

Abstract
The study of hepatitis C virus (HCV), a major cause of chronic liver disease, has been hampered by the lack of a cell culture system supporting its replication. Here, we have successfully generated infectious pseudo-particles that were assembled by displaying unmodified and functional HCV glycoproteins onto retroviral and lentiviral core particles. The presence of a green fluorescent protein marker gene packaged within these HCV pseudo-particles allowed reliable and fast determination of infectivity mediated by the HCV glycoproteins. Primary hepatocytes as well as hepato-carcinoma cells were found to be the major targets of infection in vitro. High infectivity of the pseudo-particles required both E1 and E2 HCV glycoproteins, and was neutralized by sera from HCV-infected patients and by some anti-E2 monoclonal antibodies. In addition, these pseudo-particles allowed investigation of the role of putative HCV receptors. Although our results tend to confirm their involvement, they provide evidence that neither LDLr nor CD81 is sufficient to mediate HCV cell entry. Altogether, these studies indicate that these pseudo-particles may mimic the early infection steps of parental HCV and will be suitable for the development of much needed new antiviral therapies.
Keywords: hepatitis, viral assembly, glycoproteins, receptor, neutralization
Introduction
Worldwide, several hundred million people are infected with the hepatitis C virus (HCV;* reference 1). Progression to chronic disease occurs in the majority of HCV-infected persons. Infection is associated with an increased risk for liver disease and hepato-cellular carcinoma and has become the main indication for liver transplantation. HCV infection also increases the number of complications in HIV-infected people (2). No vaccine is currently available to prevent new infections and the only treatment for chronic hepatitis C is interferon-α therapy, either alone or in combination with the guanosine analogue ribavirin. However, only ∼40% of patients respond to treatment. Clearly, novel therapeutic strategies are urgently required as the health costs for HCV-infected people are predicted to spiral dramatically in the next few decades.
Unfortunately, no efficient and reliable culture system is available to amplify the virus (3), preventing the elaboration of reliable infection assays. Recently, a model for HCV replication, based on the self-replication of engineered minigenomes in cell cultures, has been established (4, 5). Although very useful in the study of HCV genomic replication, this system does not support production of HCV particles (6). HCV virus-like particles have been generated in insect cells or, alternatively, by pseudotyping vesiculovirus or influenza virus particles with modified HCV E1 and E2 glycoproteins, harboring alterations in their transmembrane domains (7–10). However, they were only poorly infectious or not at all, and inconsistencies in the results prevented their use in functional investigations of HCV cell entry (7, 8). Therefore, new approaches are sorely needed to study HCV assembly and infection to design HCV cell entry inhibitors and to study the humoral immune response against HCV. Here, we sought to overcome these hurdles by developing infectious, genetically tagged HCV pseudo-particles harboring unmodified E1 and E2 glycoproteins. High infectivity of these particles allowed the precise investigation of HCV E1 and E2 glycoproteins and their potential receptors in cell entry, HCV host-range, and neutralization by antibodies from HCV patient sera.
Materials and Methods
HCV E1E2 Expression Constructs.
The phCMV-7a expression vector (see Fig. 1 A) encoding the E1 and E2 glycoproteins from a 1a-type HCV was generated by inserting into a nonpackageable, CMV promoter-driven expression construct (see Fig. 1 B; reference 11) a DNA fragment encoding the last 60 residues of HCV core and all of the E1 and E2 proteins that were isolated from pSKp5E1E2, a construct derived from the pTM1p5E1E2(745) plasmid (12). The phCMV-E1 expression vector (see Fig. 1 A), expressing only HCV E1 glycoprotein, was derived from phCMV-7a by adding a stop codon to the COOH terminus of E1 with primers 5′-actggacgacgcaaagctgc-3′ and 5′-cgcggatcctacgcgtcgacgccggcaaa-3′. The resulting PCR fragment was digested with BamHI and ligated into BamHI-digested phCMV-7a. The phCMV-E2 construct (Fig. 1 A), only expressing an HCV E2 glycoprotein, was obtained by fusing the NH2 terminus of E2 to the COOH terminus of the HCV core using two PCR fragments generated with the two primer pairs 5′-tgcccgcttcagccgaaacccacgtcaccggggga-3′ + 5′-gccagaagtcagatgctcaagg-3′ and 5′-tactctgagtccaaaccg-3′ + 5′-gtgacgtgggtttcggctgaagcgggcacagtcag-3′. The two PCR fragments were fused in a second-round PCR. The resulting DNA fragment was digested with BamHI and ligated into BamHI-cut phCMV-7a. Expression vectors for E1E2 glycoproteins of 1b genotype were constructed by similar strategies.
Figure 1.

HCV E1E2 and retroviral expression constructs. (A) A cDNA derived from the HCV polyprotein gene was used to express the E1E2 glycoproteins and the COOH terminus of the C protein, which provides the signal peptide for E1 (SP E1). The position of stop codons (asterisk) inserted in the expression constructs to terminate translation of the proteins is shown. The transmembrane domain (TMD) of E1 provides the signal peptide (SP E2) for the E2 glycoprotein (46). NTR, nontranslated region; IRES, internal ribosome entry site. (B) The expression constructs encoding the different components required to assemble infectious pseudo-particles are shown. The shaded boxes represent the viral genes and the marker gene (GFP) transferred to the infected cells. The open boxes show the cis-acting sequences. LTR, long terminal repeat; CMV, cytomegalovirus immediate-early promoter; PBS, primer binding site; Ψ, packaging sequence; PPT, poly-purine track; polyA, polyadenylation site. Vector particles were produced by cotransfection of plasmids harboring the packaging functions, the transfer vector and the viral glycoproteins (GP) into 293T cells. The viral GPs were the HCV E1 and E2 glycoproteins, expressed individually or as a ΔCE1E2 polyprotein (Fig. 1 A), the VSV-G, or the RD114 glycoproteins. The supernatants of transfected cells were collected during transient expression, concentrated by ultracentrifugation, and used for target cell transduction.
Packaging and Transfer Vectors Constructs.
The CMV-Gag-Pol murine leukemia virus (MLV) packaging construct, encoding the MLV gag and pol genes, and the MLV-GFP plasmid, encoding an MLV-based transfer vector containing a CMV-GFP internal transcriptional unit, were described previously (see Fig. 1 B; reference 11). The pCMVΔ8.2 (13) HIV-1 Gag-Pol packaging construct contains all HIV-1 genes except env, which encodes the envelope glycoproteins, and nef. The HPPT-EF1α-GFP (14) HIV-1–based transfer vector contains the EF1α internal promoter driving the eGFP marker gene. The gene encoding GFP in this latter vector was replaced by the nlslacZ gene, encoding a nuclear-targeted β-galactosidase, to obtain the HPPT-EF1α-nlslacZ HIV-1–based vector. The phCMV-G (11) and phCMV-RD (15) expression vectors encode the vesicular stomatitis virus (VSV) G protein and the feline endogenous virus RD114 glycoproteins, respectively.
Cells.
The following cells were grown as recommended by the American Type Culture Collection: 293T human embryo kidney cells (CRL-1573); Huh-7 human hepatocellular carcinoma (16); PLC/PRF/5 human hepatoma (CRL-8024); Hep 3B human hepatocellular carcinoma (HB-8064); HepG2 human hepatocellular carcinoma (HB-8065); A431 human epidermoid carcinoma (CRL-1555); Caco-2 human colon adenocarcinoma (HTB-37); HCT 116 human colorectal carcinoma (CCL-247); HOS human osteosarcoma (CRL-1543); HT-1080 human fibrosarcoma (CCL-121); HT-29 human colorectal adenocarcinoma (HTB-38); LoVo human colorectal adenocarcinoma (CCL-229); MCF-7 human breast adenocarcinoma (HTB-22); TE671 human rhabdomyosarcoma (CRL-8805); U118 human glioblastoma (HTB-15); Jurkat human T cell leukemia (TIB-152); CEM human lymphoblastic leukemia (CCL-119); Molt-4 human lymphoblastic leukemia (CRL-1582); Raji Burkitt's lymphoma (CCL-86); CMMT Rhesus monkey mammalian carcinoma (CRL-6299); COS-7 African green monkey fibroblasts kidney (CRL-1651); Vero African green monkey kidney (CCL-81); PG-4 feline astrocyte (CRL-2032); BHK-21 golden hamster kidney (CCL-10); Chinese hamster ovary (CCL-61); BRL 3A rat hepatocytes (CRL-1442); NIH/3T3 mouse fibroblasts (CRL-1658); and QT6 quail fibrosarcoma (CRL-1708).
Cryopreserved primary hepatocytes, isolated from human adult biopsy samples checked for absence of HBV, HCV, and HIV, were purchased from Biopredic International and cultured on collagen I–coated plates according to recommendations of the supplier. Peripheral blood mononuclear cells (PBMCs), derived from healthy blood donors, were isolated, cultivated, and infected as described previously (14, 15).
Production and Detection of Pseudo-particles.
To generate HCV pseudo-particles, 293T cells were transfected with expression vectors encoding the viral components (see Fig. 1 B), i.e., E1E2 glycoproteins, retroviral core proteins, and packaging-competent GFP- or nlslacZ-containing retroviral transfer vectors. In brief, the Gag-Pol packaging construct (8.1 µg), the transfer vector construct (8.1 µg), and the glycoprotein-expressing construct (2.7 µg) DNAs were transfected into 2.5 × 106 293T cells seeded the day before in 10-cm plates using a calcium phosphate transfection protocol (CLONTECH Laboratories, Inc.), as described previously (11). The medium (8 ml/plate) was replaced 16 h after transfection. Supernatants containing the pseudo-particles were harvested 24 h later, filtered through 0.45-µm pore-sized membranes, and used in infection assays. Purified virus samples were obtained by ultracentrifugation of 10-ml viral supernatants through a 1.5-ml 20% sucrose cushion in an SW 41 Beckman rotor (25,000 rpm, 2.5 h, 4°C). Viral pellets were suspended in 50 µl PBS. Immunoblots of producer cell lysates and purified pseudo-particles were performed as described previously (15). Fractionation of the sucrose cushion purified viral pellets was achieved by an overnight equilibrium density centrifugation in a 20–60% sucrose gradient at 35,000 rpm and 4°C in a Beckman SW 41 rotor. Fractions of 0.7 ml were collected, precipitated with TCA, and analyzed by Western blotting.
Infection Assays.
Target cells were seeded in 12-well plates at a density of 8 × 104 cells per well and incubated overnight at 37°C. Unless otherwise indicated, dilutions of viral supernatants containing the pseudo-particles were added to the cells and the plates were incubated for 3 h. The supernatants were removed and the cells were incubated in regular medium for 72 h at 37°C. The infectious titers, expressed as transducing units (TU) per milliliter, were deduced from the transduction efficiencies, determined as the percentage of GFP-positive cells measured by FACS® analysis (15). Transduction efficiency of hepatocytes was determined by counting GFP-positive cells under a UV microscope.
Results
HCV Pseudo-particles Can Be Assembled In Vitro.
HCV proteins are expressed from a single polyprotein precursor and individually released in their respective cell compartments on cleavage by cellular and viral proteases (3, 17). The envelope of HCV is thought to consist of two glycoproteins, E1 and E2, which reside in the ER, where they associate as heterodimers in their prebudding form (18, 19). Therefore, HCV E1E2 glycoproteins were expressed from a polyprotein containing the COOH terminus of the core (Fig. 1 A, C) protein, which serves as signal peptide for E1, and the E1 and E2 glycoproteins (Fig. 1 A; reference 18). This ΔCE1E2 polyprotein was expressed by transient transfection in 293T cells by using an expression construct under control of the CMV immediate-early promoter (Fig. 1 B). As shown by FACS® analysis (see Fig. 2 A) and in situ immunofluorescence (unpublished data) of permeabilized or nonpermeabilized transfected cells, the E1 and E2 glycoproteins were strongly expressed in the cell cytoplasm but could also be detected at the cell surface.
Figure 2.
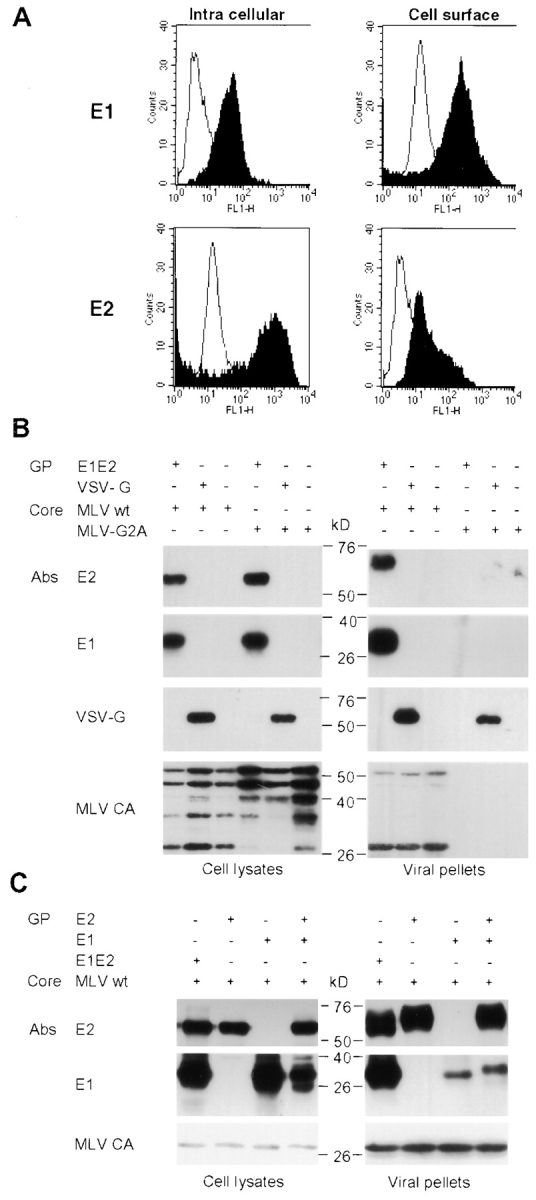
Assembly of HCV pseudo-particles. (A) Detection of E2 glycoproteins by flow cytometry. E1E2-transfected 293T cells were or were not permeabilized before staining with A4 or H53 monoclonal antibodies against E1 (28) and E2 (black areas; reference 47). Cell-bound antibodies were incubated with FITC-conjugated antibodies before FACS® analysis. The background staining was provided by staining the cells with the conjugated antibodies only (white areas). (B) Immunoblots of lysates of 293T-transfected cells and of pseudo-particles pelleted through 20% sucrose cushions are shown. Expression of E1 and E2 glycoproteins from HCV-1a genotype and of MLV core proteins was revealed in reducing and denaturing conditions with A4 and H52 monoclonal antibodies against E1 (28) and E2 (29) or with an anticapsid (MLV CA) antiserum. VSV-G expressed in control pseudo-particles was detected with the monoclonal antibody P5D4. The positions of the molecular mass markers (kD) are shown. E2 present within the viral pellets migrated slightly slower than the cell-associated forms due to modifications of the associated glycans by Golgi enzymes (not depicted). The presence of VSV-G in viral pellets generated with MLV-G2A assembly-defective core proteins is due to empty vesicles formed by VSV-G itself (48). (C) Immunoblots of lysates of 293T producer cells and of purified pseudo-particles generated with E1 or E2 expressed alone, from two separate expression units, or coexpressed in trans or in cis, from a ΔCE1E2 polyprotein (E1E2).
HCV pseudo-particles (HCVpp) were generated by assembling these full-length, unmodified E1 and E2 glycoproteins onto retroviral core proteins derived from MLV. Such pseudo-particles were obtained with E1E2 glycoproteins, derived from HCV genotypes 1a and 1b, that stand among the most prevalent and most resistant to interferon-α therapy (20). Retroviruses were chosen as platforms for assembly of HCVpp because their cores can incorporate a variety of different cellular and viral glycoproteins (15, 21, 22), and because they can easily package and integrate genetic markers into DNA of infected cells (23). HCVpp were produced by transfecting human 293T cells with three expression vectors (Fig. 1 B) encoding a ΔCE1E2 polyprotein, the MLV Gag-Pol core proteins, and a packaging-competent MLV–derived genome encoding the green fluorescent protein (GFP) marker protein. Control pseudo-particles were generated without glycoproteins, with the VSV-G glycoprotein (11), with the feline endogenous virus RD114 glycoprotein (15), and/or with assembly-defective MLV core proteins (MLV-G2A; reference 24). Analysis of immunoblots of transfected cells showed that the structural components of the pseudo-particles were readily detected at the expected molecular masses; i.e., ∼30 kD for E1, ∼60 kD for E2, ∼60 kD for VSV-G (Fig. 2, B and C) , and ∼70 kD for the RD114 glycoprotein (unpublished data). MLV core proteins were detected as Gag precursors of 65 kD that were partially processed by the MLV protease into mature core components (Fig. 2 B). Viral particles were harvested from the supernatant of transfected cells and purified by ultracentrifugation through high density sucrose cushions, a purification process that removes loosely associated glycoproteins (25, 26). The E1 and E2 glycoproteins were readily detected in the pellets of purified virions generated with the wild-type MLV core particles but not with the viral assembly-deficient MLV-G2A mutant (Fig. 2 B). Comparison of the relative levels of VSV-G or HCV glycoproteins in producer cell lysates versus viral pellets suggested efficient incorporation of E1 and E2 into viral particles. Likewise, E1 and E2 glycoproteins could efficiently assemble on retroviral core proteins derived from HIV-1 (unpublished data), raising the possibility of pathogenic interactions between the two parental viruses in vivo because coinfection of patients with HCV and HIV is prevalent (2). Further evidence that the E1 and E2 glycoproteins were properly incorporated on retroviral core particles was provided by their analysis on density gradients (unpublished data). Immunoblots of viral particles fractionated through an equilibrium density centrifugation in 20–60% sucrose gradients indicated that the same fractions contained the Gag-Pol core proteins and the envelope glycoproteins, for both HCVpp and control pseudo-particles, at a sucrose density that corresponded to that of bona fide retroviral particles. Altogether, these results indicated that transient expression of E1 and E2 in 293T cells leads to specific and efficient incorporation of HCV glycoproteins into pseudo-particles generated with retroviral cores. Although HCV envelope glycoproteins have been shown to be retained in the ER in several cell types (19), our FACS® results of transfected 293T cells show that a small fraction of E1 and E2 reach the cell surface (Fig. 2 A), where MLV budding normally occurs (24), most likely due to saturation/leakiness of the ER retention on overexpression. Therefore, our data suggest that HCVpp may bud from the cell surface and not from the ER lumen.
To further investigate the role of HCV glycoproteins in incorporation on retroviral cores, we designed expression vectors that encoded individually either E1 or E2 glycoproteins (Fig. 1 A). Individual expression or coexpression of E1 and E2 from distinct expression units in trans led to normal levels of synthesis, as compared with expression of both glycoproteins in cis from a single E1E2 polyprotein (Fig. 2 C). Moreover, HCVpp were found to incorporate similar levels of E2 glycoprotein, whether it was expressed alone, or coexpressed with E1 in cis or in trans (Fig. 2 C). Finally, incorporation of E1 expressed alone or coexpressed in trans, with E2, occurred but at reduced levels, consistent with the chaperone activity of the latter protein (19).
HCV Pseudo-particles Infect Hepatic Cells.
We assessed the infectivity of HCVpp on a panel of target cell lines. Cells were incubated for 3 h with dilutions of supernatants containing the HCVpp, washed, and cultured until expression of the GFP marker gene harbored by the virions was measured by FACS® analysis 72 h later. Because HCVpp were generated with replication-defective viral components, this procedure allowed evaluation of the specific infectivity of the pseudo-particles after a one-round infection process. The capsid–protein content of HCVpp and control pseudo-particles generated with VSV-G was identical for virions prepared simultaneously (Fig. 2, B and C), allowing direct comparison of their respective infectivity. However, consistent with previous studies comparing batches of pseudo-particles prepared independently (15), we found important variations in capsid amounts (unpublished data) despite similar infectious titers (Fig. 3 A). Thus, to minimize artifacts due to differences in the quality of HCVpp stocks, infectivity was defined as TU per milliliter rather than TU per nanogram of capsid proteins. Infectious titers of up to 3 × 105 TU/ml (mean, 1.7 × 105 ± 105; n = 6) were reproducibly obtained for the HCVpp on Huh-7 human hepato-carcinoma cells (Fig. 3 A). Upon concentration of the producer cell supernatants by ultracentrifugation, infectious titers of 2 × 106 TU/ml, on average, could be readily obtained (Fig. 3 A). The other target cell types used in the infection assays displayed weaker (PLC/PRF/5, Hep 3B, HepG2, Caco-2, HT1080, HT-29, LoVo, MCF-7, U118, 293T, and Vero) or undetectable (A431, HCT 116, HOS, TE671, Jurkat, Molt-4, CEM, Raji, CMMT, Cos-7, BHK-21, CHO, PG-4, BRL 3A, NIH/3T3, and QT6) levels of infection with the HCVpp (Fig. 3 A), although all these cells were readily infected with control pseudo-particles generated with VSV-G and with titers over 7 × 106 TU/ml (unpublished data). This suggested that all the molecules necessary for HCV entry were sufficiently expressed in the former cell types. Infectious HCVpp could be generated at comparable efficiencies with E1E2 glycoproteins derived from HCV genotypes 1a and 1b (Fig. 3 B, h and i) and/or with retroviral core proteins derived from either HIV-1 or MLV (Fig. 3 B, d and h). Incubation of HCVpp and target cells with AZT, a reverse-transcriptase inhibitor that prevents conversion of the retroviral RNA genome of the HCVpp into integration-competent proviral DNA, inhibited transduction (Fig. 3 B, j). Moreover, long-term expression of GFP could be demonstrated after serial passaging of infected cells for more than one month (unpublished data). These results indicated that infection of the target cells led to integration into the host cell DNA and stable expression of the GFP marker gene transduced by the HCVpp. Additionally, no infection could be obtained with viral particles lacking both E1 and E2 glycoproteins, or lacking core proteins, or, alternatively, when using the MLV-G2A assembly-defective core proteins (Fig. 3 B, a–c). Altogether, these results demonstrate that HCVpp harboring the E1 and E2 glycoproteins and retroviral core proteins were infectious, leading to retroviral-mediated integration of their vector genome. Finally, despite reduced levels of E1 incorporation (Fig. 2 C), HCVpp generated with E1 and E2 glycoproteins expressed in trans, from distinct vectors, were nearly as infectious as those generated with the E1E2 polyprotein construct (Fig. 3 B, g and h). However, HCVpp assembled with either E1 or E2 glycoproteins only were 500-fold less infectious (Fig. 3 B, e and f), demonstrating that both glycoproteins need to be coincorporated on HCVpp to allow efficient virus entry and infection.
Figure 3.
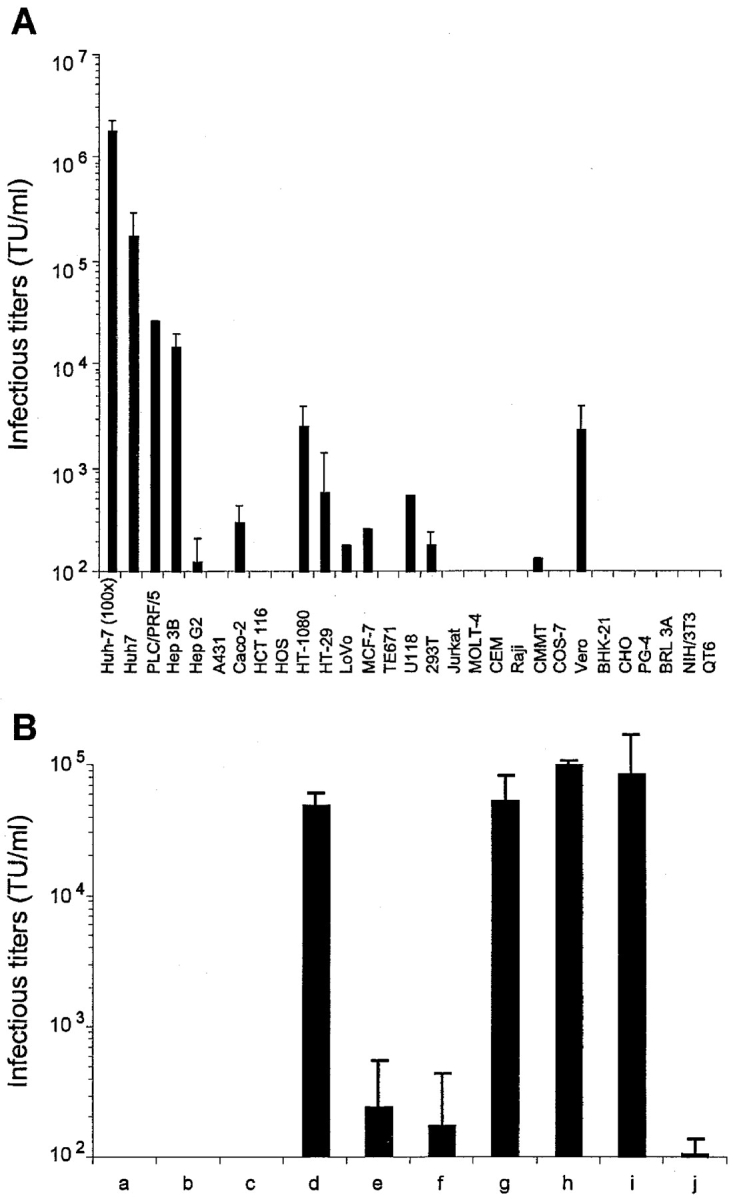
Infectivity of HCV pseudo-particles. (A) The results of experiments performed with different target cell types are displayed as TU per milliliter of supernatant (mean ± SD of up to six experiments) for HCVpp of 1a genotype. The infectivity on Huh-7 cells of HCVpp concentrated 100× by ultracentrifugation is shown. Similar results of infectivity and host-range were obtained for HCVpp of 1b genotype (not depicted). The infectivity of control pseudo-particles generated with VSV-G ranged from 7 × 106 to 2 × 107 TU/ml, depending on the target cell type (not depicted). (B) Infectivity of HCV pseudo-particles generated without E1E2 (a), without retroviral core proteins (b), with MLV-G2A assembly defective core proteins (c), with HIV-1 core proteins (d), with E1 or E2 alone (e or f, respectively), with E1 + E2 expressed in trans from two independents vectors (g), with HCV-1a E1E2 expressed from the same vector in cis (h), or with HCV-1b E1E2 (i). HCVpp were treated with 25 µM AZT (3′-azido-3′-deoxythymidine) before and during infection of target cells (j). Infectious titers (TU/ml) were determined on Huh-7 target cells and are displayed as mean ± SD of up to four experiments.
Hepatocytes represent the principal site of HCV replication in vivo, yet ex vivo studies have suggested that HCV may also infect lymphoid cells (3, 17). To address whether either cell types could be infected, we transduced human adult hepatocytes from biopsy samples and PBMCs from healthy blood donors. Because vectors derived from MLVs do not efficiently transduce these cells (14, 27), we took advantage of the possibility that HCVpp can be generated with HIV-1–derived vectors (Fig. 3 B) that allow efficient transduction of nondividing target cells (13–15, 27). Relative to infection of Huh-7 cells, these HCVpp could readily infect the primary hepatocytes derived from different donors (Fig. 4, A and B) , yet the transduction efficiencies were negatively influenced by the quality and cell culture viability of the individual biopsies. In contrast, infection of PBMCs yielded undetectable levels of transduction with the HCVpp, although control pseudo-particles generated with VSV-G could readily infect these primary cells (unpublished data), as shown previously (14, 15). Altogether, these data suggested that HCVpp seem to reproduce the tropism of infection of wild-type HCV and preferentially infect hepatocytes and hepato-carcinoma cells.
Figure 4.


Results of infection on human primary hepatocytes. Infection assays were performed with HCV pseudo-particles generated with core proteins derived from HIV-1 rather than from MLV, which do not permit transduction of nonproliferating target cells (11). (A) Photographs of human adult primary hepatocytes infected by HCVpp of genotype 1a carrying a lacZ marker gene encoding a nuclear-targeted β-galactosidase. The cells were stained 3 d after infection with X-Gal, as described previously (26). Magnification, 150×. The specificity of infection was demonstrated by the absence of transduction when target cells were infected with HIV-1–derived pseudo-particles lacking glycoproteins (pp cores) or by the reduced levels of transduction when target cells were preincubated with 30 µg/ml JS-81 anti-CD81 antibodies before infection (+JS-81). (B) Quantitative analysis of the infectivity of HCVpp on hepatocytes derived from two donors is expressed as a percentage of infectivity determined on Huh-7 cells.
HCV Pseudo-particles Are Neutralized by Antibodies and Patient Sera.
We sought to determine whether E1 and E2 and their interactions with cell-surface receptors specifically mediate infectivity of the HCVpp. We used a panel of monoclonal antibodies shown previously to specifically react against HCV-1a glycoproteins (9, 28–31). About half of these antibodies significantly inhibited (i.e., >20% inhibition) the infectivity of HCVpp. The H35 and H48 monoclonal antibodies, which recognize conformational epitopes on E2, reduced the infectivity of HCVpp of genotype 1a by up to 70% (Fig. 5 A). That neutralization was incomplete might be due to the fact that these monoclonal antibodies, which were developed and selected for binding to intracellular E1–E2 complexes, have been shown to be sensitive to post-translational modifications of E2 (30). Indeed, we found that, compared with its intracellular counterpart, E2 associated to HCVpp had undergone sugar modifications (Fig. 2, B and C), most likely as a result of its export through the cell secretory pathway (Fig. 2 A). Interestingly, none of these genotype 1a-specific antibodies could neutralize the HCVpp of genotype 1b (unpublished data) or the VSV-Gpp control pseudo-particles generated with VSV-G (Fig. 5 A). This indicated that infectivity of the HCVpp was specifically mediated by E1 and E2 glycoproteins and suggested that these pseudo-particles represent a valid model to investigate the early steps of HCV infection, i.e., receptor binding, membrane fusion, and envelope uncoating, as well as sero-neutralization.
Figure 5.

Neutralization of HCV pseudo-particles. (A) Results of neutralization of HCVpp-1a generated with HCV-1a E1E2 and with MLV core proteins preincubated before infection of Huh-7 cells with 20 µg/ml saturating concentrations of monoclonal antibodies against E1 (A4) or E2 (H31, H33, H35, H44, H48, H53, H54, H60, and H61) glycoproteins of genotype 1a. Hmix: neutralization assays with pooled antibodies. Negative control experiments were performed using no antibodies (Control) or using pseudo-particles generated with VSV-G (VSV-Gpp). Neutralization of the infectivity of these control pseudo-particles was achieved by using the VSV-G neutralizing 41A.1 monoclonal antibody. Results are expressed as the percentages of inhibition of the average infectious titers ± SD relative to incubation in the absence of antibodies. (B) Results of neutralization of HCVpp with HCV patient sera. HCVpp of genotypes 1a or 1b were preincubated for 30 min at room temperature with sera from chronically HCV-infected patients diluted 1:50 before infection of Huh-7 target cells. The genotype of HCV diagnosed in these patients is indicated in brackets. Results are expressed as percentages of inhibition of the average infectious titers ± SD relative to incubation with control sera from healthy individuals. Control experiments were performed using pseudo-particles generated with RD114 glycoproteins (RD114pp), rather than with VSV-G, which exhibits sensitivity to human complement (15). Efficient neutralization of the control pseudo-particles (not depicted) was demonstrated with a hyper-immune goat serum raised against the RD114 SU glycoprotein.
Next, we investigated whether the infectivity of HCVpp could be neutralized by sera derived from chronically HCV-infected patients. HCVpp were incubated with sera before performing infection assays using Huh-7 as target cells (Fig. 5 B). No or nonsignificant neutralization of control pseudo-particles could be detected with patient sera or with control sera derived from healthy donors. However, most, if not all, of the patient sera could neutralize the infectivity of HCVpp in contrast to sera derived from healthy donors (Fig. 5 B). Neutralization levels varied between the different patient sera from 20 to 90% inhibition of infection. Interestingly, at a 1:50 dilution, sera derived from patients infected with genotype 1b neutralized HCVpp-1a and -1b with similar efficiency, suggesting that cross-neutralization occurs (Fig. 5 B).
LDLr and CD81 Are Not the Major Cell Entry Receptors of HCV Pseudo-particles.
Both the LDL receptor (LDLr) and CD81, a member of the tetraspanin family of receptors, have been proposed as putative HCV receptors (32–34). However, recent studies have questioned the role of these molecules in HCV cell entry, despite their unequivocal capacity to bind HCV particles and/or glycoproteins (35–40). Thus, we sought to investigate the contribution of either cell-surface molecule in the early stages of HCV cell entry by performing receptor competition assays. LDLr binds apolipoprotein B within LDL and apolipoproteins B and E (ApoB and ApoE) within very low density lipoprotein (VLDL) complexes; both complexes have been found associated with HCV particles in the plasma of infected patients (36). Purified LDLs and VLDLs were not found to significantly out-compete infection of HCVpp on Huh-7 LDLr-positive cells (Fig. 6 A). When monoclonal antibodies targeted to the receptor binding sites of ApoB and ApoE were preincubated with the HCVpp and control pseudo-particles, only the anti-ApoE antibodies could inhibit, albeit weakly, the infectivity of the HCVpp of genotypes 1a (Fig. 6 A) and 1b (unpublished data). This is consistent with our observation that HCVpp could not or hardly infect LDLr-positive cell lines such as HepG2, Jurkat, CEM, Molt-4, Raji, and TE671 (Fig. 3 A). Thus, although these results do not exclude a role of LDLr in HCV entry, possibly via association of LDLs/VLDLs with HCV particles (36), they suggest that LDLr is not the major receptor of HCVpp entry. Yet this assertion should be mitigated because in contrast to wild-type HCV (36), our HCV pseudo-particles, which were generated in nonhepatic cells (Fig. 2), may not be specifically associated with LDLs and VLDLs.
Figure 6.

Cell entry receptor usage by HCV pseudo-particles. (A) Results of LDL receptor competition assays was performed with 10 µg/ml purified LDLs and VLDLs or using the monoclonal antibodies 5E11, 4G3 (anti-ApoB), or 1D7 (anti-ApoE) preincubated with the pseudo-particles before infection at the indicated concentrations (µg/ml) are shown. Mix: the 5E11, 4G3, and 1B7 antibodies were mixed at the indicated individual concentrations in the neutralization assays. (B) Results of CD81 receptor competition assays are shown. hCD81-LEL GST-fusion polypeptides were preincubated with the pseudo-particles before infection or JS-81 anti-CD81 antibodies were preincubated with the target cells before infection at the indicated concentrations (µg/ml). Results are expressed as percentages of inhibition of the infectious titers obtained on Huh-7 cells relative to titers obtained in the absence of inhibitors. Control experiments were performed using pseudo-particles generated with VSV-G (VSV-Gpp). (C) Comparative results of infection obtained on NIH/3T3, NIH/3T3-hCD81, and Huh-7 cells infected with HCVpp and with VSV-Gpp. Infectious titers (TU/ml) are displayed as mean ± SD of up to three experiments.
Recombinant GST-fusion polypeptides encompassing the large extracellular loop (LEL) of human CD81, which has been shown to bind HCV E2 (29, 34), were then used to investigate the role of hCD81 in HCVpp cell entry. The GST-fusion hCD81-LEL polypeptides, preincubated with the HCVpp, could specifically precipitate E1–E2 complexes (unpublished data), confirming their capacity to bind the E2 glycoprotein (29, 34), and were found to neutralize HCVpp infection on Huh-7 cells, yet with a relatively poor efficiency (Fig. 6 B). Inhibition ranged from 38 to 54% inhibition of infectivity and appeared to be dose-dependent. Consistently, preincubation of Huh-7 cells with JS-81 monoclonal antibody, which blocks binding of recombinant E2 to hCD81 (29), was found to inhibit infectivity of HCVpp in both Huh-7 target cells (Fig. 6 B) and human primary hepatocytes (Fig. 4, A and B). At high antibody concentrations (30 µg/ml), over 90% inhibition of infection could be obtained for HCVpp based on genotypes 1a (Fig. 6 B) as well as 1b (unpublished data). However, several target cells nonpermissive to infection by HCVpp, like TE671, Jurkat, CEM, Molt-4, and Raji cells (Fig. 3 A) were found to express high densities of hCD81 (unpublished data), indicating a lack of correlation between CD81 expression and HCVpp infection. Moreover, expression of hCD81 in nonpermissive NIH/3T3 mouse fibroblasts did not allow infection with the HCVpp (Fig. 6 C). Altogether, these results indicate that although hCD81 binds HCV E2 and might play a role in cell surface attachment of HCV, it is not sufficient by itself to allow HCVpp infection. Additional interactions between the virions and cell-surface components are likely necessary to allow HCVpp entry into cells.
Discussion
Despite 13 yr of research, no efficient and reliable cell culture system is available to amplify HCV particles, making the study of the virus as well as the design of inhibitors and vaccines very difficult. Here, we have exploited two well-documented properties of retroviruses––the capacity to incorporate foreign glycoproteins (15, 21) and the capacity to integrate and express marker genes from replication-incompetent viral particles (23)––to generate a specific, fast, and reliable in vitro infection assay based on pseudo-particles displaying unmodified E1 and E2 HCV glycoproteins. Thus, we describe for the first time the formation of highly infectious HCV pseudo-particles that may share early cell entry properties with parental HCV.
Our data indicate that the E1 and E2 glycoproteins represent the envelope glycoproteins of wild-type HCV and are both required for infection; each glycoprotein is potentially involved in separate steps of cell entry (9, 17). Production of HCV virus-like particles in insect cells has already been reported (7, 39–41). However, these particles were not secreted and their extraction from intracellular compartments yielded virus-like particle preparations that were contaminated with cellular materials. Moreover, although useful to analyze interactions with the cell surface, they were not infectious and did not permit functional investigation of the putative HCV receptors in cell entry. Pseudotyped VSV or influenza virus particles have also been engineered with chimeric E1 and/or E2 glycoproteins, whose transmembrane domains were modified to allow transport to and assembly at the cell surface (8–10, 42). However, such modifications disturb conformation and functions of the E1–E2 complexes (42), and although VSV pseudo-particles stand among candidate HCV vaccines (43), their use as a tool to investigate HCV assembly and cell entry remains controversial (8). In contrast, we found that no structural modifications of the E1E2 glycoproteins were required for their correct assembly on retroviral and lentiviral cores nor for the generation of high titer infectious HCVpp with functional E1E2 glycoproteins.
Our findings that HCVpp show a preferential tropism for hepatic cells (Figs. 3 and 4), and that they are specifically neutralized by anti-E2 monoclonal antibodies as well as sera of HCV-infected patients (Fig. 5), further support the contention that the E1–E2 complexes assembled on HCVpp mimic those displayed by parental HCV. Therefore, the HCVpp described here may represent a valid model to investigate some of the infectious properties of HCV. Although we found evidence that proposed previously that HCV receptor candidates, LDLr and CD81 (32–34), may be involved in the early steps of infection, by mediating binding of HCV glycoproteins and viral particles (9, 10, 29, 31–40, 42), we show that none of these molecules were sufficient to permit infection (Fig. 6). Indeed, some human cells that express LDLr and/or CD81 were found to be nonpermissive to infection (Fig. 3). Additionally, ectopic expression of human CD81 did not restore infection in nonpermissive murine cells (Fig. 6). One may consider that these LDLr+/CD81+ cells are nonpermissive for HCVpp infection as a result of expression of a factor that prevents infection rather than as a result of lack of expression of a critical HCV receptor component. However, we favor the latter hypothesis because if a negative factor was expressed in these cells, it should restrict infection of both the HCVpp and the control pseudo-particles, for which the only difference lies in the composition of the viral envelope. Indeed, such cells could be readily infected when the HCV-specific receptor/entry phase of infection was bypassed by using alternative glycoproteins such as VSV-G or RD114 Env (Fig. 6 C; unpublished data). These data suggest that, like many other viruses (44, 45), the different steps of HCV entry (i.e., binding, internalization, and cell penetration) are mediated by different molecules. With the HCVpp described here, it should become possible to investigate these early events of HCV infection in detail and to identify novel HCV receptors or coreceptors.
Acknowledgments
We thank S. Levy for providing the hCD81-LEL purified polypeptides, S. Abrignani for the hCD81-transfected NIH/3T3 cells, M. Andrawiss for MLV-G2A construct, and C. Bain for the HCV patient sera. We thank D. Lavillette for input at the early stage of the paper, P. Kellam, F. Penin, G. Inchauspé, and H. Gilgenkrantz for stimulating discussions, and C. Goujon, D. Olivier, and V. Delauzun for excellent technical assistance.
This work was supported by Agence Nationale pour la Recherche contre le SIDA, the European Community (contracts QLK3 1999–00859 and QLK2-1999-00356), Association pour la Recherche contre le Cancer, Réscov National Hépatite C, Institut Pasteur de Lille, CNRS, and INSERM (Action Thématique Concertée “Hépatite C”). B. Bartosch is supported by a Marie Curie Fellowship from the European Community.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; HCV, hepatitis C virus; HCVpp, HCV pseudo-particles; LDLr, low density lipoprotein receptor; MLV, murine leukemia virus; PBMC, peripheral blood mononuclear cell; TU, transducing unit; VLDL, very low density lipoprotein.
References
- 1.Lavanchy, D., R. Purcell, F.B. Hollinger, C. Howard, A. Alberti, M. Kew, G. Dusheiko, M. Alter, E. Ayoola, P. Beutels, et al. 1999. Global surveillance and control of hepatitis C. J. Viral Hepat. 6:35–47. 10847128 [Google Scholar]
- 2.Dieterich, D.T. 2002. Treatment of hepatitis C and anemia in human immunodeficiency virus-infected patients. J. Infect. Dis. 185:S128–137. [DOI] [PubMed] [Google Scholar]
- 3.Lindenbach, B.D., and C.M. Rice. 2001. Flaviviridae: the viruses and their replication. In Fields Virology, 4th ed. D.M. Knipe and P.M. Howley, editors. Lippincott Williams & Wilkins, Philadelphia. 991–1042.
- 4.Blight, K.J., A.A. Kolykhalov, and C.M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science. 290:1972–1974. [DOI] [PubMed] [Google Scholar]
- 5.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 285:110–113. [DOI] [PubMed] [Google Scholar]
- 6.Pietschmann, T., V. Lohmann, A. Kaul, N. Krieger, G. Rinck, G. Rutter, D. Strand, and R. Bartenschlager. 2002. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76:4008–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumert, T.F., S. Ito, D.T. Wong, and T.J. Liang. 1998. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J. Virol. 72:3827–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buonocore, L., K.J. Blight, C.M. Rice, and J.K. Rose. 2002. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J. Virol. 76:6865–6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flint, M., J.M. Thomas, C.M. Maidens, C. Shotton, S. Levy, W.S. Barclay, and J.A. McKeating. 1999. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol. 73:6782–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagging, L.M., K. Meyer, R.J. Owens, and R. Ray. 1998. Functional role of hepatitis C virus chimeric glycoproteins in the infectivity of pseudotyped virus. J. Virol. 72:3539–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nègre, D., P. Mangeot, G. Duisit, S. Blanchard, P. Vidalain, P. Leissner, A. Winter, C. Rabourdin-Combe, M. Mehtali, P. Moullier, et al. 2000. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 7:1613–1623. [DOI] [PubMed] [Google Scholar]
- 12.Op De Beeck, A., R. Montserret, S. Duvet, L. Cocquerel, R. Cacan, B. Barberot, M. Le Maire, F. Penin, and J. Dubuisson. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 275:31428–31437. [DOI] [PubMed] [Google Scholar]
- 13.Naldini, L., U. Blömer, P. Gallay, D. Ory, R. Mulligan, F.H. Gage, I.M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 272:263–267. [DOI] [PubMed] [Google Scholar]
- 14.Maurice, M., E. Verhoeyen, P. Salmon, D. Trono, S.J. Russell, and F.-L. Cosset. 2002. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood. 99:2342–2350. [DOI] [PubMed] [Google Scholar]
- 15.Sandrin, V., B. Boson, P. Salmon, W. Gay, D. Nègre, R.L. Grand, D. Trono, and F.-L. Cosset. 2002. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and non-human primates. Blood. 100:823–832. [DOI] [PubMed] [Google Scholar]
- 16.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858–3863. [PubMed] [Google Scholar]
- 17.Bartenschlager, R., and V. Lohmann. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631–1648. [DOI] [PubMed] [Google Scholar]
- 18.Dubuisson, J. 2000. Folding, assembly and subcellular localization of hepatitis C virus glycoproteins. Curr. Top. Microbiol. Immunol. 242:135–148. [DOI] [PubMed] [Google Scholar]
- 19.Op De Beeck, A., L. Cocquerel, and J. Dubuisson. 2001. Biogenesis of hepatitis C virus envelope glycoproteins. J. Gen. Virol. 82:2589–2595. [DOI] [PubMed] [Google Scholar]
- 20.Zein, N.N. 2000. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 13:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott, D.E. 1997. Cellular proteins in HIV virions. Rev. Med. Virol. 7:167–180. [DOI] [PubMed] [Google Scholar]
- 22.Sung, V.M., and M.M. Lai. 2002. Murine retroviral pseudotype virus containing hepatitis B virus large and small surface antigens confers specific tropism for primary human hepatocytes: a potential liver-specific targeting system. J. Virol. 76:912–917. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Nègre, D., G. Duisit, P.-E. Mangeot, P. Moullier, J.-L. Darlix, and F.-L. Cosset. 2002. Lentiviral vectors derived from simian immunodeficiency virus (SIV). Curr. Top. Microbiol. Immunol. 261:53–74. [DOI] [PubMed] [Google Scholar]
- 24.Swanstrom, R., and J.W. Wills. 1997. Synthesis, assembly, and processing of viral proteins. Retroviruses. J.M. Coffin, S.H. Hughes, and H.E. Varmus, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 263–334. [PubMed]
- 25.Kizhatil, K., A. Gromley, and L.M. Albritton. 2001. Two point mutations produce infectious retrovirus bearing a green fluorescent protein-SU fusion protein. J. Virol. 75:11881–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lavillette, D., A. Ruggieri, B. Boson, M. Maurice, and F.-L. Cosset. 2002. Relationship between SU subdomains that regulate the receptor-mediated transition from the native (fusion-inhibited) and fusion-active conformations of the murine leukemia virus glycoprotein. J. Virol. 76:9685–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen, T.H., J. Oberholzer, J. Birraux, P. Majno, P. Morel, and D. Trono. 2002. Highly efficient lentiviral vector-mediated transduction of nondividing, fully reimplantable primary hepatocytes. Mol. Ther. 6:199–209. [DOI] [PubMed] [Google Scholar]
- 28.Dubuisson, J., H.H. Hsu, R.C. Cheung, H.B. Greenberg, D.G. Russell, and C.M. Rice. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flint, M., C. Maidens, L.D. Loomis-Price, C. Shotton, J. Dubuisson, P. Monk, A. Higginbottom, S. Levy, and J.A. McKeating. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flint, M., J. Dubuisson, C. Maidens, R. Harrop, G.R. Guile, P. Borrow, and J.A. McKeating. 2000. Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J. Virol. 74:702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel, A.H., J. Wood, F. Penin, J. Dubuisson, and J.A. McKeating. 2000. Construction and characterization of chimeric hepatitis C virus E2 glycoproteins: analysis of regions critical for glycoprotein aggregation and CD81 binding. J. Gen. Virol. 81:2873–2883. [DOI] [PubMed] [Google Scholar]
- 32.Agnello, V., G. Abel, M. Elfahal, G.B. Knight, and Q.X. Zhang. 1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA. 96:12766–12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monazahian, M., I. Bohme, S. Bonk, A. Koch, C. Scholz, S. Grethe, and R. Thomssen. 1999. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 57:223–229. [DOI] [PubMed] [Google Scholar]
- 34.Pileri, P., Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A.J. Weiner, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani. 1998. Binding of hepatitis C virus to CD81. Science. 282:938–941. [DOI] [PubMed] [Google Scholar]
- 35.Allander, T., X. Forns, S.U. Emerson, R.H. Purcell, and J. Bukh. 2000. Hepatitis C virus envelope protein E2 binds to CD81 of tamarins. Virology. 277:358–367. [DOI] [PubMed] [Google Scholar]
- 36.Andre, P., F. Komurian-Pradel, S. Deforges, M. Perret, J.L. Berland, M. Sodoyer, S. Pol, C. Brechot, G. Paranhos-Baccala, and V. Lotteau. 2002. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 76:6919–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meola, A., A. Sbardellati, B. Bruni Ercole, M. Cerretani, M. Pezzanera, A. Ceccacci, A. Vitelli, S. Levy, A. Nicosia, C. Traboni, et al. 2000. Binding of hepatitis C virus E2 glycoprotein to CD81 does not correlate with species permissiveness to infection. J. Virol. 74:5933–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarselli, E., H. Ansuini, R. Cerino, R. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellnitz, S., B. Klumpp, H. Barth, S. Ito, E. Depla, J. Dubuisson, H.E. Blum, and T.F. Baumert. 2002. Binding of hepatitis C virus-like particles derived from infectious clone H77C to defined human cell lines. J. Virol. 76:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Triyatni, M., B. Saunier, P. Maruvada, A.R. Davis, L. Ulianich, T. Heller, A. Patel, L.D. Kohn, and T.J. Liang. 2002. Interaction of hepatitis C virus-like particles and cells: a model system for studying viral binding and entry. J. Virol. 76:9335–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owsianka, A., R.F. Clayton, L.D. Loomis-Price, J.A. McKeating, and A.H. Patel. 2001. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 82:1877–1883. [DOI] [PubMed] [Google Scholar]
- 42.Matsuura, Y., H. Tani, K. Suzuki, T. Kimura-Someya, R. Suzuki, H. Aizaki, K. Ishii, K. Moriishi, C.S. Robison, M.A. Whitt, and T. Miyamura. 2001. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology. 286:263–275. [DOI] [PubMed] [Google Scholar]
- 43.Rose, N.F., P.A. Marx, A. Luckay, D.F. Nixon, W.J. Moretto, S.M. Donahoe, D. Montefiori, A. Roberts, L. Buonocore, and J.K. Rose. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 106:539–549. [DOI] [PubMed] [Google Scholar]
- 44.Haywood, A. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J. Virol. 68:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klasse, P.J., R. Bron, and M. Marsh. 1998. Mechanisms of enveloped virus entry into animal cells. Adv. Drug Deliv. Rev. 34:65–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cocquerel, L., A. Op de Beeck, M. Lambot, J. Roussel, D. Delgrange, A. Pillez, C. Wychowski, F. Penin, and J. Dubuisson. 2002. Topologic changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J. 21:2893–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cocquerel, L., J.-C. Meunier, A. Pillez, C. Wychowski, and J. Dubuisson. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abe, A., S.T. Chen, A. Miyanohara, and T. Friedmann. 1998. In vitro cell-free conversion of noninfectious Moloney retrovirus particles to an infectious form by the addition of the vesicular stomatitis virus surrogate envelope G protein. J. Virol. 72:6356–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]