

Abstract
In this study we have shown that activation of arthritogenic splenocytes with antigen and agonistic anti-CD40 gives raise to a B cell population that produce high levels of interleukin (IL)-10 and low levels of interferon (IFN)-γ. Transfer of these B cells into DBA/1-TcR-β-Tg mice, immunized with bovine collagen (CII) emulsified in complete Freund's adjuvant inhibited T helper type 1 differentiation, prevented arthritis development, and was also effective in ameliorating established disease. IL-10 is essential for the regulatory function of this subset of B cells, as the B cells population isolated from IL-10 knockout mice failed to mediate this protective function. Furthermore, B cells isolated from arthritogenic splenocytes treated in vitro with anti–IL-10/anti–IL-10R were unable to protect recipient mice from developing arthritis. Our results suggest a new role of a subset of B cells in controlling T cell differentiation and autoimmune disorders.
Keywords: autoimmunity, immunoregulation, B cells, interleukin 10, CD40
Introduction
CD40 is a member of the TNF superfamily and is expressed mainly on B cells and dendritic cells (DCs)* as well as endothelial cells, macrophages, T cells, and fibroblasts (1, 2). CD40-CD154 interaction is crucial for the production of soluble factors such as cytokines (e.g., IL-6, TNF-α, IL-12, IL-10, IL-1), chemokines (e.g., macrophage inflammatory protein-1 α), and for the up-regulation of other costimulatory molecules such as CD80, CD86, and CD54 (1). The CD40 pathway plays an important role in B cell activation, inducing proliferation and isotype switching (3, 4) while the engagement of CD40 expressed on DCs drives their terminal maturation and the production of IL-12 (5). Given the broad effects mediated by the engagement of CD40 with CD154, it is hardly surprising that blocking this interaction, using for example anti-CD154 mAb, strongly impaired the activation of antigen-specific immune response, leading to reduced T cell priming, defects in effector function, and under specific circumstances to tolerance (6, 7).
Chronic collagen-induced arthritis (CCIA) is a Th1 cell–dependent chronic arthritis, which develops after immunization of DBA/1-TcR-β-Tg mice with type II collagen (CII) in CFA (8). Previous experiments from our laboratory showed that agonistic anti-CD40 surprisingly ameliorated established arthritis. After anti-CD40 treatment an increase of IL-10 production and a decrease of IFN-γ release was observed (9). These results suggested that the therapeutic effect observed after administration of anti-CD40, could have been achieved by redirecting pathogenic Th1 type response toward the “protective” Th2 type (9).
In this study, we set out to investigate the basis of this effect in detail. Although anti-CD40 treatment might work by altering DCs or B cell function, two observations led us to focus on the effect that anti-CD40 stimulation has on B cells. First, CD40 activation of DCs, especially after stimulation by bacterial components leads to IL-12 production (5); this effect is unlikely to alleviate a Th1-driven disease. Second, a number of studies have highlighted the influence that B cells have on T cell differentiation (10–12) and on the down-regulation of the IL-12 produced by DCs after B cell CD40 engagement (11, 13). One of these studies has also indicated that the influence of B cells on T cell differentiation was mediated by cytokines, such as IL-6 and IL-10, after CD40 activation (11). More recently, Harris et al. have defined subset of B effector cells secreting cytokines in a similar way to Th1 and Th2 cells (12). The same authors have also shown that the cytokines produced by B effector cells (Be1 and Be2), contribute to the in vitro differentiation of naive T cell into Th1 or Th2 type (12). Here we report that in vitro stimulation with an agonistic anti-CD40 mAb and antigen generates B cells capable of inhibiting Th1 differentiation and preventing as well as ameliorating arthritis induced by CII/CFA immunization, when injected into DBA/1-TcR-β-Tg mice. CII/anti-CD40–stimulated B cells isolated from IL-10 KO mice were unable to protect recipient mice from arthritis, suggesting that IL-10 plays a key role in this mechanism of immune suppression.
Materials and Methods
Mice.
The previously described heterozygous TCR-β Tg SWR/J mice were extensively backcrossed with DBA/1 mice in order to derive the CCIA model (8, 13). Mice from generation N15 typed for TCR-β Tg expression, were selected for the experiments. IL-10 KO in H2q background were generated by backcrossing the original IL-10 KO/H2b (14) with DBA/1 H2q mice. The mice were typed by PCR and IL-10 KO−/−H2q were further backcrossed into DBA/1. Mice from the fourth generation (IL-10 KO−/−/H2q) were used for the transfer experiments.
Antibodies.
The treatment antibodies used were FGK45, rat IgG, agonistic mAb reactive with mouse CD40; 1B1.2, rat IgG, blocking mAb reactive with mouse IL-10 receptor (IL-10R) (provided by F. Powrie, Oxford, UK); JES5–2A5, (rat IgG1) an anti–mouse IL-10 mAb; 11.B.11 (rat IgG) neutralizing IL-4; AFRC-Mac-1 (isotype control) rat IgG anti–dog chlamydomonas cell wall glycoprotein (European Collection of Animal Cell culture, Salisbury, UK). The mAbs were purified from culture supernatants by affinity chromatography, using a staphylococcal protein G column (Bioprocessing) and filter sterilized. All antibodies used throughout the experiments were below the limit of detection for LPS levels using the Limulus Amebocyte lysate test (Bio Whittaker Inc.).
The following mouse antibodies were purchased from BD Biosciences. The mAbs used for FACS® analysis were against mouse CD3 PE or FITC, mouse CD19 FITC, mouse vβ12 TcR FITC (screening). The mAbs used for the intracellular detection of cytokines were against mouse IFN-γ PE, IL-10 PE, and CD19 FITC or CD3 FITC mAbs. The coating/detection antibody pairings used for ELISA are as follows; IL-4: ID11/24G2 (American Type Culture Collection and BD Biosciences, respectively); IL-10, rat mAbs 2A5/SXC-1: and IFN-γ (R46A2/XMG1.2), American Type Culture Collection, courtesy of Dr. J. Abrams at DNAX (Palo Alto, CA).
Preparation of Collagen.
Bovine CII was purified and prepared as described previously (8). Bovine CII was solubilized by stirring overnight at 4°C in 0.1 M acetic acid, to be used for immunization, or 0.05 mM Tris-HCL, 0.2 M NaCl, pH 7.4, for in vitro stimulation of splenocyte cultures.
Induction, Assessment of Arthritis and Histological Examination.
Sex-matched DBA/1-TcR-β-Tg mice (8–12-wk-old) were immunized with 100 μg of bovine CII emulsified in CFA (Difco Laboratories). The development of arthritis was assessed daily for the duration of the experiment. The clinical severity of arthritis was graded as follows, 0 = normal, 1 = slight swelling and/or erythema, 2 = pronounced edematous swelling, 3 = pronounced edematous swelling plus light joint rigidity, 4 = laxity (8). Each limb was graded, allowing a maximal clinical score of 16 for each animal. Swelling of hind paws was recorded with a pair of calipers. All clinical evaluations were performed in a blinded manner.
Magnetic Sorting of Cells.
For the purification of B cells, single cell suspensions from spleens isolated from arthritic DBA/1-TcR-β-Tg mice were cultured at concentration of 5 × 106 cells/ml in complete medium for 48 h with: medium/isotype control (AFRC-Mac-1; 5 μg/ml); 50 μg/ml of CII /isotype control; medium/anti-CD40 (FGK45; 5 μg/ml); CII/anti-CD40. Cells were pooled from different wells and washed with complete medium; 107 cells were then resuspended in 90 μl of PBS, 0.5% of FCS, and 5 mM of EDTA. The cell suspensions were incubated for 15 min at 4°C with 10 μl of anti-CD43 magnetic beads for negative selection or CD19+ magnetic beads for positive selection (Miltenyi Biotec) per 90 μl of cell suspension. The cells were washed twice to remove unbound beads resuspended in 500 μl of buffer (PBS, 0.5% FCS, 5 mM EDTA) and purified using a magnetic activated cell-sorting (MACS) system. FACS® staining for the B cell marker CD19 was used to control cell purity. This procedure normally yielded B cell preparations which were >95% CD19. DCs were purified as previously described (15). DCs were depleted by positive selection, using CD11c microbeads (Miltenyi Biotec), from spleens isolated from arthritic mice, prior CII (50 μg/ml)/isotype control (5 μg/ml) or CII/anti-CD40 (5 μg/ml) stimulation. Briefly, spleens were isolated from at least eight arthritic DBA/1-TcR-β-Tg mice and placed in Collagenase D (7 mg/ml). Spleens were then injected with 500 μl of Collagenase D for 60 min at 37°C. The digested suspension was then passed through a steel mesh using a plunger. 108 cells were collected in a 50 ml tube, washed with buffer (PBS, 0.5% FCS, 5 mM EDTA), and resuspended in 400 μl buffer. The cell suspensions were incubated for 15 min at 4°C with 100 μl of MACS CD11c Micro-Beads (Miltenyi Biotec) per 400 μl of cell suspension. The cells were washed twice to remove unbound beads and resuspended in 500 μl of buffer (PBS, 0.5% FCS, 5 mM EDTA) for magnetic separation. FACS® analysis was used to monitor the levels of CD11c+CD11b+ cells in the negative fraction and less than 0.01% of any of these cells was detected in the negative fraction. CD3+ T cells were purified from spleen cell suspension by a positive selection procedure using anti–Thy-1.2 labeled micro-beads and MACS. This procedure normally yielded cells which were >98% CD3+ T cells. CD4+ T cells were purified by positive selection using anti-CD4+ labeled micro-beads.
Flow Cytometric Analysis of Intracellular IFN-γ and IL-10 Synthesis.
Intracellular cytokine analysis was performed as described previously (9, 12). Briefly, splenocytes or purified cells were resuspended at 5 × 106 cells/ml with medium or CII (50 μg/ml) for 48 h unless stated otherwise. PMA (50 ng/ml; Sigma-Aldrich), ionomycin (500 ng/ml; Sigma-Aldrich), and Brefeldin A (10 μg/ml; Sigma-Aldrich) were added for an additional 6 h. To detect the surface Ags, cells were washed with PBS/BSA/azide and then incubated with mouse anti-CD19 FITC-conjugated mAb. After fixing (4% paraformaldehyde) for 10 min at 4°C, cells were washed and permeabilized with PBS containing 1% BSA and 0.5% saponin (Sigma-Aldrich) for 10 min. Permeabilized cells were incubated with anti–mouse IFN-γ PE-conjugated, or anti–mouse IL-10 PE-conjugated mAbs (BD Biosciences). To demonstrate the specificity of staining, fixed/permeabilized cells were incubated with excess of unlabeled anti–IFN-γ or anti–IL-10 (BD Biosciences) before incubation with FITC anti–IFN-γ or PE anti–IL-10. The cells were acquired with the FACScan™ flow cytometer (Becton Dickinson) and analyzed by WinMdi software.
Spleen Cell Culture, Cytokines Quantification, Carboxyl Fluorescein Succinimidyl Ester Labeling, and Adoptive Transfer to DBA/1-TcR-β-Tg Mice.
Spleens were removed from arthritic DBA/1-TcR-β-Tg mice on the day of clinical onset of arthritis. Single cell suspensions were prepared and depleted of red blood cells with red cell lysis buffer (Sigma-Aldrich), washed, and cultured in RPM1 1640 containing 10% (vol/vol) heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 × 10−5 M 2-mercaptoethanol, and 20 mM l-glutamine. Splenocytes were cultured in 24-well plates (Nunc) at a density of 5 × 106 cells/ml in medium alone or with 50 μg/ml of CII and 5 μg/ml of isotype control (AFRC-Mac-1) or with 50 μg/ml CII and 5 μg/ml of anti-CD40 (FGK45) 48 h. Purified B cells, B cell–depleted fraction, or DC-depleted fractions were transferred intraperitoneally or intravenously to DBA/1-TcR-β-Tg at the time of CII/CFA immunization. Only CD19+ purified B cells, whose purity was >98%, were used in the transfer experiment.
B cells were purified by negative selection. CD43− fraction (CD19+ B cells) was labeled with the carboxyl fluorescein succinimidyl ester (CFSE) by incubation with 2 μl of CFSE (5 μM; Molecular Probes) for 8 min at 37°C in PBS, followed by quenching of the unlabeled CFSE with excess FCS and extensive washing. CFSE-labeled B cells were then remixed with the CD43+ fraction. The reconstituted splenocytes were cultured in 24-well plates at a density of 5 × 106 cells/ml in medium alone or with CII/isotype control (AFRC-Mac-1) or with CII/anti-CD40 (FGK45) for 48 h. 107 CFSE-labeled B cells were transferred intraperitoneally to DBA/1-TcR-β-Tg at the time of CII/CFA immunization. 3 d after the in vivo transfer, spleens were collected and cultured with medium or CII for 48 h. CFSE-labeled B cells were sorted on a FACSVantage™ (Becton Dickinson). 5 × 105 CFSE-sorted B cell were culture for 6 h with PMA (50 ng/ml) and ionomycin (500 ng/ml). Supernatants were analyzed for cytokine secretion by ELISA. All populations were 98% pure on reanalysis. For cytokines quantification splenocytes or LNCs were cultured for 24 to 72 h. ELISA measured cytokine productions as described previously (16).
Statistical Analysis.
For the statistical analysis of the data, the Mann-Whitney U test and the Fisher exact test were applied to analyze clinical results. Unpaired t tests were applied on cytokine quantification experiments. P < 0.05 was considered significantly different.
Results
Adoptive Transfer of Anti-CD40–treated Splenocytes Prevented the Evolution of Arthritis.
We have previously shown that in vitro anti-CD40 mAb (hereafter anti-CD40) stimulation of splenocytes from arthritic mice prevented the transfer of arthritis into SCID mice (9). Here we have examined whether the transfer of anti-CD40–treated in vitro splenocytes to DBA/1-TcR-β-Tg mice, at the time of CII in CFA immunization, could also inhibit disease progression. Spleens were isolated from DBA/1-TcR-β-Tg arthritic mice, and restimulated in vitro for 48 h with CII/anti-CD40 or with CII/isotype control (referred to as control). 5 × 106 splenocytes were transferred at the time of CII in CFA immunization, intraperitoneally to DBA/1-TcR-β-Tg mice. A group of mice was also left untreated. The results reported in Fig. 1 a show that while 100% of DBA/1-TcR-β-Tg mice injected with control splenocytes developed severe arthritis, transfer of splenocytes challenged in vitro with anti-CD40 remarkably suppressed disease evolution in 80% of the recipient mice (Fig. 1 a). The remaining 20% developed a significantly milder arthritis compared with the control or untreated group (P < 0.001; Fig. 1 b).
Figure 1.

Transfer of anti-CD40–stimulated splenocytes inhibits arthritis in DBA/1-TcR-β Tg mice. Spleen cells isolated from mice with established arthritis were cultured in vitro for 48 h with CII/isotype control or CII/anti-CD40. 5 × 106 cells were transferred to DBA/1-TcR-β Tg mice on the day of CII/CFA immunization. A group of mice was left untreated. (a) Incidence of arthritis; groups of mice were compared by statistical analysis using the Fisher exact test. (b) Arthritis severity; data are expressed as mean ± SE. Groups of mice were compared by statistical analysis using the nonparametric Mann-Whitney U test. Data are representative of three experiments.
Anti-CD40–mediated Protection Is B Cell Dependent.
We next investigated whether the protective effect induced after anti-CD40 treatment was either B cell or DC dependent. B cells or DCs were depleted by immune-magnetic positive selection, before in vitro CII/anti-CD40 or CII/isotype control stimulation. After 48 h of incubation, 5 × 106 total or B cell–depleted splenocytes were transferred to DBA/1-TcR-β-Tg mice at the time of CII/CFA immunization. Consistent with the results shown above, 70% of recipient mice treated with anti-CD40 total splenocytes in Fig. 2 a, and 30% in Fig. 2 b remained disease free until the end of the experiment, while the remaining mice (30% in experiment a and 70% in b) showed very mild signs of inflammation. The picture changed when B cells were depleted before anti-CD40 stimulation. In this case, 100% of the recipient mice developed arthritis within 27 d from CII immunization (Fig. 2 a). Interestingly, a significantly earlier day of onset was also observed in mice transferred with control treated B cell–depleted splenocytes, compared with those treated with total splenocytes (P < 0.05; Fig. 2 a). Next we evaluated the impact of depletion of DCs, before in vitro anti-CD40 stimulation. 100% of mice transferred with 5 × 106 cells control DC-depleted splenocytes developed arthritis. However, the disease onset was delayed compared with mice treated with total splenocytes (Fig. 2 b). In contrast, only 25% of mice treated with DC depleted, before anti-CD40 stimulation, splenocytes developed arthritis while the remaining 75% remained disease free until the end of the experiment (Fig. 2 b). These results suggest that B cells, but not DCs, are involved in the antiinflammatory effect induced by anti-CD40 observed in the in vivo transfer experiments.
Figure 2.

Are B cells the key players in the antiinflammatory effect mediated by in vitro stimulation with agonistic anti-CD40? B cells or DCs were depleted by positive selection from splenocytes isolated from arthritic DBA/1-TcR-β Tg mice. Undepleted or depleted splenocytes were stimulated with CII/isotype control, CII/anti-CD40. After 48 h incubation, 5 × 106 nondepleted or depleted cells were transferred intraperitoneally, at the time of CII/CFA immunization, to DBA/1-TcR-β Tg mice. (a and b) Incidence of arthritis. Mice groups were compared by statistical analysis using the Fisher exact test. Data are representative of three experiments.
B Cells from Anti-CD40 Challenged Splenocytes Produce CII-specific IL-10.
B cells, in a fashion similar to T cells, can differentiate into polarized subsets of Be 1 or Be 2 that are capable of secreting unique cytokines (12). Therefore, we next investigate whether B cells activated with CII and anti-CD40 released detectable levels of IFN-γ and IL-10. Arthritogenic splenocytes were cultured in vitro with or without CII and also with isotype control or anti-CD40 for 48 h. B cells were enriched by negative selection and the release of cytokines was detected by intracellular staining as described in Materials and Methods. Upon in vitro control antibody stimulation, 11.06% of B cells produced IFN-γ, while 7.54% produced IL-10. After stimulation with anti-CD40, the number of IFN-γ–producing B cells was reduced to 2.63% while the numbers of IL-10+ B cells were increased to 22.15% (Fig. 3 a). We also measured the levels of IL-4 production under the same conditions and we found no differences between the different groups (unpublished data). IFN-γ and IL-10 release by purified B cells, after PMA and ionomycin stimulation, were measured by ELISA. As shown in Fig. 3 b, B cells purified from control splenocytes produce IFN-γ and a lower amount of IL-10. On the contrary, B cells isolated from anti-CD40–stimulated splenocytes produce IL-10 and little IFN-γ. In the absence of CII stimulation the levels of IFN-γ become undetectable in all groups, while a small amount of IL-10 was released in the B cells isolated from anti-CD40 stimulated splenocytes (<500 pg; unpublished data).
Figure 3.


Generation of B cells with a regulatory phenotype is controlled by endogenous IL-10. Arthritogenic splenocytes were cultured with CII/isotype control, or CII/anti-CD40 for 48 h. B cells were then purified by negative selection and cultured with PMA plus ionomycin in the presence of Brefeldin A for an additional 6 h. (a) The intracellular levels of IFN-γ and IL-10 were measured using PE-conjugated mAb against IFN-γ or IL-10 and the purity of B cells was controlled using anti-CD19 FITC mAb. (b) Supernatant was collected from purified B cells stimulated in vitro with PMA plus ionomycin and the secreted cytokines were measured by ELISA. Data show mean ± SE of triplicate wells, and are representative of four experiments. Data were compared by statistical analysis using the unpaired t test. (c) B cells were purified from arthritogenic splenocytes isolated from mice immunized with CII/CFA or with CFA alone. Negatively purified B cells were cultured with or without CII anti-CD40, anti-CD40 + anti-κ for 72 h. Data are representative of three experiments that gave similar results.
To further test if IL-10 production required antigen stimulation, B cells were negatively purified from arthritogenic splenocytes isolated from mice immunized with CII/CFA or with CFA alone. To ensure B cell receptor cross-linking, plates were precoated with CII. Negatively purified B cells were added to CII-coated or uncoated plates and cultured for 72 h in the presence of anti-CD40 and anti-κ. The results reported in Fig. 3 c show that B cells (from CII/CFA immunized mice) incubated with antigen alone do not produce detectable levels of IL-10. Stimulation with anti-CD40 and CII only marginally increased the production of IL-10 while higher levels were detected upon culture with anti-CD40 and anti-κ. B cells isolated from spleens of mice immunized with CFA alone were unable to respond to anti-CD40 and CII stimulation; however, high levels of IL-10 were measured upon anti-CD40 and anti-κ stimulation. Therefore, we can conclude that the production of IL-10 required BCR engagement as well as CD40 signal.
B Cells Isolated from Anti-CD40–stimulated Splenocytes Prevent the Induction of Arthritis.
We next evaluated whether B cells, isolated from anti-CD40–treated splenocytes, can protect recipient mice from developing arthritis. Splenocytes isolated from DBA/1-TcR-β-Tg arthritic mice were cultured for 48 h with medium/anti-CD40, CII/anti-CD40, or medium/isotype control, and CII/isotype control. 5 × 105 B cells were purified and transferred intra-peritoneally to syngeneic mice at the day of CII/CFA immunization. More than 99% of purified B cells were CD19+, and less than 1% expressed T cell (CD3; Fig. 4, e and f) , or myeloid (Mac-1) specific markers (unpublished data). Fig. 4 a shows that only 20% of mice treated with B cells isolated from CII/anti-CD40–treated splenocytes, developed arthritis and the disease severity was significantly milder compared with that observed in the untreated group (P < 0.001; Fig. 4 b). 100% of mice transferred with B cells isolated from CII/isotype control-treated splenocytes developed disease by day 50 after immunization; however, this group of mice showed a delay in the onset of arthritis and a significantly milder disease compared with the untreated mice (P < 0.01 Fig. 4 b). This finding suggests that B cells with “protective” phenotype might be present during an antigen-mediated inflammatory response and their activity can be amplified by CD40 stimulation. The protective effect mediated by anti-CD40–stimulated B cells was found to be antigen dependent as B cells purified from anti-CD40–stimulated splenocytes cultured without CII, fail to confer protection to the recipient mice (Fig. 4, c and d).
Figure 4.



Transfer of B cells isolated from anti-CD40 activated splenocytes prevents chronic arthritis development. (a–c) DBA/1-TcR-β-Tg splenocytes were stimulated in vitro for 48 h with isotype control/CII; isotype control alone or with anti-CD40/CII or anti-CD40 alone. B cells were enriched by negative selection, using an anti-CD43 magnetic beads conjugated antibody, and 5 × 105 B cells were transferred intraperitoneally, at the time of CII/CFA immunization, to syngeneic mice. Percentage of mice with arthritis. Mice groups were compared by statistical analysis using the Fisher exact test. (b–d) Severity of disease. The data represent the mean of (n) mice ± SE and they are representative of five experiments. Groups of mice were compared by statistical analysis using the nonparametric Mann-Whitney U test. The purity of B cells, isolated from (e) control or (f) anti-CD40–treated splenocytes, was checked before transfer, by staining the negative fraction with FITC-conjugated anti-CD19 and PE-conjugated anti-CD3 mAbs. (c and d) DBA/1-TcR-β-Tg splenocytes were stimulated in vitro for 48 h with isotype control or with anti-CD40 alone. B cells were enriched by negative selection, using an anti-CD43 magnetic beads conjugated antibody, and 5 × 105 B cells were transferred intraperitoneally, at the time of CII/CFA immunization, to syngeneic mice. Percentage of mice with arthritis. (g) B cells were enriched by negative selection, using an anti-CD43 magnetic beads conjugated antibody, from DBA/1-TcR-β-Tg arthritogenic splenocytes. Purified B cells were stimulated in vitro for 48 h with isotype control/CII; or with anti-CD40/CII. 5 × 106 B cells were transferred intraperitoneally, at the time of CII/CFA immunization, to syngeneic mice. A group of mice was left untreated. Percentage of mice with arthritis. Data are representative of four experiments.
To explore whether other cells within the splenic pool were necessary to induce their secreting IL-10 phenotype, B cells were purified by negative selection from arthritogenic splenocytes and cultured in vitro with CII and anti-CD40 or CII and isotype control for 48 h. Under this experimental condition 100% of the recipient mice treated either with B cells from CII/isotype control or CII/anti-CD40 treated splenocytes, developed severe arthritis (Fig. 4 g). No protection from arthritis was observed in mice transferred with B cells cultured in medium alone and anti-CD40 or with isotype control (unpublished data).
Transfer of CII/anti-CD40 Stimulated B Cells into Immunized Mice Inhibits IFN-γ Response and Up-regulates IL-10 Production.
To test how the B cell transfer was influencing the cytokines profile to auto-antigen in the host mice, we assessed the recall responses to CII of LNCs and splenocytes taken from B cell–treated mice.
Fig. 5 a shows that LNCs isolated from mice treated with control or anti-CD40 stimulated B cells responded in vitro to CII restimulation with a similar magnitude to those of untreated mice. This observation suggests that transfer of B cells into immunized mice does not appear to affect CII-induced T cell activation assessed by proliferation.
Figure 5.
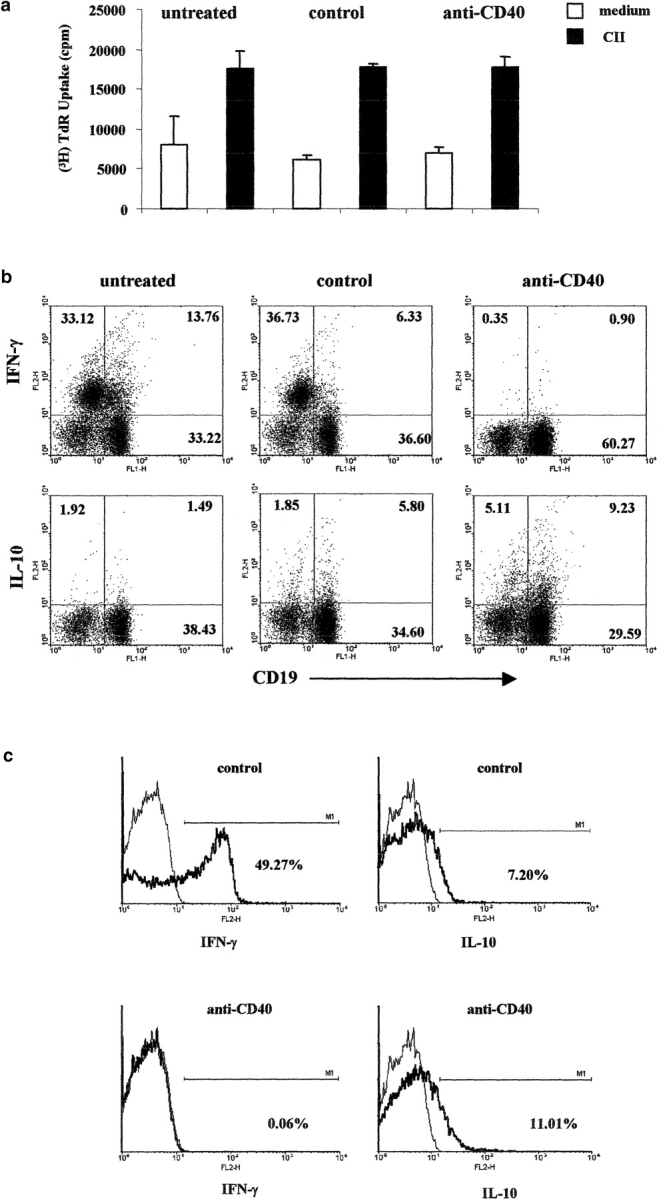
Treatment with anti-CD40–stimulated B cells, inhibits IFN-γ production, and increases the release of IL-10 in CII immunized DBA/1-TcR-β-Tg mice. Six DBA/1-TcR-β-Tg mice were injected intraperitoneally with B cells isolated from isotype control or anti-CD40 in vitro challenged splenocytes. A group of six mice were left untreated. 20 d after transfer draining lymph nodes were isolated, and cultured in medium alone or in the presence of CII for 72 h. (a) The proliferation of LNCs was measured in medium alone or in response to CII; data show mean ± SE of triplicate wells, and are representative of four experiments. Data were compared by statistical analysis using the Unpaired t test. (b) Splenocytes, were cultured with CII/isotype control, or CII/anti-CD40 for 48 h PMA, ionomycin and Brefeldin A were added for an additional 6 h. The intracellular levels of IFN-γ and IL-10 were measured using PE-conjugated mAb against IFN-γ or IL-10. Splenocytes were stained with FITC-conjugated mAb against CD19, to define the percentage of B cells synthesizing cytokines. Data are representative of four experiments. (c) Lymph nodes were isolated from the same group of mice and cultured with CII/isotype control, or CII/anti-CD40 for 48 h. T cells were purified by positive selection using anti–Thy-1.2 magnetic beads antibodies and cultured for an additional 6 h with PMA, ionomycin, and Brefeldin A. The intracellular levels of IFN-γ and IL-10 were measured using and PE-conjugated mAb against IFN-γ or IL-10.
Spleens were isolated from the same group of mice and the levels of IFN-γ and IL-10 produced by B cells were detected by intracellular staining after CII in vitro restimulation. Cytokine profiles revealed an inhibition in the number of IFN-γ + cells and an up-regulation of the expression of IL-10–producing B cells in the CII/anti-CD40–stimulated B cell–treated mice compared with CII/isotype control B cells and untreated (Fig. 5 b). The number of IFN-γ+ cells was undetectable in the absence of CII in the control B cells group, while 5% of B cells produce IL-10 in medium alone in the anti-CD40 B cells group (unpublished data). Mice treated with control B cells showed a reduced number of IFN-γ–producing cells and a modest increase of IL-10+ B cells compared with the untreated group (Fig. 5 b). To confirm that B cells transfer was influencing Th1 differentiation, we assessed the recall response to CII of T cells purified from LNCs isolated from control and anti-CD40 B cell–treated mice. Consistently with the results reported in Fig. 4 B, CD3+ cells expressing IFN-γ (49.27%) were very high in arthritic control mice. In contrast, Th1 differentiation was completely suppressed in mice treated with anti-CD40–stimulated B cells (Fig. 5 c). Similar percentages of IL-10+ CD3+ expressing cells were observed in the control or in the anti-CD40–stimulated B cells group (Fig. 5 c).
Transferred B Cells Produce IL-10 Ex Vivo.
We next investigate whether the B cells adoptively transferred into syngeneic mice were able to produce IL-10 in vivo. Ten million CFSE-labeled B cells were transferred to recipient mice. 3 d later, CFSE+ B cells were purified by sterile sorting from spleens isolated from transferred mice, and IFN-γ and IL-10 levels were measured by ELISA after in vitro stimulation as described in Materials and Methods. The results reported in Fig. 6 a show that higher levels of CII-specific IFN-γ were detected in CFSE+ B cells isolated from animals transferred with CII/isotype control B cells compared with anti-CD40-stimulated B cells group (P < 0.0002). In contrast, the latter B cells produce significantly higher levels of IL-10 compared with CII/isotype control B cells (P < 0.001; Fig. 6 b). These results therefore suggest that CII/anti-CD40 stimulated B cells are able to produce IL-10 in vivo.
Figure 6.

In vivo cytokine production by anti-CD40 stimulated B cells reisolated from spleen of treated mice. B cells were purified by negative selection from arthritic splenocytes and labeled with CFSE. The labeled fraction was reconstituted with the CD43+-positive fraction and cultured with CII/isotype control or with CII/anti-CD40 for 48 h. 107 CFSE+ B cells were then transferred to immunized DBA/1-TcR-β-Tg mice after purification by negative selection. 3 d after the transfer, 5 × 106 splenocytes were incubated in medium alone or with CII. 48 h later CFSE labeled B cells were sorted and stimulated for a 6 h with ionomycin and PMA. The release of (a) IFN-γ and (b) IL-10 was analyzed by ELISA. The data (mean ± SE of triplicate wells) are representative experiments, using six to eight mice per group. Each experiment was repeated twice with similar results. Data were compared by statistical analysis using the unpaired t test.
IL–10 Is Essential for the In Vivo Protective Effect of CII/anti-CD40–stimulated B Cells.
To determine whether the IL-10 produced by CII/anti-CD40 stimulated B cells is directly involved in the suppression of arthritis, IL-10 KO (H2b) mice were backcrossed into DBA/1 (H2q) mice and the B cells from the IL-10 KO (IL-10KO/H2q) or wild-type (IL-10+/H2q) mice were used for the transfer studies after CII/anti-CD40 in vitro stimulation. Unlike B cells isolated from the wild-type group, CII/anti-CD40–stimulated B cells isolated from IL-10 KO mice were unable to protect wild-type recipient from developing arthritis (Fig. 7 , a and b). Additionally, in vivo neutralization of IL-10 activity, by administration of neutralizing anti–IL-10 and anti–IL-10r mAbs, also abolished the protective effect assert by anti-CD40 B cell transfer (unpublished data), supporting the hypothesis that B cells, throughout the provision IL-10, interfere with the induction of arthritis.
Figure 7.

B cells isolated from IL-10 KO/H2q mice fail to differentiate into a “regulatory” phenotype. Spleens were isolated from arthritic IL-10 KO/H2q mice or from the control IL-10 wild-type mice. Splenocytes were cultured for 48 h with CII/isotype control, CII/anti-CD40. B cells were enriched by negative selection, using an anti-CD43 magnetic beads–conjugated antibody, and 5 × 105 cells were transferred intraperitoneally, at the time of CII/CFA immunization, to IL-10 wild-type mice. Arthritis evolution and severity was measured as described previously. Data shows incidence of arthritis (a) and mean of clinical score ± SE (b), and are representative of three experiments. Groups of mice were compared by statistical analysis using the Fisher exact test.
IL-10 and CD4+ T Cells Are Essential for the Generation of B Cells which Can Suppress Arthritis.
To establish whether IL-10 is involved in the generation of the B cells able to suppress arthritis, splenocytes were cultured with CII/anti-CD40 or CII/isotype control in the presence of neutralizing anti–IL-10R mAb (17) or anti–IL-4 mAb. B cells were purified by negative selection and 5 × 105 cells were transferred to immunized DBA/1-TcR-β-Tg mice. Inhibition of IL-10 during in vitro stimulation with CII/anti-CD40, abrogates the protective effect mediated by anti-CD40–stimulated B cells in recipient mice (Fig. 8 a). Thus, 100% of the recipient mice treated with B cells isolated from splenocytes costimulated with CII/anti-CD40 and anti-IL-10R mAb developed arthritis within 36 d of immunization. In contrast, neutralization of IL-4 did not affect the protective effect of anti-CD40 (Fig. 8 a).
Figure 8.

(a) Generation of B cells with “protective” phenotype is controlled by endogenous IL-10. Splenocytes isolated from DBA/1-TcR-β-Tg were cultured with CII/isotype control, or CII/anti-CD40 for 48 h alone or in the presence of neutralizing anti–IL-10R mAb (1B1.2) or anti–IL-4 mAb (11B11). 5 × 105 B cells were purified and transferred intraperitoneally to DBA/1-TcR-β-Tg mice at the time of CII/CFA immunization. Data are representative of two experiments. Groups of mice were compared by statistical analysis using the Fisher exact test. (b) Antigen-specific CD4+ T cells also play a crucial role in the generation of B cells with “protective” phenotype. CD4+ T cells were depleted by positive selection from splenocytes isolated from arthritic DBA/1-TcR-β Tg mice. Undepleted or depleted splenocytes were stimulated with CII/isotype control, CII/anti-CD40. After 48 h incubation, 5 × 106 nondepleted or depleted cells were transferred intraperitoneally, at the time of CII/CFA immunization, to DBA/1-TcR-β Tg mice. Incidence of arthritis. Mice groups were compared by statistical analysis using the Fisher exact test. Data are representative of three experiments.
The lack of protection observed in mice treated with B cells directly isolated from arthritogenic splenocytes followed by CII/anti-CD40 in vitro stimulation (Fig. 4 g) suggest that other “signals” in addition to anti-CD40 engagement are required for the fully differentiation of B cells with “protective” phenotype. CD4+ T cells were depleted, by positive selection, from arthritogenic splenocytes before in vitro CII/anti-CD40 or CII/isotype control stimulation. After 48 h incubation, 5 × 106 CD4+ T cell–depleted splenocytes were transferred to recipient mice. A group of mice was also treated with nondepleted splenocytes challenged in vitro with CII/anti-CD40. Consistently with the results reported in Fig. 2, 75% of mice treated with nondepleted splenocytes treated with CII/anti-CD40 remained disease free over 35 d after immunization. In contrast, 100% of mice treated with B cells differentiated in the absence of CD4+ T cells developed severe arthritis (Fig. 8 b).
Therapeutic Effect of IL-10–producing B Cells on the Development of Established Arthritis.
In the following experiments we have tested the therapeutic potential of the anti-CD40 B cell transfer as a possible approach for the amelioration of established arthritis. In the first experiment, CII/CFA immunized mice were randomly assigned to a treatment group and transferred with 5 × 105 B cells (intravenously), isolated from splenocytes treated in vitro with either CII/control antibody or CII/anti-CD40, on the first day of disease. The treatment was repeated on day 5 and 10 after disease onset. A group of mice was also left untreated. Transfer of anti-CD40 challenged B cell was found to be effective in altering the course of disease when transferred after initiation of arthritis (Fig. 9 a; P < 0.002 versus antibody control or untreated group). In contrast, transfer of B cell isolated from splenocytes treated with antibody control produced no sustained therapeutic effect on the progression of CCIA.
Figure 9.

Therapeutic effect of anti-CD40–treated B cells on established arthritis. B cells were isolated from CII/anti-CD40 or CII/isotype control stimulated arthritogenic splenocytes and enriched by negative selection. DBA/1-TcR-β-Tg mice were treated (intravenously) at the day of disease onset and every 5 d (three doses in total) with 5 × 105 B cells in the experiment (a) and at day 5 and 10 in experiment (b). The severity of arthritis is reported as clinical score. Mice groups were compared by statistical analysis using two-sided Student's t test.
In the next experiment we aimed to assess whether anti-CD40 B cells treatment is also efficient in mice with established disease. DBA/1-TcR-β-Tg mice immunized with CII and CFA were treated at day 5 and 10 after disease onset. The results reported in Fig. 9 b show that the transfer of anti-CD40 challenged B cells but not antibody control B cells, significantly improved the severity of arthritis (P < 0.02 compared with untreated). However the efficacy of the treatment was found to be less effective compared with previous experiment where mice were treated during the very early phase of arthritis.
Discussion
We show here that B cells producing IL-10 arises after stimulation of arthritogenic splenocytes with antigen (CII) and agonistic anti-CD40 mAb. These IL-10–secreting B cells when transferred to syngenic mice control the pathogenic Th1 response (8), inhibit the onset of CCIA, and treat ongoing disease.
How These B Cells Work In Vivo?
The development of CIA in DBA/1 mice involves the induction of CII specific Th1 cells (16, 18), as supported by the beneficial effect observed after neutralization of IFN-γ or IL-12 during the early phase of disease (19, 20). On the other hand IL-10−/− mice showed an exacerbated disease compared with the wild-type, suggesting that this cytokine is involved in the down-regulation of the autoimmune response initiated by CII in CFA immunization (21). In this context CIA is ameliorated by administration of rIL-10 (22); however, the effect is transitory, and disease flares after interruption of the treatment. On the contrary, transfer of B cells producing IL-10 provides a long lasting effect. We have also generated preliminary results suggesting that the transfer of B cells can ameliorate established arthritis and therefore this approach might represent a very attractive strategy to treat autoimmune diseases, as the IL-10 can be delivered by the B cells in an antigen-specific fashion to the crucial site of priming or inflammation.
The obvious explanation of this data is that the IL-10 made by these B cells inhibits the inflammatory cascade initiated by CII/CFA immunization. This could be achieved throughout a constant supply of IL-10 by transferred B cells that may induce the production of other antiinflammatory soluble factors (i.e., TGF-β) or the differentiation of cells with “regulatory” activity.
IL-10 plays an important immuno-regulatory role in the homeostasis of the immune system. The antiinflammatory activities of IL-10 include inhibition of antigen-specific T cell proliferation and the down-regulation of costimulatory molecules on the APCs, which are directly involved in the differentiation of Th1 response (23, 24). In addition IL-10 directly suppresses the release of the inflammatory cytokines produced by macrophages and monocytes (25). In our model, the histological analysis of the protected paws showed a lack of recruitment of inflammatory cells in the synovium, suggesting that B cells, via the production of IL-10, are involved in the down-regulation of the inflammatory cascade initiated after CII/CFA immunization. We have taken a further step in showing that the protection was IL-10 dependent. Indeed, neutralization of IL-10 after transfer inhibited the protective effect assert by B cells producing IL-10 (unpublished data). Whether this is due to the disruption of the inflammatory cascade induced by pathogenic Th1 cells, or due to the lack of migration of the pathogenic T cell itself in the joints is not known. A similar scenario is well described in IBD models for colitis, where regulatory CD4+CD25+ T cells mediate their protective effect by releasing IL-10 and TGFβ (26). In this model, the importance of IL-10 was confirmed by the finding that CD4+CD25+ T cells from IL-10 KO mice did not prevent IBD, whereas CD4+CD25+ T cells from IL-10+ mice did (17). Similarly, anti-CD40–stimulated B cells isolated from IL-10 KO mice did not prevent arthritis development, while anti-CD40–stimulated B cells isolated from IL-10 wild-type mice significantly reduced the severity of disease in the recipient mice. Therefore, all our in vivo and in vitro results point to B cells as the crucial role of IL-10 in the down-regulation of the inflammatory cascade initiated after immunization.
It has been previously shown that CD40 engagement is one of the early events in a cascade of signals passing back and forth between T cells and APCs and that can augment activation or modulate the response, for instance by pushing Th2 differentiation or the generation of T regulatory cells (27). In our model it could be argued that the production of IL-10 by B cells might induce the differentiation of Tr1 regulatory CD4+ T cells (28). However, this seems unlikely in this system as T cells purified after in vitro anti-CD40 stimulation failed to protect the recipient mice from disease.
Recent in vitro studies have suggested a model in which Th2 differentiation is controlled by cytokines released by a subset of B cells (11, 12). In this in vitro model the ability of “B effector” 2 cells to promote Th2 differentiation in vitro it appears to be due through the production of IL-4. While we have not found IL-4 to be made by B cells, in our system we do show that B cells producing cytokines, in this case IL-10, strongly affect the polarization of T helper cell differentiation, resulting in the inhibition of antigen-specific Th1 response. These data show that the dialogue between B and T cells during an (auto)-immune response is not one sided and demonstrate that B cells have a strong impact in conditioning T cell differentiation.
A potential “protective” role of B cells in autoimmune diseases was previously suggested by Wolf et al. using an EAE model (29), showing more severe disease in B cell–deficient mice. Recently work by Fillatreau et al. has extended this by investigating the development of EAE in bone marrow chimeras mice in which B cells, but not T cells or APCs, are deficient in IL-10 production. Such chimeric mice exhibit a more severe EAE disease course without going into the remission phase that is normally seen in wild-type mice (or control chimeras). This prolonged disease is correlated with high levels of IFN-γ without any accompanying loss of the IL-4 response. Similarly, chimeras in which only B cells are deficient in CD40, also exhibit the more severe EAE, indicating that CD40 activation of B cells precedes IL-10 production (30). These results are in complete agreement with our findings, and strengthen the conclusion that upon CD40 engagement B cells differentiate into a B regulatory population and that their generation is also IL-10 dependent.
The observation that a subset of B cell acquires a regulatory function after anti-CD40 stimulation reinforces the concept that CD40 might have a role in a negative feedback loop that in this model would lead to the control of Th1 differentiation by B cells. However, the fact that direct stimulation of purified B cells with antigen and anti-CD40 failed to trigger the differentiation of B cells into a regulatory phenotype suggest that more complex cell/cell interactions are required for their differentiation. In particular the results shown in Fig. 8 b suggest that during the induction phase there is a cross-talk between B cells and CD4+ T cells. Previous data have shown that in vitro BCR engagement and CD40 stimulation are required for the production of IL-10 by B cells (11). However, in our system these coupled signals alone do not appear to be sufficient for the generation of B cell with a “protective” phenotype. This could be due to limiting amounts of IL-10 released in this experimental condition, compared with those produced by B cells isolated from arthritogenic splenocytes cultured with CII/anti-CD40. We know that IL-10 is also important for the generation of such B cells as addition of anti–IL-10r/anti–IL-10 during in vitro stimulation abrogates their development. Therefore, we could envisage that CD4+ T cells either directly via the release of soluble factors (i.e., factors inducing IL-10 or IL-10 itself) or indirectly by cell–cell contact is necessary for the development of high level cytokine secreting B “effector” cell. Thus, our data, together with those recently published describing a role for IL-10–producing B cells in regulating Th2-driven chronic intestinal inflammation, introduce a “regulatory” function for B cells in T cell dependent chronic inflammatory disorders (31).
In summary, our study shows that antigen-specific B cells, activated in the presence of T cells and additionally CD40 engagement, produce IL-10 and that these B cells play an important immunoregulatory role in a model of arthritis. More consideration to the cytokines specifically released by B cells and to their effect on T cells differentiation is needed to understand the mechanisms that regulate antigen-specific immune responses in chronic inflammatory disorders. These results also shed light on how in vivo anti-CD40 treatment may ameliorate arthritis by inducing the generation of B cells capable of controlling the proinflammatory Th1 type response via the release of IL-10. Our study, by showing how to induce this subset of B cell in CCIA, will help in the study of their role in health and disease and might provide the basis for novel therapeutic approaches for rheumatoid arthritis.
Acknowledgments
We thank P. Warden, M. Medghalci, and M. Sanchez for breeding the animals.
This project has been supported by the Wellcome Trust UK, grant no. 054620 and 068629. Claudia Mauri is funded by The Wellcome Trust. The Arthritis and Rheumatism Campaign, UK, supports The Kennedy Institute of Rheumatology.
Footnotes
Abbreviations used in this paper: CCIA, chronic collagen-induced arthritis; CFSE, carboxyl fluorescein succinimidyl ester; CII, type II collagen; DC, dendritic cell.
References
- 1.Banchereau, J., F. Bazan, D. Blanchard, F. Briere, J.P. Galizzi, C. van Kooten, Y.J. Liu, F. Rousset, and S. Saeland. 1994. The CD40 antigen and its ligand. Annu. Rev. Immunol. 12:881–922. [DOI] [PubMed] [Google Scholar]
- 2.Hollenbaugh, D., N. Mischel-Petty, C.P. Edwards, J.C. Simon, R.W. Denfeld, P.A. Kiener, and A. Aruffo. 1995. Expression of functional CD40 by vascular endothelial cells. J. Exp. Med. 182:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu, Y.J., D.E. Joshua, G.T. Williams, C.A. Smith, J. Gordon, and I.C. MacLennan. 1989. Mechanism of antigen-driven selection in germinal centres. Nature. 342:929–931. [DOI] [PubMed] [Google Scholar]
- 4.Rousset, F., E. Garcia, and J. Banchereau. 1991. Cytokine-induced proliferation and immunoglobulin production of human B lymphocytes triggered through their CD40 antigen. J. Exp. Med. 173:705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cella, M., D. Scheidegger, K. Palmer-Lehmann, P. Lane, A. Lanzavecchia, and G. Alber. 1996. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 184:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Kooten, C., and J. Banchereau. 2000. CD40-CD40 ligand. J. Leukoc. Biol. 67:2–17. [DOI] [PubMed] [Google Scholar]
- 7.Graca, L., K. Honey, E. Adams, S.P. Cobbold, and H. Waldmann. 2000. Cutting edge: anti-CD154 therapeutic antibodies induce infectious transplantation tolerance. J. Immunol. 165:4783–4786. [DOI] [PubMed] [Google Scholar]
- 8.Mauri, C., C.Q. Chu, D. Woodrow, L. Mori, and M. Londei. 1997. Treatment of a newly established transgenic model of chronic arthritis with nondepleting anti-CD4 monoclonal antibody. J. Immunol. 159:5032–5041. [PubMed] [Google Scholar]
- 9.Mauri, C., L.T. Mars, and M. Londei. 2000. Therapeutic activity of agonistic monoclonal antibodies against CD40 in a chronic autoimmune inflammatory process. Nat. Med. 6:673–679. [DOI] [PubMed] [Google Scholar]
- 10.Stockinger, B., T. Zal, A. Zal, and D. Gray. 1996. B cells solicit their own help from T cells. J. Exp. Med. 183:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skok, J., J. Poudrier, and D. Gray. 1999. Dendritic cell-derived IL-12 promotes B cell induction of Th2 differentiation: a feedback regulation of Th1 development. J. Immunol. 163:4284–4291. [PubMed] [Google Scholar]
- 12.Harris, D.P., L. Haynes, P.C. Sayles, D.K. Duso, S.M. Eaton, N.M. Lepak, L.L. Johnson, S.L. Swain, and F.E. Lund. 2000. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 1:475–482. [DOI] [PubMed] [Google Scholar]
- 13.Mori, L., H. Loetscher, K. Kakimoto, H. Bluethmann, and M. Steinmetz. 1992. Expression of a transgenic T cell receptor beta chain enhances collagen-induced arthritis. J. Exp. Med. 176:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 15.Guery, J.C., F. Ria, and L. Adorini. 1996. Dendritic cells but not B cells present antigenic complexes to class II-restricted T cells after administration of protein in adjuvant. J. Exp. Med. 183:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mauri, C., R.O. Williams, M. Walmsley, and M. Feldmann. 1996. Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur. J. Immunol. 26:1511–1518. [DOI] [PubMed] [Google Scholar]
- 17.Asseman, C., S. Mauze, M.W. Leach, R.L. Coffman, and F. Powrie. 1999. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Germann, T., J. Szeliga, H. Hess, S. Storkel, F.J. Podlaski, M.K. Gately, E. Schmitt, and E. Rude. 1995. Administration of interleukin 12 in combination with type II collagen induces severe arthritis in DBA/1 mice. Proc. Natl. Acad. Sci. USA. 92:4823–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler, D.M., A.M. Malfait, R.N. Maini, F.M. Brennan, and M. Feldmann. 1999. Anti-IL-12 and anti-TNF antibodies synergistically suppress the progression of murine collagen-induced arthritis. Eur. J. Immunol. 29:2205–2212. [DOI] [PubMed] [Google Scholar]
- 20.Malfait, A.M., D.M. Butler, D.H. Presky, R.N. Maini, F.M. Brennan, and M. Feldmann. 1998. Blockade of IL-12 during the induction of collagen-induced arthritis (CIA) markedly attenuates the severity of the arthritis. Clin. Exp. Immunol. 111:377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson, A.C., A.S. Hansson, K.S. Nandakumar, J. Backlund, and R. Holmdahl. 2001. IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J. Immunol. 167:3505–3512. [DOI] [PubMed] [Google Scholar]
- 22.Walmsley, M., P.D. Katsikis, E. Abney, S. Parry, R.O. Williams, R.N. Maini, and M. Feldmann. 1996. Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum. 39:495–503. [DOI] [PubMed] [Google Scholar]
- 23.Howard, M., and A. O'Garra. 1992. Biological properties of interleukin 10. Immunol. Today. 13:198–200. [DOI] [PubMed] [Google Scholar]
- 24.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 25.Fiorentino, D.F., A. Zlotnik, T.R. Mosmann, M. Howard, and A. O'Garra. 2001. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147:3815–3822. [PubMed] [Google Scholar]
- 26.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 27.Kumanogoh, A., X. Wang, I. Lee, C. Watanabe, M. Kamanaka, W. Shi, K. Yoshida, T. Sato, S. Habu, M. Itoh, et al. 2001. Increased T cell autoreactivity in the absence of CD40-CD40 ligand interactions: a role of CD40 in regulatory T cell development. J. Immunol. 166:353–360. [DOI] [PubMed] [Google Scholar]
- 28.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 29.Wolf, S.D., B.N. Dittel, F. Hardardottir, and C.A. Janeway, Jr. 1996. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 184:2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fillatreau, S., C.H. Sweenie, M.J. McGeachy, D. Gray, and S.M. Anderton. 2002. B cells regulate autoimmunity by provision of IL-10. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3:944–950. [DOI] [PubMed] [Google Scholar]
- 31.Mizoguchi, A., E. Mizoguchi, H. Takedatsu, R.S. Blumberg, and A.K. Bhan. 2002. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 16:219–230. [DOI] [PubMed] [Google Scholar]