

Abstract
Activated insulin-specific CD8+ T cells (IS-CD8+ cells) home to the pancreas, destroy β cells, and cause rapid diabetes upon transfer into diabetes-prone NOD mice. Surprisingly, they also cause diabetes in mouse strains that are free of preexistent inflammation. Thus, we hypothesized that islet-specific homing may be in part dependent on IS-CD8+ cells' recognition of the cognate major histocompatibility complex (MHC)/peptide complexes presented by pancreatic endothelial cells, which acquire the antigen (insulin) from β cells. In fact, islet-specific homing was abrogated in mice that lack MHC class I expression, or presentation of the specific peptide, or have impaired insulin secretion. Moreover, we found that IS-CD8+ cells directly recognized pancreatic endothelial cells in islet organ cultures. Triggering of IS-CD8+ cells' T cell receptor (TCR) led to activation of integrins expressed by these cells. In addition, chemokines, particularly SLC (CCL21), were also required for IS-CD8+ cells' adhesion to endothelial monolayers and for successful homing in vivo. Thus, signaling through TCR and chemokine receptors work in concert to assure firm adhesion of T cells to the pancreatic endothelium. The antigen cross-presentation ability of endothelia may therefore contribute to the specificity of homing of activated T lymphocytes to the tissues where antigens are generated by other cell types.
Keywords: autoimmunity, diabetes, chemokines, CD8 T cells, homing
Introduction
T cells cause autoimmune diabetes. For this process both CD4+ and CD8+ cells are required (1–3). In NOD/LtJ (NOD) mice, CD8+ cells are necessary for initiation of spontaneous diabetes, as NOD mice lacking expression of MHC class I are protected from the disease (4). Insulin-specific CD8+ T cells found within the infiltrates in the pancreata of prediabetic NOD mice (5) recognize a peptide from insulin B chain amino acids 15–23 (InsB15–23)* in the context of the MHC class I molecule, H2-Kd and are potent inducers of diabetes (6). However, in order to destroy pancreatic islets, insulin-specific CD8+ T cells (IS-CD8+ cells) must first home into the islets. T cells follow certain rules when homing to the secondary lymphoid organs, specialized compartments such as intestinal epithelium and skin, and also to sites of inflammation (7, 8). Inflammation is undoubtedly a significant factor in lymphocyte homing, as it stimulates leukocyte adhesion to vascular endothelium (9). It is obviously very important to know whether homing of CD8+ T cells to the site of an immune response against a pathogen is driven solely by nonspecific adhesion mechanisms, or whether T cell receptor specificity contributes to the process. The same question can justly be asked about T cells that mount autoimmune responses against organ-specific self-antigens. It has been previously suggested that the antigen-presenting function of endothelial cells could contribute to lymphocyte homing (10, 11). We found that IS-CD8+ cells were able to induce diabetes in H2-Kd–positive mouse strains other than NOD. This suggested to us that preexistent inflammation in the pancreas is dispensable for the homing of the activated IS-CD8+ cells. Thus, we sought to investigate whether cross-presentation of the specific antigen by pancreatic endothelium is one of the requirements for the homing of these cells into the islets.
Materials and Methods
Mice.
C57BL/6J (B6), C57BL/6-TgN(ACTbEGFP)1Osb (B6-GFP), NOD/LtJ (NOD), DBA/2J, BALB/cJ, (C3H/HeJxDBA/2J) F1 (C3D2), (C57BL/6JxDBA/2J) F1 (B6D2), NOD.129P2(B6)-B2mtm1Unc/J (NOD.β2m-KO), NOD.129S7 (B6)-Rag1tm1Mom (NOD.Rag1-KO), NOD.B10Sn-H2 b, NOD.NON-H2 nb1, B6.NOD-D17Mit21-D17Mit10 (H2 g7, Idd1) (B6.NOD-H2 g7), and B10.BR-H2 k H2-T18a/SgSnJ (B10.BR) mice were obtained from The Jackson Laboratory. C57BL/6-Ins2 Akita (B6Akita/ +) mice and B6Akita/ + backcrossed to NOD for N2–3 (NOD.B6Akita/ +) were a gift of Dr. Leonard Shultz (The Jackson Laboratory, Bar Harbor, ME). All animals were housed in a specific pathogen-free research facility.
CD8+ T Cell Clones.
Insulin-specific, Kd-restricted T cells, IS-CD8+ cells of the TGNFC8 clone (5) were maintained in vitro as described previously (6) in Click's medium (Irvine Scientific) supplemented with 5% FCS (Sigma-Aldrich), 2 × 10−5 M β2-mercaptoethanol (Bio-Rad Laboratories), 20 mM penicillin-streptomycin mixture (Life Technologies), 3 mg/ml l-glutamine (Life Technologies), and 5 U/ml mouse recombinant IL-2. Cells were stimulated every 3 wk by irradiated (2,000 Rad) NOD-derived pancreatic islets, or by NOD splenocytes loaded with 10 μg/ml of the synthetic insulin B chain (InsB15–23, LYLVCGERG) peptide produced by Research Genetics. Pancreatic islets were isolated by a collagenase digestion method, and hand-picked in Hanks' solution (Life Technologies) after purification on a Histopaque 1119 (Sigma-Aldrich) gradient (12). Control Db-restricted T cell clones LPa/2R-1 and B/L specific for the peptides derived from minor histocompatibility antigen H3a (alleles H3aa and H3ab, respectively; reference 13) were provided by Dr. Derry Roopenian (The Jackson Laboratory). Kd-restricted anti-listeriolysin peptide (LLO91–99) CD8+ T cell clone L12.3 (14) was a gift from Dr. Eric Pamer (Memorial Sloan-Kettering Cancer Center, New York, NY).
Induction of Diabetes.
IS-CD8+ cells were washed with PBS, counted, and injected intravenously at 107 cells per animal into irradiated (725 Rad, 24 h in advance) recipients. Both males and females 5–8 wk of age were used. Diabetes was detected by daily monitoring for 21 d of glucose levels in urine, using Diastix reagent strips (Bayer Corp.).
Fluorescent Tracing and Morphometric Analysis.
For trafficking studies, IS-CD8+ and control T cells were incubated at 107 cells/ml for 30 min at 37°C in the dark in complete medium containing 5% FCS and 0.0075 mg/ml of fluorescent dye didodecyl-tetramethylindocarbocyanine perchlorate (DiI; Molecular Probes), then washed three times with PBS. For analysis of short-term proliferation and adhesion studies IS-CD8+ cells were stained in vitro with fluorescent dye carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) as described (12). 107 of labeled IS-CD8+ cells were injected intravenously into irradiated (725 Rad, 24 h in advance) animals. Mice were killed at time points indicated, and their spleens, pancreata, lymph nodes, peripheral blood, and lungs were removed, lymphocytes were isolated as described (5), stained with allophycocyanin-conjugated anti-CD8 (53–6.7) antibody (BD Biosciences), and subjected to FACS® analysis. Alternatively, organs were fixed in 0.1 M periodate-lysine-paraformaldehyde phosphate buffer, sucrose-saturated, and freeze-molded in OCT compound (Sakura Finetek Inc.). 7-μm-thick cryostat sections of the entire pancreas were obtained at 60 μm intervals using a Leica CM1900 cryotom (Leica). Distribution of DiI-labeled CD8+ cells within the islets was examined using a fluorescent microscope DMLB (Leica). At least 100 islets per mouse were examined. Cells were counted within areas relevant to a given islet (see Fig. 1 for representative image). Each IS-CD8+ cell was ascribed a specific position: either at the islet entrance (attached to the capillary wall or located in the isthmus) or inside the islet.
Figure 1.

Preexistent inflammation is not required for IS-CD8+ cells homing into pancreatic islets. (A) IS CD8+ cells cause rapid diabetes in mice that express H2-Kd. Recipient mice were irradiated with 725 Rad and injected with 107 IS-CD8+ T cells. Diabetes was detected by measuring urine glucose, and confirmed by blood glucose measurement. (B) Cryosections of pancreata from NOD and DBA/2J mice injected with 107 DiI-labeled IS-CD8+ cells and killed 24 h later. Islet boundaries are marked by a dotted line. For morphometric analysis labeled cells were counted within an area relevant to a given islet (white rectangle). Cells within islet boundaries were considered to be “inside,” cells within rectangle but outside of islet boundaries were considered to be “at the entrance.” (C) IS-CD8+ cells can be easily detected in disaggregated pancreatic islets from NOD.Rag1-KO mice 24 h after injection. T cells were labeled with CFSE. Numbers indicate percentage of CFSE+ CD8+ cells in total live cell populations within scatter gates containing (but not limited to) the lymphocyte fraction. (D) Pancreatic islets of NOD.β2m-KO mice contain no CFSE+ IS-CD8+ cells 24 h after injection. Gates as in C.
Monoclonal Antibodies and FACS® Analysis.
IS-CD8+ cells were stained in FACS® buffer (PBS with 1% FCS and 0.1% NaN3) with anti-CD11a (2D7), anti-CD18 (C71/16), anti-CD31 (MEC13.3), anti-CD44 (1M7.8.1), anti-CD49d (SG31), anti-CD62L (MEL-14), anti-CD54 (3.E.2), anti-CD103 (2-E7), anti-LPAM (DATK-32), mAbs (all BD Biosciences); anti-CD29 mAb (Chemicon Corp.); anti CD49a (Ha 31/8), anti-CD49b (Ha 1/29) mAbs (Biogen Corp.), and anti–E-cadherin (DECMA-1) mAb (Sigma-Aldrich). This was followed by staining with appropriate FITC- or PE-conjugated secondary antibodies (BD Biosciences). Staining with mouse P-selectin-human Ig fusion protein (BD Biosciences), and mouse ELC-human Ig fusion protein (gift of Dr. Ulrich von Andrian, Harvard University, Boston, MA) was followed by goat anti–human Ig-FITC conjugate (Sigma-Aldrich). IS-C8+ cells were counterstained with PE or FITC-conjugated anti-CD8 antibody (Sigma-Aldrich). For genotyping of H2k/g7 and H2d/g7 segregants of (C3D2)F1xNOD origin, mouse PBL were stained with FITC-conjugated anti-H2-Kk (36.7.5 or 16–3-1) antibodies (BD Biosciences), in combination with Red-316-conjugated anti-CD4 (BRL) and PE-conjugated anti-CD8 antibodies (BD Biosciences). Multi-color analysis was performed using a FACScan™ flow cytometer and using CELLQuest™ software (Becton Dickinson).
Chromium Release Cytotoxicity Assay.
Pancreatic islets or monolayers of cultured aortal endothelial cells were dispersed into single-cell suspension by incubation in cell-dissociation buffer (Sigma-Aldrich), labeled with 100 μCi of Na2 51CrO4 (ICN Pharmaceuticals) in 200 μl of complete Click's medium containing 5% FCS for 2 h at 37°C, washed three times, and cocultured for 8–12 h in 96-well plates (104 targets per well in 200 μl of Click's medium with 5% FCS) with effector IS-CD8+ cells at different effector-to-target ratios with different concentrations of InsB15–23 peptide. To confirm the cytotoxic potential of IS-CD8+ cells, labeled P815 mastocytoma (H2d) cells were loaded with different concentrations of InsB15–23 peptide and used as targets in the 4 h cytotoxicity assay. Specific cytotoxicity was based on the measurement of 51Cr release in 100 μl aliquots of cell-free supernatant using a gamma-counter (Wallac) and calculated using the formula: specific cytotoxicity (%) = ([experimental release − spontaneous release]/[maximum release − spontaneous release]) × 100%.
Immunohistochemistry.
Cryostat sections of fresh-frozen pancreas were cut 7-μm thick, immediately fixed in cold acetone (Fisher Scientific; 3 min at –20°C), air-dried for 2 h, and stained for 1 h at room temperature with mAbs against adhesion molecules: anti-VCAM-1 (429), anti-MadCAM (MECA-367), anti-PECAM (MEC 13.3), anti-CD34 (RAM34), anti-PNAd (MECA-79), anti-ICAM-1 (3.E.2), anti- ICAM-2 (3C4); endothelial cells marker: anti-CD105 (MJ7/18) (all BD Biosciences), and polyclonal antibodies against the β cell marker Glut-2 (whole anti-Glut-2 rabbit serum, gift from Dr. Bernard Thorens, University of Lausanne, Switzerland), and polyclonal goat anti-mouse antibodies against chemokine anti-SLC (R&D Systems). Staining with corresponding FITC- or Rhodamine-labeled secondary antibodies (Jackson ImmunoResearch Laboratories) followed. After the final wash, slides were mounted in Vectashield mounting medium (Vector Laboratories) and examined using fluorescent microscopy.
Pancreatic Islet Cultures and Cytotoxicity Assays.
Pancreatic islets were cultured in Click's medium supplemented with 10% FCS and 0.1 mg/ml of Endothelial Cell Growth Supplement (ICN Pharmaceuticals) in 8-well chambered glass slides (Nunc), precoated with 2% gelatin (Sigma-Aldrich). 7 d later, IS-CD8+ cells were added to the half of the chambers containing islet cultures for 12 h, while the other half was left unaltered. Live cultures were stained for 1 h at room temperature with affinity-purified rabbit anti–Glut-2 polyclonal antibodies and rat anti-CD105 (MJ7/18) or anti-CD31 (MEC13.3) mAbs followed by two 5-min washes and addition of corresponding secondary antibodies. After the final wash, chambers were detached, and slides were mounted in Vectashield medium and immediately examined. CD105+ or CD31+ cells in each well were counted, and the following formula was used to measure specific cytotoxicity: specific cytotoxicity (%) = (mean # of CD105+ cells in intact wells − mean # of CD105+ cells in the wells exposed to IS-CD8+): ([mean # of CD105+ cells in intact chambers]) × 100%. Mixed NOD and B6-GFP islet organ cultures were exposed to IS-CD8+ cells or left intact, stained for CD105, and after the final wash were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 min at +4°C. Slides mounted in Vectashield medium were examined under the fluorescent microscope. The ratio of GFP+CD105+ cells to CD105+ cells was determined for each well. At least 200 cells/well of 3–4 wells per experiment were counted.
Fibronectin Binding Assay.
IS-CD8+ cells harvested four days after IL-2 stimulation were used for the soluble fibronectin (FBN) binding assay performed as described (15). Briefly, 5 × 106 IS-CD8+ cells were preincubated with 5 μg/ml of biotinylated 145–2C11 (anti-CD3) antibody (BD Biosciences) in Click's medium containing 5% FBS for 30 min on ice, washed two times in PBS buffer, and cross-linked with 5 μg/ml of avidin-PE conjugate (BD Biosciences) in the presence of 100 μg/ml of FITC-labeled FBN or FITC-labeled BSA (Sigma-Aldrich) as a control. FITC-labeling was performed using the Fluorotag FITC conjugation kit (Sigma-Aldrich) according to the manufacturer's instructions. Purified R1–2 (anti–VLA-4) and M17/4 (anti–LFA-1) antibodies were added at a concentration of 5 μg/ml during the addition of soluble FBN. Upon addition of labeled FBN-FITC or BSA-FITC, cells were incubated for 30 min on ice followed by transfer to a 37°C water bath for 10 min. After two washes with ice-cold FACS® buffer, cells were analyzed immediately by FACS®.
Primary Vascular Endothelium Cell Culture.
Isolation and culture of vascular endothelium from mouse aorta was performed as described (16). Briefly, neutralized collagen extracellular matrix was aliquoted into 24-well plates, allowed to gel at 37°C for 60 min, and equilibrated overnight with complete endothelial cell medium consisting of complete Click's medium with 20% FCS, and 50 μg/ml of Endothelial Cell Growth Supplement (Becton Dickinson). The segments of thoracic aorta from adult NOD mice were placed endothelial-side down onto the collagen gel. 36 h later 1 ml of complete endothelial cell medium was carefully added. After 3–5 d, the collagen gel was digested with a 0.3% collagenase H (Sigma-Aldrich) solution and the released cells were transferred to a T25 tissue culture flask. After reaching confluence, cells were detached by incubation with cell-dissociation buffer (Sigma-Aldrich), and split at a 1:3 ratio. Passage 2 to 5 cells were used for the experiments.
Parallel Plate Flow Chamber and Adhesion Assay.
Aortal endothelial cells were plated on 35-mm Costar tissue culture dishes and allowed to reach confluence. The cells were then treated for 2–3 h with 100 μM of InsB15–23 or LLO91–99 peptides in complete endothelial cell medium. In some experiments anti-Kd (SF 1–1.1) blocking mAbs were added for 20 min before the adhesion assay. The adhesion of IS-CD8+ cells under shear stress was examined with a parallel plate flow chamber obtained from Glycotech following the manufacturer's instructions and as described previously (17–19). A flow chamber with a 5-mm-wide gasket was used. Negative flow pressure was generated and controlled with an automated syringe pump (Braintree Scientific). IS-CD8+ cells were labeled with CFSE as described above, pretreated in some experiments with 100 ng/ml of mouse recombinant SLC (R&D Systems) in Click's medium for 30 min, and injected at a concentration of 3 × 106/ml into the flow chamber. After IS-CD8+ cells were allowed to settle on the plate for the indicated length of time, the flow rate was increased stepwise every 10 s in increments of 1.28 ml/min (∼4 dynes/cm2 shear stress) to a maximum of 7.7 ml/min (corresponding to ∼24 dynes/cm2). Lymphocyte adhesion was visualized with a Leica inverted fluorescent microscope and recorded using a Spot-RT digital camera (Diagnostic Instruments). Images were converted into digital movies and analyzed using Metamorph software (Universal Imaging Corp.). Labeled IS-CD8+ cells were counted in frames separated by equal time intervals from the start of the stress. Adhesion of cells in each frame was determined as a fraction (%) of the initial number of cells in the frame preceding the start of flow.
Treatment with Pertussis Toxin.
For treatment with Pertussis toxin (PTx), IS-CD8+ cells were harvested, resuspended at 107 cells/ml in complete medium containing 100 ng/ml of PTx (Sigma-Aldrich), and incubated for 2 h at 37°C. The final 30 min of incubation were combined with DiI-labeling. Treated cells were washed and injected into irradiated hosts or cultured under standard conditions in vitro.
Treatment with Anti-SLC.
Irradiated (725 Rad, 24 h in advance) NOD mice were injected intravenously with 30–50 μg of polyclonal anti-SLC antibody (R&D Systems) or control goat Ig. DiI-labeled IS-CD8+ cells were injected 1 h later. Mice were killed at times indicated, and morphometric analysis of the pancreata was performed.
Pancreatic Corrosion Casting.
Corrosion casts of pancreatic microcapillaries were produced as described (20) with modifications. Briefly, mice were killed by overdose of anesthesia, perfused with 250 ml of warm PBS containing 1 U/ml of heparin and green food dye to ensure perfusion quality. Resin was prepared before injection by mixing Mercox, methyl methacrylate (Polysciences Inc.), and Catalyst at a 4:1:0.2 ratio and injected with a 30 g needle into the abdominal aorta near the branching of the upper mesenteric artery. Mercox and Catalyst were purchased as a kit from Ladd Research Industries. In 5–10 min, pancreata with polymerized resin were excised, placed in 54°C water for 1–2 h, incubated overnight with 2 mg/ml of pronase (Roche Diagnostics) in 50 mM Tris buffer with 20 mM EDTA, 2% SDS, pH 8.0, washed in H2O, and immersed in 50% KOH solution at 50°C for 24 h. Casts washed with hot running water were dried, mounted, gold-coated, and examined by SEM.
Online Supplemental Material.
Supplemental Fig. S1 showing that irradiation has no effect on pancreatic homing of IS-CD8+ cells, supplemental Fig. S2 illustrating that homing to the islets depends upon T cell specificity, supplemental Fig. S3 showing that accumulation of IS-CD8+ cells in the islets is not due to local proliferation, and supplemental Fig. S4 showing that activation of IS-CD8+ cells through the TCR leads to activation of integrins are available at http://www.jem.org/cgi/content/full/jem.20021378/DC1.
Results
Homing of the IS-CD8+ Cells to the Pancreatic Islets Does Not Require Preexistent Inflammation.
To address the question of whether the homing of IS-CD8+ cells into the pancreas requires an ongoing inflammation that induces expression of adhesion molecules and chemokines, we performed adoptive transfer experiments. IS-CD8+ cells were injected intravenously into NOD/LtJ, BALB/cJ, and DBA/2J mice and caused rapid diabetes in all strains tested (Fig. 1 A). All of these mice express MHC class I Kd molecules. Strains that do not express Kd were previously shown to be resistant (6). We also performed a direct in vivo tracing experiment. For that, IS-CD8+ cells were labeled with a fluorescent dye, DiI, and injected intravenously into NOD and DBA/2J recipients. 24 h later the pancreata were fixed and cryostat sections analyzed. Labeled IS-CD8+ cells were readily detected in the islets in both NOD and DBA/2J mice (Fig. 1 B). In addition, we injected labeled IS-CD8+ cells into NOD.Rag1-KO mice, which lack T and B lymphocytes, and are inflammation-free. Fig. 1 C shows that IS-CD8+ cells could be easily found in the islets of NOD.Rag1-KO mice.
As we routinely irradiate recipient mice to create a “niche” for donor cells and to minimize host response to incompatible donor cells, we tested whether irradiation affects homing to the islets. There was no difference in homing of IS-CD8+ cells into the islets of irradiated and nonirradiated NOD.Rag1-KO mice (online supplemental Fig. S1). Hence, irradiation was not responsible for the “activation” of endothelium in inflammation-free animals. Thus, IS-CD8+ cells can home to the islets that do not have prior inflammation and their homing is quite tissue-specific. IS-CD8+ cells express cellular adhesion molecules CD31, CD44, and CD54; integrins CD11a (αL chain), CD103 (αIEL chain), CD49b (α2 chain), CD49d and CD29 (α4 integrin VLA-4), and CD18 (β2 integrin LFA-1), as well as P-selectin ligands (not shown). l-selectin, LPAM, E-cadherin, and CD49a molecules were absent from IS-CD8+ cells' surface (not shown). Low levels of ligands for LFA-1 are known to be constitutively expressed by the endothelium of the islets, while the VLA-4 ligand VCAM-1 is inducible (21–23). Thus, integrins and/or P-selectin ligands are likely to contribute to the early stages of adhesion to the pancreatic endothelium engaging constitutively expressed counterparts present in the absence of inflammation.
Homing to the Islets Requires MHC Class I Expression.
As inflammation was not necessary for the initial steps in pancreatic homing of IS-CD8+ T cells, we tested whether homing of IS-CD8+ T cells could be antigen-driven. For that, NOD mice lacking MHC class I molecules (NOD.β2m-KO mice), were injected with labeled IS-CD8+ cells and their pancreata examined (Fig. 1 D). There was hardly any significant number of IS-CD8+ cells in the isolated islets 24 h after injection. To show that pancreas was the only organ in the NOD.β2m-KO mice where IS-CD8+ cells refused to home, compared with NOD mice, we analyzed cryostat sections of several tissues from NOD and NOD.β2m-KO mice injected with DiI-labeled IS-CD8+ cells (Fig. 2) . This systematic analysis of tissue distribution of the IS-CD8+ cells revealed that the difference between the two recipients was obvious only in the pancreatic islets. Was MHC class I–dependent homing indeed peptide specific? Evidence for peptide specificity came from the analysis of mice that expressed the H2k MHC haplotype along with expression of the H2-Kd molecule. Such mice are resistant to IS-CD8+ cell-induced diabetes (see online supplemental Table S1), and resistance was linked to the lack of specific homing of activated IS-CD8+ cells to the islets in H-2k/g7 animals (Fig. 3 A, a): 24 h after IS-CD8+ cell injection H-2k-positive islets contained ∼16-fold fewer labeled cells. Further analysis revealed that the presence of H2 k (or closely linked genes) caused the lack of presentation of InsB15–23 peptide by Kd molecules. This was shown by testing of the direct recognition of islet β cells from H2k/d animals by IS-CD8+ cells. Islets from C3D2 (H2k/d) and control B6D2 (H2b/d) mice were dispersed to single cell suspensions, labeled with 51Cr, and exposed to IS-CD8+ cells.
Figure 2.
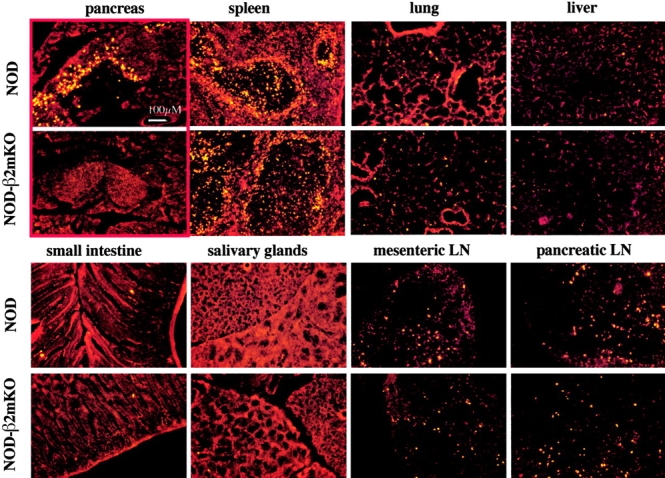
MHC class I expression affects homing of IS-CD8+ cells to the pancreatic islets, but not to other tissues. Cryostat sections of the indicated organs isolated 24 h after injection of the DiI-labeled IS-CD8+ cells. The red color of the tissue is autofluorescence, DiI stained cells are bright yellow. Most typical examples of sections are shown. The red frame highlights the difference in homing of the labeled cells to the pancreata of NOD and NOD.β2m-KO mice.
Figure 3.

Lack of specific MHC class I–peptide complexes affects homing of IS-CD8+ cells. (A) In the presence of H2k IS-CD8+ cells fail to migrate into islets (a), or kill islet cells (b). Both properties can be explained by the lack of Kd-InsB15–23 complexes. (a) Labeled IS-CD8+ cells were injected into (C3D2XNOD)F1 mice with either H2k/g7 (filled bar) or H2d/g7 (clear bar) MHC alleles. Morphometric analysis of the pancreata was performed 24 h later. Combined data from two experiments represent mean numbers of IS-CD8+ cells per islet (within white rectangle in Fig. 1)±SE. n, number of mice per group. *, difference vs. H2d/g7 mice is significant by Student's paired t test (P < 0.05). (b) IS-CD8+ cells kill islet cells isolated from H2b/d B6D2 mice, but not the islet cells from H2k/d C3D2 mice. Addition of exogenous InsB15–23 peptide (done at a 50:1 effector-to-target ratio only) restored killing of H2k/d islet cells to the level of killing of H2b/d islets. Data from a representative experiment. (B) Morphometric analysis of pancreata from β2m-deficient, and -sufficient NOD mice for the presence of labeled IS-CD8+ cells at different times after injection. Each cell was ascribed its position as shown in Fig. 1. n, number of mice of each genotype analyzed at each time point. Data show mean numbers of IS-CD8+ cells per islet detected at the indicated location ± SE. *, difference between NOD and NOD.β2m-KO is significant by Student's t test (P < 0.01).
β cells from mice positive for H2k were resistant to lysis (Fig. 3 A, b). However, when the InsB15–23 synthetic peptide was added exogenously, lysis was restored (Fig. 3 A, b), arguing that the block of lysis was neither due to a general malfunction of Kd molecules, nor to activation of inhibitory receptors on the CTL. The only reasonable explanation for this finding was that products of genes linked to H2 k were prohibiting the formation of functional Kd/insB15–23 complexes. Thus, the absence of specific antigenic complexes, and not the general deficiency in MHC class I (found in NOD.β2m-KO mice), is critical for the lack of specific homing of IS-CD8+ cells to the pancreas.
To confirm that homing to the islets is not a property of any activated T cell, we also used T cell clones with different specificities. DiI-labeled cells of LPa/2R-1 CD8+ clone (13) reactive to Db complex with a peptide from minor histocompatibility antigen H3aa which is widely expressed in NOD mice were not detectable in the pancreatic islets 2 or 24 h after injection, while the numbers of the LPa/2R-1 cells in the other tissues such as lungs (online supplemental Fig. S2) were comparable to the numbers of IS-CD8+ cells. Similarly, two other CD8+ T cell lines, Db-restricted H3ab-specific clone B/L (recognizing H3ab allele not expressed in NOD mice; reference 13; not shown), and L12.3, Kd–restricted anti-listeriolysin clone (14) migrated to spleens but not to pancreata of NOD recipients (online supplemental Fig. S2).
Thus, homing of IS-CD8+ cells appears to be both antigen-driven and pancreas-specific. However, IS-CD8+ cells need to cross the endothelial cell layer in order to penetrate the islets. Taking in consideration that homing also occurs in the absence of local inflammation, we hypothesized that endothelial cells present MHC–insulin peptide complexes to T cells, stimulating their adhesion and penetration into the islets.
Endothelial Cells from Pancreatic Islets Can Be Recognized by IS-CD8+ Cells In Vivo and In Vitro.
To test this hypothesis, we further used NOD.β2m–KO mice as recipients of DiI-labeled IS-CD8+ T cells. Their islets were analyzed at different time points after injection of IS-CD8+ cells (Fig. 3 B). Morphometric analysis allows not only a calculation of the numbers of infiltrating cells, but also reveals their localization. The numbers of cells accumulated in different locations reflect the process of T cell homing. As expected, the numbers of IS-CD8+ cells found in the MHC class I–deficient islets 24 h after injection were extremely small compared with those found in the islets of MHC-sufficient NOD mice. Most importantly, very few T cells were found to be located at the islet vascular isthmus in MHC class I–negative NOD mice. If their firm adhesion to the endothelium was MHC class I independent, we would be able to detect IS-CD8+ cells in the vascular isthmus of NOD.β2m–KO mice, which was not the case. It was possible, however, that accumulation of IS-CD8+ cells in the islets of NOD mice was exclusively due to a secondary activation of endothelium caused by recognition of Kd–insulin peptide complexes in the islets themselves, which would be impossible in β2m-negative mice. That would lead to fast “passing-through” of IS-CD8+ cells in β2m-negative islets, and 24 h after injection we would be simply unable to detect that. However, we were able to detect IS-CD8+ cell accumulation at the entrance rather early (1.5 h after injection) in NOD mice (Fig. 3 B). Importantly, such accumulation preceded penetration into the islets. In contrast, at no time points could we detect accumulation of IS-CD8+ cells in β2m-negative mice at the islet isthmus or inside the islets. These results argue that not only the islet penetration, but also the adhesion to endothelium, were abolished by elimination of MHC class I expression. Moreover, accumulation with time of IS-CD8+ cells at the isthmi and inside the islets is not due to local proliferation, but due to additional recruitment from the bloodstream. 24 h after injection we found no explicit proliferation of CFSE-labeled IS-CD8+ cells isolated from the islets compared with IS-CD8+ cells isolated from the spleens (online supplemental Fig. S3).
To further support the hypothesis that endothelial cells can function as APC for IS-CD8+ cells, we proceeded to show that endothelial cells in the pancreatic islets could be directly recognized by IS-CD8+ cells. For that we used islet organ cultures. On day 7 of the islet organ culture IS-CD8+ cells were added for 12 h. Immediately after that, the live cultures were stained with antibodies against a β cell marker, glucose transporter 2 (Glut-2), and an endothelial marker CD105 (endoglin). Glut-2+ and CD105+ cells are clearly nonoverlapping populations in islets of the intact pancreas or in isolated islets in culture (Fig. 4 A). The numbers of remaining CD105+ cells were counted in islet cultures exposed or not to IS-CD8+ cells. In the cultures of MHC class I–sufficient NOD or B6.NOD-H2 g7 (congenic strain with H2g7 on B6 background) islets the majority of the Glut-2+ cells were destroyed by IS-CD8+ cells. Importantly, most of the CD105+ cells were also destroyed. In NOD.β2m-KO or B6 islet cultures, on the other hand, Glut-2+ and CD105+ cells remained intact, indicating that the killing of CD105+ cells (or CD31+ cells in some experiments, not shown) was specific and that B6 genetic background did not matter for susceptibility to specific killing (Fig. 4 B, a). Moreover, when NOD endothelial cells were isolated from nonislet source (aorta) and cultured without insulin-producing β cells, they were insensitive to IS-CD8+ cells cytotoxicity unless exogenous Ins-B15–23 peptide was added (Fig. 4 B, b). However, the possibility existed that in NOD islet cultures the recognition by IS-CD8+ cells of their primary targets, the antigen-expressing β cells, led to secretion of cytotoxic cytokines that killed endothelial (CD105+) cells nonspecifically. This would not have occurred in NOD.β2m-KO or B6 islet cultures, as MHC class I Kd molecules were not present, or in aortal endothelial cells. To address this caveat, we cultured a mixture of the islets isolated from NOD and B6-GFP mice, which ubiquitously express green fluorescent protein (GFP). These mixed islet cultures were exposed to IS-CD8+ cells and stained with anti-CD105 (Fig. 4 C). The ratio of cells double-expressing GFP and CD105+ to the total number of CD105+ cells was measured by direct cell counting using fluorescent microscopy. The results revealed that NOD-derived GFP−CD105+ cells were eliminated, while B6-derived GFP+CD105+ cells remained intact (Fig. 4 C, c). That suggested that CD105+cells must express the cognate MHC–peptide complex to be destroyed by IS-CD8+ cells.
Figure 4.

Endothelial cells from islet organ cultures are killed by IS-CD8+ cells in antigen-specific fashion. (A) β cells and endothelial cells can clearly be distinguished both in the native islets (a) and in in vitro culture (b) by staining with anti-Glut-2 (green) and anti-CD105 (red). (c) Nomarsky (DIC) image of islet culture (same field as in b). 10× and 20× lenses were used in a and b, c, respectively. (B) CD105+ cells in the islet cultures are destroyed by IS-CD8+ cells in vitro. (a) Specific killing of endothelial cells in islet organ cultures from NOD, NOD.β2m-KO, B6.NOD-H2g7, and B6 mice by IS-CD8+ cells added for 12 h on day 7 of islet culture was calculated using the formula: Specific cytotoxicity (%) = [(mean # of CD105+ cells in intact wells − mean # of CD105+ cells in the wells exposed to IS-CD8+ cells)/mean # of CD105+ cells in intact chambers] × 100%. CD105+ cells (3–4 parallel wells per experiment) were counted using a fluorescent microscope. Combined data from three independent experiments. *, difference vs. NOD is significant (P < 0.005); **, difference vs. B6.NOD-H2g7 is significant (P < 0.005) by Student's paired t test. (b) Endothelial cells isolated from NOD aorta were killed by IS-CD8+ cells in 51Cr release assay only when exogenous Ins B15–23 peptide (0.1 μM) was added. Addition of 0.1 μM of control Kd-binding LLO91–99 peptide had no effect. Data from a representative experiment. (C) IS-CD8+ cells do not kill bystander Kd-negative CD105+ cells in the presence of Kd-sufficient targets. Islets from NOD and B6-GFP mice were mixed, cultured in vitro for 7 d, then exposed to IS-CD8+cells for 12 h or left intact, stained for CD105, fixed, and analyzed by fluorescent microscopy. Representative images of NOD and B6-GFP mixed islet cultures before (a), and after exposure to IS-CD8+cells (b). 40× objective was used. (c) Formal estimation of the results shown in a and b. Specific loss of GFP-negative, CD105+, Kd-sufficient NOD cells was determined by the proportion (%) of GFP+ CD105+ cells among total CD105+ cells in mixed NOD and B6-GFP islet cultures left intact or exposed to IS-CD8+ cells. Combined data from three independent experiments. *, difference is significant (P < 0.05) by Student's t test.
How is the insulin peptide-Kd complex acquired by endothelium? High local concentration of insulin suggests that secreted protein may be taken up by endothelial cells and then cleaved to peptides. If that was true, β cells that make insulin (and insulin peptide-Kd complexes) but have impaired insulin secretion, would be susceptible to lysis by CTL but inaccessible for them because the endothelium is not cross-presenting the antigen. Support for this hypothesis came from the analysis of IS-CD8+ cells trafficking into the islets of NOD.B6Akita/ + mice homozygous for H2g7 and heterozygous for the dominant mutation designated Ins2 Akita in the insulin 2 gene. This point mutation (Cys96Tyr) affects processing and secretion of both insulin 1 and insulin 2 proteins in a dominant fashion, so that insulin release is greatly diminished and diabetes develops very early (24, 25). Labeled IS-CD8+ cells were found to home significantly less efficiently (tested at 2 and 24 h after injection) to the islets of mice bearing the Ins2 Akita mutation than to the islets of their wild-type littermates (Fig. 5 A). Very importantly, β cells isolated from mutant mice were sensitive to IS-CD8+ cells in vitro (Fig. 5 B). Because of the substantial depletion of β cells from the pancreatic islets of Ins2 Akita mice (24), the level of cytotoxicity for mutant islets was lower than that observed for wild-type islets (Fig. 5 B). Nevertheless, IS-CD8+ cells were clearly capable of direct recognition of the islet cells of NOD.B6Akita/ + mice. Thus, IS-CD8+ cells were able to recognize β cells but stopped short of adhesion to the islet vasculature. This argues that secretion of insulin, the protein donor of antigenic peptide, is essential for presentation of the Kd-Ins15–23 complex by endothelial cells, and suggests that generation of MHC–insulin peptide complexes depends on degradation of insulin by endothelial cells.
Figure 5.

Secretion of insulin is a prerequisite for homing of IS-CD8+ cells. Labeled IS-CD8+ cells were injected into NOD.B6Akita/ + or NOD.B6+/ + wild-type littermates. All mice were homozygous for H2g7. (A) Morphometric analysis of pancreata was performed at indicated times after injection. Data represent mean numbers of IS-CD8+ cells per islet±SE. n, number of mice per group. Difference between groups is significant, as *P = 0.04, **P = 0.03 by Student's t test. (B) Pancreatic islet cells isolated from NOD.B6Akita/ + and NOD.B6+/ + were sensitive to direct lysis by IS-CD8+ effector cells, showing that the cognate MHC–peptide complex was produced by β cells and expressed on their surface. B6Akita/ + - Kd-negative control. Data from a representative experiment.
Chemokine Receptors Are Involved in IS-CD8+ Cell Homing.
Although signaling through TCR leads to activation of integrins on the T cell surface (references 26–28; we found that it was also true for the particular IS-CD8+ cell clone that we were using, see online supplemental Fig. S4), it is unlikely that this mechanism alone could account for the strength of interactions with the endothelium needed for complete arrest of T cell rolling. Integrin activation may be also triggered through G-protein–coupled chemokine receptors (29–32). As homing of lymphocytes usually depends heavily on the recognition of tissue-specific chemokines (33), we sought to address the question of whether chemokine receptors are involved in trafficking of IS-CD8+ cells to the pancreatic islets. We started with treatment of IS-CD8+ cells with PTx, a potent inhibitor of G-protein–coupled receptor signaling. IS-CD8+ cells treated with PTx or PBS were stained with DiI and injected into NOD mice. The cells treated with PTx did not lose their viability and responded normally to InsB15–23 peptide stimulation 24 h after treatment (not shown). The presence of IS-CD8+ cells in the pancreatic islets was assessed 24 h after injection by analysis of cryostat sections (Fig. 6 A). PTx-treated IS-CD8+ cells were practically absent from the islets, suggesting the possibility that chemokine receptors were involved in the homing of IS-CD8+ cells. The multitude of chemokines and their receptors complicates the task of identifying of a specific pair. However, when we examined lymphoid organs of NOD mice injected with PTx-treated IS-CD8+ cells, we detected a clue to the identity of the chemokine that could assist in trafficking to the pancreatic islets. In our experiments, the presence of labeled IS-CD8+ cells in the spleen always serves as an internal control for the quality of the transfer. When NOD mice were injected with IS-CD8+ cells treated with PTx, we found that the distribution of these cells in the spleen was different from the distribution of untreated cells: PTx-treated cells avoided T cell zones (Fig. 6 B, a and b). Homing of T cells to the T cell zones of the secondary lymphoid organs is controlled by a pair of chemokines, CCL21 (also known as 6Ckine, Exodus-2, SLC) and CCL19 (MIP-3β, Exodus-3, ELC; 34, 35). SLC expression in the T cell zones of spleen by immunofluorescence is shown in Fig. 6 B, c. As both SLC (CCL21) and ELC (CCL19) chemokines have been shown to trigger β2 integrin affinity and mobility changes promoting LFA-1–mediated lymphocyte adhesion (36, 37), we asked whether these chemokines were involved in the homing of IS-CD8+ cells to the pancreatic islets.
Figure 6.

Chemokines play an important role in IS-CD8+ cells homing to the islets. (A) Treatment of IS-CD8+ cells with PTx which blocks signaling through chemokine receptors, abolishes their homing to the islets. IS-CD8+ cells treated with PTx in vitro were labeled and injected into NOD mice. In 24 h untreated cells penetrated into islets (a), while cells treated with PTx did not (b). (c) Morphometric analysis shows quantitative measurement of homing of PTx-treated vs. nontreated IS-CD8+ cells. n, number of mice per group. *P < 0.003 by Student's t test. (B) Unlike untreated IS-CD8+ cells (a), PTx-treated IS-CD8+ cells lost ability to home to T cell zones in the spleen (b), suggesting that SLC may be involved. (c) SLC (CCL-21) expression in the splenic T cell zones revealed by staining with specific anti-SLC antibodies (green fluorescence). Serves as positive control to D. (C) SLC is a primary chemokine responsible for IS-CD8+ cells homing to the islets. Pretreatment of recipient mice with anti-SLC serum abolished IS-CD8+ cells homing to the islets. Data represent mean number of IS-CD8+ cells per islet ± SE detected at indicated times after injection. n, number of mice per group. *P = 0.002, **P = 0.001 by Student's t test. (D) SLC expression was found in the isthmus regions of the islets in NOD, DBA/2J, and C3D2 mice, indicating that SLC is not the sole factor determining homing of IS-CD8+ cells. Negative control included treatment with normal goat serum and secondary labeled antibodies. Islet boundaries are marked by a dotted red line.
First, we blocked SLC function by treating mice with anti-SLC antibodies in vivo, followed by the transfer of labeled IS-CD8+ T cells. Cryostat sections of the pancreata from mice injected with anti-SLC were compared with those from mice injected with normal goat Ig by counting labeled cells per islet (Fig. 6 C). Anti-SLC antibodies had a profound effect on the IS-CD8+ T cells' ability to adhere to the islet endothelium (2 and 24 h after injection) and to penetrate the islets (24 h after injection). Clearly, the homing of IS-CD8+ T cells decreases if SLC is neutralized. The antibodies did not cause any loss of injected cells as their numbers in peripheral blood and spleens were similar (not shown).
Second, we stained cryostat sections of the pancreata from different mice with anti-chemokine antibodies. We found that SLC was expressed in the isthmus of the islets (Fig. 6 D). Most of the SLC-positive cells are localized to lymphatics and lack endothelial markers (not shown), but SLC is easily and highly diffusible: intradermal injection of SLC affected homing of T cells to the regional lymph nodes (38), and ectopic overexpression in β cells (39, 40) recruited lymphocytes to the islets. Importantly, SLC expression was found in the islets of diabetes-prone NOD mice, as well as in the islets of mice with no spontaneous diabetes and no preexistent inflammation, i.e., DBA/2J (H2d) and C3D2 (H2kxd) mice. Of the latter two strains, DBA/2J mice were sensitive to the transfer of IS-CD8+ T cells, while C3D2 mice were resistant.
Two important conclusions can be drawn from these findings: (a) chemokines, namely SLC, play a critical role in IS-CD8+ T cells homing to the islets of Langerhans, but (b), SLC expression does not correlate with inflammation and is not sufficient per se for the homing of IS-CD8+ T cells, and is likely to act in concert with recognition of an islet-specific peptide presented by endothelial cells.
Triggering of TCR and Chemokine Receptors of IS-CD8+ Cells Cooperate in Strengthening T Cell Adhesion to the Endothelium.
To test whether activation of IS-CD8+ cells through their TCR would lead to a detectable activation of integrins on their surface, we stimulated IS-CD8+ cells with anti-CD3 antibodies and immediately stained them with FITC-labeled fibronectin. Anti-CD3 stimulation led to increased binding of FBN-FITC to IS-CD8+ cells (online supplemental Fig. S4 A). FBN binds to α4 integrins with high affinity when they are activated (41–43). This binding, along with the low binding seen in IS-CD8+ cells before anti-CD3 exposure, was completely blocked by antibodies to VLA-4 integrin (online supplemental Fig. S4 B), but not by antibodies to LFA-1, which does not bind FBN. Thus, activation of T cells through their antigen-specific receptors led to the activation of integrins. These results clearly support the idea that recognition of specific MHC-peptide complexes may participate in the homing of IS-CD8+ cells, enhancing adhesion between T cells and endothelium. We applied an “adhesion under shear stress” approach to gain further support for our hypothesis that MHC–peptide recognition is important for T cell adhesion to endothelial cells and, hence, for homing. For that, the monolayers of aortal endothelium from NOD mice were used as targets for adherence of IS-CD8+ cells in the Glycotech flow chamber. T cells were allowed to adhere to the endothelial cells, and then the flow of tissue culture medium with several increments of pressure was initiated. It became clear that precultivation of endothelial monolayers with the InsB15–23 peptide (but not without the peptide or with an irrelevant Kd-binding peptide) led to increased adhesion of the T cells (Fig. 7 A). However, to achieve adhesion significantly different from that in the no-peptide control, T cells had to be allowed to stay in contact with endothelial cells for 300 s before the start of flow. The adhesion, however, became stronger when T cells were pretreated with SLC before the test: the time necessary for adhesion was reduced to 90 s, the fraction of adherent cells was larger, and more of them stayed attached longer than T cells that were not exposed to SLC. This adhesion was specific, as it occurred only in the presence of specific peptide, and it was possible to suppress it by addition of antibodies against H-2Kd molecules to the monolayers before the test (Fig. 7 B). The pretreatment with SLC also increased the baseline adhesion in the absence of specific peptide, but the cooperation of peptide and SLC made the specific adhesion stronger.
Figure 7.

Expression of specific MHC–peptide complex induces adhesion of IS-CD8+ cells to endothelium and works in concert with SLC. (A) After 300 s of adhesion to aortal endothelial cell monolayers pretreated or not with InsB15–23 peptide, IS-CD8+ cells were exposed to shear stress of the flow of tissue culture medium implemented in 4 dyne/cm2 increments with 10 s intervals. *, difference in percentage of adherent IS-CD8+ cells is significant for each pair of points of equal shear stress applied (P < 0.05 by Student's t test). (B) IS-CD8+ cells were pretreated with 100 ng/ml of SLC and allowed to adhere to endothelial monolayers expressing Kd complexes with InsB15–23 or LLO91–99 for 90 s. In parallel experiments anti-Kd monoclonal antibodies were added to monolayers expressing Kd complexes with InsB15–23. *, difference in percentage of adherent IS-CD8+ cells is significant for each pair of points of equal shear stress applied (P < 0.05 by Student's t test). n, number of measurements.
Thus, cooperation of multiple mechanisms regulating T cell adhesion to the endothelium is required for their specific homing. The two mechanisms described above are both complementary and essential for this process.
Discussion
Homing of activated T cells to sites where they are most needed (where pathogens are present) or where they are most harmful (in autoimmunity) are likely to be controlled by similar mechanisms. Using T cells that recognize an antigen uniquely produced by a specialized tissue, the islets of Langerhans in the pancreas, we were able to analyze the interactions required for the islet-specific homing of CD8+ T cells. Homing is a multistep process that allows T cells to cross into tissues through the endothelium of microcapillary vessels. Each of these steps (initial tethering, slow rolling, activation-dependent arrest, and diapedesis) is controlled by different interactions of surface molecules and multiple signaling pathways triggered by such interactions (30, 33).
Some aspects of T cell trafficking to the pancreas, such as the role of integrins and their ligands, have been addressed previously. It has been shown that LFA-1/ICAM-1, VLA-1/VCAM-1, and α4 integrin/MadCAM interactions are important for the homing of diabetogenic T cells to the pancreas (21, 44, 45). It is widely believed (and rightly so) that inflammation that induces many adhesion molecules facilitates the homing of T cells. However, when we introduced IS-CD8+ T cells into mice with no preexistent inflammation, the mice rapidly developed diabetes, and labeled IS-CD8+ cells were found in their islets (Fig. 1). This was taken as evidence that no inducible molecules are required for the initiation of homing of IS-CD8+ to the islets. Interestingly, IS-CD8+ cells were never found in salivary glands of NOD mice (Fig. 2), although NOD mice typically develop inflammation of salivary glands. Thus, inflammation by itself cannot determine the homing of IS-CD8+ T cells.
To explain the inflammation-independent organ-specific homing of IS-CD8+ cells, we put forward a hypothesis that endothelium can cross-present the antigen and thus participate in T cell adhesion in an antigen-specific fashion. We found experimental evidence to support this hypothesis: first, homing to the pancreas but not to other organs depended on expression of MHC class I and of the specific peptide (Figs. 2 and 3); second, IS-CD8+ cells up-regulated itegrin avidity upon stimulation of their TCR (online supplemental Fig. S4) and demonstrated adhesion to endothelial cells under shear stress in the presence of specific peptide (Fig. 7); third, endothelial cells from pancreatic islets could be directly recognized in in vitro cytotoxicity assay (Fig. 4). This direct recognition did not occur due to the presence of insulin in the tissue culture medium, as aortal endothelium cultivated in the same medium was not recognized by IS-CD8+ cells (Fig. 4 B, b). Thus, pancreatic endothelial cells have a necessary machinery to acquire insulin from β cells and produce the cognate peptide. The mode of acquisition of the Kd-InsB15–23 complexes by endothelial cells is not yet established. However, the observation that IS-CD8+ cells have a reduced ability to home into the islets of NOD.B6Akita/ + mice (Fig. 5) suggested that the peptide was generated by endothelial cells from secreted insulin rather than acquired as a peptide or an MHC–peptide complex produced by β cells. It is possible, however, that in NOD.B6Akita/ + mice other properties of endothelium are affected by insulin deficiency reducing T cell adhesion.
The need in secreted insulin for peptide presentation suggests that the peptide is generated by endosomal rather than proteosomal degradation. This scenario is not unprecedented, as evidence in favor of alternatives to the conventional MHC class I peptide processing and presentation pathway is accumulating (46–48). These studies have shown that MHC class I molecules could be found in classical MHC class II compartments, and that the TAP-independent and proteosome-independent pathways can lead to presentation of peptides by MHC class I. The fact that endothelial cells can serve as APC has been established previously (10, 11, 49). Hepatic endothelial cells were found capable of presenting complexes of MHC class I molecules with peptides derived from an injected foreign soluble protein (50). However, cross-presentation of peptides derived from endogenously produced protein has not been described. Local concentration of insulin is very high amounting to 7% of total islet protein (51). Insulin produced locally in such high concentrations may be broken down into peptides by endothelial cells. We do not know yet whether endothelium is destroyed by IS-CD8+ cells in vivo, or it is activated to facilitate T cell adhesion and diapedesis. This is an important issue and a matter of future studies. Our own data (Fig. 3 B) showed that secondary activation of endothelium is occurring with time, leading to an increase of the numbers of IS-CD8+ cells found in the isthmus.
Firm adhesion of IS-CD8+ cells is also controlled by G-protein–coupled chemokine receptors. We have found that SLC is the chemokine responsible for homing of IS-CD8+ T cells (Fig. 6 C). Its expression was detected in the pancreas by immunostaining (Fig. 6 D), but it was present not only in NOD mice, which may have an ongoing inflammation, but also in DBA/2J mice (with no preexistent inflammation) and in C3D2 mice, in which IS-CD8+ T cells do not home to the islets. It is very likely that at advanced stages of diabetes development, SLC expression in the pancreas of NOD mice increases enhancing the infiltration by T cells (52, 53). However, our data showed that constitutive presence of SLC in the pancreas in diabetes-free mice is required for IS-CD8+ homing, but by itself is not sufficient for successful homing. Similarly, in the in vitro adhesion under shear stress assay, SLC treatment of T cells slightly increased their adhesion to the endothelium without the cognate peptide. Nevertheless, the best adhesion was achieved when MHC/peptide complexes and SLC worked in concert (Fig. 7 B). A model describing additive effects of antigen recognition and activation of the chemokine receptor CCR7 on the surface of IS-CD8+ cells is shown in Fig. 8 . CCR7 expression by IS-CD8+ cells was confirmed by staining with ELC-Ig fusion protein, and by real-time RT-PCR (not shown). Thus, SLC-triggered integrin activation may be aided by TCR-mediated activation of integrins (26, 36, 41). We found that TCR-mediated integrin activation was also a property of IS-CD8+ cells, as the avidity of VLA-4 toward fibronectin increased after stimulation with anti-TCR antibody (online supplemental Fig. S4). Other integrins (e.g., LFA-1) are likely to be activated in IS-CD8+ cells in a similar fashion after TCR engagement. Homing specificity is maintained by fulfillment of both requirements (presence of appropriate peptide and an appropriate chemokine receptor): a CCR7-positive T cell clone LPa/2R-1 with different specificity did not show any significant accumulation in the pancreatic islets 2 or 24 h after injection (online supplemental Fig. S2). The results show that pancreatic endothelium is not randomly attracting any T cells that are capable of sensing SLC, but only those that have appropriate specificity (Fig. 8).
Figure 8.

A model showing how endothelial cells in the pancreas contribute to islet-specific homing of diabetogenic T cells. (A) The vast microcapillary network within the islet of Langerhans revealed by corrosion casting technique suggests an immense surface of interaction between insulin-producing cells and endothelial cells. (B) T cell invasion of the islets requires firm adhesion to endothelial cells. This can be achieved by activation of integrins on the T cell via two independent, but cooperating pathways: via chemokine receptors sensing chemokines deposited on the endothelial cell surface (nonspecific component), and via the TCR (specific component). The latter requires presentation of pancreatic antigens such as insulin by endothelial cells. Although it is yet unclear how the cross-presentation occurs, the intact secretion of insulin by β cells is needed.
Our study suggests that endothelial cells may play a more prominent role in the trafficking of activated T cells than is currently appreciated, providing antigen-driven specificity of homing. It remains to be established how general this principle is. While insulin is a secreted protein, it is unclear whether similar mechanisms may work for nonsecreted antigens. However, two recent studies (49, 54) suggest that endothelium (at least in transplantation models) can cross-present nonsecreted antigens. It is difficult to underestimate the significance of such a mechanism for the development of autoimmune diabetes. Because IS-CD8+ cells were found to be a predominant fraction of the islet-infiltrating cell population in NOD mice very early in the pathogenesis of diabetes (5) they may provide assistance to other T cells in the penetration of islets through activation of the local endothelium. The presence of SLC and activating signals from IS-CD8+ cells may lead to the initial accumulation of lymphocytes at the vascular entrance (known as periinsulitis), which later develops into accumulation of lymphocytes within the islets (insulitis) and damage to the insulin-producing cells (diabetes).
Acknowledgments
We thank Andrew Tcherepanov and Irene Visintin for technical assistance, Lesley Bechtold for excellent assistance with SEM and the SEM image, Greg Martin for help with image analysis, Dan Shaeffer for help with real-time RT-PCR, and Drs. M. Dee Gunn and Nadejda Logunova for helpful discussions. We are also grateful to Dr. Bernard Thorens for the anti-Glut-2 polyclonal serum, to Dr. Ulrich von Andrian for the ELC-Ig chimeric protein, to Dr. Eric Pamer for the T cell line L12.3, to Dr. Derry Roopenian and Greg Christianson for the LPa/2R-1 and B/L T cell clones, and to Dr. Leonard Shultz for the kind gift of NOD.B6Akita/ + mice.
This work was supported by National Institutes of Health grant IDDK53561, JDRFI grants 166 and 546 to A.V. Chervonsky, and by a Postdoctoral Fellowship from JDRFI to A.Y. Savinov. F.S. Wong is a Wellcome Trust Senior Research Fellow in Clinical Science.
This online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: CFSE, carboxyfluorescein diacetate succinimidyl ester; DiI, didodecyl-tetramethylindocarbocyanine perchlorate; FBN, fibronectin; GFP, green fluorescent protein; InsB15-23, insulin B chain peptide amino acids 15-23; LLO91-99, listeriolysin peptide, amino acids 91–99; IS-CD8+ cells, insulin-specific CD8+ T cells; PTx, pertussis toxin.
References
- 1.Tisch, R., and H.O. McDevitt. 1996. Insulin-dependent diabetes mellitus. Cell. 85:291–297. [DOI] [PubMed] [Google Scholar]
- 2.Delovitch, T.L., and B. Singh. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 7:727–738. [DOI] [PubMed] [Google Scholar]
- 3.Wong, F.S., and C.A. Janeway, Jr. 1999. The role of CD4 vs. CD8 T cells in IDDM. J. Autoimmun. 13:290–295. [DOI] [PubMed] [Google Scholar]
- 4.Serreze, D.V., E.H. Leiter, G.J. Christianson, D. Greiner, and D.C. Roopenian. 1994. Major histocompatibility complex class I-deficient NOD-B2m null mice are diabetes and insulitis resistant. Diabetes. 43:505–509. [DOI] [PubMed] [Google Scholar]
- 5.Wong, F.S., J. Karttunen, C. Dumont, L. Wen, I. Visintin, I.M. Pilip, N. Shastri, E.G. Pamer, and C.A. Janeway, Jr. 1999. Identification of a MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 5:1026–1031. [DOI] [PubMed] [Google Scholar]
- 6.Wong, F.S., I. Visintin, L. Wen, R.A. Flavell, and C.A. Janeway, Jr. 1996. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J. Exp. Med. 183:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell, D.J., C.H. Kim, and E.C. Butcher. 2001. Separable effector T cell populations specialized for B cell help or tissue inflammation. Nat. Immunol. 2:876–881. [DOI] [PubMed] [Google Scholar]
- 8.Sebastiani, S., P. Allavena, C. Albanesi, F. Nasorri, G. Bianchi, C. Traidl, S. Sozzani, G. Girolomoni, and A. Cavani. 2001. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J. Immunol. 166:996–1002. [DOI] [PubMed] [Google Scholar]
- 9.Osborn, L. 1990. Leukocyte adhesion to endothelium in inflammation. Cell. 62:3–6. [DOI] [PubMed] [Google Scholar]
- 10.Marelli-Berg, F.M., L. Frasca, L. Weng, G. Lombardi, and R.I. Lechler. 1999. Antigen recognition influences transendothelial migration of CD4+ T cells. J. Immunol. 162:696–703. [PubMed] [Google Scholar]
- 11.Pober, J.S., M.S. Kluger, and J.S. Schechner. 2001. Human endothelial cell presentation of antigen and the homing of memory/effector T cells to skin. Ann. NY Acad. Sci. 941:12–25. [DOI] [PubMed] [Google Scholar]
- 12.Savinov, A.Y., F.S. Wong, and A.V. Chervonsky. 2001. IFN-gamma affects homing of diabetogenic T cells. J. Immunol. 167:6637–6643. [DOI] [PubMed] [Google Scholar]
- 13.Zuberi, A.R., G.J. Christianson, L.M. Mendoza, N. Shastri, and D.C. Roopenian. 1998. Positional cloning and molecular characterization of an immunodominant cytotoxic determinant of the mouse H3 minor histocompatibility complex. Immunity. 9:687–698. [DOI] [PubMed] [Google Scholar]
- 14.Busch, D.H., I. Pilip, and E.G. Pamer. 1998. Evolution of a complex T cell receptor repertoire during primary and recall bacterial infection. J. Exp. Med. 188:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods, M.L., C. Cabanas, and Y. Shimizu. 2000. Activation-dependent changes in soluble fibronectin binding and expression of beta1 integrin activation epitopes in T cells: relationship to T cell adhesion and migration. Eur. J. Immunol. 30:38–49. [DOI] [PubMed] [Google Scholar]
- 16.Kreisel, D., A.S. Krupnick, W.Y. Szeto, S.H. Popma, D. Sankaran, A.M. Krasinskas, K.M. Amin, and B.R. Rosengard. 2001. A simple method for culturing mouse vascular endothelium. J. Immunol. Methods. 254:31–45. [DOI] [PubMed] [Google Scholar]
- 17.Daniels, M.A., L. Devine, J.D. Miller, J.M. Moser, A.E. Lukacher, J.D. Altman, P. Kavathas, K.A. Hogquist, and S.C. Jameson. 2001. CD8 binding to MHC class I molecules is influenced by T cell maturation and glycosylation. Immunity. 15:1051–1061. [DOI] [PubMed] [Google Scholar]
- 18.Walcheck B., J. Kahn, J.M. Fisher, B.B. Wang, R.S. Fisk, D.G. Payan, C. Feehan, R. Betageri, K. Darlak, A.F. Spatola, and T.K. Kishimoto. 1996. Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature. 380:720–723. [DOI] [PubMed] [Google Scholar]
- 19.Walcheck, B., K.L. Moore, R.P. McEver, and T.K. Kishimoto. 1996. Neutrophil-neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation of P-selectin in vitro. J. Clin. Invest. 98:1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto, K., J.-I. Miyagawa, T. Hanafusa, N. Itoh, A. Miyazaki, C. Nakawaga, S. Tarui, N. Kono, and Y. Matsuzawa. 1992. Endothelial and microvascular abnormalities in the islet of non-obese diabetic mice: transmission and scanning electron microscopic studies. Biomed. Res. 13:259–267. [Google Scholar]
- 21.Baron, J.L., E.P. Reich, I. Visintin, and C.A. Janeway, Jr. 1994. The pathogenesis of adoptive murine autoimmune diabetes requires an interaction between alpha 4-integrins and vascular cell adhesion molecule-1. J. Clin. Invest. 93:1700–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanninen, A., C. Taylor, P.R. Streeter, L.S. Stark, J.M. Sarte, J.A. Shizuru, O. Simell, and S.A. Michie. 1993. Vascular addressins are induced on islet vessels during insulitis in nonobese diabetic mice and are involved in lymphoid cell binding to islet endothelium. J. Clin. Invest. 92:2509–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michie, S.A., H.K. Sytwu, H.O. McDevitt, and X.D. Yang. 1998. The roles of alpha 4-integrins in the development of insulin-dependent diabetes mellitus. Curr. Top. Microbiol. Immunol. 231:65–83. [DOI] [PubMed] [Google Scholar]
- 24.Kayo, T., and A. Koizumi. 1998. Mapping of murine diabetogenic gene mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and beta cell development during the perinatal period. J. Clin. Invest. 101:2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, J., T. Takeuchi, S. Tanaka, S.-K. Kubo, T. Kayo, D. Lu, K. Takata, A. Koizumi, and T. Izumi. 1999. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Modymouse. J. Clin. Invest. 103:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dustin, M.L., and T.A. Springer. 1989. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 341:619–624. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths, E.K., C. Krawczyk, Y.Y. Kong, M. Raab, S.J. Hyduk, D. Bouchard, V.S. Chan, I. Kozieradzki, A.J. Oliveira-Dos-Santos, A. Wakeham, et al. 2001. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science. 293:2260–2263. [DOI] [PubMed] [Google Scholar]
- 28.Peterson, E.J., M.L. Woods, S.A. Dmowski, G. Derimanov, M.S. Jordan, J.N. Wu, P.S. Myung, Q.H. Liu, J.T. Pribila, B.D. Freedman, et al. 2001. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science. 293:2263–2265. [DOI] [PubMed] [Google Scholar]
- 29.Campbell, J.J., J. Hedrick, A. Zlotnik, M.A. Siani, D.A. Thompson, and E.C. Butcher. 1998. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 279:381–384. [DOI] [PubMed] [Google Scholar]
- 30.von Andrian, U.H., and C.R. Mackay. 2000. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 31.Berlin, C., R.F. Bargatze, J.J. Campbell, U.H. von Andrian, M.C. Szabo, S.R. Hasslen, R.D. Nelson, E.L. Berg, S.L. Erlandsen, and E.C. Butcher. 1995. Alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 80:413–422. [DOI] [PubMed] [Google Scholar]
- 32.Butcher, E.C., M. Williams, K. Youngman, L. Rott, and M. Briskin. 1999. Lymphocyte trafficking and regional immunity. Adv. Immunol. 72:209–253. [DOI] [PubMed] [Google Scholar]
- 33.Moser, B., and P. Loetscher. 2001. Lymphocyte traffic control by chemokines. Nat. Immunol. 2:123–128. [DOI] [PubMed] [Google Scholar]
- 34.Gunn, M.D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L.T. Williams, and H. Nakano. 1999. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ngo, V.N., H.T. Tang, and J.G. Cyster. 1998. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J. Exp. Med. 188:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Constantin, G., M. Majeed, G. Giagulli, L. Piccio, J.Y. Kim, E.C. Butcher, and C. Laudanna. 2000. Chemokines trigger immediate beta 2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 13:759–769. [DOI] [PubMed] [Google Scholar]
- 37.Stein, J.V., A. Rot, Y. Luo, M. Narasimhaswamy, H. Nakano, M.D. Gunn, A. Matsuzawa, E.J. Quackenbush, M.E. Dorf, and U.H. von Andrian. 2000. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J. Exp. Med. 191:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baekkevold, E.S., T. Yamanaka, R.T. Palframan, H.S. Carlsen, F.P. Reinholt, U.H. von Andrian, P. Brandtzaeg, and G. Haraldsen. 2001. The CCR7 ligand ELC (CCL19) is transcytosed in high endothelial venules and mediates T cell recruitment. J. Exp. Med. 193:1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen, S.C., G. Vassileva, D. Kinsley, S. Holzmann, D. Manfra, M.T. Wiekowski, N. Romani, and S.A. Lira. 2002. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J. Immunol. 168:1001–1008. [DOI] [PubMed] [Google Scholar]
- 40.Fan, L., C.R. Reilly, Y. Luo, M.E. Dorf, and D. Lo. 2000. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J. Immunol. 164:3955–3959. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu, Y., G.A. Van Seventer, K.J. Horgan, and S. Shaw. 1990. Regulated expression and binding of three VLA (beta 1) integrin receptors on T cells. Nature. 345:250–253. [DOI] [PubMed] [Google Scholar]
- 42.Elices, M.J., L. Osborn, Y. Takada, C. Crouse, S. Luhowskyj, M.E. Hemler, and R.R. Lobb. 1990. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 60:577–584. [DOI] [PubMed] [Google Scholar]
- 43.Faull, R.J., N.L. Kovach, J.M. Harlan, and M.H. Ginsberg. 1994. Stimulation of integrin-mediated adhesion of T lymphocytes and monocytes: two mechanisms with divergent biological consequences. J. Exp. Med. 179:1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabien, N., I. Bergerot, J. Orgiazzi, and C. Thivolet. 1996. Lymphocyte function associated antigen-1, integrin alpha 4, and L-selectin mediate T-cell homing to the pancreas in the model of adoptive transfer of diabetes in NOD mice. Diabetes. 45:1181–1186. [DOI] [PubMed] [Google Scholar]
- 45.Hanninen, A., I. Jaakkola, and S. Jalkanen. 1998. Mucosal addressin is required for the development of diabetes in nonobese diabetic mice. J. Immunol. 160:6018–6025. [PubMed] [Google Scholar]
- 46.Gromme, M., F.G. Uytdehaag, H. Janssen, J. Calafat, van R.S. Binnendijk, M.J. Kenter, A. Tulp, D. Verwoerd, and J. Neefjes. 1999. Recycling MHC class I molecules and endosomal peptide loading. Proc. Natl. Acad. Sci. USA. 96:10326-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellino, F., P.E. Boucher, K. Eichelberg, M. Mayhew, J.E. Rothman, A.N. Houghton, and R.N. Germain. 2000. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J. Exp. Med. 191:1957–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norbury, C.C., M.F. Princiotta, I. Bacik, R.R. Brutkiewicz, P. Wood, T. Elliott, J.R. Bennink, and J.W. Yewdell. 2001. Multiple antigen-specific processing pathways for activating naive CD8+ T cells in vivo. J. Immunol. 166:4355–4362. [DOI] [PubMed] [Google Scholar]
- 49.Kreisel, D., A.S. Krupnick, A.E. Gelman, F.H. Engels, S.H. Popma, A.M. Krasinskas, K.R. Balsara, W.Y. Szeto, L.A. Turka, and B.R. Rosengard. 2002. Non-hematopoietic allograft cells directly activate CD8+ T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat. Med. 8:233–239. [DOI] [PubMed] [Google Scholar]
- 50.Limmer, A., J. Ohl, C. Kurts, H.G. Ljunggren, Y. Reiss, M. Groettrup, F. Momburg, B. Arnold, and P.A. Knolle. 2000. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 12:1348–1354. [DOI] [PubMed] [Google Scholar]
- 51.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 52.Hjelmstrom, P., J. Fjell, T. Nakagawa, R. Sacca, C.A. Cuff, and N.H. Ruddle. 2000. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am. J. Pathol. 156:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luther, S.A., T. Lopez, W. Bai, D. Hanahan, and J.G. Cyster. 2000. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 12:471–481. [DOI] [PubMed] [Google Scholar]
- 54.Valujskikh, A., O. Lantz, S. Celli, P. Matzinger, and P.S. Heeger. 2002. Cross-primed CD8(+) T cells mediate graft rejection via a distinct effector pathway. Nat. Immunol. 3:844–851. [DOI] [PubMed] [Google Scholar]