expression (Fig. 7 A). Stimulation with the immunostimulatory CpG oligodeoxynucleotide 1668 (CpG-ODN) or Poly-(I:C) induced the strongest CCL17 expression (Fig. 7 A). In contrast, the level of CCL17 production after treatment with TNF-α was relatively low. More than 50% of the CCL17+ DCs participated in IL-12 production upon treatment with CpG-ODN, LPS, or PAM3Cys as determined by intracellular staining with an anti–IL-12p40/p70-specific antibody (Fig. 7 B). Thus, CCL17 synthesis is strongly induced in response to various TLR ligands and correlates with increased production of IL-12.
Figure 7.

CCL17 expression is induced by various TLR ligands and correlates with IL-12 production. (A) Purified BM-derived DCs from CCL17E/+ mice were cultured for 15 h in the presence of the TLR ligands PAM3Cys, Poly-(I:C), LPS, and CpG-ODN or the proinflammatory cytokine TNF-α and EGFP expression was analyzed by flow cytometry. (B) Flow cytometric analysis of intracellular IL-12 production and EGFP expression in the same cultures as described in panel A. Representative data from one of three experiments are shown.
CCL17 Expression in the Spleen Cannot Be Induced by Microbial Challenge.
CCL17 expression upon microbial stimulation in vivo was quantified in various organs 15 h after intraperitoneal LPS injection. CCL17+ DCs were threefold more frequent among DCs of mLNs compared with those of saline injected mice (Fig. 8 A). Notably, the increase in frequency of CCL17+ DCs in PPs and lung was even more profound (6–7-fold) and associated with higher levels of CCL17 expression as compared with the other organs investigated (Fig. 8 B). Similar results were obtained upon injection of reporter mice with CpG-ODN (unpublished data), known to induce an array of inflammatory cytokines in vivo (31). Although up-regulation of MHC class II molecules and costimulatory molecules such as CD40 and CD86 on splenic DCs upon intraperitoneal stimulation confirmed activation of DCs in vivo, the expression of CCL17 in the spleen did not exceed background levels in contrast to the mLN (see Fig. 8 C, and unpublished data). Intravenous application of these stimuli led to the same results (unpublished data). To investigate whether CCL17 expression in the spleen was induced during acute bacterial infection we infected mice with Listeria monocytogenes which has been shown to lead to splenic DC maturation (32). The absence of CCL17 expression in the inflamed spleens 4 d after bacterial challenge is striking (Fig. 8 C), considering that at this time point listeria colonize the spleen and induce a severe inflammatory response (33), accompanied by splenomegaly (unpublished data).
Figure 8.

Accumulation of CCL17+ DCs upon LPS stimulation in various lymphoid organs, but absence of CCL17-expressing cells in the spleen during inflammatory responses. (A) Flow cytometric analysis of mLNs of unstimulated C57BL/6, control, and LPS-stimulated CCL17E/+ mice. DCs were prepared from collagenase digested mLNs from untreated mice or 15 h after LPS stimulation and enriched by MACS. Expression of CD11c, MHC class II (I-Ab), and CD86 were determined. The percentages of cells are indicated in each quadrant. Representative data from one of four independent experiments are shown. (B) MACS enriched DCs of PPs (top) and lung (bottom) of C57BL/6 control mice, unstimulated CCL17E/+ mice, and CCL17E/+ mice 15 h after LPS stimulation were gated as CD11c+ cells and analyzed for CCL17 expression. Values shown in the histograms represent the percentage of CCL17-positive cells. One representative of three experiments is shown. (C) Confocal analysis of cryostat sections of mLN (top) and spleen (bottom) from CCL17E/+ mice 15 h after injection of LPS or CpG-ODN, or 4 d after infection with Listeria monocytogenes, as indicated. (D) CCL17 expression is not induced in splenic DC of CCL17E/+ mice after in vitro stimulation. Flow cytometric analysis was performed on MACS purified splenic DCs after incubation with GMCSF and LPS, PAM3Cys, CpG-ODN, or αCD40 for 24 h. Cells were analyzed for CD11c and EGFP expression.
To exclude that diminished accessibility of splenic DCs for circulating bacterial compounds or activation-induced DC emigration might be the cause for the absence of CCL17 in this environment, we cultured splenic DCs for 24 h in the presence of inflammatory mediators, proven to up-regulate CCL17 expression in BM-derived DCs (see Fig. 7). Remarkably, the level of EGFP/CCL17 expression was not increased by splenic DCs exposed to LPS, PAM3Cys, CpG-ODN, or αCD40 (Fig. 8 D) and CCL17 protein was undetectable in CpG-ODN stimulated splenic DC supernatants by ELISA (unpublished data), demonstrating that DCs located in the spleen fail to produce CCL17 after maturation.
Discussion
Using an in vivo EGFP reporter system, we have elucidated the expression and function of the chemokine CCL17 and its regulation after stimulation with microbial components. Analysis of CCL17E/+ mice demonstrated that CCL17+ DCs belong to the most mature CD8− CD11b+DEC205+ DC population which is part of the so called myeloid-related DC population and includes the descendants of LCs of the skin (34). CCL17 expression may be induced by environmental antigenic stimulation, as CCL17+ DCs are preferentially located in skin or gut draining LNs and at mucosal surfaces, albeit the presence of a high proportion of CCL17+ DCs in the neonatal thymus as well as GM-CSF–stimulated BM cultures suggests that CCL17 may also be expressed constitutively.
In PPs, CD11b+ DCs are anatomically restricted to the subepithelial dome representing the main entry site of intestinal antigens and microbes. From there they migrate to the interfollicular region and accomplish T cell contact after microbial challenge (35, 36). Interestingly, CCL17+ DCs locate to the same areas in PPs, indicating that CCL17 is highly expressed in those intestinal DCs which are actively involved in the uptake of luminal antigens. In addition, an alternative route of bacterial uptake in the intestine was proposed in which DCs localized in the LP penetrate the gut epithelium by opening tight junctions (37). Subepithelial localization of CCL17+ cells in the basal crypts of the colonic LP (Fig. 2) points to a potential role of CCL17 in this kind of antigen uptake. The identification of CCL17+ DCs at important antigen entry sites into the intestinal immune system is in line with the fact that intestinal CCL17 expression is up-regulated in the context of inflammatory bowel disease (38). Up-regulation of CCL17 through receptors of the innate immune system was particularly evident upon application of ligands for TLR 1/2, 3, 4, or 9, like Pam3Cys, Poly-(I:C), LPS, or CpG-ODN (reference 39; Fig. 7). As it is known that systemic application of LPS induces migration of DCs from nonlymphoid tissues into lymphoid organs (40), it is likely that enhanced recruitment of peripheral DCs accounts at least in part for the absolute increase in CCL17-expressing DCs in LNs and PPs.
An outstanding result of the present analysis is that splenic DCs, in sharp contrast to DCs isolated from other lymphoid organs, fail to express CCL17 in response to microbial stimuli. This finding is further strengthened by the fact that CCL22, the second known CCR4 ligand, is also undetectable in the spleen following intravenous LPS injection (41). It is unlikely that circulating LPS is not accessible to splenic DCs, as these cells expressed other activation markers upon systemic LPS stimulation and also did not respond with CCL17 synthesis after in vitro stimulation (Fig. 8). A more likely explanation for the lack of CCL17 expression in the spleen is that splenic DCs are in a distinct differentiation stage which prohibits CCL17/CCL22 production. It is conceivable that CCL17+ DCs represent migratory cells which preferentially home to LNs but not to the spleen as was shown for epidermal LCs (42). Alternatively, CCL17 expression may be actively suppressed by the splenic microenvironment or specifically induced by cell types which are absent from the spleen, e.g., epithelial cells (43).
It is an intriguing question why the production of CCR4 ligands which preferentially attract activated/memory T cells (9, 11, 13, 15) should be undesirable in the spleen, considering the well known capacity of splenic DCs for antigen presentation and T cell priming (1). Splenectomized patients have an increased susceptibility to infections with encapsulated bacteria (44) which is mostly due to a deficiency of protective bacterial polysaccharide specific antibodies of the thymus-independent (TI)-2 type (45). Rapid production of TI-2 antibodies is of major importance when the host has to eliminate pathogens quickly in the case of hematogenic spread of bacterial or viral infections (46), and is best induced in the marginal zone of the spleen as a filter of blood-borne antigens (47–50). Consequently, it may even be detrimental to simultaneously trigger TI immune responses and to attract effector/memory T cells into the spleen because of a possible incompatibility of the corresponding cytokine responses (51). Therefore, the differential function of splenic versus LN DCs regarding CCL17/CCL22 production may ascertain the development of the appropriate type of immune response to systemic versus local infections.
The ability of CCL17+ DCs to produce IL-12 and to induce proliferation as well as IFN-γ production of antigen-specific T cells (Figs. 4 and 7) argues for an immunostimulatory function of these cells. In comparison to CCL17− BM- or LN-derived DCs the CCL17+ DC subset represents the more potent APC population. In line with these results, there is a marked reduction in the T cell–dependent CHS response toward reactive haptens in CCL17E/E mice as compared with CCL17E/+ control mice, suggesting that LC inefficiently attract hapten-specific effector T cells in the absence of CCL17. However, it may also be possible that CCL17 is also involved in regulating the migration of LCs themselves, as it was reported in human that LCs express CCR4 in inflamed skin (52). Cross-priming of viral antigens after intradermal immunization has recently been demonstrated to be mediated by CD8− skin-derived DCs (53) which phenotypically resemble CCL17+ DCs in cLNs. In addition, subcutaneous immunization with a tumor cell vaccine which requires cross-priming by host APCs was shown to be of much higher efficiency than intrasplenic immunization (54). The important role of LNs in the development of cutaneous hypersensitivity responses (42) as well as efficient antiviral immune responses (55) was further underlined by immunization of mouse mutants which are congenitally deficient in the development of peripheral LNs or the spleen.
Our studies provide the first evidence that CCL17 production by recipient cells plays a crucial role in the development of allograft rejection, as the survival of fully MHC-disparate cardiac grafts is markedly enhanced in CCL17E/E recipient mice in a chronic transplantation model. Prior studies also indicated that the interaction of chemokines with their respective receptors is important in graft rejection (56, 57). However, allograft survival time in mice deficient for receptors for DC-derived chemokines was significantly increased only in the presence of immunosuppression induced by Cyclosporin A (CsA; reference 58). Given recent evidence for a participation of CD4 T cells and NK cells in the induction of chronic transplant rejection (25, 59), diminished attraction of CCR4-bearing effector cells to the graft or local draining LNs may be a potential mechanism for enhanced transplant acceptance in the absence of CCL17.
Additional data on the functional role of CCL17 have been obtained by treatment of mice with CCL17-specific antibodies, by which the development of antigen-specific asthmatic lung reactions (60) as well as Propionibacterium acnes induced liver pathology (61) could be inhibited. Unexpectedly, however, CCR4-deficient mice were not protected from allergic airway inflammation but rather showed a marked resistance to LPS-induced septic shock syndromes (21) thus challenging the common view that CCL17 and CCL22 preferentially promote T cell responses with a Th2 bias (13, 15, 17). We and others have previously shown that both Th2 and Th1 cells can be attracted to CCL17 or CCL22 (9, 11), and CCR4 expression was detected on ex vivo isolated human CD4+ memory T cells carrying Th1 as well as Th2 markers (16). Taken together, a functional role of CCL17 as a chemoattractant of effector/memory T cells is evident but this may not be restricted to Th2 cells only. Expression of CCR4 has also been detected on the CD25+ regulatory T cell subset (62) suggesting that CCL17 may possibly also participate in feedback inhibition of ongoing immune responses.
In conclusion, we have demonstrated that the splenic DC population of mice differs from the majority of CD11b+ DCs derived from other lymphoid organs and skin or mucosal surfaces regarding the production of the T cell attractant chemokine CCL17. This result indicates that chemokine usage by DCs is distinctly regulated in secondary lymphoid organs depending on positioning of the organ in central versus peripheral immune surveillance.
Acknowledgments
We are grateful to M. Schiemann and N. Wiechmann for technical help; to B. Kyewski for providing Dep-TCR transgenic mice; and to S. Scheu, H. Hochrein, S. Seewaldt, C. Berek, R. Jack, and K. Pfeffer for advice and comments on the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft through SFB 576, SFB 548, and by the Volkswagen Foundation. I. Lieberam was supported by a PhD scholarship of the Boehringer Ingelheim Fonds.
J. Alferink and I. Lieberam contributed equally to this work.
J. Alferink's present address is Institute for Molecular Medicine and Experimental Immunology, University of Bonn, D-53105 Bonn, Germany.
I. Lieberam's present address is Howard Hughes Medical Institute, Department of Biochemistry and Molecular Biophysics, Center for Neurobiology and Behavior, Columbia University, New York, NY 10032.
Footnotes
Abbreviations used in this paper: BM, bone marrow; CHS, contact hypersensitivity; cLN, cutaneous lymph node; CRP, C-reactive protein; DC, dendritic cell; DNFB, dinitrofluorobenzene; EGFP, enhanced green fluorescent protein; LC, Langerhans cell; LP, lamina propria; MACS, magnetic-activated cell sorting; mLN, mesenteric lymph node; ODN, oligodeoxynucleotide; PFA, paraformaldehyde; PP, Peyer's patch; TLR, Toll-like receptor; TRITC, tetramethylrhodamine-5-(and-6-)-isothiocyanate.
References
- 1.Steinman, R.M. 1991. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9:271–296. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 3.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 4.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 5.Cyster, J.G. 1999. Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J. Exp. Med. 189:447–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Austyn, J.M. 1996. New insights into the mobilization and phagocytic activity of dendritic cells. J. Exp. Med. 183:1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraal, G., E. van Wilsem, and J. Breve. 1993. The phenotype of murine Langerhans cells from skin to lymph node. In Vivo. 7:203–206. [PubMed] [Google Scholar]
- 8.Huang, F.P., N. Platt, M. Wykes, J.R. Major, T.J. Powell, C.D. Jenkins, and G.G. MacPherson. 2000. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lieberam, I., and I. Förster. 1999. The murine beta-chemokine TARC is expressed by subsets of dendritic cells and attracts primed CD4+ T cells. Eur. J. Immunol. 29:2684–2694. [DOI] [PubMed] [Google Scholar]
- 10.Ross, R., X.L. Ross, H. Ghadially, T. Lahr, J. Schwing, J. Knop, and A.B. Reske-Kunz. 1999. Mouse langerhans cells differentially express an activated T cell-attracting CC chemokine. J. Invest. Dermatol. 113:991–998. [DOI] [PubMed] [Google Scholar]
- 11.Schaniel, C., F. Sallusto, C. Ruedl, P. Sideras, F. Melchers, and A.G. Rolink. 1999. Three chemokines with potential functions in T lymphocyte-independent and -dependent B lymphocyte stimulation. Eur. J. Immunol. 29:2934–2947. [DOI] [PubMed] [Google Scholar]
- 12.Cyster, J.G. 1999. Chemokines and cell migration in secondary lymphoid organs. Science. 286:2098–2102. [DOI] [PubMed] [Google Scholar]
- 13.Imai, T., M. Nagira, S. Takagi, M. Kakizaki, M. Nishimura, J. Wang, P.W. Gray, K. Matsushima, and O. Yoshie. 1999. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 11:81–88. [DOI] [PubMed] [Google Scholar]
- 14.Luther, S.A., and J.G. Cyster. 2001. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2:102–107. [DOI] [PubMed] [Google Scholar]
- 15.Sallusto, F., A. Lanzavecchia, and C.R. Mackay. 1998. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today. 19:568–574. [DOI] [PubMed] [Google Scholar]
- 16.Andrew, D.P., N. Ruffing, C.H. Kim, W. Miao, H. Heath, Y. Li, K. Murphy, J.J. Campbell, E.C. Butcher, and L. Wu. 2001. C-C chemokine receptor 4 expression defines a major subset of circulating nonintestinal memory T cells of both Th1 and Th2 potential. J. Immunol. 166:103–111. [DOI] [PubMed] [Google Scholar]
- 17.Andrew, D.P., M.S. Chang, J. McNinch, S.T. Wathen, M. Rihanek, J. Tseng, J.P. Spellberg, and C.G. Elias, III. 1998. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J. Immunol. 161:5027–5038. [PubMed] [Google Scholar]
- 18.Campbell, J.J., G. Haraldsen, J. Pan, J. Rottman, S. Qin, P. Ponath, D.P. Andrew, R. Warnke, N. Ruffing, N. Kassam, et al. 1999. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 400:776–780. [DOI] [PubMed] [Google Scholar]
- 19.Imai, T., M. Baba, M. Nishimura, M. Kakizaki, S. Takagi, and O. Yoshie. 1997. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J. Biol. Chem. 272:15036–15042. [DOI] [PubMed] [Google Scholar]
- 20.Romagnani, S. 2002. Cytokines and chemoattractants in allergic inflammation. Mol. Immunol. 38:881–885. [DOI] [PubMed] [Google Scholar]
- 21.Chvatchko, Y., A.J. Hoogewerf, A. Meyer, S. Alouani, P. Juillard, R. Buser, F. Conquet, A.E. Proudfoot, T.N. Wells, and C.A. Power. 2000. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J. Exp. Med. 191:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karasuyama, H., and F. Melchers. 1988. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur. J. Immunol. 18:97–104. [DOI] [PubMed] [Google Scholar]
- 23.Klein, L., T. Klein, U. Ruther, and B. Kyewski. 1998. CD4 T cell tolerance to human C-reactive protein, an inducible serum protein, is mediated by medullary thymic epithelium. J. Exp. Med. 188:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross, R., C. Gillitzer, R. Kleinz, J. Schwing, H. Kleinert, U. Forstermann, and A.B. Reske-Kunz. 1998. Involvement of NO in contact hypersensitivity. Int. Immunol. 10:61–69. [DOI] [PubMed] [Google Scholar]
- 25.Maier, S., C. Tertilt, N. Chambron, K. Gerauer, N. Huser, C.D. Heidecke, and K. Pfeffer. 2001. Inhibition of natural killer cells results in acceptance of cardiac allografts in CD28−/− mice. Nat. Med. 7:557–562. [DOI] [PubMed] [Google Scholar]
- 26.Kraal, G., M. Breel, M. Janse, and G. Bruin. 1986. Langerhans' cells, veiled cells, and interdigitating cells in the mouse recognized by a monoclonal antibody. J. Exp. Med. 163:981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang, W., W.J. Swiggard, C. Heufler, M. Peng, A. Mirza, R.M. Steinman, and M.C. Nussenzweig. 1995. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 375:151–155. [DOI] [PubMed] [Google Scholar]
- 28.Ruedl, C., P. Koebel, M. Bachmann, M. Hess, and K. Karjalainen. 2000. Anatomical origin of dendritic cells determines their life span in peripheral lymph nodes. J. Immunol. 165:4910–4916. [DOI] [PubMed] [Google Scholar]
- 29.Merad, M., L. Fong, J. Bogenberger, and E.G. Engleman. 2000. Differentiation of myeloid dendritic cells into CD8alpha-positive dendritic cells in vivo. Blood. 96:1865–1872. [PubMed] [Google Scholar]
- 30.Orosz, C.G., E. Wakely, S.D. Bergese, A.M. VanBuskirk, R.M. Ferguson, D. Mullet, G. Apseloff, and N. Gerber. 1996. Prevention of murine cardiac allograft rejection with gallium nitrate. Comparison with anti-CD4 monoclonal antibody. Transplantation. 61:783–791. [DOI] [PubMed] [Google Scholar]
- 31.Sparwasser, T., E.S. Koch, R.M. Vabulas, K. Heeg, G.B. Lipford, J.W. Ellwart, and H. Wagner. 1998. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 28:2045–2054. [DOI] [PubMed] [Google Scholar]
- 32.Liu, T., H. Nishimura, T. Matsuguchi, and Y. Yoshikai. 2000. Differences in interleukin-12 and -15 production by dendritic cells at the early stage of Listeria monocytogenes infection between BALB/c and C57 BL/6 mice. Cell. Immunol. 202:31–40. [DOI] [PubMed] [Google Scholar]
- 33.Poston, R.M., and R.J. Kurlander. 1991. Analysis of the time course of IFN-gamma mRNA and protein production during primary murine listeriosis. The immune phase of bacterial elimination is not temporally linked to IFN production in vivo. J. Immunol. 146:4333–4337. [PubMed] [Google Scholar]
- 34.Henri, S., D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman, and K. Shortman. 2001. The dendritic cell populations of mouse lymph nodes. J. Immunol. 167:741–748. [DOI] [PubMed] [Google Scholar]
- 35.Iwasaki, A., and B.L. Kelsall. 1999. Mucosal immunity and inflammation. I. Mucosal dendritic cells: their specialized role in initiating T cell responses. Am. J. Physiol. 276:G1074–G1078. [DOI] [PubMed] [Google Scholar]
- 36.Iwasaki, A., and B.L. Kelsall. 2001. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer's patch dendritic cells. J. Immunol. 166:4884–4890. [DOI] [PubMed] [Google Scholar]
- 37.Rescigno, M., M. Urbano, B. Valzasina, M. Francolini, G. Rotta, R. Bonasio, F. Granucci, J.P. Kraehenbuhl, and P. Ricciardi-Castagnoli. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:361–367. [DOI] [PubMed] [Google Scholar]
- 38.Jugde, F., M. Alizadeh, C. Boissier, D. Chantry, L. Siproudhis, S. Corbinais, E. Quelvennec, F. Dyard, J.P. Campion, M. Gosselin, et al. 2001. Quantitation of chemokines (MDC, TARC) expression in mucosa from Crohn's disease and ulcerative colitis. Eur. Cytokine Netw. 12:468–477. [PubMed] [Google Scholar]
- 39.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675–680. [DOI] [PubMed] [Google Scholar]
- 40.De Smedt, T., B. Pajak, E. Muraille, L. Lespagnard, E. Heinen, P. De Baetselier, J. Urbain, O. Leo, and M. Moser. 1996. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 184:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang, H.L., and J.G. Cyster. 1999. Chemokine up-regulation and activated T cell attraction by maturing dendritic cells. Science. 284:819–822. [DOI] [PubMed] [Google Scholar]
- 42.Rennert, P.D., P.S. Hochman, R.A. Flavell, D.D. Chaplin, S. Jayaraman, J.L. Browning, and Y.X. Fu. 2001. Essential role of lymph nodes in contact hypersensitivity revealed in lymphotoxin-alpha-deficient mice. J. Exp. Med. 193:1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soumelis, V., P.A. Reche, H. Kanzler, W. Yuan, G. Edward, B. Homey, M. Gilliet, S. Ho, S. Antonenko, A. Lauerma, et al. 2002. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 3:673–680. [DOI] [PubMed] [Google Scholar]
- 44.Hansen, K., and D.B. Singer. 2001. Asplenic-hyposplenic overwhelming sepsis: postsplenectomy sepsis revisited. Pediatr. Dev. Pathol. 4:105–121. [DOI] [PubMed] [Google Scholar]
- 45.Amlot, P.L., and A.E. Hayes. 1985. Impaired human antibody response to the thymus-independent antigen, DNP-Ficoll, after splenectomy. Implications for post-splenectomy infections. Lancet. 1:1008–1011. [DOI] [PubMed] [Google Scholar]
- 46.Ochsenbein, A.F., D.D. Pinschewer, B. Odermatt, A. Ciurea, H. Hengartner, and R.M. Zinkernagel. 2000. Correlation of T cell independence of antibody responses with antigen dose reaching secondary lymphoid organs: implications for splenectomized patients and vaccine design. J. Immunol. 164:6296–6302. [DOI] [PubMed] [Google Scholar]
- 47.Kraal, G. 1992. Cells in the marginal zone of the spleen. Int. Rev. Cytol. 132:31–74. [DOI] [PubMed] [Google Scholar]
- 48.Wardemann, H., T. Boehm, N. Dear, and R. Carsetti. 2002. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J. Exp. Med. 195:771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guinamard, R., M. Okigaki, J. Schlessinger, and J.V. Ravetch. 2000. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 1:31–36. [DOI] [PubMed] [Google Scholar]
- 50.Balazs, M., F. Martin, T. Zhou, and J. Kearney. 2002. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 17:341–352. [DOI] [PubMed] [Google Scholar]
- 51.Garg, M., A.M. Kaplan, and S. Bondada. 1994. Cellular basis of differential responsiveness of lymph nodes and spleen to 23-valent Pnu-Imune vaccine. J. Immunol. 152:1589–1596. [PubMed] [Google Scholar]
- 52.Katou, F., H. Ohtani, T. Nakayama, K. Ono, K. Matsushima, A. Saaristo, H. Nagura, O. Yoshie, and K. Motegi. 2001. Macrophage-derived chemokine (MDC/CCL22) and CCR4 are involved in the formation of T lymphocyte-dendritic cell clusters in human inflamed skin and secondary lymphoid tissue. Am. J. Pathol. 158:1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruedl, C., T. Storni, F. Lechner, T. Bachi, and M.F. Bachmann. 2002. Cross-presentation of virus-like particles by skin-derived CD8(-) dendritic cells: a dispensable role for TAP. Eur. J. Immunol. 32:818–825. [DOI] [PubMed] [Google Scholar]
- 54.Cayeux, S., Z. Qin, B. Dorken, and T. Blankenstein. 2001. Decreased generation of anti-tumor immunity after intrasplenic immunization. Eur. J. Immunol. 31:1392–1399. [DOI] [PubMed] [Google Scholar]
- 55.Karrer, U., A. Althage, B. Odermatt, C.W. Roberts, S.J. Korsmeyer, S. Miyawaki, H. Hengartner, and R.M. Zinkernagel. 1997. On the key role of secondary lymphoid organs in antiviral immune responses studied in alymphoplastic (aly/aly) and spleenless (Hox11(−)/−) mutant mice. J. Exp. Med. 185:2157–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hancock, W.W., W. Gao, K.L. Faia, and V. Csizmadia. 2000. Chemokines and their receptors in allograft rejection. Curr. Opin. Immunol. 12:511–516. [DOI] [PubMed] [Google Scholar]
- 57.Gerard, C., and B.J. Rollins. 2001. Chemokines and disease. Nat. Immunol. 2:108–115. [DOI] [PubMed] [Google Scholar]
- 58.Gao, W., K.L. Faia, V. Csizmadia, S.T. Smiley, D. Soler, J.A. King, T.M. Danoff, and W.W. Hancock. 2001. Beneficial effects of targeting CCR5 in allograft recipients. Transplantation. 72:1199–1205. [DOI] [PubMed] [Google Scholar]
- 59.Shirwan, H. 1999. Chronic allograft rejection. Do the Th2 cells preferentially induced by indirect alloantigen recognition play a dominant role? Transplantation. 68:715–726. [DOI] [PubMed] [Google Scholar]
- 60.Kawasaki, S., H. Takizawa, H. Yoneyama, T. Nakayama, R. Fujisawa, M. Izumizaki, T. Imai, O. Yoshie, I. Homma, K. Yamamoto, and K. Matsushima. 2001. Intervention of thymus and activation-regulated chemokine attenuates the development of allergic airway inflammation and hyperresponsiveness in mice. J. Immunol. 166:2055–2062. [DOI] [PubMed] [Google Scholar]
- 61.Yoneyama, H., A. Harada, T. Imai, M. Baba, O. Yoshie, Y. Zhang, H. Higashi, M. Murai, H. Asakura, and K. Matsushima. 1998. Pivotal role of TARC, a CC chemokine, in bacteria-induced fulminant hepatic failure in mice. J. Clin. Invest. 102:1933–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iellem, A., M. Mariani, R. Lang, H. Recalde, P. Panina-Bordignon, F. Sinigaglia, and D. D'Ambrosio. 2001. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 194:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]