

Over half a million people in the United States have systemic lupus. Most are women in their childbearing years. They usually have arthritis and scarring rashes, and about half have kidney involvement that may progress to renal failure. With time, there is an increasing likelihood that the brain will be involved, often resulting in loss of cognitive ability, seizures, and psychosis. Although the clinical features are highly variable, the patients are unified by the constant presence of autoimmunity to nucleoproteins, particularly to chromatin and small nuclear RNA-protein particles such as the U series of snRNPs that mediate premessenger RNA processing, and the Ro antigen (1). The resulting autoantibodies account for much of the tissue injury. For example, anti-DNA antibodies are harmful because they form immune complexes with extra cellular DNA and trigger an Arthus type of tissue injury, and sometimes they cross-react with cell surface proteins to exert a direct cytotoxic effect (2). Because of their central role in pathogenesis as well as their diagnostic usefulness, much effort has focused on understanding the mechanisms that account for these autoantibodies (3).
Among many early clues, it was noted that IFN might have pathogenic importance in lupus. For example IFN levels are typically elevated in patients with lupus, and IFN accelerates disease in NZB/W lupus mice (4, 5). Subsequently, it became apparent that IFN treatment of patients with neoplastic and viral diseases regularly induces fever, lethargy, and arthralgias much like the symptoms found among patients with active lupus. Occasionally, these patients develop antinuclear antibodies and a small number go on to express overt lupus (6–8). Finally, it was reported that IFN-α in sera from patients with lupus causes monocytes to differentiate into effective antigen presenting cells that are hypothesized to enhance formation of autoreactive T cells (9).
This line of inquiry is extended now in two reports in this issue. The paper by Santiago-Raber et al. demonstrates that type I IFNs are essential for most of the autoimmune phenotype in NZB lupus mice (10). The paper by Bennett et al. shows that peripheral blood mononuclear cells from pediatric patients with lupus regularly have alterations in gene transcription that are attributable to changes induced by IFN-α and the effects of accelerated granulopoiesis (11). Here, I will suggest that the emerging insights concerning IFN-α, dendritic cell (DC) biology, and apoptosis provide a rationale for autoimmunity in humans with lupus that is focused on nucleoproteins. A mechanism is proposed in which DCs both generate and respond to IFN-α in a way that enhances presentation of selected nuclear constituents to T helper cells.
The NZB mouse strain studied by Santiago-Raber et al. normally exhibits severe Coomb's positive hemolytic anemia, anti-DNA antibodies, and glomerulonephritis. This group has now developed a transgenic strain of NZB mice that lack the α chain of the IFN-α/β receptor (12). They report that these mice have a near normal phenotype with very little hemolytic anemia, glomerulonephritis, or autoantibody production compared with littermate controls. This work makes it clear that type I IFNs have an essential role in this mouse model of lupus. Similar work demonstrates that IFN-α determines the level of autoimmunity that develops in C57/B6 lpr/lpr mice (13). It will be of interest to learn if the same is true in other models such as NZB/W mice that have a more severe lupus-like phenotype. Caution is warranted in extrapolating directly from these mouse models to human lupus, as signaling pathways for IFN differ to some extent between mouse and man (14).
Direct evidence that IFN-α is the principal cytokine affecting peripheral blood mononuclear cells in lupus is provided by the study of Bennett et al. This group reports that genes regulated by IFN-α are up-regulated in pediatric patients with lupus. Interestingly, several of the augmented genes encode for proteins that play direct roles in pathogenesis and tissue injury in lupus such as one of the Ro autoantigens, two inhibitors of the complement cascade, and several genes related to apoptosis. Conceivably the products of these genes contribute respectively to enhanced levels of autoantigen, impaired clearance of immune complexes, and altered sensitivity to cell death signals. Other up-regulated genes encode proteins associated with DC maturation such as DC-LAMP, TAP1, and CD83 consistent with the earlier finding that IFN-α induces peripheral blood monocytes to transform into myeloid DCs and promotes their maturation (9). These studies were performed on the complex mixture of cells in the peripheral blood. The fact that the IFN signature is reproduced when PBMCs are incubated in culture with IFN-α argues strongly that the changes observed in patients is caused by IFN-induced alterations in gene transcription and is not secondary to shifts in these cell populations.
A finding in this latter study that is likely to astonish rheumatologists is the presence of a population of large granular cells that bear phenotypic markers of immature granulocytes. Apparently, these cells escaped our attention previously. Perhaps, because many of them are so immature that nuclear condensation has not occurred, we have mistaken them for monocytes. Possibly they are more prominent among patients in the pediatric age group. Associated with their presence, a number of genes related to granulopoiesis are dramatically overexpressed. In contrast to the IFN-α signature, neither these genes nor the large granular cells themselves are responsive to treatment with corticosteroids. It is unclear if and how these cells participate in the pathophysiology of lupus, and further information about the mechanisms that drive their production is needed. In any case at least, enhanced turnover of granulocytes is likely to add to the burden of material that must be cleared by phagocytic cells.
Generation of IFN in Lupus.
The two studies discussed here imply that excessive amounts of IFN α are reliably produced in lupus and that this cytokine could have a powerful effect on determining the disease process. But what might the source be? We know that IFN is produced by a wide variety of cells and secreted in response to viral and bacterial infection. Plasmacytoid DCs stand out as a major possibility because of their capacity to produce unusually large amounts of this cytokine (15–17). These cells secrete IFN-α in response to viral infections and when exposed to sera of patients with lupus (18).
Recent data indicate that DNA-anti-DNA complexes in lupus sera can provide an activation signal for IFN α secretion through the toll-like receptor (TLR) 9, the same TLR that responds to bacterial DNA sequences containing nonmethylated CpG motifs (19, 20). There are two limitations here. First, the DNA motifs that can trigger TLR 9 are characteristic of bacterial DNA but relatively uncommon in the mammalian genome, and what is there, is usually methylated so that it does not trigger the receptor. Second, the immune complexes from lupus sera require both the DNA and the Fc component of the anti-DNA antibody but there is scant evidence that plasmacytoid DCs express the types of Fc receptors that sensitize cells to activation. Possibly the complexes from lupus sera contain DNA fragments of bacterial or viral origin or the antibody somehow enriches nonmethylated CpG motifs from mammalian DNA as it resides in the extra cellular environment. Moreover, immune complexes in lupus sera may trigger other so-called myeloid DCs or other cells to produce IFN-α. It is noteworthy also that immune complexes containing DNA activate B cells by cross-linking the B cell antigen receptor with TLR 9 (21). It remains to be determined if this activation is associated with IFN-α secretion. Finally, immune complexes containing small RNAs such as the Ro RNAs potentially activate toll 3 receptors that are expressed on myeloid DCs since these RNAs have a double stranded structure that might mimic the double stranded viral RNAs normally recognized by this receptor. Quite possibly several of the mechanisms mentioned above are operative within individual patients with active lupus.
DCs in Lupus.
It has recently become evident that DCs can play a pivotal role in determining the balance between responsiveness and tolerance in the immune system in an antigen specific manner (22). One proposal is that immature DCs constantly circulate through the tissues of the body and load themselves with “self” molecules and other harmless environmental substances. Upon interaction with T cells, they deliver a signal that results in the deletion, anergy and/or regulation of cells that recognize the material they carry. This ability has been established most clearly for the so-called myeloid lineage of DCs (22–24). It remains to be determined whether DCs derived from monocytes ever have a phase during which they are able to tolerize T cells. It is clear however, that these cells are potent stimulators of naive T cells (9).
It is not surprising that cells with these abilities should attract the attention of investigators interested in lupus (15). Thus far, what has been learned includes the fact that so called myeloid DCs in blood tend to be reduced in number but fluctuate widely over time in individual patients (25). In contrast, plasmacytoid DCs are more consistently reduced in the circulation, presumably because they have become activated and migrated into peripheral lymphoid tissues and sites of inflammation. In fact, they have been observed to accumulate in lupus skin lesions and nasal mucosa of patients with allergies (26, 27). One study reports that lupus monocytes have a deficit in differentiation into DCs but the significance of this observation remains unclear (28). An important finding in mice, is that overexpression of CD40L in the basal layers of the epidermis accelerates DC maturation and migration to regional lymph nodes (29). The result is severe dermatitis with both T cell and B cell autoimmunity to local skin antigens and humoral autoimmune responses to nuclear antigens including DNA. This model supports the idea that chronic DC activation leads to autoimmunity and demonstrates that the antigens they deliver to T cells correspond to the site of activation. The fact that DC maturation signals in the skin induce autoimmunity to DNA as well as to local skin-specific antigens likely reflects the wide spread availability of chromatin throughout the bodies tissues. Along this line, DCs that have ingested apoptotic cells in culture induce antinuclear antibodies in BALB/c mice and accelerate autoimmunity in NZB/W mice (30). Also, the autologous mixed lymphocyte response by T cells appears to be largely dependent upon DCs presenting antigens derived from apoptotic elements (31, 32). Taken together, these observations indicate that chronic activation of DCs induces immune responses to nucleoproteins in mice. They are in line with observations in humans that antinuclear antibodies frequently accompany a broad range of chronic infectious diseases where DC maturation is likely to occur.
Setting the Focus on Nucleoproteins.
The work of Rosen et al. indicate that many of the lupus autoantigens are early targets for caspase digestion and are selectively incorporated into blebs on apoptotic cells (33). This material induces antinuclear antibodies when injected into several different strains of mice (34), presumably because it is processed and presented to T cells by immunostimulatory DCs. This mechanism is likely to account for T cell priming. The question then is how does the process surmount tolerance barriers in the B cell compartment. Perhaps an important clue is that in a number of cases, early and possibly initial B cell responses in both lupus prone mice and patients are directed against epitopes that exist only on intact nucleoprotein particles such as the interface structures of histone H2A-H2B (35–37). Presumably, such complex structures are labile and have little ability to tolerize B cells within the bone marrow. This concept accounts for a population of autoreactive B cells that need only T cell help to respond and to utilize epitope spreading and hypermutation mechanisms to generate the broad array of autoantibodies to nucleoproteins that mediate the disease.
Whether or not lupus results from these DC–T cell and T cell–B cell interactions is clearly determined genetically. For example, in mice over 35 different single gene mutations are associated with a lupus phenotype that always includes the presence of antinuclear and anti-chromatin antibodies (38). Many of these genes encode proteins that regulate apoptosis, determine clearance of apoptotic cells, or involve mechanisms of negative selection in central tolerance (39). For example, mice deficient in the gene encoding mer, a receptor on macrophages required for up-take of apoptotic cells, develop systemic autoimmunity that includes production of antibodies to nuclear autoantigens. Presumably, impaired macrophage clearance of cellular debris enhances the amount of apoptotic material that is loaded into DCs and possibly augments the amount of material that can stimulate DC maturation. Single gene mutations in humans and mice that are consistently associated with lupus include defects in the early complement components, most notably C1q (40) and possibly DNase I (41). Both are examples of gene defects that probably augment the exposure of DCs to nuclear autoantigens through up-take of immune complexes. However, in most patients with lupus and in several classical animal models such as NZB/W mice, multiple genes interact to create a predisposition for the disease (42). Presumably these genes act in concert to achieve an effect on T and B cell signaling and autoantigen presentation that equates with the single gene defects discussed above.
Perpetuating the Response.
These considerations suggest a pathway that can account for immune responses to nucleoproteins, as illustrated in Fig. 1 . Any event that triggers secretion of excessive amounts of IFN-α such as infection and UV skin injury (known activators of lupus [43]), can set the process in motion. The resulting IFN-α promotes the differentiation of monocytes into immunostimulatory DCs as shown earlier (9). This process is likely to be enhanced by immune complexes delivering sensitizing signals through FcRIII receptors on these cells (44, 45) and genetic alterations that increase the flux of chromatin into DCs as mentioned above. Other cytokines such as TNF-α are also involved as well. The augmentation of matured DCs presenting nucleoprotein peptides now favors priming of naive T cells and potentially impairs generation of T regulatory and T suppressor cells. In any event, the T cell help required to support autoreactive B cells is now available and the latter generate the immune complexes that appear to trigger IFN-α secretion. This process may require an increase in the source of apoptotic cells or a reduction in macrophage clearance of these cells. Tissue injury from immune complexes and cytotoxic T cells is one source. It is tempting also to speculate that rapid turnover of polymorphonuclear leukocytes in lupus, as implied by the finding of excessive numbers of early granulocyte forms in the study by Bennett and colleagues, represents another that might serve to perpetuate the disease.
Figure 1.
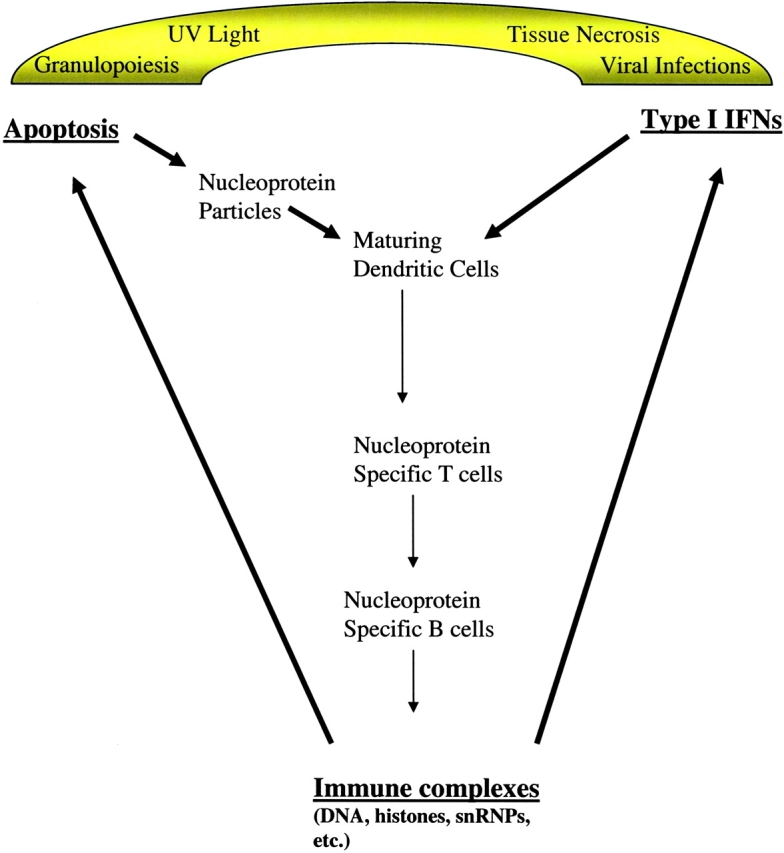
Hypothetical model for the role of IFN-α in propagating autoimmunity to chromatin. An exogenous event such as a viral infection triggers IFN-α secretion, thus simultaneously augmenting apoptosis, monocyte maturation, and T cell priming. In an individual with a permissive genetic make-up, T cell responses to chromatin components such as histones are induced providing the necessary help for specific B cell responses to components such as DNA. Once initiated, the response is perpetuated endogenously by the ability of immune complexes based on anti-DNA antibodies (and possibly other specificities) to stimulate secretion of IFN-α by plasmacytoid DCs. Cell injury secondary to these complexes results in apoptotic cell debris that contributes to enhanced loading of nucleoproteins into DCs. Accelerated granulopoiesis may contribute to the burden of this material and represent an important source of nucleoprotein autoantigens as these cells turn over.
Opportunities for Intervention.
This scheme suggests several therapeutic strategies to interrupt this vicious cycle. They include blockade of IFN-α either by preventing its secretion from specific cell sources or by neutralization in the extra cellular environment. Benefit may also be achieved by enhancing destruction of nuclear autoantigens with agents such as DNase I as well as strategies that augment macrophage scavenging. It may also be possible to find ways to regulate DC maturation to limit excessive stimulation of autoreactive T cells.
The model presented here rationalizes the occurrence of antinuclear antibodies in many settings when DCs are called upon for immune defenses and develops a hypothesis for how this process might be propagated in individuals with permissive genetic backgrounds. It also suggests that the elusive trigger for lupus that has been so widely anticipated may often be any factor that can generate sufficient IFN-α to set the disease in motion. We must be cautious as we harness an increasing number of cytokines for therapeutic purposes because these molecules play such an important role in balancing self-tolerance and immune protection.
Acknowledgments
The author is deeply grateful to Drs Ralph Steinman and Betty Diamond for their careful critiques of the manuscript and thanks Drs Peter Albersheim, Kara Bickham, Joe Craft, Shao-Lee Lin, Christian Münz, Jeffrey Ravetch, and Matthew Scharff for helpful advice and discussion during the preparation of this work.
References
- 1.Hardin, J.A. 1986. The lupus autoantigens and the pathogenesis of systemic lupus erythematosus. Arthritis Rheum. 29:457–460. [DOI] [PubMed] [Google Scholar]
- 2.Beger, E., B. Deocharan, M. Edelman, B. Erblich, Y. Gu, and C. Putterman. 2002. A peptide DNA surrogate accelerates autoimmune manifestations and nephritis in lupus-prone mice. J. Immunol. 168:3617–3626. [DOI] [PubMed] [Google Scholar]
- 3.Rovere, P., M.G. Sabbadini, F. Fazzini, A. Bondanza, V.S. Zimmermann, C. Rugarli, and A.A. Manfredi. 2000. Remnants of suicidal cells fostering systemic autoaggression. Apoptosis in the origin and maintenance of autoimmunity. Arthritis Rheum. 43:1663–1672. [DOI] [PubMed] [Google Scholar]
- 4.Hooks, J.J., H.M. Moutsopoulos, S.A. Geis, N.I. Stahl, J.L. Decker, and A.L. Notkins. 1979. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 301:5–8. [DOI] [PubMed] [Google Scholar]
- 5.Steinberg, A.D., S. Baron, and N. Talal. 1969. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc. Natl. Acad. Sci. USA. 63:1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ytterberg, S.R., and T.J. Schnitzer. 1982. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 25:401–406. [DOI] [PubMed] [Google Scholar]
- 7.Ronnblom, L.E., G.V. Alm, and K.E. Oberg. 1991. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann. Intern. Med. 115:178–183. [DOI] [PubMed] [Google Scholar]
- 8.Ioannou, Y., and D.A. Isenberg. 2000. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 43:1431–1442. [DOI] [PubMed] [Google Scholar]
- 9.Blanco, P., A.K. Palucka, M. Gill, V. Pascual, and J. Banchereau. 2001. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 294:1540–1543. [DOI] [PubMed] [Google Scholar]
- 10.Santiago-Raber, R. Baccala, K.M. Haraldsson, D. Choubey, T.A. Stewart, D.H. Kono, and A.N. Theofilopoulos. 2003. Type I interferon receptor deficiency reduces Lupus-like disease in NZB mice. J. Exp. Med. 197:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pestka, S., J.A. Langer, K.C. Zoon, and C.E. Samuel. 1987. Interferons and their actions. Annu. Rev. Biochem. 56:727–777. [DOI] [PubMed] [Google Scholar]
- 13.Braun, D., P. Geraldes, and J. Demengeot. 2003. Type I interferon controls the onset and severity of autoimmune manifestations in lpr mice. J. Autoimmun. In press. [DOI] [PubMed] [Google Scholar]
- 14.Farrar, J.D., J.D. Smith, T.L. murphy, S. Leung, G.R. Stark, and K.M. Murphy. 2000. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat. Immunol. 1:65-69. [DOI] [PubMed] [Google Scholar]
- 15.Ronnblom, L., and G.V. Alm. 2002. The natural interferon-alpha producing cells in systemic lupus erythematosus. Hum. Immunol. 63:1181–1193. [DOI] [PubMed] [Google Scholar]
- 16.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 17.Siegal, F.P., N. Kadowaki, M. Shodell, P.A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y.J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science. 284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 18.Cederblad, B., S. Blomberg, H. Vallin, A. Perers, G.V. Alm, and L. Ronnblom. 1998. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-alpha-producing cells. J. Autoimmun. 11:465–470. [DOI] [PubMed] [Google Scholar]
- 19.Vallin, H., A. Perers, G.V. Alm, and L. Ronnblom. 1999. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J. Immunol. 163:6306–6313. [PubMed] [Google Scholar]
- 20.Vallin, H., S. Blomberg, G.V. Alm, B. Cederblad, and L. Ronnblom. 1999. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin. Exp. Immunol. 115:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leadbetter, E.A., I.R. Rifkin, A.M. Hohlbaum, B.C. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 22.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawiger, D., K. Inaba, Y. Dorsett, K. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gill, M.A., P. Blanco, E. Arce, V. Pascual, J. Banchereau, and A.K. Palucka. 2002. Blood dendritic cells and DC-poietins in systemic lupus erythematosus. Hum. Immunol. 63:1172–1180. [DOI] [PubMed] [Google Scholar]
- 26.Wollenberg, A., M. Wagner, S. Gunther, A. Towarowski, E. Tuma, M. Moderer, S. Rothenfusser, S. Wetzel, S. Endres, and G. Hartmann. 2002. Plasmacytoid dendritic cells: a new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J. Invest. Dermatol. 119:1096–1102. [DOI] [PubMed] [Google Scholar]
- 27.Farkas, L., K. Beiske, F. Lund-Johansen, P. Brandtzaeg, and F.L. Jahnsen. 2001. Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 159:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinbach, F., F. Henke, B. Krause, B. Thiele, G.R. Burmester, and F. Hiepe. 2000. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann. Rheum. Dis. 59:283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehling, A., K. Loser, G. Varga, D. Metze, T.A. Luger, T. Schwarz, S. Grabbe, and S. Beissert. 2001. Overexpression of CD40 ligand in murine epidermis results in chronic skin inflammation and systemic autoimmunity. J. Exp. Med. 194:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bondanza, A., V.S. Zimmermann, G. Dell'Antonio, E. Dal Cin, A. Capobianco, M.G. Sabbadini, A.A. Manfredi, and P. Rovere-Querini. 2003. Cutting edge: dissociation between autoimmune response and clinical disease after vaccination with dendritic cells. J. Immunol. 170:24–27. [DOI] [PubMed] [Google Scholar]
- 31.Chernysheva, A.D., K.A. Kirou, and M.K. Crow. 2002. T cell proliferation induced by autologous non-T cells is a response to apoptotic cells processed by dendritic cells. J. Immunol. 169:1241–1250. [DOI] [PubMed] [Google Scholar]
- 32.Amel Kashipaz, M.R., M.L. Huggins, R.J. Powell, and I. Todd. 2002. Human autologous mixed lymphocyte reaction as an in vitro model for autoreactivity to apoptotic antigens. Immunology. 107:358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casciola-Rosen, L., G. Anhalt, and A. Rosen. 1994. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 179:1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mevorach, D., J.L. Zhou, X. Song, and K.B. Elkon. 1998. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med. 188:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jafri, F., J.A. Hardin, and W.S. Dynan. 2001. A method to detect particle-specific antibodies against Ku and the DNA-dependent protein kinase catalytic subunit in autoimmune sera. J. Immunol. Methods. 251:53–61. [DOI] [PubMed] [Google Scholar]
- 36.Amoura, Z., S. Koutouzov, and J.C. Piette. 2000. The role of nucleosomes in lupus. Curr. Opin. Rheumatol. 12:369–373. [DOI] [PubMed] [Google Scholar]
- 37.Takeda, Y., K.S. Wise, G. Wang, G. Grady, E.V. Hess, G.C. Sharp, W.S. Dynan, and J.A. Hardin. 1998. Human autoantibodies recognizing a native macromolecular structure composed of Sm core proteins in U small nuclear RNP particles. Arthritis Rheum. 41:2059–2067. [DOI] [PubMed] [Google Scholar]
- 38.Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. [DOI] [PubMed] [Google Scholar]
- 39.Shlomchik, M.J., J.E. Craft, and M.J. Mamula. 2001. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 1:147–153. [DOI] [PubMed] [Google Scholar]
- 40.Mitchell, D.A., M.C. Pickering, J. Warren, L. Fossati-Jimack, J. Cortes-Hernandez, H.T. Cook, M. Botto, and M.J. Walport. 2002. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J. Immunol. 168:2538–2543. [DOI] [PubMed] [Google Scholar]
- 41.Yasutomo, K., T. Horiuchi, S. Kagami, H. Tsukamoto, C. Hashimura, M. Urushihara, and Y. Kuroda. 2001. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28:313–314. [DOI] [PubMed] [Google Scholar]
- 42.Sobel, E.S., L. Morel, R. Baert, C. Mohan, J. Schiffenbauer, and E.K. Wakeland. 2002. Genetic dissection of systemic lupus erythematosus pathogenesis: evidence for functional expression of Sle3/5 by non-T cells. J. Immunol. 169:4025–4032. [DOI] [PubMed] [Google Scholar]
- 43.Cooper, G.S., M.A. Dooley, E.L. Treadwell, E.W. St Clair, and G.S. Gilkeson. 2002. Risk factors for development of systemic lupus erythematosus: allergies, infections, and family history. J. Clin. Epidemiol. 55:982–989. [DOI] [PubMed] [Google Scholar]
- 44.Regnault, A., D. Lankar, V. Lacabanne, A. Rodriguez, C. Thery, M. Rescigno, T. Saito, S. Verbeek, C. Bonnerot, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Fcγ receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J. Exp. Med. 189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalergis, A.M., and J.V. Ravetch. 2002. Inducing tumor immunity through the selective engagement of activating Fcγ receptors on dendritic cells. J. Exp. Med. 195:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]