

Abstract
Fibrin, a product of the blood coagulation cascade, accompanies many type 1 immune responses, including delayed-type hypersensitivity, autoimmunity, and graft rejection. In those settings, fibrin is thought to exacerbate inflammation and disease. Here, we evaluate roles for coagulation during infection with Toxoplasma gondii, a pathogen whose control requires robust type 1 immunity. We establish that fibrin prevents infection-stimulated blood loss, thereby performing a protective function that is essential for survival. Remarkably, fibrin does not simply protect against vascular damage caused directly by the infectious agent, but rather, protects against hemorrhage evoked by interferon-γ, a critical mediator of type 1 immunity. This finding, to our knowledge, is the first to document a beneficial role for coagulation during type 1 immunity, and suggests that fibrin deposition protects host tissue from collateral damage caused by the immune system as it combats infection.
Keywords: cellular immunity, interferon type II, toxoplasma, blood coagulation, hemorrhage
Introduction
During the coagulation cascade, fibrin is generated by the action of thrombin on fibrinogen. Although best appreciated for its roles in vascular hemostasis, coagulation leading to fibrin deposition also frequently accompanies type 1 immune responses, including delayed-type hypersensitivity (1–3), autoimmunity (4–10), and graft rejection (11–13). Prior studies indicate that fibrin, and other coagulant products, can exacerbate inflammation and disease in those settings (1–16). Coagulation also frequently accompanies infectious disease, and apparently can exacerbate pathology in that context as well (16). Indeed, administration of activated protein C, a natural anticoagulant protein, reduces septic mortality in humans (17). While these and other studies have documented pathological roles for coagulation during immunity and infection, beneficial functions for immune-associated coagulation have yet to be established.
Toxoplasma gondii is a widely distributed, obligate intracellular, protozoan parasite (18, 19). Natural infection is acquired by ingestion of encysted organisms, which are released in the digestive tract, invade intestinal tissues, and then disseminate. In mouse models, T. gondii infection elicits a robust type 1 immune response, which eradicates most parasites within 2 wk of infection. Nevertheless, some organisms escape immune surveillance, encyst, and enter a dormant state from which they can emerge if immunity wanes, as frequently occurs in humans suffering from the acquired immunodeficiency syndrome.
Here, we evaluate roles for fibrin in the context of the strong type 1 immune response prompted by T. gondii infection. In contrast to prior studies of immune-associated fibrin deposition, we demonstrate that coagulation performs a critical protective function in the setting of T. gondii infection by suppressing hemorrhage evoked by IFN-γ, a critical mediator of type 1 immunity. Thus, while coagulant products have the potential to function pathologically, our data indicate that immune-associated coagulative responses can also protect host tissue from collateral damage caused by the immune system as it combats infection.
Materials and Methods
Mice.
6–10-wk-old male mice were used for all experiments. C57BL/6 and IFN-γ–deficient mice were purchased from Taconic and The Jackson Laboratory, respectively. Fibrinogen-deficient mice (reference 20; backcrossed seven generations to C57BL/6 mice) were generously supplied by Jay Degen (Children's Hospital Medical Center, Cincinnati, OH) and were bred at the Trudeau Institute Animal Breeding Facility. Animals were housed in a specific pathogen-free facility and cared for according to the Trudeau Institute Animal Care and Use Committee guidelines.
Infections.
T. gondii strain ME49 was originally provided by Jack Remington (Palo Alto Medical Foundation, Palo Alto, CA). ME49 cysts were obtained from brains of chronically infected C57BL/6 mice, and infections were initiated by peroral administration of 10 cysts in 0.1 ml of diluted brain suspension using a 19-gauge gavage needle. Sham-infected mice received similarly diluted brain suspensions from uninfected mice. Where indicated, 2 mg/L warfarin (3-(α-acetonylbenzyl)-4-hydroxy-coumarin; Sigma-Aldrich) was added to drinking water and replenished every 48 h. The anti–IFN-γ mAb XMG1.2 and control rat IgG1 mAb HRPN were Protein A–purified from culture supernatants. Each contained <1 EU/mg as determined by Limulus amebocyte lysate assay.
Measurements of Immune Parameters.
IFN-γ protein levels in sera were determined by sandwich ELISA (BD Biosciences). Nitric oxide levels in plasma were determined as described (21), except that plasma was diluted fourfold in water and filtered (10000 MWCO; Amicon) before analysis. Tissue levels of mRNA encoding IFN-γ, TNF-α, IL-10, inducible nitric oxide synthase (iNOS), and the T. gondii p30 gene were measured by real-time PCR (PerkinElmer) and normalized to levels of β-2-microglobulin (β2m). For T. gondii, standard curves were prepared by adding known numbers of ME49 tachyzoites (obtained from peritoneal exudate fluid of T. gondii–infected IFN-γ–deficient mice) to uninfected liver tissue, and then preparing cDNA. Real-time PCR primer and probe sequences were previously described (22), supplied by Daniel Douek (NIH Vaccine Research Center, Bethesda, MD), or designed using Primer Express software (PerkinElmer). Except for p30, each primer combination amplifies cDNA, but not genomic DNA. p30-forward TTTCCGAAGGCAGTGAGACG, p30-reverse GGATCCGATGCCATAGCG, p30-probe TTGCCGCGCCCACACTGATG); IFN-γ-forward CATTGAAAGCCTAGAAAGTCTGAATAAC, IFN-γ-reverse TGGCTCTGCAGGATTTTCATG, IFN-γ-probe TCACCATCCTTTTGCCAGTTCCTCCAG; TNF-α-forward CATCTTCTCAAAATTCGAGTGACAA, TNF-α-reverse TGGGAGTAGACAAGGTACAACCC, TNF-α-probe CACGTCGTAGCAAACCACCAAGTGGA; IL-10-forward GAAGACCCTCAGGATGCGG, IL-10-reverse ACCTGCTCCACTG-CCTTGCT, IL-10-probe TGAGGCGCTGTCATCGATTT-CTCCC; iNOS-forward CAGCTGGGCTGTACAAACCTTC, iNOS-reverse CATTGGAAGTGAAGCGTTTCG, iNOS-probe CGGGCAGCCTGTGAGACCTTTGA; β2m-forward TGACCGGCTTGTATGCTATC, β2m-reverse CAGTGTGAGCCAGGATATAG, β2m-probe TATACTCACGCCACCCACCGGAGAA.
Measurement of Fibrin Levels.
Fibrin levels within tissue samples were quantified essentially as described previously (23, 24). Tissue was harvested from anticoagulated mice (500 U heparin, intravenously), weighed, and stored at –70°C. Samples were then homogenized in 10 mM sodium phosphate (pH 7.5) containing 5 mM EDTA, 100 mM ε-aminocaproic acid, 10 U/ml aprotinin, 10 U/ml heparin, and 2 mM phenylmethanesulfonyl fluoride. After overnight rocking at 4°C, insoluble fibrin was pelleted (10,000 g, 10 min), washed (10 mM sodium phosphate, 5 mM EDTA), extracted twice with 3 M urea at 37°C, and then solubilized in reducing SDS-PAGE buffer at 65°C. Samples were normalized based upon the initial tissue weight. Fibrin levels were then quantified by Western blotting, using biotinylated anti-β fibrin mAb 350 (2 ng/ml; American Diagnostica, Inc.) followed by rabbit anti-biotin (1:20,000; DakoCytomation), anti–rabbit horseradish peroxidase polymer (1:40; DakoCytomation), and chemiluminescent detection (Santa Cruz Biotechnology, Inc.). To generate fibrin for standard curves, purified mouse fibrinogen (Sigma-Aldrich) was treated with human thrombin (Enzyme Research Laboratories).
Statistics.
Statistical analyses were performed using the program Instat 3.0 (GraphPad Software), using either Mann-Whitney or Student's t tests, as appropriate.
Results
Upon peroral infection with 10 cysts of avirulent strain ME49 T. gondii, C57BL/6 mice mount a prototypical type 1 immune response, characterized by robust IFN-γ production, which soon contains parasite replication (Fig. 1 ; references 19 and 25). We found that infection with T. gondii also prompted a transient coagulative response. Fibrin was never detectable within tissues of uninfected mice. However, as reported for many other conditions that elicit type 1 immunity (1–13), we detected markedly elevated levels of fibrin in T. gondii–infected mice (Fig. 1). Fibrin deposition within hepatic and intestinal tissues peaked at day 8 after infection, correlating kinetically with peaks in both parasite burdens and hallmarks of type 1 immunity (Fig. 1, and unpublished data).
Figure 1.
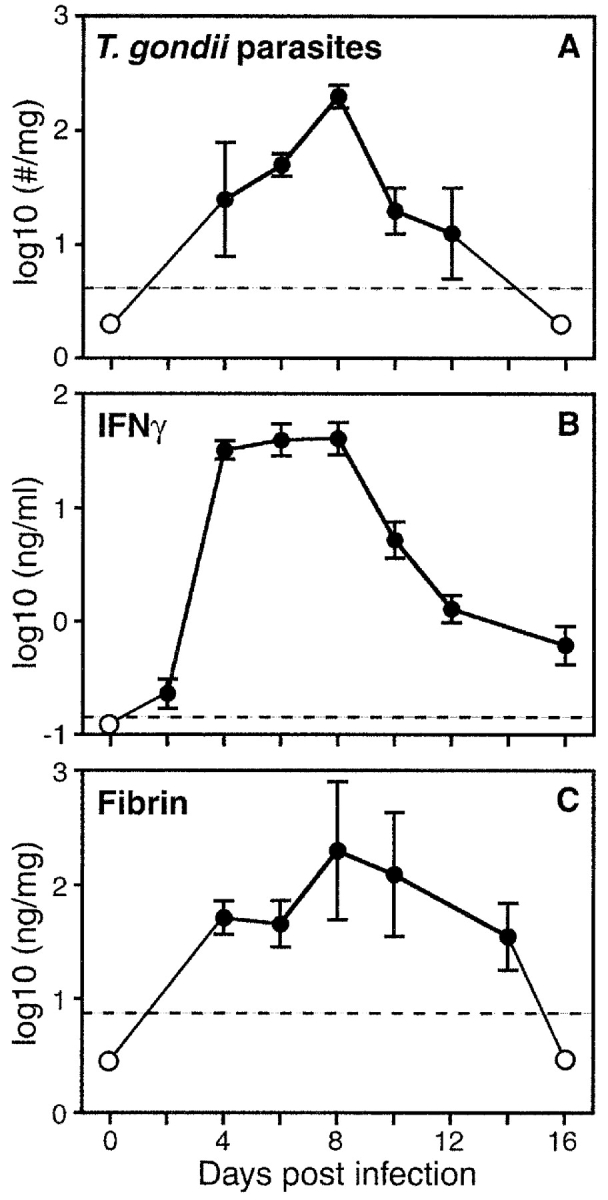
Fibrin formation accompanies T. gondii infection. Wild-type C57BL/6 mice were infected with T. gondii (10 strain ME49 cysts, peroral). On the indicated days, we measured parasite burdens within liver tissue (A), IFN-γ protein levels in sera (B), and fibrin deposition within liver tissue (C). Data depict the averages ± SD of five mice per time point. Dashed lines indicate levels of detection, and open circles depict points below that level. T. gondii infection stimulated significant increases in liver fibrin deposition (P < 0.01), peaking at day 8 d after infection, and correlating with peak parasite burdens and IFN-γ levels.
To evaluate the function of this infection-stimulated coagulative response, we treated mice with warfarin, an orally available pharmaceutical used clinically for the long-term anticoagulation of thrombosis-prone humans. Supplementing drinking water with 2 mg/L warfarin did not impair the growth or survival of uninfected mice, but was clinically effective, as it suppressed T. gondii–stimulated fibrin deposition to below the detection limit of our assay (unpublished data). Strikingly, warfarin-treated mice acutely succumbed to T. gondii infection, dying around the time of the peak coagulative response of untreated animals (Fig. 2 A). That finding suggested that coagulation might function protectively during T. gondii infection.
Figure 2.

Fibrin functions protectively during T. gondii infection. (A) Wild-type C57BL/6 mice were infected with T. gondii. Where indicated, mice were anticoagulated with warfarin. Anticoagulated mice succumbed to T. gondii infection significantly earlier than did untreated mice (P < 0.01; n = 5 mice per group). (B) Fibrinogen-deficient (Fib KO) mice and littermate control fibrinogen-heterozygous (Fib Het) were infected with T. gondii. Fibrinogen-deficient mice succumbed to T. gondii infection significantly earlier than did littermate control mice (P < 0.01; n = 5 mice per group). Similar results to those shown in A and B were independently replicated in two subsequent experiments.
To specifically evaluate protective roles for fibrin during T. gondii infection, we examined gene-targeted fibrinogen-deficient mice (20), which cannot produce fibrin. We found that fibrinogen-deficient mice also succumbed acutely to T. gondii infection (Fig. 2 B). As infection prompted fibrin deposition in normal mice (Fig. 1), and acute mortality in both warfarin-treated and fibrinogen-deficient mice (Fig. 2), we conclude that fibrin performs a critical protective function during T. gondii infection.
Fibrin may protect against infection-triggered mortality via a number of plausible mechanisms. As levels of fibrin and parasitemia correlated (Fig. 1), we considered the possibility that coagulation might function to physically limit parasite dissemination (14). However, parasite numbers within liver, intestine, Peyer's patch, spleen, and brain were not significantly increased in fibrinogen-deficient mice (Fig. 3 , and unpublished data).
Figure 3.

Mice lacking fibrin exhibit neither increased parasite burdens nor suppressed anti-parasite immunity. Fibrinogen-deficient (white bars) and littermate control fibrinogen-heterozygous mice (black bars) were infected with T. gondii. Parasite numbers (p30 mRNA in liver), IFN-γ (protein in sera, mRNA in liver), nitric oxide (nitrite/nitrate in sera), inducible nitric oxide synthase (iNOS, mRNA in liver), IL-10 (mRNA in liver), and TNF-α (mRNA in liver) were assayed on days 4 and 8 after infection. There were no significant differences in the levels of any of the measured parameters. The data shown was compiled from four independent experiments. Bars depict average ± SD of 8 mice per group (Day 0), 4 mice per group (Day 4), or 14 to 16 mice per group (Day 8).
We next assessed whether fibrin might function protectively by affecting immunity. Aberrant levels of IL-12, IFN-γ, TNF-α, IL-10, nitric oxide, or neutrophils can impair survival of T. gondii-infected mice (19). However, using ELISA, real-time PCR, and/or histologic assays, we never observed significant differences in levels of those mediators when comparing T. gondii-infected fibrinogen-deficient and littermate control mice (Fig. 3, and unpublished data). Thus, fibrin probably does not function protectively during T. gondii infection by influencing levels of critical immune mediators.
In experimental models, acute mortality during T. gondii infection can result from either a failure to control parasite numbers, or excessive immunopathology (19). During the course of our studies we noted exacerbated gross pathology in liver tissue of T. gondii–infected fibrinogen-deficient mice (Fig. 4 B). Microscopic examination revealed intense infiltration of mixed inflammatory cells and areas of hepatocellular necrosis in livers of T. gondii–infected control mice (Fig. 4 C). In fibrinogen-deficient mice, infiltrates tended to be more diffuse, and necrosis was more extensive and frequently associated with hemorrhage (Fig. 4 D).
Figure 4.

Exacerbated hepatic pathology in T. gondii-infected, fibrin-deficient mice. Representative liver lobes harvested at day 8 after T. gondii infection of littermate control fibrinogen-heterozygous (A) and fibrinogen-deficient (B) mice. Hemorrhagic foci are evident in livers of T. gondii–infected fibrinogen-deficient mice. (C and D) Micrographs (200×) depicting hematoxylin and eosin stained tissue samples isolated from T. gondii–infected mice. Note that areas of hepatocellular necrosis, which are characteristic of T. gondii infection in fibrinogen-sufficient mice (arrow in C), are largely replaced by hemorrhagic pathology in fibrinogen-deficient mice (arrow in D).
To quantitatively assess whether infection-stimulated fibrin formation protects against hemorrhagic events, we evaluated hematologic parameters in T. gondii–infected mice (Fig. 5) . In wild-type mice, we observed significant reductions in hematocrits, red cell numbers, and hemoglobin levels during the course of T. gondii infection, first evident around day 8 after infection and peaking around day 10 (Fig. 5 A, and unpublished data). Compared with control heterozygous mice, T. gondii–infected fibrinogen-deficient mice displayed significantly greater reductions in hematocrits, red cell counts, and hemoglobin levels at day 8 after infection (all P < 0.05; Fig. 5 B, and unpublished data). Treatment of wild-type mice with warfarin similarly exacerbated infection-associated red cell loss (unpublished data). Evaluation of peripheral blood smears revealed elevated numbers of reticulocytes, suggesting that red cell loss likely reflected infection-stimulated hemorrhage, rather than decreased erythropoiesis. Together, our histologic and hematologic observations indicate that coagulation protects against hemorrhage during T. gondii infection.
Figure 5.


Fibrin protects against infection-stimulated, IFN-γ–mediated hemorrhagic immunopathology. (A) Wild-type C57BL/6 mice were infected with T. gondii. On the indicated days, blood was collected and hematocrits, red cell numbers (unpublished data), and hemoglobin levels (unpublished data) were determined using a Coulter Counter (Beckman Coulter). Compared with uninfected mice, each of those parameters were significantly reduced by day 10 after infection (P < 0.001; n = 5 mice per group). (B) Fibrinogen-deficient and littermate control heterozygous mice were infected with T. gondii or were sham infected. On day 8 after infection, hematocrits were significantly reduced in T. gondii–infected control mice (*P < 0.02; n = 5 mice per group). Compared with infected control mice, T. gondii–infected fibrinogen-deficient mice displayed even greater hematocrit reductions (**P < 0.005; n = 5 mice per group). (C) Wild-type C57BL/6 and C57BL/6-backcrossed IFN-γ–deficient mice were infected with T. gondii. 8 d later, hematocrits were significantly reduced in T. gondii–infected wild-type control mice (*P < 0.005; n = 5 mice per group), but not in the IFN-γ–deficient mice. (D) Wild-type C57BL/6 received a single injection (1 mg, intraperitoneal) of control rat IgG1 mAb or anti–IFN-γ mAb (αIFNγ). The next day, mice were sham infected or infected with T. gondii. 8 d later, hematocrits were significantly reduced in T. gondii–infected control mAb-treated mice (*P < 0.008; n = 5 mice per group), but not in the anti-IFN-γ–treated mice. (E) Fibrinogen-deficient and littermate control mice received control rat IgG1 mAb or anti-IFN-γ mAb (1 mg, intraperitoneal), and were infected with T. gondii the following day. Hematocrits were measured at day 8 after infection. In both sets of mice, anti-IFN-γ mAb treatment significantly protected against hematocrit reductions (*P < 0.01; n = 4 mice per group). Notably, the hematocrits of anti-IFN-γ mAb-treated fibrinogen-deficient and littermate control mice were not significantly different. Similar results to those shown were replicated in two (A) or more (B–E) independent experiments.
We next assessed how infection evokes a coagulative response. T. gondii infects all nucleated cells (18), and thus has the potential to directly damage vascular cells, thereby causing hemorrhage and activating a coagulative response. However, parasite-stimulated immunopathology could also prompt hemorrhage leading to coagulation, as levels of IFN-γ and its potentially destructive downstream product, nitric oxide, both peak around the time of the acute death of coagulation-impaired T. gondii–infected animals (Fig. 1, and unpublished data). Notably, IFN-γ also up-regulates cellular expression of procoagulant molecules (26–28), thereby potentially amplifying fibrin production.
To decisively evaluate roles for IFN-γ in T. gondii–stimulated hemorrhagic pathology, we infected gene-targeted IFN-γ–deficient mice. Prior studies established that IFN-γ is a major mediator of resistance to T. gondii, as its neutralization leads to increased parasite burdens and acute mortality (25). Indeed, compared with wild-type mice infected in parallel, we found that T. gondii parasite numbers were elevated more than 1,000-fold in IFN-γ–deficient mice at day 8 after infection (P < 0.008; n = 5; wild-type 77 ± 36 parasites/mg liver tissue, IFN-γ–deficient = 79,000 ± 52,000 parasites/mg liver tissue). Nevertheless, hematocrits, red cell counts, and hemoglobin levels were not significantly reduced in T. gondii–infected IFN-γ–deficient mice (Fig. 5 C, and unpublished data). We observed similar results upon depletion of IFN-γ in wild-type C57BL/6 mice using a specific neutralizing mAb (Fig. 5 D). As parasite burdens were elevated, but red cell loss was not, we conclude that parasites do not directly cause significant hemorrhage. Rather, our data suggest that fibrin functions to suppress hemorrhagic pathology caused by IFN-γ, a product of type 1 immunity.
To formally establish that fibrin functions to suppress IFN-γ–stimulated hemorrhagic pathology, we evaluated IFN-γ–depleted, T. gondii–infected, fibrinogen-deficient mice. We found that neutralization of IFN-γ in fibrinogen-deficient mice suppressed the T. gondii–stimulated gross liver pathology (unpublished data) and hematocrit reductions (Fig. 5 E; P < 0.01; n = 4). Importantly, hematocrits in IFN-γ–depleted, T. gondii–infected, fibrinogen-deficient, and littermate control mice were not significantly different, establishing that the exacerbated hemorrhagic pathology observed in fibrinogen-deficient mice, like the lesser pathology observed in wild-type mice, is caused by IFN-γ.
Numerous prior studies concluded that immune-associated fibrin deposition exacerbates inflammation and pathology. Studies using factors isolated from snake venoms that transiently generate a fibrinogen-deficient state suggested pathological roles for fibrin in animals models of glomerulonephritis (4), arthritis (7), encephalomyelitis (9, 10), and transplant rejection (12). Studies using mice with impaired fibrin degrading capacities (i.e., gene-targeted plasminogen- or plasminogen activator-deficient mice) confirmed such pathological roles for fibrin during glomerulonephritis and arthritis (6–8). Studies using fibrinogen-deficient mice revealed pathological roles for fibrin(ogen) during atherosclerosis (29), and demonstrated that fibrin can up-regulate expression of inflammatory cytokines and chemokines in vivo (30). Finally, studies using anticoagulated or congenitally fibrinogen-deficient subjects established that fibrin also has inflammatory properties in humans (2, 3).
In contrast to those prior reports, our studies demonstrate that coagulation can also function protectively during immunity. Specifically, we found that fibrin can prevent infection-stimulated blood loss, thereby performing a protective function that is essential for survival. Remarkably, fibrin does not simply protect against vascular damage caused directly by the infectious agent, but rather, protects against hemorrhage evoked by IFN-γ, a critical mediator of type 1 immunity. Notably, IFN-γ also stimulates expression of genes that favor and/or stabilize fibrin deposition (26–28), suggesting that type 1 immunity may have evolved means to locally up-regulate coagulant activity, thereby preemptively protecting against its own destructive power.
Our observations indicate that the pathological potential of immune-associated coagulation is counter-balanced by a critical protective function. Such findings help to explain why type 1 immunity frequently elicits fibrin deposition (1–13), and suggest that clinicians should be cautious in ascribing pathological assessments to immune-associated fibrin deposits. While the inflammatory activities of fibrin and other coagulant products may function pathologically in certain contexts (1–16), immune-associated coagulative responses can also function beneficially, by protecting against immunopathology.
Notably, our data do not exclude the possibility that fibrin may also function protectively through direct effects on immunity and/or inflammation. In comparison to littermate control mice, there were no significant differences in pathogen dissemination or levels of immune mediators in T. gondii–infected fibrinogen-deficient mice (Fig. 3). However, the robust type 1 immune response that accompanies T. gondii infection may have masked immune-related impacts of fibrin-deficiency. Indeed, while own prior work demonstrated that thrombin can stimulate fibrin-dependent secretion of inflammatory cytokines/chemokines in vivo (30), subsequent studies found that endotoxin- or T cell–stimulated secretion of those same cytokines/chemokines is fibrin-independent (unpublished data). Thus, we suspect that infection-stimulated immunity may well mask fibrin's inflammatory activities during T. gondii infection.
Our data suggest that fibrin-unrelated coagulation deficiencies, whether genetic or pharmacologic, may also exacerbate immunopathology. As such, we are now exploring the generality of our findings. We are also working to define precisely how IFN-γ evokes pathology necessitating a protective coagulative response. We anticipate that those studies may suggest novel therapies for pathologies characterized by extensive fibrin deposition, including transplantation and many forms of autoimmunity, and may also impact treatments for the numerous virulent human pathogens that elicit deleterious coagulative and/or hemorrhagic responses.
Acknowledgments
We are grateful to Drs. Robert North, Troy Randall, Wayne Hancock, and Terri Laufer for critical reading of this manuscript. We also wish to thank Paula Lanthier and Jean Brennan for technical assistance, and Pamela Scott Adams for help with real-time PCR. We are indebted to the employees of the Trudeau Institute Animal Breeding and Maintenance Facilities for dedicated care of the mice used for these studies.
This work was supported by PHS Grant AI46571 (L.L. Johnson) and funds from Trudeau Institute.
References
- 1.Nelson, D.S. 1965. The effects of anticoagulants and other drugs on cellular and cutaneous reactions to antigen in guinea pigs with delayed-type hypersensitivity. Immunology. 9:219–234. [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards, R.L., and F.R. Rickles. 1978. Delayed hypersensitivity in man: effects of systemic anticoagulation. Science. 200:541–543. [DOI] [PubMed] [Google Scholar]
- 3.Colvin, R.B., M.W. Mosesson, and H.F. Dvorak. 1979. Delayed-type hypersensitivity skin reactions in congenital afibrinogenemia lack fibrin deposition and induration. J. Clin. Invest. 63:1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holdsworth, S.R., N.M. Thomson, E.F. Glasgow, and R.C. Atkins. 1979. The effect of defibrination on macrophage participation in rabbit nephrotoxic nephritis: studies using glomerular culture and electronmicroscopy. Clin. Exp. Immunol. 37:38–43. [PMC free article] [PubMed] [Google Scholar]
- 5.Neale, T.J., P.G. Tipping, S.D. Carson, and S.R. Holdsworth. 1988. Participation of cell-mediated immunity in deposition of fibrin in glomerulonephritis. Lancet. 2:421–424. [DOI] [PubMed] [Google Scholar]
- 6.Kitching, A.R., S.R. Holdsworth, V.A. Ploplis, E.F. Plow, D. Collen, P. Carmeliet, and P.G. Tipping. 1997. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J. Exp. Med. 185:963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Busso, N., V. Peclat, K. Van Ness, E. Kolodziesczyk, J. Degen, T. Bugge, and A. So. 1998. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J. Clin. Invest. 102:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang, Y.H., P. Carmeliet, and J.A. Hamilton. 2001. Tissue-type plasminogen activator deficiency exacerbates arthritis. J. Immunol. 167:1047–1052. [DOI] [PubMed] [Google Scholar]
- 9.Paterson, P.Y. 1976. Experimental allergic encephalomyelitis: role of fibrin deposition in immunopathogenesis of inflammation in rats. Fed. Proc. 35:2428–2434. [PubMed] [Google Scholar]
- 10.Inoue, A., C.S. Koh, K. Shimada, N. Yanagisawa, and K. Yoshimura. 1996. Suppression of cell-transferred experimental autoimmune encephalomyelitis in defibrinated Lewis rats. J. Neuroimmunol. 71:131–137. [DOI] [PubMed] [Google Scholar]
- 11.Dvorak, H.F., M.C. Mihm, Jr., A.M. Dvorak, B.A. Barnes, E.J. Manseau, and S.J. Galli. 1979. Rejection of first-set skin allografts in man. The microvasculature is the critical target of the immune response. J. Exp. Med. 150:322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, J., R. Munda, P. Glas-Greenwalt, M.A. Weiss, V.E. Pollak, and J.W. Alexander. 1983. Prolongation of survival of a heart xenograft by defibrination with ancrod. Transplantation. 35:620–622. [PubMed] [Google Scholar]
- 13.Labarrere, C.A., D.R. Nelson, and W.P. Faulk. 1998. Myocardial fibrin deposits in the first month after transplantation predict subsequent coronary artery disease and graft failure in cardiac allograft recipients. Am. J. Med. 105:207–213. [DOI] [PubMed] [Google Scholar]
- 14.Edwards, R.L., and F.R. Rickles. 1992. The role of leukocytes in the activation of blood coagulation. Semin. Hematol. 29:202–212. [PubMed] [Google Scholar]
- 15.Degen, J.L. 1999. Hemostatic factors and inflammatory disease. Thromb. Haemost. 82:858–864. [PubMed] [Google Scholar]
- 16.Esmon, C.T. 2001. Role of coagulation inhibitors in inflammation. Thromb. Haemost. 86:51–56. [PubMed] [Google Scholar]
- 17.Bernard, G.R., J.L. Vincent, P.F. Laterre, S.P. LaRosa, J.F. Dhainaut, A. Lopez-Rodriguez, J.S. Steingrub, G.E. Garber, J.D. Helterbrand, E.W. Ely, and C.J. Fisher, Jr. 2001. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med. 344:699–709. [DOI] [PubMed] [Google Scholar]
- 18.McCabe, R.E., and J.S. Remington. 1990. Toxoplasma gondii. 3rd ed. Principles and Practices of Infectious Diseases. G.L. Mandell, R.G. Douglas, and J.E. Bennett, editors. Churchill Livingstone, Inc., New York. 2090–2103.
- 19.Denkers, E.Y., and R.T. Gazzinelli. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11:569–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suh, T.T., K. Holmback, N.J. Jensen, C.C. Daugherty, K. Small, D.I. Simon, S. Potter, and J.L. Degen. 1995. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 9:2020–2033. [DOI] [PubMed] [Google Scholar]
- 21.Verdon, C.P., B.A. Burton, and R.L. Prior. 1995. Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP+ when the Griess reaction is used to assay for nitrite. Anal. Biochem. 224:502–508. [DOI] [PubMed] [Google Scholar]
- 22.Overbergh, L., D. Valckx, M. Waer, and C. Mathieu. 1999. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 11:305–312. [DOI] [PubMed] [Google Scholar]
- 23.Weiler-Guettler, H., P.D. Christie, D.L. Beeler, A.M. Healy, W.W. Hancock, H. Rayburn, J.M. Edelberg, and R.D. Rosenberg. 1998. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J. Clin. Invest. 101:1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth, S.S., E.D. Reis, H. Vaananen, W. Zhang, and B.S. Coller. 2001. Variable protection of beta 3-integrin-deficient mice from thrombosis initiated by different mechanisms. Blood. 98:1055–1062. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki, Y., M.A. Orellana, R.D. Schreiber, and J.S. Remington. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science. 240:516–518. [DOI] [PubMed] [Google Scholar]
- 26.Moon, D.K., and C.L. Geczy. 1988. Recombinant IFN-gamma synergizes with lipopolysaccharide to induce macrophage membrane procoagulants. J. Immunol. 141:1536–1542. [PubMed] [Google Scholar]
- 27.Schwager, I., and T.W. Jungi. 1994. Effect of human recombinant cytokines on the induction of macrophage procoagulant activity. Blood. 83:152–160. [PubMed] [Google Scholar]
- 28.Hamilton, J.A., G.A. Whitty, K. Last, A.K. Royston, P.H. Hart, and D.R. Burgess. 1992. Interleukin-4 suppresses plasminogen activator inhibitor-2 formation in stimulated human monocytes. Blood. 80:121–125. [PubMed] [Google Scholar]
- 29.Lou, X.J., N.W. Boonmark, F.T. Horrigan, J.L. Degen, and R.M. Lawn. 1998. Fibrinogen deficiency reduces vascular accumulation of apolipoprotein(a) and development of atherosclerosis in apolipoprotein(a) transgenic mice. Proc. Natl. Acad. Sci. USA. 95:12591–12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szaba, F.M., and S.T. Smiley. 2002. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood. 99:1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]