

Abstract
In a transgenic model of multi-stage squamous carcinogenesis induced by human papillomavirus (HPV) oncogenes, infiltrating CD4+ T cells can be detected in both premalignant and malignant lesions. The lymph nodes that drain sites of epidermal neoplasia contain activated CD4+ T cells predominantly reactive toward Staphylococcal bacterial antigens. HPV16 mice deficient in CD4+ T cells were found to have delayed neoplastic progression and a lower incidence of tumors. This delay in carcinogenesis is marked by decreased infiltration of neutrophils, and reduced activity of matrix metalloproteinase-9, an important cofactor for tumor progression in this model. The data reveal an unexpected capability of CD4 T cells, whereby, proinflammatory CD4+ T cells, apparently responding to bacterial infection of dysplastic skin lesions, can inadvertently enhance neoplastic progression to invasive cancer.
Keywords: mice, transgenic, cancer, inflammation, lymphocyte
Introduction
All tumors of epithelial origin are composed of multiple cell types that include both transformed genetically altered cells and several nontransformed cell types, including fibroblasts, endothelial cells, macrophages, neutrophils, mast cells, and lymphocytes. We have studied a transgenic mouse model of epithelial carcinogenesis wherein the human papillomavirus type 16 (HPV16)* early region genes are expressed in the basal keratinocytes under the control of the human keratin 14 promoter (1). The K14-HPV16 mice show multistage development of invasive squamous cell carcinoma (SCC) of the epidermis (2). The pathway is characterized by a phenotypically normal epidermis at birth, followed by the development of epidermal hyperplasia beginning at one month of age, and subsequent focal progression to highly angiogenic dysplasias between 3 and 6 mo of age (3, 4). The dysplasias are characterized by mast cell and neutrophil infiltration of the reactive stroma, and by increased density and altered architecture of the vasculature. By 1 yr of age, ∼50% of transgenic mice develop invasive carcinomas, of which ∼30% metastasize to regional lymph nodes.
A protease, matrix metalloproteinase-9 (MMP-9), has been revealed as a functional contributor to squamous epithelial carcinogenesis in K14-HPV16 mice; and remarkably, bone marrow-derived inflammatory cells are a sufficient source of the MMP-9 for its demonstrable contributions to malignant progression (5). The realization that a protease supplied by infiltrating immune cells is fueling neoplastic progression in mouse models of skin and pancreatic islet cancer (5, 6) comes amidst increasing experimental evidence linking inflammation and cancer (7). Inflammation has been predominantly viewed as beneficial to the host and deleterious to tumors (8), although a variety of circumstantial evidence has long suggested the possibility that inflammatory responses can enhance carcinogenesis (9). Recent studies using gene knockout mice and other tools to probe functional roles have substantiated the capability of immune cells to enhance cancer phenotypes, in tumor models involving orthotopically transplanted neoplastic cells, chemically induced cancers, as well as in transgenic mouse models (10–13).
In this report, we investigated the abundance and possible roles of CD4+ and CD8+ T lymphocytes during epithelial carcinogenesis in K14-HPV16 mice. CD4+ T cells were found to be infiltrating both premalignant skin lesions and SCCs. We initially hypothesized that the T cell infiltration was intended to antagonize malignant progression by an immune surveillance mechanism. To test this hypothesis, we crossed the K14-HPV16 mice to mice with homozygous deletions of the genes encoding the CD4 or CD8 coreceptors. Unexpectedly, the data revealed that infiltrating CD4+ T cells were functionally promoting epithelial carcinogenesis and that their predominant reactivity was toward Staphylococcal antigens, not the HPV16 oncogenes, likely in response to extensive Staphylococcus colonization of the neoplastic epidermis.
Materials and Methods
K14-HPV16 Transgenic, CD4 Null, and CD8 Null Mice.
The K14-HPV16 transgenic mice (1) and the characterization of neoplastic stages based on hematoxylin and eosin staining and keratin intermediate filament expression for histologic examination have been reported previously (2). Histologic grading criteria of low versus high grade hyperplastic lesions consisted of examination of epidermal thickness, presence of hyperchromatic nuclei in suprabasal epidermis, and presence of an intact granular cell layer containing prominent keratohyalin granules and flattened cellular morphology. Histologic grading criteria for low grade dysplastic lesions consisted of greater than twofold increase in the thickness of the epidermis with basal and spinous keratinocytes present throughout the epidermis, focal loss of keratohyalin granules, and appearance of keratinocytes with rounded morphology throughout entire suprabasal compartment. High grade dysplastic lesions were positively scored based on presence of an epidermis exclusively containing basal and spinous keratinocytes with hyperchromatic nuclei, complete loss of keratohyalin granule containing cells, and extensive rete ridging of the basement membrane. The K14-HPV16 mice were maintained in the FVB/n background (FVB/n; The Jackson Laboratory). The generation of CD4 homozygous null animals (14) and CD8α homozygous null animals (15) has been reported. CD4+/− mice were bred 14 generations into FVB/n (FVB/n; The Jackson Laboratory) before intercrossing with HPV16 transgenic mice (N21 FVB/n), in turn producing CD4-deficient HPV16 mice (−/−; n = 142). CD8α+/− mice were bred five generations into FVB/n before intercrossing with HPV16 transgenic mice (N21 FVB/n), in turn producing CD8α-deficient (−/−; n = 119) and CD8α-proficient (+/−; n = 39) HPV16 mice. To minimize potential effects of genetic drift, the crosses to maintain the HPV16 line, and the CD8+/−, and CD4+/− backcrosses to FVB/n were all performed at the same time using a common FVB/n stock obtained from the The Jackson Laboratory.
Tissue Preparation and Histology.
For paraffin sections, tissue pieces from transgenic animals were immersion-fixed in 3.75% paraformaldehyde and PBS or 10% zinc-buffered formalin followed by dehydration through graded alcohols and xylene, and embedded in paraffin. 5-μm thick paraffin sections were cut using a Leica 2135 microtome. Sections were deparaffinized and rehydrated through an alcohol series then subjected to histochemical or immunohistochemical staining as described previously (2). 5-μM sections were stained with hematoxylin and eosin (H&E) for histological analysis. For frozen sections, tissue was embedded without fixation in OCT (Tissue-Tek) and frozen on dry ice. 10-μm thick frozen sections were cut using a Leica CM1900 cryostat. Sections were air-dried, fixed in acetone, and subjected to immunohistochemical or immunofluorescence staining.
Immunohistochemistry and Immunofluorescence Microscopy.
Paraffin sections were used for MMP-9, PCNA, keratin 14, keratin 10, keratin 8, pan-keratin, and vimentin substrate immunohistochemistry. Dilution used for rabbit anti-MMP-9 (a gift of Dr. Zena Werb; reference 16) was 1:1,000, 1:100 for mouse anti-PCNA (19A2, BioGenex), 1:2,500 for rabbit anti-keratin 14 (MK14, BabCo), 1:2,500 for rabbit anti-keratin 10 (MK10, BabCo), 1:50 for rat anti-K8 (TROMA 1, a gift of Dr. Rolf Kemler, Max-Planck Institute for Immunobiology, Freiburg, Germany), 1:200 for rabbit anti-pan-keratin (Biogenex), and 1:50 rabbit anti-vimentin (Vim3B4; DakoCytomation) in blocking solution containing PBS pH 7.4, 2.5% goat serum, and 1% BSA. Frozen sections were used for CD4 and CD8α immunohistochemistry. Dilution used for rat anti–mouse CD8α (53–6.7; BD Biosciences) and rat anti–mouse CD4 (H129.19; BD Biosciences) was 1:200 in a blocking solution containing PBS pH 7.4 and 0.25% blocking reagent (NEN Life Science Products). Incubation with primary antibody was overnight at 4°C. After incubation with a biotinylated secondary antibody (goat anti–rabbit IgG, goat anti–rat IgG, or goat anti–mouse IgM, 1:200; Pierce Chemical Co.) for 30 min at ambient temperature, antigens were revealed with 3,3′-diaminobenzidine (DAB; Sigma-Aldrich) according to the manufacturer's instructions. Sections were counterstained in 1% methyl green, dehydrated in iso-butanol and xylene, mounted in Cytoseal 60 (Stephens Scientific), and visualized with Nomarski optics. All immunolocalization experiments were repeated three times on multiple tissue sections and included negative controls for determination of background staining, which was negligible. All images were digitally captured on a Nikon Microphot-FX microscope equipped with a DC-330 CCD color camera (DAGE-MTI) and imaged using Improvision OpenLab software. Frozen sections were used for immunofluorescence staining. Dilution used for rabbit anti-MMP-9 was 1:1,000, 1:200 for rat anti–mouse Mac-1 FITC (CD11b, M1/70; BD Biosciences), and 1:100 for rat anti–mouse CD4 (H129.19; BD Biosciences) in blocking solution containing PBS and 0.25% blocking reagent (NEN Life Science Products). Incubation with primary antibodies was 1 h at ambient temperature followed by incubation with a Cy3 conjugated donkey anti–rabbit IgG (Jackson Immunochemicals) secondary antibody at 1:200 and Cy2 conjugated donkey anti–rat IgG (Jackson Immunochemicals) for 1 h at ambient temperature. Sections were mounted in VectaShield containing DAPI (Vector Laboratories) and images were digitally captured on a Carl Zeiss Microimaging, Inc. Axioskop 2 plus equipped with a Hamamatsu-Orca digital camera and imaged using Improvision OpenLab software.
Flow Cytometry and Cell Sorting.
To assess total cell number and the activation status of lymph node T cells, brachial lymph nodes were removed and transferred to PBS. Single-cell suspensions were generated using a glass homogenizer and filtered through a 70-μm nylon filter (Falcon). Total cell counts were performed using a Coulter Counter. Lymph node cells were stained with either rat anti–mouse CD4 FITC (H129.19; BD Biosciences) or CD8α FITC (53–6.7; BD Biosciences) at a dilution of 1:20 and rat anti–mouse CD69 PE (H1.2F3; Caltag Laboratories) at a dilution of 1:10 in PBS and 1% BSA (Intergen) (PBS/BSA). Cells were incubated for 20 min at 4°C and washed twice with PBS/BSA. A total of 20,000 gated events were collected per sample on a Becton Dickinson FACSCaliber™. All samples were stained with rat IgG Fitc and rat IgG PE controls to determine background staining for setting gates. Values represent the mean ± SE of the percentage of CD4+ or CD8+ T cells expressing CD69 (four or more mice per group). To quantify and sort infiltrating cells from skin or tumors, tissue pieces were manually minced using a scalpel, followed by a 13 min enzymatic digestion with 2.5 mg/ml Collagenase Type II (GIBCO BRL), 2.5 mg/ml Collagenase Type IV (GIBCO BRL), and 0.5 mg/ml DNase (Sigma-Aldrich) in PBS/BSA at 37°C. The digest was quenched by adding of DMEM (GIBCO BRL) containing 10% of FBS (heat inactivated; GIBCO BRL) and was filtered through a 70-μm nylon filter (Falcon). After one wash in PBS/BSA, the cells were incubated for 10 min in rat anti–mouse CD16/CD32 monoclonal antibody (BD Biosciences) at a 1:10 dilution in PBS/BSA. For analysis, rat anti–mouse CD4 FITC (1:20), rat anti–mouse CD8α FITC (1:20), rat anti–mouse Mac-1 FITC (1:40), or rat anti–mouse Gr-1 FITC (1:60) were diluted in PBS/BSA, incubated for 20 min at 4°C, and washed twice in PBS/BSA. A total of 50,000 gated events were collected per sample on Becton Dickinson FACSCalibur™. Values represent the mean ± SE of the percentage of total cells in the tissue preparation (three or more mice per group). For cell sorting, 7-AAD (BD Biosciences), a viability marker, was added at a dilution of 1:10 to the cells to ensure that only live cells were collected. The CD4+ T cell, Mac-1+/Gr-1+ and “other” cell populations were sorted and collected under RNase free conditions, using a high-speed fluorescently activated cell sorter (FACSVantage™; Becton Dickinson).
Antigens.
A lysate of Staphylococcus aureus (SANSORBIN Cells; Calbiochem) was prepared by sonication of cells in 50 mM Tris-HCl (pH 7.4) and 150 mM NaCl. The S. aureus lysate was passed through a sterile 0.22-μm filter and protein concentration determined by Bradford assay (Bio-Rad Laboratories) using a BSA standard. Endotoxin levels were measured by Limulus ameobocyte lysate assay (BioWhittaker) and Staphylococcus aureus lysates contained 38.4 endotoxin units (EU)/ml. NH2-terminal histidine-tagged HPV16-E7 protein was cloned into a pET vector and expressed in E. coli strain BL21(DE3) (Novagen; bacterial stocks a generous gift from Dr. Roland Tisch, University of North Carolina at Chapel Hill, Chapel Hill, NC). Bacteria were solubilized in 6 M Guanidine-HCl and purified over a Ni-NTA Superflow column (QIAGEN). The column was washed with 1% Triton X-100/6 M Guanidine-HCl to remove endotoxin. HPV16-E7 was eluted from the column with 6 M Guanidine-HCl, 50 mM imidazole (pH 4.2) and dialyzed into PBS/20% glycerol. HPV16-E7 preparations contained <0.1 endotoxin units (EU)/ml. Ovalbumin (Sigma-Aldrich) was used as a control for HPV16-E7. Concanavalin A (Sigma-Aldrich) was used as a positive control.
Cytokine Assays.
Cervical lymph nodes were removed under sterile conditions, disrupted using glass homogenizers, washed with PBS, and quantified by hemocytometer. Lymph node cells were cultured in 96-well half-well flat-bottom microculture plates (Falcon) with 1.0 × 106 cells/well in 0.2 ml HL-1 serum free media (BioWhittaker) and the indicated antigen. Class II MHC blocking antibody (M5/114) (American Type Culture Collection) and isotype control (BD Biosciences) were used at 50 mg/ml in the indicated cultures. Cultures were set up with replicates in triplicate and incubated at 37°C in 5% CO2/95% air (5% CO2) for 48 h. The supernatants were analyzed for IFN-γ, IL-4, and IL-3 by ELISA. Capture antibodies, cytokine standards, biotinylated detecting antibodies, and streptavidin-alkaline phosphatase were obtained from BD Biosciences. IFN-γ capture mAb (R46-A2) was coated at 2 μg/ml. Biotinylated IFN-γ detecting mAb (XMG1.2) was used at 0.5 μg/ml. IL-4 capture mAb (BVD4–1D11) was coated at 0.5 μg/ml. Biotinylated IL-4 detecting mAb (BVD6–24G2) was used at 0.5 μg/ml. IL-3 capture mAb (MP2–8F8) was coated at 2 μg/ml. Biotinylated IL-3 detecting mAb (MP2–43D11) was used at 0.25 μg/ml. Streptavidin-alkaline phosphatase was used at a 1:2,000 dilution. Reference curves were generated for each assay using recombinant IFN-γ, IL-4, and IL-3 as standards. Optical density of ρ−nitrophenyl phosphate (Sigma-Aldrich) substrate was determined at 405 nm on a Thermomax microplate reader (Molecular Devices). Values represent the mean ± SD of triplicates.
Substrate Zymography.
Tissue samples representing distinct ages and SCCs from HPV16 and HPV16/CD4−/− mice were weighed and then homogenized (1:4 weight to volume) in lysis buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% NP-40, 0.5% deoxycholate, 0.1% SDS. Soluble and insoluble extracts were separated by centrifugation and subsequently stored at –80°C. Equivalent amounts of soluble extract were analyzed by gelatin zymography (17) on 10% SDS-polyacrylamide gels copolymerized with substrate (1 mg/ml of gelatin) in sample buffer (10% SDS, 0.25 M Tris-HCl, 0.1% Bromophenol Blue, pH 6.8). After electrophoresis, gels were washed three times for 30 min in 2.5% Triton X-100, three times for 15 min in ddH2O, incubated overnight at 37°C in 50 mM Tris-HCl, 10 mM CaCl2 (pH 7.6), and then stained in 0.5% Coomassie Blue and destained in 20% methanol/10% acetic acid. Negative staining indicates the location of active protease bands. Exposure of proenzymes within tissue extracts to SDS during gel separation procedure leads to activation without proteolytic cleavage (18). Zymography was done on tissue samples from mice of each genotype taken from 1 mo (n = 5), 3 mo (n = 5), and 6 mo (n = 10) of age and SCCs (n = 10). Data shown in Fig. 6 C are representative of results obtained from 6-mo-old mice (n = 10) and SCC-bearing animals (n = 10).
Figure 6.

MMP-9 is expressed by Mac-1+ cells not CD4+ T cells. (A) RT-PCR analysis of cell populations isolated from an HPV16 tumor by FACS®. Total RNA from CD4+, Mac-1+/Gr-1+, and unlabelled cells was made and reverse transcribed into cDNA. PCR for MMP-9 (35 cycles), CD4 (35 cycles), and L19 (housekeeping gene; 31 cycles) was performed on the cDNA from the three populations. The product was run on a 1.5% agarose gel and the results are shown with +RT and –RT displayed for each population of cells. (B) Immunolocalization of MMP-9 (red) and Mac-1 (green) in a tumor from an HPV16 mouse. MMP-9 and Mac-1 are found to be colocalizing (yellow) on many cells within the tumor. The inset shows a higher magnification of several cells staining for both MMP-9 and Mac-1. Bar: 20 μm; 10 μm (inset). (C) Immunolocalization of MMP-9 (red) and CD4 (green) in a tumor from an HPV16 mouse. No colocalization of MMP-9 and CD4 was observed. Bar: 7 μm.
RT-PCR.
Total RNA was made from CD4 and Mac-1/Gr-1 sorted cells and unsorted cells using an RNeasy kit (QIAGEN). Total RNA was reverse transcribed by using random hexamers (GIBCO BRL) and M-MLV reverse transcriptase (GIBCO BRL). Specific primers were used to amplify cDNAs: MMP-9 sense, 5′-CCCTGGAACTCACACGACATC-3′; MMP-9 antisense, 5′-TCACACGCCAGAAGAATTTGC-3′; CD4 sense, 5′-GAGGTTCGCCTTCGCAGTTTGAT-3′; CD4 antisense, 5′-GGTGCAGTTCCAGAAGTCGCT-3′. Primers for the ribosomal protein L19 (L19 sense, 5′-CTGAAGGTCAAAGGGAATGTG-3′; L19 antisense, 5′-GGACAGAGTCTTGATGATCTC-3′) were included in each experiment as an internal control. 35 cycles (MMP-9 and CD4) and 31 cycles (L19) were performed with a PTC-200 thermocycler (MJ Research) as follows: 30 s at 95°, 30 s at 55°, 30 s at 72°. PCR products were run on 1.5% agarose gels. Gels were photographed using an IS-1000 Digital Imaging System (Alpha Innotech Corp.).
Statistical Analysis.
Flow cytometric data, immunohistochemistry data, and cytokine assays were analyzed using an unpaired t test (InStat v1.12; GraphPad Software). Pre-malignant pathology was analyzed using a chi-squared test for trend (InStat v3.0; GraphPad Software). Tumor-free survival was analyzed using a log-rank test (Prizm 3.0A; GraphPad Software).
Results
T Cells Infiltrate Dysplasias and Squamous Carcinomas.
To determine if T lymphocytes were associated with the distinctive stages of epidermal carcinogenesis, we analyzed dysplastic (premalignant) epidermis and tumors (invasive squamous cell carcinomas, SCCs) from K14-HPV16 mice for the presence of T cells by immunohistochemistry. Using a monoclonal antibody specific to CD4, we observed CD4+ T cell infiltration in dysplastic skin (Fig. 1 A) and tumors (Fig. 1 B). However, little to no T cell infiltration was detected in more benign hyperplastic lesions (data not depicted) or in control skin from nontransgenic mice (Fig. 1 C). Immunohistochemistry to detect CD8+ T cells did not reveal an appreciable infiltration of hyperplastic and dysplastic lesions, whereas a low level infiltrate was detected in SCCs (unpublished data).
Figure 1.

T cells infiltrate sites of neoplasia in HPV16 transgenic mice. (A–C) Immunolocalization of CD4 (brown staining) in skin sections counterstained with methyl green from HPV16 dysplastic ear skin (A), a HPV16 Grade II SCC (B), and a normal nontransgenic ear (C). The epidermal (e), dermal (d), malignant tumor keratinocytes (t), and tumor stroma (s) regions of the tissues are indicated. The epithelial basement membrane is marked with a dashed line. Bar: 10 μm (A, C); 30 μm (B). (D) Tissue digests from normal and HPV16 transgenic skin were analyzed by flow cytometry for their CD4+ or CD8+ T cell content of as a percentage of total cells. Normal nontransgenic skin was obtained from 2-mo-old FVB/n mice, dysplastic skin was obtained from 6–8-mo-old HPV16 mice, and tumors were obtained from 8–14-mo-old HPV16 mice. We collected 50,000 events per sample from normal nontransgenic ear skin (blue, n = 4), HPV16 dysplastic ear skin (orange, n = 5), and HPV16 tumors (yellow, n = 7). Results are shown as mean percentages ± standard error of the mean.
To provide a quantitative measure of the degree of infiltration, we analyzed nontransgenic skin, dysplastic skin, and tumors by flow cytometry. Single cell suspensions were generated by subjecting tissue to mechanical disruption followed by enzymatic digestion of the extracellular matrix. As expected from immunohistochemical analysis, we observed more CD4+ T cells than CD8+ T cells in both dysplastic skin and solid tumors (Fig. 1 D)
Lymph Nodes Draining Sites of Epidermal Neoplasia Contain Activated CD4+ T Cells Reactive to Bacterial Antigens.
In an effort to characterize the T cell responses observed in premalignant dermis, we analyzed T cells from the draining lymph nodes. The brachial lymph nodes that were draining premalignant lesions of the epidermis showed a significant increase in total cellularity (nontransgenic 4.1 × 106 cells vs. HPV16 40.8 × 106 cells; P = 0.0079, unpaired t test). Furthermore, we analyzed brachial lymph node cells for evidence of T cell activation by determining the fraction of cells expressing the early activation marker CD69. In HPV16 mice with tumors, ∼24% of the CD4+ T cells in the draining lymph nodes were expressing CD69, in contrast to ∼7% CD69+CD4+ T cells in control nontransgenic mice (P < 0.0001, unpaired T test; Fig. 2 A). We observed a minimal increase in activated CD8+ T cells, from ∼5% CD69+CD8+ cells in nontransgenic control skin to ∼9% in tumor-bearing HPV16 mice (P = 0.0095, Wilcoxon test; Fig. 2 A).
Figure 2.
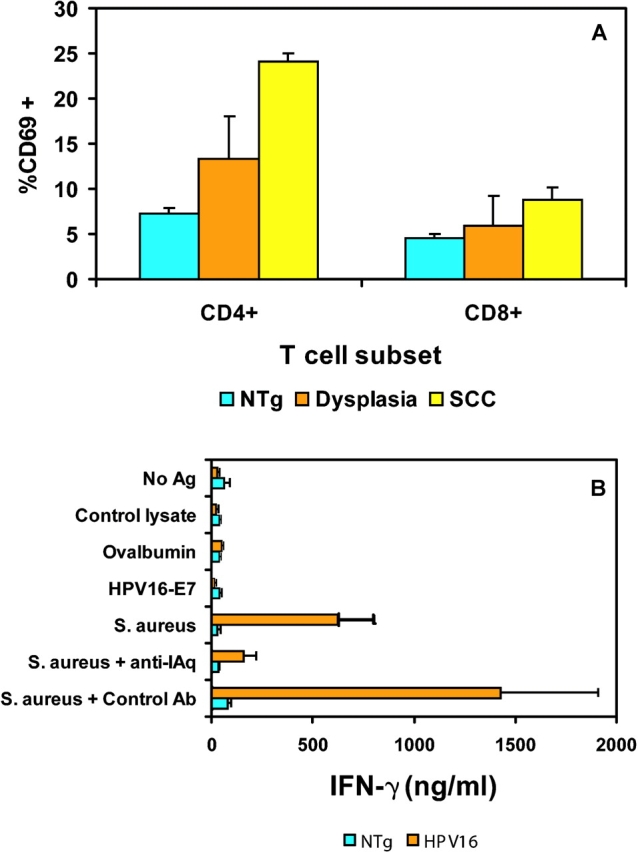
Activated CD4+ T cells are reactive to Staphylococcus aureus. (A) Brachial LNCs from normal nontransgenic mice (blue, n = 6), HPV16 mice with epidermal dysplasias (orange, n = 4), and HPV16 mice with tumors (yellow, n = 4) were analyzed by flow cytometry for the percent of gated CD4+ or CD8+ T cells expressing CD69. We collected 10,000 gated events per sample. Results are shown as mean percentages ± standard error of the mean. (B) Cytokine assay of cervical LNCs from 6–8-mo-old normal nontransgenic mice (blue, n = 5) or HPV16 mice (orange, n = 5) were prepared as a single cell suspension and cultured in the presence of medium, 100 μg/ml ovalbumin (2.8 μM), 40 μg/ml HPV16-E7 (2.8 μM), a control lysate, or 20 μg/ml S. aureus lysate. To determine if the response was class II restricted, cultures stimulated with the S. aureus lysate were treated with a class II MHC blocking antibody (M5/114; P = 0.0127; unpaired t test) or isotype control (P = 0.0521; unpaired t test) and compared with S. aureus lysate stimulated cells. LNCs (1.0 × 106/well) were incubated for 48 h in HL-1 medium. LNCs were stimulated with 0.25 μg/ml Con A as a positive control. All mice displayed similar Con A responses (unpublished data). Supernatants were collected and analyzed for IFN-γ production by ELISA. The results shown are mean ± standard deviation of triplicate cultures from a representative experiment.
We surmised that the activated T cells could be responding to ‘tumor antigens’ expressed in the neoplasms in particular the HPV16 oncoproteins encoded by the K14-HPV16 transgene. Indeed, the E6 and E7 oncoproteins are well-characterized tumor antigens in human cervical cancer (19). However, in many cases, self-tolerance has been established toward transgene products in mice resultant to expression during embryogenesis, obviating immunoreactivity, even in the case of tumorigenesis (20, 21). Therefore, we first asked whether the K14-HPV16 mice were tolerant of the E7 gene product, by immunizing nontransgenic and HPV16 mice with recombinant HPV16-E7 protein. Both control and HPV16 transgenic mice demonstrated CD4+ T cell reactivity, indicating the transgenic mice were nontolerant and capable of developing an immune response to E7 (unpublished data). Furthermore, the HPV16 mice developed spontaneous autoantibodies against E7, further supporting the lack of self-tolerance induction toward the transgenic oncoprotein (unpublished data). Therefore, we asked whether the reactive lymph nodes in the HPV16 transgenic mice contained T cells specific for E7. Lymph node cells from cervical nodes draining dysplastic skin lesions were stimulated with recombinant HPV16-E7, and supernatants analyzed for IFN-γ release (Fig. 2 B). Surprisingly, the HPV16 lymph node cells did not show a measurable response toward the HPV16-E7 protein (Fig. 2 B), indicating that their abundance was low, albeit apparently sufficient to support the observed humoral response.
Because HPV-E7–responsive T cells were evidently rare and thus could not readily explain the abundance of activated T cells in the reactive lymph nodes, we considered other explanations. We suspected that the neoplastic, hyperkeratotic skin was impaired in its barrier function, and might be allowing fortuitous bacterial infections to take hold, evoking an immune response. Indeed, when we evaluated both nontransgenic normal skin and dysplastic transgenic skin biopsies for bacteria, rampant infection was detected only in lesional skin; predominant amongst the bacteria were gram-positive Staphylococcus aureus and Staphylococcus epidermidis (Table I ). Therefore, we asked whether the reactive draining lymph nodes had developed T cell responsiveness to Staphylococcus aureus proteins, using bacterial lysates as an antigenic source in standard T cell assays (Fig. 2 B). A proinflammatory IFN-γ response by lymph node T cells was observed in response to the S. aureus lysate in HPV16 mice, but not in naive nontransgenic mice. Furthermore, this response was inhibited by including a class II MHC (I-Aq) blocking antibody, indicating that this was a CD4+ T cell response toward a class II–restricted Staphylococcal antigen. To determine the cytokine profile of the CD4+ T cell response, the supernatants were also tested for IL-3 and IL-4 release. There was no detectable IL-3 or IL-4 production under any of the conditions tested (unpublished data). Thus, the Staphylococcus-reactive CD4+ T cells displayed a Th1 proinflammatory phenotype.
Table I.
Microbial Analysis of Skin from HPV16 Mice
| Genotype | Staphylococcus aureus | Staphylococcus epidermidis |
|---|---|---|
| HPV16 | ++++ | ++++ |
| HPV16 | ++++ | ++++ |
| HPV16 | ++++ | ++ |
| HPV16/CD4−/− | +++ | − |
| HPV16/CD4−/− | +++ | − |
| HPV16/CD4−/− | +++ | − |
| NTg | + | − |
| NTg | + | + |
| NTg | + | + |
Ear and chest skin from normal non-transgenic FVB/n (NTg) and HPV16 mice between 8 and 12 mo of age was removed under sterile conditions and submitted to IDEXX Veterinary Services (Sacramento, CA) for identification and quantification of aerobic bacteria present within the tissue by semiquantitative limiting dilution analysis. Results shown are from ear skin in which both Staphylococcus aureus and Staphylococcus epidermidis were detected. The data shown is on a scale of one to four (+). All other aerobic microbial species were one (+) or lower. Equivalent infection was observed in chest skin.
Genetic Deletion of CD4+ T Cells Decreases the Incidence of Carcinomas.
To assess the functional significance of the observed T cell infiltrate in premalignant lesions during neoplastic progression, we took a genetic approach, using mice carrying homozygous disruptions of either the CD4 or CD8α genes (14, 15). An ancillary motivation was to assess the possibility that immune surveillance might underlay the slow time to progression from angiogenic dysplasias, and the 50% penetrance of that progression to invasive squamous cell carcinomas. We backcrossed the CD4-null mutation to n14 in the FVB/n mouse strain before generating HPV16/CD4−/− mice (n = 142), whose phenotype was monitored over a one year time-course, revealing significant differences. HPV16/CD4−/− mice showed a marked delay in the kinetics of progression to squamous carcinomas by a defined endpoint of 12 mo of age (P = 0.0107, log-rank test; Fig. 3 A). At 6 mo of age, only ∼53% of HPV16/CD4−/− mice manifested focal dysplasias, characterized by loss of terminal differentiation and increased proliferation of keratinocytes; by contrast, dysplasia is observed in ∼92% of control HPV16 animals (P = 0.0175, chi-squared test for trend; Fig. 3 B). Assessing another parameter of neoplasia, we determined the proliferation index of premalignant lesions from HPV16/CD4−/− mice by immunohistochemical analysis of tissue sections for up-regulation of PCNA (proliferating cell nuclear antigen). We also observed a modest decrease in proliferation index in HPV16/CD4−/− mice (38.4 ± 1.8%) as compared with HPV16/CD4+/+ control mice (42.8 ± 2.4%; P = 0.1339; unpaired t test). The HPV16/CD4−/− mice also exhibited a reduced incidence of squamous carcinomas by the 12-mo endpoint. Only 41% of HPV16/CD4−/− mice developed tumors, in contrast to the 52% incidence in CD4-competent HPV16 mice.
Figure 3.
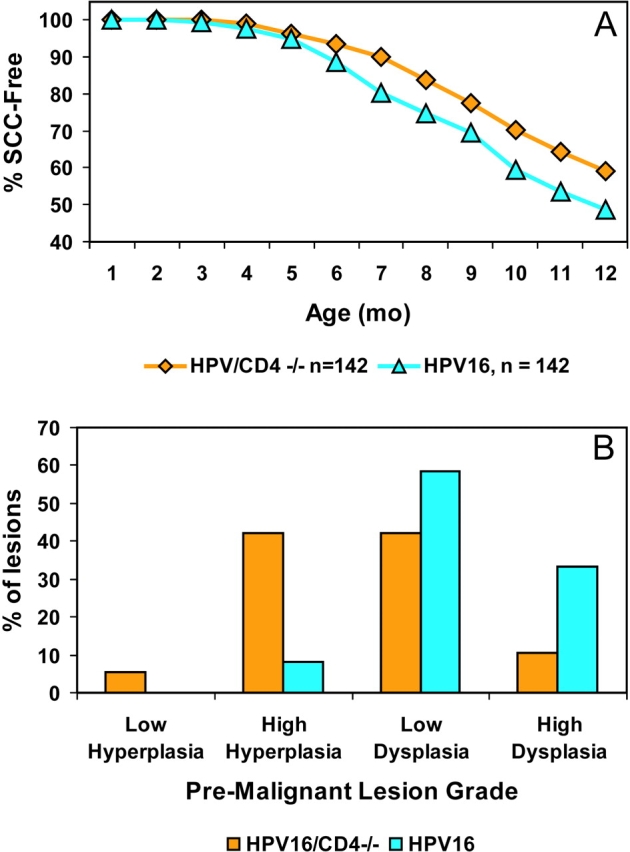
HPV16/CD4−/− mice show delayed tumorigenesis. (A) Percentage of SCC-free transgenic mice in two cohorts: HPV16/CD4+/+, n = 142 (FVB/n N14-N20; blue triangles) and HPV16/CD4−/−, n = 142 (FVB/n N14; orange diamonds). HPV16/CD4+/+ mice exhibit an overall tumor incidence of 52%. In contrast, the incidence of tumors in HPV16/CD4−/− mice was reduced to 41% (Fisher's Exact test, P = 0.0954; log-rank test, P = 0.0107). (B) A shift toward lower grade premalignant lesions in HPV16/CD4−/− mice. 6-mo-old ear skin from HPV16/CD4+/+ mice (blue, n = 12) and HPV16/CD4−/− (orange, n = 19) was collected and graded based on criteria previously described (reference 2) and summarized in Materials and Methods. HPV16/CD4-deficient mice exhibited an altered spectrum of premalignant skin lesions biased toward less malignant lesions (P = 0.0175, chi-squared test for trend).
Tumors arising in immune competent HPV16 mice display a spectrum of carcinoma grades, based on characteristic epithelial differentiation markers (5). The anatomical distribution and average number per mouse (∼1.3) of tumors in HPV16/CD4−/− mice was comparable to the immune competent HPV16 mice (unpublished data).
In a comparable analysis, CD8α+/− mice were backcrossed five generations into FVB/n and then intercrossed with HPV16 mice to produce HPV16 mice that lacked CD8+ T cells. We aged a cohort of CD8−/− HPV mice for 1 yr, comparing their neoplastic phenotype to CD8+/− HPV littermates. The absence of CD8+ T cells slightly delayed the temporal appearance of invasive cancers (n = 119; P = 0.0011, log rank test; unpublished data); however, in contradistinction to the situation when the CD4+ T cells were missing, the absence of CD8+ T cells did not alter the overall incidence of invasive cancers arising by the defined endpoint of 12 mo, either in comparison to CD8+/− littermates or historical control CD8+/+ HPV mice. Thus, there was no evidence for CD8 T cell mediated immune surveillance in the form of an more aggressive tumor phenotype in their absence, but rather a suggestion of weak immune enhancement of tumor progression mediated by CD8+ T cells.
Infiltration of Dysplasias by Innate Immune Cells Is Decreased in HPV16/CD4−/− Mice.
Previous studies in this model revealed an important role for bone marrow–derived inflammatory cells (mast cells, macrophages, and neutrophils) in supplying MMP-9, itself a demonstrably important functional contributor to squamous carcinogenesis in HPV16 mice (4, 5). Recognizing that CD4+ T cells can recruit both macrophages and neutrophils into tissue sites and activate them therein, we analyzed HPV16/CD4−/− mice for evidence of changes in the inflammatory infiltrate. A significant inflammatory infiltrate is characteristic both in neoplastic skin of 6-mo-old immune competent HPV16 mice as well as in invasive tumors. Therefore, we compared dysplastic skin lesions from age matched 6-mo-old mice and tumors from HPV16/CD4−/− and HPV16/CD4+/+ mice for differences in inflammatory cell populations. Single cell suspensions were generated by subjecting tissue to mechanical disruption followed by enzymatic digestion of the extracellular matrix (ECM). To quantify the inflammatory infiltrate, we used flow cytometry to assess the infiltration of cells expressing the cell surface molecules Gr-1 (Ly-6G) and Mac-1 (CD11b). Gr-1 is expressed on neutrophils and Mac-1 (CD11b) is expressed on granulocytes, macrophages, dendritic cells, and natural killer cells.
There was a significant decrease in the number of inflammatory cells expressing Mac-1 In lesional skin from 6-mo-old HPV16/CD4−/− mice compared with their CD4 proficient counterparts (P = 0.0136, unpaired t test; Fig. 4) . HPV16/CD4+/+ mice had a Mac-1+ cell infiltrate representing ∼30% of the total cells within the skin, whereas HPV16/CD4−/− mice showed a ∼15% Mac-1+ infiltrate. We observed a similar decrease in Gr-1+ cell infiltrates in CD4-null dysplasias (P = 0.0425, unpaired t test; Fig. 4). In contrast, when solid tumors from HPV16/CD4−/− mice were analyzed for either Mac-1+ or Gr-1+ cell infiltration, no significant difference was observed in comparison to immune competent HPV16/CD4+/+ mice (P = 0.6047 for Mac-1, P = 0.7520 for Gr-1, unpaired t test; Fig. 4). Tumors from both HPV16/CD4+/+ and HPV16/CD4−/− mice contained ∼20% infiltration by Mac+ and Gr-1+ cells.
Figure 4.

Reduced inflammatory infiltrate in HPV16/CD4−/− skin. Tissue digests from HPV16/CD4+/+ (blue) and HPV16/CD4−/− (orange) 6-mo-old skin (n = 5) and SCCs (n = 3) were analyzed by flow cytometry for their content of Mac-1+ or Gr-1+ cells as a percentage of total cells. We collected 25,000 events per sample. Results are shown as mean percentages ± standard error of the mean. A ∼50% reduction (*) in the numbers of Mac-1+ (P = 0.0136; unpaired t test) and Gr-1+ (p = 0.0425; unpaired t test) cells was observed in 6-mo-old ear skin from HPV16/CD4−/− mice as compared with HPV16/CD4+/+ mice. No reduction in Mac-1+ or Gr-1+ infiltrate was observed in SCCs from HPV16/CD4−/− mice (Mac-1, P = 0.6047; Gr-1, P = 0.7520; unpaired t test).
Fewer MMP-9–expressing Cells and Decreased MMP-9 Activity in HPV16/CD4−/− Mice.
Previous studies have shown that activity of the matrix metalloproteinase MMP-9 is induced during neoplastic progression in HPV16 mice (5). Furthermore, functional assessment of MMP-9 in HPV16/MMP-9−/− mice revealed its activity to be important, in that absence of MMP-9 resulted in delayed neoplastic progression and a lower incidence of squamous carcinomas. Since we observed a decrease in the inflammatory infiltrate in HPV16/CD4−/− mice, biopsies of premalignant 6-mo-old skin and tumors from HPV16/CD4−/− mice were analyzed for MMP-9 expression by immunohistochemistry. In lesional skin of HPV16/CD4−/− mice, we observed a ∼60% decrease in the number of cells expressing MMP-9 as compared with HPV16/CD4+/+ mice (P = 0.0087, unpaired t test; Fig. 5 A). HPV16/CD4+/+ mice had ∼1,200 MMP-9+ cells per mm2; whereas HPV16/CD4−/− mice had ∼500 MMP-9+ cells per mm2. This difference is consistent with the ∼50% decrease in the inflammatory infiltrate detected by flow cytometry. In contrast, tumors from HPV16/CD4−/− mice showed no decrease in number of MMP-9+ cells compared with control HPV16 mice (Fig. 5 B), much as they showed no difference in the overall abundance of infiltrating immune cells (Fig. 4). Tumors from both genotypes showed ∼1,800 MMP-9+ cells/mm2. Thus, the immunohistochemical analysis indicated a selective decrease in the abundance of MMP-9+ cells in premalignant dysplastic skin lesions but not squamous carcinomas of CD4-null HPV mice.
Figure 5.
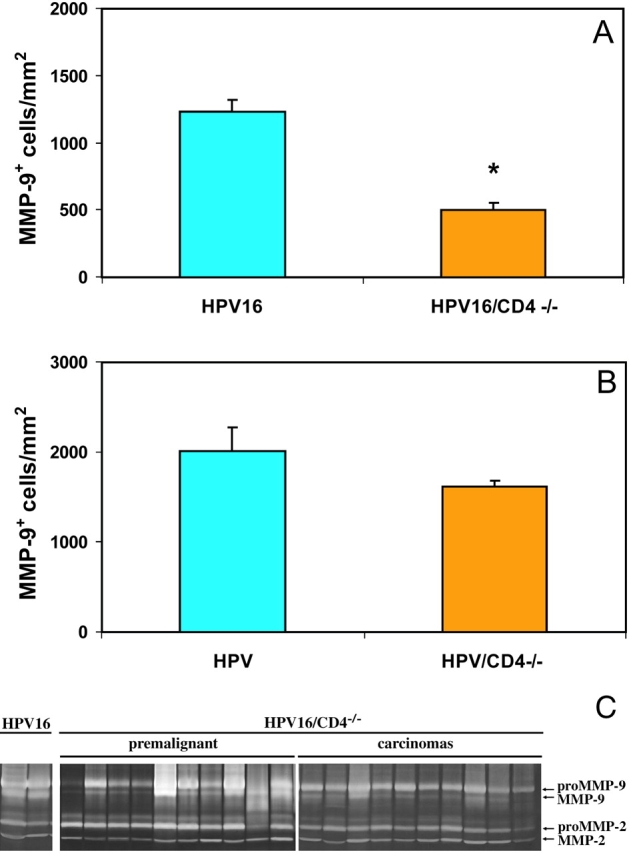
Reduced numbers of MMP-9+ cells infiltrating HPV16/CD4−/− skin. Immunolocalization and quantification of MMP-9+ cells in HPV16/CD4+/+ and HPV16/CD4−/− 6-mo-old skin (A) and SCCs (B). MMP-9+ cells/mm2 was obtained by counting the total number of brown cells (MMP-9) and dividing that by the total area in a field subjacent to the epidermis for a total of five 20× microscope fields per mouse for five mice per group (n = 5). A ∼60% reduction (*) in the MMP-9+ cells/mm2 was observed in 6-mo-old skin from HPV16/CD4−/− mice relative to HPV16/CD4+/+ mice (A; P = 0.0087, unpaired t test), but no significant difference in infiltrating cells was observed in SCCs from HPV16/CD4−/− mice (B; P = 0.5476, Wilcoxon test). (C) Gelatin-substrate zymography of skin biopsy lysates from HPV16/CD4+/+ controls, HPV16/CD4−/− 4–6-mo-old ear skin (premalignant) and carcinomas. Arrows indicate location of enzymatic activity corresponding to pro- and active forms of MMP-9 and MMP-2.
To determine if the decrease in MMP-9+ cell infiltrate in premalignant HPV16/CD4−/− skin affected tissue MMP-9 enzyme activity, we analyzed lysates from neoplastic skin obtained from HPV16/CD4−/− mice of varying ages (and stages of disease progression) for proteolytic activity using gelatin substrate zymography. As previously reported (5), the pro-enzyme and active forms of MMP-9 are not found in normal FVB/n skin (unpublished data). Both the pro-enzyme and active forms of MMP-9 were detected at similarly low levels in skin lysates from 1- and 3-mo-old HPV16/CD4−/− and control HPV16 mice (unpublished data). As expected, both forms of MMP-9 were found at markedly higher levels in 82% of 4–6-mo-old HPV16 skin lysates containing angiogenic dysplasias (n = 66) and in 95% of solid tumors (n = 26; Fig. 5 C). In contrast, only ∼30% of HPV16/CD4−/− mice showed similarly high levels of active MMP-9 in neoplastic lesions from age-matched 4–6-mo-old mice (Fig. 5 C), and only ∼20% of squamous carcinomas displayed abundant levels of active MMP-9 (Fig. 5 C).
MMP-9 Is Not Expressed by Infiltrating CD4+ T Cells.
MMP-9 has previously been detected by immunostaining in granulocytes, macrophages, and mast cells found in association with neoplastic lesions in HPV16 mice, using morphological criteria to identify the immune cell types. This pattern was consistent with the finding that bone marrow from wild-type MMP-9+/+ mice restored the characteristic HPV16 phenotype when transferred to HPV16/MMP-9−/− mice (5). To determine if CD4+ T cells infiltrating tumors were also producing MMP-9, we used fluorescence activated cell sorting to fractionate CD4+ T cells and Gr-1+/Mac-1+ cells from a single cell suspension prepared from tumors. Total RNA was isolated from the sorted cell populations and used to prepare cDNA for PCR analysis using oligonucleotide primers specific to MMP-9, CD4, and L19 (a housekeeping gene). MMP-9 mRNA was only detected in the Gr-1/Mac-1+ and unlabelled (OC) populations (Fig. 6 A). After 35 cycles of PCR, no MMP-9 product was detected in the CD4+ T cell population. In contrast, a prominent PCR product was detected in the Gr-1/Mac-1+ cell-derived cDNA. As a positive control for the fractionation method, the CD4+ T cell population was tested for the expression of CD4 mRNA and found to be positive.
The results of the RT-PCR analysis indicated that MMP-9 mRNA was expressed in the Gr-1+/Mac-1+ cells, not CD4+ T cells. We sought to confirm these results by analyzing MMP-9 protein expression by immunostaining of tissue sections. HPV16 tumors were assessed for MMP-9 and either Mac-1 or CD4 colocalization with specific antibodies. As expected, we observed significant colocalization of MMP-9 and Mac-1 on cells within squamous carcinomas (Fig. 6 B). In contrast, no colocalization of MMP-9 and CD4 were observed (Fig. 6 C). These results are consistent with the finding that MMP-9 mRNA is expressed in Mac-1+/Gr-1+ cells, but not CD4+ T cells. From these data, we can conclude that CD4+ T cells participate in the recruitment of MMP-9+ inflammatory cells into neoplastic lesions, but do not themselves produce the MMP-9 that contributes to the malignant progression.
Discussion
This study has revealed an unexpected role for CD4+ T cells in the regulation of squamous carcinogenesis. Using CD4 gene knockout mice that lack traditional helper T cells, we found that CD4+ T cells were enhancing the progression of epidermal hyperplasias to dysplasias via facilitating the recruitment of neutrophils (and potentially other Mac-1+ leukocytes). These innate immune cell types are in turn demonstrably important for angiogenesis and neoplastic cell proliferation through their provision of MMP-9. While T cells were found inside solid tumors, their absence did not impact tumor growth, suggesting that their ‘helper’ functions were most important for the premalignant phase of squamous carcinogenesis. Remarkably, CD4+ T cell reactivity was predominantly directed toward antigens expressed by the Staphylococcal bacteria infecting the dysplastic skin lesions, suggesting that infections of neoplastic skin evoke an inflammatory response that inadvertently enhances malignant progression.
A Link between Infection and Cancer.
The etiologies of several forms of human cancer are associated with infection by parasites, viruses, or bacteria (for a review, see reference 22). In some instances, the pathogen provides oncogenes that can directly transform the target cell, in a rather straightforward causal relationship (23, 24). By contrast, another class of pathogens does not appear to directly transform the target cell, but instead, induces a state of chronic inflammation that can ultimately lead to genetic damage in the target tissue, resulting in transformation of the cells and tumor development. There is evidence that the chronic immune response to hepatitis B virus (HBV) or hepatitis C virus (HCV) contributes to the transformation of hepatocytes, remodeling of the stroma, and progression to hepatocellular carcinoma (25). Helicobacter pylori infection of gastric epithelium is associated with the development of gastric lymphomas (maltomas) and gastric adenocarcinomas (for a review, see reference 26). H. pylori infection of gastric epithelial cells initiates a host immune response by induction of IL-8 expression (27). IL-8 recruits inflammatory cells, including proinflammatory Th1 cells and B cells (28). A state of chronic inflammation sustained by H. pylori results in an environment enriched for DNA damaging reactive oxygen and nitrogen species. This can lead to the transformation of either the gastric epithelium or the infiltrating B cells. Elimination of H. pylori infection using antibiotics can reverse the premalignant changes associated with the inflammatory response (29).
We have documented gram-positive Staphylococcus infections of the dysplastic epidermis from HPV16 mice with a concomitant S. aureus–reactive T cell response detectable in the draining lymph nodes. The high proportion of activated CD4+ T cells and lymphadenopathy would suggest a state of chronic inflammation that is similar to what is observed in the gastric epithelium during H. pylori infection. Furthermore, when CD4+ T cell responses were eliminated in HPV16/CD4−/− mice, a delay to malignant progression and decrease in the incidence of SCCs was observed. While no link has been observed between Staphylococcus infection and squamous cancer in humans, the observation has relevance and implications for modeling human cancers associated with a chronic immune response to a pathogen. In two different mouse models of colon cancer, spontaneous adenocarcinomas were prevented when the mice were housed under germ-free conditions (30, 31); in the later example, when Helicobacter hepaticus bacteria was reintroduced into the mice, tumors again developed (31). Similarly, we are in the process of transferring HPV16 mice into a germ-free notobiotic environment to clarify the functional importance of bacterial infection for squamous carcinogenesis of the skin.
Absence of Immune Surveillance.
The HPV16 mice develop multifocal angiogenic dysplasia by 6 mo with 100% penetrance, and yet only one invasive carcinoma emerges on average in 50% of the mice by a defined endpoint of 1 yr of age. We hypothesized that immune surveillance could be restricting the appearance of invasive carcinomas (32), particularly when we detected spontaneous anti-E7 autoantibodies indicative of an anti-HPV immune response. Several epidemiological studies have demonstrated that the incidence and severity of HPV-associated cervical or anogenital neoplasias are higher in HIV-positive immunocompromised humans (33, 34). Furthermore, a similar increase in nonmelanoma skin cancer is observed in transplant patients receiving pharmacological immunosuppression (35). Thus, when we crossed in the CD4 and CD8 gene knockouts, we expected earlier appearance of tumors, increased numbers of tumors per animal, and a higher incidence of tumors in the population (above the 50% seen in wild-type HPV mice). Instead, absence of CD8+ T cells caused a slight delay in the appearance of tumors, whereas absence of CD4+ T cells both delayed the temporal appearance and reduced the incidence of tumors.
The HPV mice have an unusual form of ‘split tolerance’ toward HPV E7 (and E6) oncoproteins. We have demonstrated humoral nontolerance and autoreactivity in the form of spontaneous and inducible antibody responses toward E7, in data to be presented elsewhere. As for the cytotoxic T cell compartment, the H-2q class I MHC alleles present in the FVB/n strain are incapable of presenting E7 or E6 epitopes (36), and thus CD8 T cells are immunologically ignorant of these oncoproteins. There is no information available regarding antigenicity of the E1 – E5 proteins that are presumably also expressed in addition to E6 and E7 in the K14-HPV16 mice, which carry the entire HPV16 early region. In any event, there is no evidence that immune surveillance underlies the long latency and incomplete penetrance of cancer observed in the HPV16 model; however, we leave open the possibility that immune surveillance could become a factor if full immune recognition in the CD8 compartment was achieved. To that end, we have begun the process of rendering the HPV16 mice congenic for the H-2b MHC but otherwise inbred into the FVB/n background that conveys susceptibility for the invasive phenotype, such that we can further probe the potential of immune surveillance as a biological mechanism. Moreover, the clue that CD8 T cells might have weak tumor-enhancing activity is complicated by the incomplete backcrossing into FVB/n of the CD8 knockout locus, which in principle could be cosegregating a closely linked modifier locus; thus definitive analysis of the roles, if any, of CD8 T cells awaits more complete inbreeding of the CD8 locus, and its combination with the H-2b MHC in the context of the FVB/n background in HPV transgenic mice.
One might similarly attribute the absence of immune surveillance seen when comparing CD4+/+ and CD4−/− animals trivially to the genetic ignorance of cytotoxic T cells toward E6/7 peptides or to polymorphic modifier genes cosegregating with the CD4-ko gene. Other data, however, suggest that neoplastic enhancement and lack of effective surveillance by CD4+ T cells is a genuine attribute of this immune cell type in this particular organ, the skin. When K14-HPV16 mice are treated with sustained low dose estrogen, cervical carcinogenesis ensues (37, 38). If HPV16 females are rendered CD4 deficient via the same crosses (and backcross generations) as studied in this report, a much different tumor phenotype is seen: cervical carcinomas develop in greater number and are larger in the HPV16 mice that lack CD4+ T cells, indicating that in the cervix, CD4+ T cells antagonize rather than enhance carcinogenesis, consistent with an immune surveillance role (unpublished data). Thus we conclude that CD4+ T cells can have both the predicted ability to interfere with tumor progression as well as the unexpected ability to functionally enhance carcinogenesis via helping innate immune cell type supply pro-angiogenic MMP-9; these distinct roles can be seen in different organs of otherwise genetically identical mice.
T Cells, Innate Immunity, and Cancer.
An emerging view of cancer is that tumors are manifestations of a complex multicellular disease, involving genetic mutations in neoplastic cells in concert with the recruitment of supporting cells, including fibroblasts, endothelial cells, and inflammatory cells, that functionally contribute to the tumor's capabilities (39). In this report, we document the development of a significant inflammatory infiltrate during neoplastic progression in HPV16 mice. We detect both CD4+ T lymphocytes (1–5%) and Mac-1+/Gr-1+ cells (20–30%) within premalignant lesions and cancers. Both macrophages and neutrophils of the innate immune system are represented in the Mac-1+/Gr-1+ infiltrate. Mast cell infiltration of premalignant dermis underlying dysplastic epidermis and the contribution of mast cells to malignant progression has been previously documented (4). Several types of cancer are associated with chronic inflammation of both infectious and noninfectious origin (for a review, see reference 40). Esophageal carcinoma is associated with chronic inflammation of the lower esophagus caused by stomach acid reflux (41). Mesothelioma is associated with a chronic inflammatory response to inhaled asbestos fibers (42). Thus, whether induced by an infectious agent, as with H. pylori infection, or other tissue injury, the feature common to these forms of malignancy is a state of chronic inflammation associated with immune cell infiltration.
Recent work in a transgenic mouse model of mammary cancer (PyMT mice) established an important role for macrophages in the development of invasive carcinomas and pulmonary metastasis (12). In PyMT mice rendered deficient in macrophage colony stimulating factor–1 (CSF-1), the neoplastic epithelial cells failed to recruit mature macrophages into the neoplastic tissue, resulting in decreased malignancy. We observe a similar effect in HPV16/CD4−/− mice, in that premalignant lesions displayed a significant decrease in the leukocyte infiltrate associated with delayed progression, and a decreased incidence of carcinomas. Infiltrating leukocytes are known to express and release an array of cytokines, chemokines, growth factors, free radicals, and proteases. Among these, MMP-9 expressed by bone marrow–derived cells has been previously shown to contribute to malignant progression in the skin of HPV16 mice (5). Now we report that, MMP-9 activity is significantly lower in the neoplastic lesions of HPV16/CD4−/− mice. Thus, elimination of CD4+ T cells reduced infiltration by macrophages and neutrophils, resulting in a decrease in MMP-9 within the premalignant lesions. CD4+ T cells have previously been documented to produce MMP-9 when stimulated by a phorbol ester (43); in contradistinction, we found no evidence of MMP-9 mRNA in CD4+ T cells fractionated from tumors by flow cytometry. Rather, MMP-9 mRNA and protein were detectable in similarly fractionated cells of the Mac-1+ lineage.
Perspective on Tumor Immunity versus Immune Enhancement.
The realization that CD4+ T cells can enhance (as opposed to antagonize) tumor progression raises an important question for experimental strategies aimed at inducing antigen-specific tumor immunity: do all antitumor immune responses include both destructive and enhancing functionalities, balanced to varying degrees? In one recent study aimed to induced tumor immunity, the opposite resulted: T cell–mediated enhancement of benign papilloma development was observed in two-step skin tumor model involving Harvey ras transgenic mice treated with PMA, in that preimmunization to elicit an anti-ras immune response enhanced (rather than protected against) papilloma formation (10). On the other hand, immunization of transgenic mice developing prostate cancer with an irradiated tumor cell vaccine significantly reduced the incidence of prostate tumors (44). Furthermore, we have immunized K14-HPV16 female transgenic mice developing cervical cancer (37) with an HPV16-E7 vaccine, and reduced the incidence of cervical cancer (unpublished data). These latter examples argue that efficacious tumor antigen-specific immunity can be induced; one wonders, nevertheless, whether the efficacy observed in each of these cases might have been attenuated by concurrent immune enhancement? Our perspective is that successful immunotherapies in the future may well combine tumor antigen-specific immune activators with selective inhibitors of the tumor-enhancing effects of inflammation, biasing the immune response solely for destruction of cancerous lesions.
Acknowledgments
We thank Nigel Killeen, Lewis Lanier, Simon Foote, Jay Levy, and Mallika Singh for discussions and insightful comments on the manuscript; Roland Tisch and Zena Werb for reagents; Mallika Singh for advice and assistance, and Jin-Sae Rhee and Lidiya Korets for technical assistance.
This work was supported by grants from the National Cancer Institute.
Footnotes
Abbreviations used in this paper: HPV16, human papillomavirus type 16; MMP-9, matrix metalloproteinase-9; SCC, squamous cell carcinoma.
References
- 1.Arbeit, J.M., K. Munger, P.M. Howley, and D. Hanahan. 1994. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J. Virol. 68:4358–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coussens, L.M., D. Hanahan, and J.M. Arbeit. 1996. Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am. J. Pathol. 149:1899–1917. [PMC free article] [PubMed] [Google Scholar]
- 3.Smith-McCune, K., Y.H. Zhu, D. Hanahan, and J. Arbeit. 1997. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14-HPV16 transgenic mice. Cancer Res. 57:1294–1300. [PubMed] [Google Scholar]
- 4.Coussens, L.M., W.W. Raymond, G. Bergers, M. Laig-Webster, O. Behrendtsen, Z. Werb, G.H. Caughey, and D. Hanahan. 1999. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 13:1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coussens, L.M., C.L. Tinkle, D. Hanahan, and Z. Werb. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 103:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergers, G., R. Brekken, G. McMahon, T.H. Vu, T. Itoh, K. Tamaki, K. Tanzawa, P. Thorpe, S. Itohara, Z. Werb, and D. Hanahan. 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balkwill, F., and A. Mantovani. 2001. Inflammation and cancer: back to Virchow? Lancet. 357:539–545. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber, H. 1999. Tumor immunology. Fundamental Immunology. W.E. Paul, editor. Lippincott Raven Press, New York. 1247–1280.
- 9.Prehn, R.T. 1994. Stimulatory effects of immune reactions upon the growths of untransplanted tumors. Cancer Res. 54:908–914. [PubMed] [Google Scholar]
- 10.Siegel, C.T., K. Schreiber, S.C. Meredith, G.B. Beck-Engeser, D.W. Lancki, C.A. Lazarski, Y.X. Fu, D.A. Rowley, and H. Schreiber. 2000. Enhanced growth of primary tumors in cancer-prone mice after immunization against the mutant region of an inherited oncoprotein. J. Exp. Med. 191:1945–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girardi, M., D.E. Oppenheim, C.R. Steele, J.M. Lewis, E. Glusac, R. Filler, P. Hobby, B. Sutton, R.E. Tigelaar, and A.C. Hayday. 2001. Regulation of cutaneous malignancy by gammadelta T cells. Science. 294:605–609. [DOI] [PubMed] [Google Scholar]
- 12.Lin, E.Y., A.V. Nguyen, R.G. Russell, and J.W. Pollard. 2001. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 193:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coussens, L.M., and Z. Werb. 2001. Inflammatory cells and cancer: think different! J. Exp. Med. 193:F23–F26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahemtulla, A., W.P. Fung-Leung, M.W. Schilham, T.M. Kundig, S.R. Sambhara, A. Narendran, A. Arabian, A. Wakeham, C.J. Paige, R.M. Zinkernagel, et al. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 353:180–184. [DOI] [PubMed] [Google Scholar]
- 15.Fung-Leung, W.P., M.W. Schilham, A. Rahemtulla, T.M. Kundig, M. Vollenweider, J. Potter, W. van Ewijk, and T.W. Mak. 1991. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 65:443–449. [DOI] [PubMed] [Google Scholar]
- 16.Behrendtsen, O., C.M. Alexander, and Z. Werb. 1992. Metalloproteinases mediate extracellular matrix degradation by cells from mouse blastocyst outgrowths. Development. 114:447–456. [DOI] [PubMed] [Google Scholar]
- 17.Herron, G.S., Z. Werb, K. Dwyer, and M.J. Banda. 1986. Secretion of metalloproteinases by stimulated capillary endothelial cells. I. Production of procollagenase and prostromelysin exceeds expression of proteolytic activity. J. Biol. Chem. 261:2810–2813. [PubMed] [Google Scholar]
- 18.Talhouk, R.S., J.R. Chin, E.N. Unemori, Z. Werb, and M.J. Bissell. 1991. Proteinases of the mammary gland: developmental regulation in vivo and vectorial secretion in culture. Development. 112:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornelison, T.L. 2000. Human papillomavirus genotype 16 vaccines for cervical cancer prophylaxis and treatment. Curr. Opin. Oncol. 12:466–473. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan, D. 1990. Transgenic mouse models of self-tolerance and autoreactivity by the immune system. Annu. Rev. Cell Biol. 6:493–537. [DOI] [PubMed] [Google Scholar]
- 21.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 22.Goedert, J.J. 2000. Infectious Causes of Cancer. Humana Press, Totowa, New Jersey.
- 23.Munger, K., M. Scheffner, J.M. Huibregtse, and P.M. Howley. 1992. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 12:197–217. [PubMed] [Google Scholar]
- 24.Kaye, K.M., K.M. Izumi, and E. Kieff. 1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA. 90:9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamoto, Y., L.G. Guidotti, C.V. Kuhlen, P. Fowler, and F.V. Chisari. 1998. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 188:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst, P.B., and B.D. Gold. 2000. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 54:615–640. [DOI] [PubMed] [Google Scholar]
- 27.Crabtree, J.E., J.I. Wyatt, L.K. Trejdosiewicz, P. Peichl, P.H. Nichols, N. Ramsay, J.N. Primrose, and I.J. Lindley. 1994. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J. Clin. Pathol. 47:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Elios, M.M., M. Manghetti, F. Almerigogna, A. Amedei, F. Costa, D. Burroni, C.T. Baldari, S. Romagnani, J.L. Telford, and G. Del Prete. 1997. Different cytokine profile and antigen-specificity repertoire in Helicobacter pylori-specific T cell clones from the antrum of chronic gastritis patients with or without peptic ulcer. Eur. J. Immunol. 27:1751–1755. [DOI] [PubMed] [Google Scholar]
- 29.Wotherspoon, A.C., C. Doglioni, T.C. Diss, L. Pan, A. Moschini, M. de Boni, and P.G. Isaacson. 1993. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 342:575–577. [DOI] [PubMed] [Google Scholar]
- 30.Kado, S., K. Uchida, H. Funabashi, S. Iwata, Y. Nagata, M. Ando, M. Onoue, Y. Matsuoka, M. Ohwaki, and M. Morotomi. 2001. Intestinal microflora are necessary for development of spontaneous adenocarcinoma of the large intestine in T-cell receptor beta chain and p53 double-knockout mice. Cancer Res. 61:2395–2398. [PubMed] [Google Scholar]
- 31.Engle, S.J., I. Ormsby, S. Pawlowski, G.P. Boivin, J. Croft, E. Balish, and T. Doetschman. 2002. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 62:6362–6366. [PubMed] [Google Scholar]
- 32.Burnet, F.M. 1970. The concept of immunological surveillance. Prog. Exp. Tumor Res. 13:1–27. [DOI] [PubMed] [Google Scholar]
- 33.Fowler, M.G., S.L. Melnick, and B.J. Mathieson. 1997. Women and HIV. Epidemiology and global overview. Obstet. Gynecol. Clin. North Am. 24:705–729. [DOI] [PubMed] [Google Scholar]
- 34.Palefsky, J.M. 2000. Anal squamous intraepithelial lesions in human immunodeficiency virus-positive men and women. Semin. Oncol. 27:471–479. [PubMed] [Google Scholar]
- 35.Fung, J.J., A. Jain, E.J. Kwak, S. Kusne, I. Dvorchik, and B. Eghtesad. 2001. De novo malignancies after liver transplantation: a major cause of late death. Liver Transpl. 7:S109–S118. [DOI] [PubMed] [Google Scholar]
- 36.Dunn, L.A., M. Evander, R.W. Tindle, A.L. Bulloch, R.L. de Kluyver, G.J. Fernando, P.F. Lambert, and I.H. Frazer. 1997. Presentation of the HPV16E7 protein by skin grafts is insufficient to allow graft rejection in an E7-primed animal. Virology. 235:94–103. [DOI] [PubMed] [Google Scholar]
- 37.Arbeit, J.M., P.M. Howley, and D. Hanahan. 1996. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. USA. 93:2930–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elson, D.A., R.R. Riley, A. Lacey, G. Thordarson, F.J. Talamantes, and J.M. Arbeit. 2000. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res. 60:1267–1275. [PubMed] [Google Scholar]
- 39.Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell. 100:57–70. [DOI] [PubMed] [Google Scholar]
- 40.O'Byrne, K.J., and A.G. Dalgleish. 2001. Chronic immune activation and inflammation as the cause of malignancy. Br. J. Cancer. 85:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jankowski, J.A., N.A. Wright, S.J. Meltzer, G. Triadafilopoulos, K. Geboes, A.G. Casson, D. Kerr, and L.S. Young. 1999. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am. J. Pathol. 154:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Britton, M. 2002. The epidemiology of mesothelioma. Semin. Oncol. 29:18–25. [DOI] [PubMed] [Google Scholar]
- 43.Weeks, B.S., H.W. Schnaper, M. Handy, E. Holloway, and H.K. Kleinman. 1993. Human T lymphocytes synthesize the 92 kDa type IV collagenase (gelatinase B). J. Cell. Physiol. 157:644–649. [DOI] [PubMed] [Google Scholar]
- 44.Hurwitz, A.A., B.A. Foster, E.D. Kwon, T. Truong, E.M. Choi, N.M. Greenberg, M.B. Burg, and J.P. Allison. 2000. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 60:2444–2448. [PubMed] [Google Scholar]