

Abstract
Full activation of naive T cells requires both engagement of the T cell antigen receptor (TCR; signal 1) and costimulatory signaling by CD28 (signal 2). We previously identified two types of rat CD28-specific monoclonal antibodies (mAbs): “conventional,” TCR signaling–dependent costimulatory mAbs and “superagonistic” mAbs capable of inducing the full activation of primary resting T cells in the absence of TCR ligation both in vitro and in vivo. Using chimeric rat/mouse CD28 molecules, we show that the superagonists bind exclusively to the laterally exposed C′′D loop of the immunoglobulin-like domain of CD28 whereas conventional, costimulatory mAbs recognize an epitope close to the binding site for the natural CD80/CD86 ligands. Unexpectedly, the C′′D loop reactivity of a panel of new antibodies raised against human CD28 could be predicted solely on the basis of their superagonistic properties. Moreover, mouse CD28 molecules engineered to express the rat or human C′′D loop sequences activated T cell hybridomas without TCR ligation when cross-linked by superagonistic mAbs. Finally, biochemical analysis revealed that superagonistic CD28 signaling activates the nuclear factor κB pathway without inducing phosphorylation of either TCRζ or ZAP70. Our findings indicate that the topologically constrained interactions of anti-CD28 superagonists bypass the requirement for signal 1 in T cell activation. Antibodies with this property may prove useful for the development of T cell stimulatory drugs.
Keywords: costimulation, CD28, T cells, lymphocyte activation, receptor structure
Introduction
Ligation of the T cell surface receptor CD28 is the most powerful way to costimulate resting T cells, turning nonproductive or even anergizing signals that result from TCR stimulation alone, into fully activating signals (1, 2). In experimental systems, the natural ligands of the TCR, the cognate MHC–peptide complexes, and of CD28, the B7 family members CD80 and CD86, can be replaced by mAbs, which bind to and cross-link the TCR and CD28, respectively.
The mechanism by which TCR- and CD28-derived signals are integrated are not completely understood. In addition to a downstream convergence of signaling cascades (3), current models postulate a cross talk between the two receptors at the cell membrane. Thus, costimulation triggers polarized transport of membrane domains rich in signaling molecules toward the T cell APC contact area (4, 5). Furthermore, CD28 colocalizes with the TCR at the center of the mature immunological synapse (also called the central supramolecular activation cluster [c-SMAC];* references 6–8), wherein CD28 engagement is proposed to enhance and sustain early signaling by the TCR (4, 9, 10). Together, these findings suggest that costimulation promotes mature synapse formation and that in the synapse itself, CD28 amplifies TCR signaling. More recent studies indicate, however, that synapse formation per se occurs independently of costimulation (6) and that the mature immunological synapse (with CD28 at the c-SMAC) is formed after TCR signaling has largely subsided (11). Thus, TCR stimulation (signal 1) now appears to create a microenvironment, the c-SMAC, which favors the interaction of CD28 with its ligands, leading to secondary signaling (i.e., signal 2; reference 6).
Based on experiments using mAbs raised against CD28, we previously proposed a two-step mechanism for costimulation (12). Specifically, we showed that rat CD28-specific mAbs fall into two functionally distinct categories: “conventional” mAbs, which are agonists only in the presence of coincident TCR ligation, and “superagonists,” which fully activate primary resting T cells in vitro and in vivo without signal 1 (12). Because the availability of the epitope recognized by superagonistic mAbs is strongly up-regulated by TCR stimulation (13), we hypothesized that superagonistic CD28-specific mAbs may recognize and recruit a distinct subset of CD28 molecules that are more signaling competent. Thus, similar to the mechanism now proposed as a likely scenario at the immunological synapse, these results suggested a mechanism for costimulation in which TCR stimulation (signal 1) alters the molecular environment of CD28, favoring secondary signaling by the clustered, signaling-competent subset of CD28 molecules (signal 2).
If the clear-cut functional differences between conventional and superagonistic CD28-specific mAbs are a reflection of the dynamic changes in supramolecular receptor reorganization during T cell activation, a precise definition of the binding sites of conventional and superagonistic mAbs on the CD28 molecule should provide clues about this physiological process. In this report, we have mapped the binding sites of superagonistic and conventional rat and human CD28-specific mAbs. We show that the superagonists bind to the laterally exposed C”D loop of the extracellular Ig-like domains of the CD28 homodimer whereas conventional anti-CD28 mAbs bind at or close to the B7 binding site, therein suggesting mechanisms for the differential effects of costimulatory and superagonistic mAbs.
We have previously shown in the rat model that superagonistic CD28-specific mAbs are highly potent stimulators of T cell proliferation in vivo without apparent toxicity (12, 14). The identification of the conserved structural basis of “direct” CD28-driven T cell activation in rodents and humans by superagonistic ligands should now facilitate the therapeutic exploitation of mitogenic and anti-apoptotic CD28 signaling in immunopathologies characterized by T cell deficiencies.
Materials and Methods
Antibodies.
The generation of the mAbs JJ316 and JJ319 has been previously described (12, 15). The other anti–rat CD28 mAbs used in this study originated from these published experiments but had not yet been characterized in detail. Anti–mouse CD28 (clone 37.51; reference 16), mouse anti–phosphotyrosine (clone 4G10), and mouse anti–human CD3 (clone UCHT1) was purchased from BD Biosciences. Mouse anti–human TCRζ (clone 6B10.2) and rabbit polyclonal antibodies to nuclear factor (NF)-κB p50, c-rel, and USF-2 were from Santa Cruz Biotechnology, Inc., and mouse anti–human ZAP-70 (clone 3.3.1; reference 17) was provided by A.D. Beyers (University of Stellenbosch, Matieland, South Africa). The generation of the anti–rat TCR mAb R73 was previously described (18).
Generation of mAbs Specific for Human CD28.
BALB/c mice were immunized repeatedly intraperitoneally with recombinant human CD28-Fc fusion protein and a CD28-transfected B lymphoma cell line. 3 d before cell fusion, animals were boosted intravenously. Fusion of spleen cells from immunized mice and X63Ag8.653 myeloma cells was performed by polyethylene glycol treatment according to standard protocols (15). Routinely, hybridoma cells secreting CD28-specific mAbs were subcloned once. Superagonistic anti-CD28 mAbs were subcloned twice.
Animals.
Lewis rats and C57BL/6 mice were bred at the Institute for Virology and Immunobiology, University of Würzburg. Mesenteric lymph nodes were taken from young adult animals and single cell suspensions were used for FACS® analysis and T cell proliferation assays.
Cloning of DNA Constructs.
The rat CD28 cDNA (15, 19) was inserted into the cloning vector Bluescript pBS KS (−). Sequence analysis revealed that amino acid (aa) at position 85 is identical to the homologue mouse CD28 aa in contrast to the data bank (NCBI protein) information. Mouse CD28 cDNA was amplified from whole mouse lymph node cDNA using 5-GCGTCGACGGCCCTCATCAGAACAATGAC-3′ as the sense primer and 5-GGAAGCTTCCCTGTCAGGGGCGGTACGCT-3′ as the antisense primer and then cloned into the pCR 3-uni-expression vector (Invitrogen) or the cloning vector Bluescript pBS KS (−).
To generate the r/mCD28 1–37, the EcoNI/XhoI mouse CD28 cDNA fragment was cloned into the corresponding EcoNI/XhoI site present in the rCD28 sequence in pBS KS (−). The resulting chimeric construct was then cloned into the expression vector pCR 3-uni. The chimeric construct m/rCD28 1–37 was generated by inserting the EcoNI/XhoI rat CD28 cDNA fragment into the corresponding EcoNI/XhoI site present in mCD28 in pBS KS (−) and then cloned into the pCR 3-uni expression vector.
For the m/r CD28 1–66 construct, the rat part of the chimeric molecule was amplified by PCR using 5′-TCGCTCGAATGCCGAGTTCAACTGTGATGG-3′ as the sense and 5′-TTTTGCTCGAGCCCTGTCAGGGGCGGTACG-3′ as the antisense primer and cloned into the Mva1269I/XhoI site present in mCD28 in pBS KS (−). The chimeric construct was then subcloned into the expression vector pHβAPr-1-neo, as this vector exhibited a higher transfection efficiency in L929 cells compared with pCR 3-uni.
All point mutants were generated using the Quick Change kit (Stratagene) according to the manufacturer's instructions. The following primers were used: mCD28 A64V, E65G: sense 5′-CCCAGTTTCGCTCGAATGTGGGGTTCAACTGCGACGGG-3′, antisense 5′-CCCGTCGCAGTTGAACCCCACATTCGAGCGAAACTGGG-3′; mCD28 S62P, A64V, E65G: sense 5′-CCAGTTTCGCCCAAATGTGGGGTTCAA-3′, antisense 5′-TGGAACCCCACATTTGGGCGAAACTGG-3′; mCD28 S62P, A64V, E65G, D71N: sense 5′-AACTGCGACGGGAATTTCGACAA-3′, antisense 5′-TTGTCGAAATTCCCGTCGCAGTT-3′; mCD28 S62P, A64V, E65G, D71N, R109K: sense 5′-GACAACGAGAAGAGCAATGGAACT-3′, antisense 5′-AGTTCCATTGCTCTTCTCGTTGTC-3′; mCD28 S62P, A64V, E65G, D71N, R109K, F98V: sense 5′-TGCAAAATTGAGGTCATGTACCCTC-3′, antisense 5′-GAGGGTACATGACCTCAATTTTGCA-3′; mCD28 S62P, A64V, E65G, D71N, T125A, S127T: sense 5′-TCTTTGTCATGCTCAGACGTCTCCTAAGC-3′, antisense 5′-GCTTAGGAGACGTCTGAGCATGACAAAGA-3′; mCD28(h) F60V, R61T, N63K, A64T, E65G: sense 5′-CCCAGTTTCGCTCGAAAACGGGGTTCAACTGCGA-3′, antisense 5′-TCGCAGTTGAACCCCGTTTTCGAGCGAAACTGGG-3′, as well as sense 5′-TATCAGCCCCAGGTTTACTCGAAAACGG-3′, antisense 5′-CCGTTTTCGAGTAAACCTGGGGCTGATA-3′. All point mutants were cloned into the expression vector pHβAPr-1-neo. The correct generation of all chimeric constructs and point mutants was verified by sequencing.
The constructs mCD28 S62P, A64V, E65G and mCD28(h) F60V, R61T, N63K, A64T, E65G were amplified using the primers sense 5′-GCGCCAATTGCCCTCATCAGAACAATGAC-3′ and antisense 5′-ATTTCGGATCCTGTCAGGGGCGGTACGCT-3′ and then cloned into the EcoRI/BamHI site of the retroviral pczCFG5 IZ vector. This is a derivative of the described pEYZ/MCS retroviral vector (20) in which the open reading frame coding for the EYFP-zeocin fusion protein was removed and an IRES/zeomycin cassette introduced instead. As control, a construct was used that contained the extracellular domain of rat CD28 coupled to the transmembrane and cytoplasmic domain of mouse CD28 (hereafter referred to as extracellular WT rat CD28). For this, the mouse part of the molecule was amplified using the primers sense 5′-TGCTCAGACGTCTCCTAAGCTGTTTT-3′, antisense 5′-GGAAGCTTCCCTGTCAGGGGCGGTACGCT-3′ and the PCR product was cloned into the AatII/HindIII site of rat CD28 in Bluescript pBS KS (−). The chimeric construct was cloned into the EcoRI site of pczCFG5 IZ.
Expression of Constructs in Cell Lines.
The constructs in the expression vectors pCR 3-uni or pHβApr-1-neo were stably transfected into L929 cells using lipofectamin (Life Technologies) according to the manufacturer's recommendations. Transfected cells were selected using RPMI 1640 medium (5% FCS; Life Technologies) containing G418 (Pan Biotech GmbH) at a final concentration of 1 mg/ml. After 2 wk of selection the percentage of cells expressing the appropriate construct ranged between 30 and 90%. If the transfection efficiency was low (as the case for m/r CD28 1–66, mCD28 S62P, A64V, E65G and mCD28 S62P, A64V, E65G, D71N, R109K), the transfected cells were stained with anti-CD28 and positive cells were sorted using a FACSVantage™ cell sorter (Becton Dickinson) and further propagated in RPMI 1640 medium containing G418.
The TCR− 58 mouse T cell hybridoma (21) was first retrovirally transduced with a rat myelin basic protein-specific TCR (provided by T. Herrmann, University of Würzburg, Würzburg, Germany, and Herrmann, T., personal communication). The CD28 constructs in the retroviral vector pczCFG5 IZ were then introduced into these cells using retroviral infection. Recombinant retroviral vector supernatants were generated by cotransfection with vectors encoding VSV-G and pHIT 60 as previously described (22, 23). >90% of the cells were positive for the expression of the appropriate molecules when tested by FACS® analysis.
FACS® Analysis.
For FACS® analysis, 5 × 105 cells were resuspended in PBS/0.1% BSA/0.02% NaN3 and incubated for 30 min with 0.5 μg purified antibody (for JJ316 and JJ319) or 100 μl culture supernatant (for all other anti–rat CD28 mAb) on ice followed by development with PE-labeled donkey anti–mouse IgG (Dianova). Flow cytometric analysis was performed with a FACScan™ flow cytometer (Becton Dickinson) and CellQuest™ software using logarithmic amplification of fluorescence signals from scatter-gated live cells.
T Cell Proliferation Assays.
Rat T cells were purified from lymph node cell suspensions by nylon wool passage. Routinely, T cells were enriched to >90% by this method whereas B cells and monocytes were efficiently depleted and made up <1% of the cell suspension. 105 purified T cells were used for the proliferation assay using either costimulatory conditions (immobilized anti-TCR mAb R73 and soluble anti-CD28 mAb) or mitogenic conditions (immobilized sheep anti–mouse Ig plus anti-CD28; references 12 and 13). Proliferation was determined by [3H]thymidine incorporation.
Human T cells from freshly drawn peripheral blood were obtained from ficoll-purified PBMC and nylon wool passage. Routinely, purity was >90%. Cells were adjusted to 5 × 105/ml and cultured in RPMI 1640 medium containing 10% autologous serum in 200 μl final volume in 96-well plates (Costar) that had previously been coated with 40 μg/ml donkey anti–mouse Ig antiserum (Dianova), followed by extensive washing and incubation with 0.003 μg/ml anti-CD3 mAbs (costimulation) or without anti-CD3 mAbs (direct stimulation) for 15 min on ice. After washing, soluble anti-CD28 mAbs were added to the cultures at a final concentration of 3 μg/ml (see Fig. 4, A and B) or as indicated (see titration in Fig. 4 C). As positive control, cells were stimulated with 5 μg/ml PHA and 200 U/ml IL-2. After 72 h, proliferation was measured by [3H]thymidine incorporation.
Figure 4.

Stimulation of human T cells by superagonistic anti-CD28 mAb in the presence and absence of TCR triggering. Freshly isolated human T cells from PBMC were cultured at 5 × 105/ml in plates containing (A) or lacking (B) immobilized anti-CD3 mAb (suboptimal concentration of 0.003 μg/ml) and soluble anti-CD28 mAb7.3B6 (a representative costimulatory mAb), 9D7 or 5.11A1 (3 μg/ml; two novel superagonistic mAb). (C) Titration of anti-CD28 mAb in the absence of anti-CD3 mAb. (A–C) Cells were cultured for 3 d before the addition of [3H]thymidine. Data are representative for four (A and B) or two (C) independent experiments.
Stimulation of 58 T Hybridoma Cells and IL-2 Measurement.
58 T hybridoma cells (21) transfected with either extracellular WT rat CD28, mCD28 S62P, A64V, E65G or mCD28(h) F60V, R61T, N63K, A64T, E65G were stimulated with either plate-coated anti-TCR mAb, a mitogenic anti-CD28 mAb (JJ316 for rat, 5.11A1 for human), a nonmitogenic anti-CD28 mAb (JJ319 for rat, 37.51 for the chimeric molecules), both at final concentrations of 10, 5, 2.5, and 1 μg/ml, or left untreated. After 2 d, the IL-2 content in the supernatant was tested using an Opti EIA mouse IL-2 detection kit (BD Biosciences) according to the manufacturer's instructions.
Modeling of the Mouse CD28 Structure.
The human CD28 monomer was modeled on the murine CTLA-4 structure (24) using the sequence alignment derived by Metzler et al. (25) and the program O (26). The dimer was constructed on the basis of the CTLA-4 homodimer observed in the crystals of the complex of human CTLA-4 and CD80 (26). Fig. 3 was drawn using BOBSCRIPT (27).
Figure 3.

Depiction of the epitopes for superagonistic and conventional CD28-specific mAbs in a three-dimensional model of the extracellular part of human CD28. The model of the CD28 monomer was derived by computer calculation from the X ray crystallographic structure of murine CTLA-4 using the sequence alignment derived by Metzler et al. (reference 25). The dimer was constructed on the basis of the CTLA-4 homodimer observed in the crystals of the complex of CTLA-4 and CD80, and is shown with the membrane-proximal region at the top. The MYPPPY motif (aa 99–104) critical for B7 binding is indicated in green, the adjacent aa 98 residue critical for binding of the conventional rat and mouse CD28-specific mAb is highlighted in yellow, and the C′′D loop responsible for the binding of superagonistic rat and human CD28-specific mAb (aa 60–65) is indicated in red.
Stimulation, Immunoprecipitation, and Preparation of Nuclear Extracts.
T cells obtained by leukopheresis were purified to ∼90% purity by nylon wool passage. 108 cells were incubated for 1 h at 4°C with 5 μg/ml conventional (7.3B6) or mitogenic anti-CD28 (5.11A1) antibodies, or a mixture of 5 μg/ml anti-CD3 (OKT3) and 0.5 μg/ml conventional anti-CD28 antibodies. Cells were washed, incubated for 30 min at 4°C with 10 μg/ml rat anti–mouse IgG, and incubated for 0.5 min at 37°C before the addition of NP-40 lysis buffer (final concentration 1% NP-40, 25 mM Tris, pH 7.5, 140 mM NaCl, 5 mM EDTA, 1 mM pefabloc, 5 mM iodoacetamide, 1 mM Na3VO4, 1 mM NaF). Lysates were centrifuged (14,000 rpm for 10 min at 4°C) and the supernatant was added to protein G–Sepharose precoated with 5 μg ZAP-70 and TCRζ antibody. Beads were incubated with rotation for 2 h at 4°C, washed four times with lysis buffer, and samples were resolved by SDS-PAGE and Western blotted. Membranes were probed with anti-phosphotyrosine antibody and reprobed with antibodies to TCRζ and ZAP-70 to ensure comparable loading. For preparation of nuclear extracts, 2 × 107 cells were stimulated for 20 h on sheep anti–mouse IgG-coated plates as previously described (13) and the protocol of Schreiber et al. (28) was followed. Protein concentration was measured using the Bradford assay (Bio-Rad Laboratories) and 6 μg protein per lane was resolved by SDS-PAGE, Western blotted, and probed with antibodies to c-Rel and USF-2.
Results
Activity of Conventional and Superagonistic Rat CD28-specific mAbs.
We had previously isolated nine mouse IgG1/κ mAbs to rat CD28 and characterized the functional activity of two representative clones: JJ319 is a conventional, i.e., TCR stimulation–dependent mAb whereas JJ316 is a superagonistic, i.e., autonomously mitogenic CD28-specific antibody (12, 15). To perform structure function analyses with a broader panel of mAbs, the activity of all nine antibodies was tested on freshly isolated rat lymph node T cells in the presence and absence of TCR ligation. As shown in Fig. 1 , all nine antibodies were potent costimulators of T cell proliferation in cultures containing immobilized TCR-specific mAbs. As expected, the majority of these mAbs (seven out of nine) were completely ineffective if signal 1, i.e., TCR stimulation, was omitted. However, two of these antibodies, the previously described superagonist JJ316 and the mAb 5S38.17, triggered strong T cell proliferation without TCR engagement. Thus, the nine rat CD28-specific mAbs analyzed fell into two categories: conventional (7) and superagonistic (2).
Figure 1.

Costimulation and direct stimulation of primary rat T cells by rat CD28-specific mAb. (A) Nylon wool–purified rat lymph node T cells (105/well) were stimulated by anti-TCR mAb R73 immobilized via sheep anti–mouse IgG and soluble rat CD28-specific mAb. An isotype-matched irrelevant mAb served as negative control for the CD28-specific mAb. On day 3, the cells were pulsed with 0.5 μCi [3H]thymidine per well for 16 h followed by harvesting and β counting. (B) As in A but without anti-TCR mAb. One representative experiment from two with the identical outcome is shown.
Separation of Epitopes Recognized by Conventional and Superagonistic CD28-specific mAbs.
The extracellular domains of mouse and rat CD28 differ at only nine aa positions (Fig. 2 A). Because our mouse mAbs to rat CD28 did not bind to mouse CD28 (unpublished data), mouse/rat CD28 chimeras were generated and tested for mAb binding. Initially, the prototypic mAbs, JJ319 (conventional) and JJ316 (superagonistic), were used for binding studies and the hamster anti–mouse CD28 mAb 37.51 (16) was included as an expression control for constructs that had lost binding of rat CD28-specific mAbs.
Figure 2.

Epitope mapping of a superagonistic (JJ316) and conventional (JJ319) rat CD28-specific mAb. (A) Comparison of extracellular portions of mouse and rat CD28. Positions of aa differences are indicated by numbers. (B) Binding of superagonistic and conventional mAbs to rat/mouse CD28 chimeras. Mouse sequences are represented in white and rat sequences in black. Positions of swapped aa are indicated on the left. All constructs were stably expressed in L929 fibroblast cells. FACS® histograms are shown for superagonistic (JJ316) and conventional (JJ319) rat and for the conventional mouse CD28-specific mAb 37.51 (solid lines) and an isotype-matched control mAb (dotted lines). At least two independent experiments were performed and a representative experiment is shown.
To narrow down the region containing the epitope(s) recognized by JJ319 and JJ316, chimeric proteins were generated in which either the aminoterminal one third (aa 1–37) of extracellular mouse CD28 was replaced with the corresponding region of the rat molecule (r/m CD28 1–37) or vice versa (m/r CD28 1–37). In addition, a chimeric molecule was generated in which the aminoterminal half (aa stretch 1–66) of extracellular rat CD28 was replaced with the corresponding mouse sequence (m/r CD28 1–66). In each case, the transmembrane region and the cytoplasmic tail were derived from the backbone molecule as defined by the extracellular region closest to the transmembrane domain (Fig. 2). L929 fibroblast cells were transfected with these chimeric molecules, resulting in expression rates ranging from 30–90%, and binding of CD28-specific mAbs was determined by flow cytometry.
As shown in Fig. 2 B, rows 1–3, superagonist (JJ316) binding mapped to the stretch of aa 37–66 located in the middle of the extracellular rat sequence whereas the epitope recognized by the conventional mAb JJ319 is located between aa 66 and the transmembrane domain. Furthermore, the conventional mouse CD28-specific mAb 37.51 binds to an epitope in the orthologous region (downstream of aa 66) of mouse CD28.
The Superagonistic mAb JJ316 Recognizes the C′′D Loop of Rat CD28.
Within the stretch of aa 37–66, defined above as critical for superagonist mAb binding, mouse and rat CD28 differ at three positions (aa 62, 64, and 65), all of which are located within the C′′D loop of the proposed CD28 structure as modeled on the CTLA-4 crystal structure (Fig. 3) . Stepwise introduction of these three rat-specific aa into mouse CD28 confirmed the requirement for the complete rat-specific C′′D sequence for full superagonist binding (Fig. 2 B, rows 4 and 5).
The Conventional CD28-specific mAbs JJ319 and 37.51 Bind Near the B7 Binding Site.
To also define the epitope seen by the conventional mAb JJ319, single aa that differ between rat and mouse CD28 in the region between aa 66 and the transmembrane region were additionally mutated. First, aspartic acid was changed to asparagine at position 71. This mutation failed, however, to restore binding of JJ319 (not depicted). The same was true when another mouse-specific aa, arginine at position 109, was changed to lysine as found in rat CD28 (Fig. 2 B, row 6). In contrast, when the remaining mouse-specific aa phenylalanine at position 98 was changed to valine, the binding of JJ319 was indistinguishable from that of JJ316 (Fig. 2 B, row 7). Therefore, valine at position 98 is critical for the epitope recognized by the conventional rat CD28-specific mAb JJ319.
Interestingly, the binding of the conventional anti–mouse CD28 mAb 37.51 was inversely related to that of the rat-specific mAb JJ319. Thus, 37.51 binding was lost when at position 98, the mouse-specific aa phenylalanine was changed to valine (Fig. 2 B, row 7). In an additional construct (not depicted), the remaining two extracellular mouse-specific aa that are located in close proximity to the transmembrane region (aa 125/127) were mutated to the rat sequence instead of aa 98. These mutations did not result in binding of the rat CD28-specific mAb JJ319 or in loss of binding of the mouse CD28-specific mAb 37.51. Therefore, these two conventional mAbs recognize an epitope at or near aa 98 and thus close to the B7 binding site (29).
Correlation between Epitope Binding and Mitogenicity of mAbs.
To test whether the structure function relationship established for the superagonistic mAb JJ316 and the conventional mAbs JJ319 and 37.51 can be generalized, the second superagonist and the remaining six conventional rat CD28-specific mAbs (Fig. 1) were tested for binding to the various mouse/rat hybrid molecules. Only one antibody, 5S38.17, reacted with L929 cells expressing mouse CD28 with the grafted rat C′′D loop but did not bind to the m/r CD28 1–66 molecule. It therefore reproduces the binding characteristics of JJ316 (Table I). The other six mAbs, defined as conventional by function, also displayed the same binding characteristics as the conventional mAb JJ319, including a requirement for the rat-specific valine at position 98. Thus, both superagonistic mAbs recognized the C′′D loop whereas all seven conventional mAbs isolated bind to an epitope near the B7 binding site.
Table I.
Reactivity of Rat CD28-specific mAb with Mouse and Rat CD28 as well as Different Chimeric and Mutant CD28 Molecules
| Antibody | MouseCD28 | RatCD28 | m CD28S62P, A64V,E65G | m/r CD281-66 | m CD28 S62P, A64V,E65G, D71N, R109K | m CD28 S62P, A64V,E65G, D71N, R109K, F98V |
|---|---|---|---|---|---|---|
| control | − | − | − | − | − | − |
| 37.51 | +++ | − | +++ | − | +++ | − |
| JJ316 | − | +++ | +++ | − | +++ | +++ |
| JJ319 | − | +++ | − | +++ | − | +++ |
| 5S28 | − | ++ | − | ++ | − | ++ |
| 5S38.17 | − | ++ | ++ | − | ++ | ++ |
| 5S247 | − | +++ | − | +++ | − | +++ |
| 5G40/2 | − | +++ | − | +++ | − | +++ |
| 5G87 | − | ++ | − | ++ | − | ++ |
| 5G111 | − | ++ | − | ++ | − | ++ |
| 5S35 | − | +++ | + | +++ | − | +++ |
+++, very strong reactivity in FACS® analysis; ++, strong reactivity; +, weak reactivity; −, no reactivity. As controls, an isotype-matched irrelevant mAb and the mouse CD28-specific 37.51 mAb were included.
Human CD28-specific Superagonistic mAbs Also Bind to the C′′D Loop.
Because superagonistic CD28 stimulation provides an attractive approach to stimulatory immunotherapy, a set of novel human CD28-specific mAbs was generated and tested for TCR-dependent and -independent induction of proliferation of primary human T cells. Initial screening of 24 CD28-specific mAbs identified two superagonists, 9D7 and 5.11A1, which were purified and functionally tested in parallel with a representative conventional anti-CD28 mAb, 7.3B6. As shown in Fig. 4 , the clear-cut functional difference between conventional and superagonistic mAbs described above for rat CD28 was reproduced for the human system. Thus, in the absence of TCR/CD3 ligation, mAbs 9D7 and 5.11A1 induced strong proliferation in freshly isolated human T cells whereas the stimulatory activity of mAb 7.3B6 was strictly dependent on the presence of immobilized anti-CD3 mAb.
Although mouse and rat CD28 share 93% sequence identity in the extracellular part of the molecule, human CD28 is only 65% identical to the mouse sequence. Accordingly, instead of mapping the epitopes for all human CD28-specific mAbs, we directly tested the hypothesis that the C′′D loop identified as the epitope for the superagonistic rat CD28-specific mAb is also the target for the novel human CD28-specific superagonistic mAb. As shown in Fig. 5 , this was indeed the case. Thus, grafting of the human C′′D loop to the mouse backbone by mutating 5 aa rendered mouse CD28 fully reactive with the superagonistic mAbs 9D7 and 5.11A1, but not with the conventional mAb 7.3B6.
Figure 5.

Epitope mapping of superagonistic human CD28-specific mAb. Binding of two superagonistic (9D7 and 5.11A1) and a conventional (7.3B6) human CD28-specific mAb to L 929 fibroblast cells expressing full-length human CD28 (top), full-length mouse CD28 (middle), and a chimeric human/mouse CD28 molecule with a humanized C”D loop (bottom). Human sequences are shown in black and mouse sequences in white. FACS® histograms are shown for 9D7, 5.11A1, and 7.3B6 (solid lines), and for an isotype-matched control mAb (dotted lines). At least two independent experiments were performed and a representative experiment is shown.
mAb Binding to Grafted Rat or Human C′′D Loops Results in Superagonistic Responses.
Next, we wanted to know whether in the heterologous environment of mouse CD28, engagement of the C′′D loop by human or rat CD28-specific mAbs is sufficient to induce a superagonistic response. To this end, mouse CD28 molecules in which the C′′D loop had been changed either to the rat sequence by three or to the human sequence by five aa exchanges were introduced into the mouse T cell hybridoma 58 (21), previously transfected with a rat TCR. As a positive control, a chimeric CD28 protein was expressed in which the whole extracellular part consisted of rat CD28 and the rest of the molecule was mouse CD28.
When these transfectants were stimulated via the TCR, they produced substantial amounts of IL-2 (Fig. 6) . Importantly, IL-2 production was also efficiently induced when the cells were exclusively stimulated via CD28 using superagonistic anti-CD28 mAbs in a dose-dependent manner. Ligation of these chimeric CD28 molecules by conventional anti-CD28 (JJ319 for rat CD28, the mouse CD28-specific mAb 37.51 for the C′′D loop grafted molecules) did not induce IL-2 production at any concentration tested. This result clearly shows that targeting mAb binding to the C′′D loop of the mouse CD28 molecule leads to a superagonistic response even in this heterologous setting.
Figure 6.

Activation of T cell hybridoma cells via superagonistic CD28-specific mAb. The 58 T cell hybridoma line supplemented with a rat TCR was retrovirally transduced with mouse CD28 containing the following replacements: (A) the whole extracellular part of rat CD28, (B) the rat C′′D loop, or (C) the human C′′D loop. The cell lines were left unstimulated or were stimulated via immobilized anti-TCR mAb R73, superagonistic CD28-specific mAb (JJ316 for rat, 5.11A1 for human) at the indicated final concentrations, ranging from 10 to 1 μg/ml, or conventional CD28-specific mAb (JJ319 for rat, 37.51 for the chimeric molecules). After 2 d, supernatants were tested for the presence of mouse IL-2 by ELISA. Because stimulation with conventional CD28-specific mAb did not result in substantial IL-2 production at any concentration tested, only results using the highest concentration (10 μg/ml) are presented. One of at least three independent experiments with similar results is shown.
Superagonistic CD28 Signaling Induces NF-κB Activation, but not TCRζ nor ZAP-70 Phosphorylation.
To further explore the mechanism of superagonistic CD28 signaling, purified peripheral human T cells were stimulated by costimulation or using superagonistic or conventional CD28-specific mAbs alone. No increase in phosphorylation of TCRζ and ZAP-70 was seen after superagonistic CD28 stimulation whereas as expected, phosphorylation was readily detectable by conventional costimulation via TCR plus CD28-specific mAbs (Fig. 7 A). This indicates that superagonistic CD28 stimulation does not address the proximal signaling machinery of the TCR. Many genes transcribed during T cell activation, e.g., the IL-2 gene, are regulated by transcription factors of the NF-κB family that are activated in a costimulation-dependent manner (3). After stimulation with superagonistic but not conventional CD28-specific mAbs alone, nuclear translocation of c-Rel (Fig. 7 B) and p50 (not depicted) was detected, as was the case for T cell costimulation via the TCR plus CD28. Therefore, the NF-κB pathway is also activated by superagonistic CD28 stimulation without TCR engagement. These data confirm and extend our previous observations concerning superagonistic CD28 signaling in the rat system (13).
Figure 7.
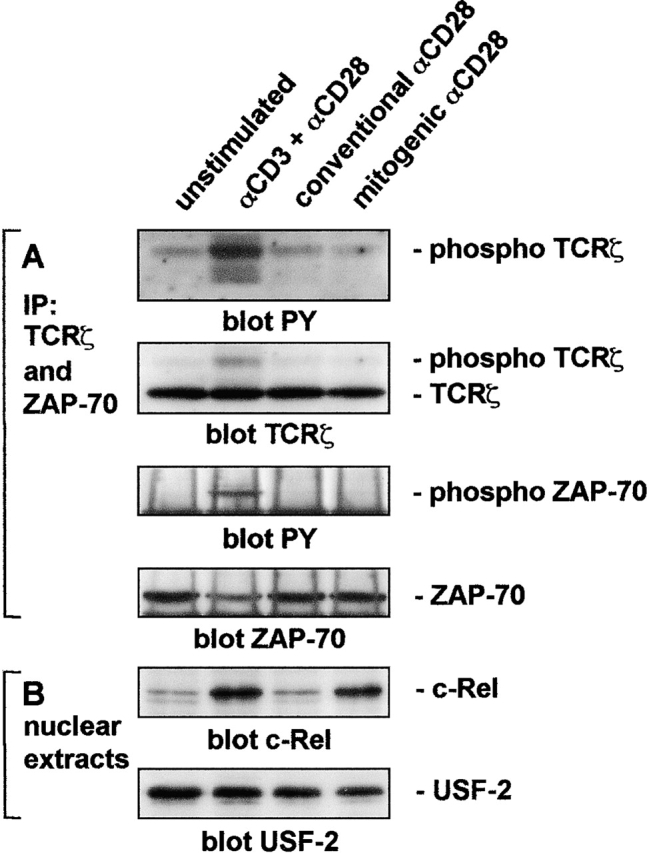
Mitogenic anti-CD28 antibody stimulation activates NF-κB without inducing tyrosine phosphorylation of TCRζ or ZAP-70. (A) T cells were preincubated with the indicated stimulating antibodies for 1 h at 4°C, washed, incubated with 10 μg/ml rat anti–mouse IgG for 30 min at 4°C, and incubated for 0.5 min at 37°C before the addition of NP-40 lysis buffer. Proteins were precipitated with antibodies to ZAP-70 and TCRζ precoupled to protein G–Sepharose, resolved by SDS-PAGE, Western blotted, and probed with antiphosphotyrosine antibody. Membranes were reprobed with antibodies to TCRζ and ZAP-70 to ensure comparable loading. (B) Nuclear extracts from the same cells stimulated with the same primary antibodies and incubated for 20 h on sheep anti–mouse IgG-coated plates were resolved by SDS-PAGE, Western blotted, and probed with antibodies to c-Rel or USF-2 as a loading control.
Discussion
Soon after the first mAbs were raised against leukocyte cell surface molecules it was realized that they could be used to alter the course of immune responses, potentially in a therapeutic setting (30–32). The majority of these antibodies block immune functions or augment them when used in conjunction with other reagents. The much smaller subset of antibodies that activate leukocytes autonomously are defined here as superagonists. Our present findings show that the costimulatory receptor, CD28, can be triggered by superagonists binding to a defined structural element of the homodimer distinct from the CD80/CD86 binding site. The evolutionary conservation of this structure–activity relationship is striking: identification of the superagonistic epitope of rat CD28 permitted an exact prediction of its human counterpart based on its location.
According to a model of the human CD28 monomer based on the CTLA-4 structure (24), residues 60–65 forming the superagonist epitope are located between the C′′ and D strands at the “edge” of the V-set domain (Fig. 3). CTLA-4 is the best current model for the CD28 homodimer and according to this, the C′′D loop maps to the “outside” of the dimer where it protrudes from the membrane-proximal part of the V-set domain (Fig. 3). In contrast to the superagonists, the “classical” costimulatory CD28-specific mAbs are critically dependent on the species-specific aa side chain at position 98 (valine and phenylalanine in the rat and mouse CD28 structures, respectively), adjacent to the ligand binding MYPPPY motif (positions 99–104; Fig. 3; reference 29).
What is the mechanistic basis for triggering by superagonistic C′′D loop–specific mAbs and why are costimulatory mAbs ineffective? Four distinct mechanisms can be considered.
Preferential Engagement of Signaling-competent CD28.
HLA class II molecules are proposed to exist in functionally distinct microclusters identified by mAbs dependent for binding on preexisting oligomerization of their targets (33). The possibility that superagonistic and conventional mAbs bind to CD28 molecules in different states of oligomerization is suggested by our previous work showing that the binding kinetics of superagonists seem to be slower than those of conventional CD28-specific mAbs, suggesting that heterogeneity exists within the population of CD28 molecules (12). The slow on-rate of superagonists was dramatically enhanced by simultaneous TCR stimulation, which was in turn dependent on cytoskeletal rearrangements (as shown by its sensitivity to cytochalasin D; reference 13). In the framework of the “microcluster hypothesis,” this suggests that TCR ligation enhances CD28 clustering and thereby facilitates superagonist binding to preactivated CD28 molecules. Given that the majority of CD28 molecules present on the surface of resting T cells have little lateral mobility and that TCR engagement appears to direct a mobile fraction toward the central zone of the immunological synapse along with the TCR leading to costimulatory signaling by CD28 (6), it is possible that only the easily mobilized CD28 molecules become signaling competent, and then only when clustered. If this is the case, and the accessibility of the mAbs to clustered CD28 molecules is highly epitope dependent, it is conceivable that the clusters will be differentially mobilized by the two groups of mAbs.
Differential Cross-Linking.
In humans, CD28 is only the third T cell surface molecule or complex found to be capable of fully activating T cells upon cross-linking with antibody alone. For the TCR–CD3 complex (34, 35), the antibodies generally need to be immobilized to achieve full activation. CD2 is distinct insofar as it requires pairs of antibodies specific for separate epitopes, but immobilization of the antibodies is not then required (36, 37), suggesting that the simultaneous binding of antibodies to two separate epitopes generates a particularly high degree of cross-linking and strong signaling. The CD80 homodimer is also proposed to link bivalent CTLA-4 partners on the opposing cell surface, allowing stable, large scale lattice formation through periodic CTLA-4–CD80 interactions (38). This is expected to potentiate inhibitory signaling by CTLA-4, and cross-linking of CD28 by the superagonistic mAbs might be both structurally and functionally analogous. The positioning of the C′′D loop on the outside of the Ig-like domains may favor bivalent mAb binding and periodic lattice formation while precluding one on one binding (i.e., the binding of both arms of the antibody to a single CD28 homodimer). The costimulatory mAbs, on the other hand, might be incapable of inducing this level of cross-linking. The CD28 homodimer binds single CD80 and CD86 molecules in contrast to CTLA-4, which is bivalent (39). Given the proximity of the epitope of the costimulatory mAbs to the ligand binding site (Fig. 3), it is possible that at the cell surface, the simultaneous binding of two costimulatory mAbs or two ligand molecules is precluded by similar mechanisms. We note, however, that the CD28 homodimer is generally bivalent for antibody binding (39).
Proximity Effects.
Overall, the simple cross-linking hypothesis does not completely explain the stimulatory effects because, as for CD2 (37), ligation with nonactivating mAbs followed by cross-linking with polyclonal anti-Fc antibody fails to fully activate T cells. Moreover, the activating and nonactivating mAbs in both systems clearly form distinct activity groups defined by shared epitopes, which is not predicted by the cross-linking model. The observation that the effects of the antibodies are highly position dependent suggests that the cell surface molecules may in fact be functionally asymmetric, or “sided.” A similar effect may account for the agonistic and antagonistic properties of anti-TCR antibodies, given the absence of any correlation with binding avidity, although the epitopes of the anti-TCR mAbs have not been mapped (40). In this case, the superagonistic mAbs bind in a position approximately orthogonal to that of the costimulatory mAbs (Fig. 3). It is possible that the costimulatory and superagonistic antibodies cross-link to the same degree and that the position of the epitope bound by the cross-linking antibody influences the proximity of intracellular effector molecules, such as kinases, attached to the cytoplasmic domains of the cross-linked molecules, thereby altering down-stream signaling processes. This requires that the geometric positioning of the cytoplasmic domain, relative to the extracellular domain, is “fixed” (i.e., that the two are unable to rotate separately). Supporting this concept, we note that the agonist and antagonist signaling properties of synthetic erythropoietin (EPO) mimics are exquisitely sensitive to the geometry of EPO receptor ligation (41).
Antibody-induced Conformational Changes.
Conformational changes have been invoked to explain the differential effects of antibodies, most notably in the case of the TCR (40). Synonymous, ligand-induced changes in the α/β subunits of the TCR capable of driving signaling have been ruled out by structural studies, however (42). Although signaling roles for conformational changes in the CD3 subunits of the TCR have been proposed (43, 44), in neither case has the native ligand been shown to induce analogous effects. Perhaps the best examples of conformational change-based mechanisms involve subunit organizational changes proposed for integrin heterodimers (45), the epidermal growth factor receptor (46, 47), and the EPO receptor homodimer (48). Leaving aside the problem of how antibodies would induce structural changes on this scale, it is very unlikely that similar intersubunit rearrangements could be propagated to the cell interior given that the relationship between the monomers is fixed by an interchain–disulphide bond at the base of the extracellular domain of CD28.
Overall, we favor the view that the superagonistic effects of certain antibodies (e.g., anti-CD28 mAbs), or combinations thereof (e.g., anti-CD2 mAbs), depend in the first instance on the formation of cross-linked, periodic arrays of those surface molecules capable of inducing signaling and secondarily, on the topology of binding. CTLA-4 and CD80, which interact with relatively high affinity, are now thought to engage in qualitatively similar interactions (38, 49, 50). This combination of properties is unprecedented at the cell surface, suggesting that uniquely stable, inhibitory signaling complexes are necessary to overturn ongoing activation signals (38).
The present data clearly suggest that the cross-linking of CD28 into periodic arrays in a similar manner generates powerful activating signals circumventing TCR-dependent signaling. Two distinct mechanisms appear to have evolved to prevent the nonspecific, polyclonal activation of T cells by CD28 via such a process. First, CD28 is rendered subservient and consequential to the TCR by virtue of the dependence of costimulatory signaling on immunological synapse formation, which is itself dependent on TCR signaling (6). Second, CD28 is incapable of forming extremely stable, avidity-driven ligand interactions similar to those of CTLA-4, not only because the interactions of CD28 are much weaker than those of CTLA-4, but also because CD28 is monovalent and CD86 is monomeric (39). These observations underscore the functional importance of structural distinctions between CD28 and CTLA-4 and between CD80 and CD86.
Finally, we and others have shown that the superagonists initiate downstream signaling events including NFAT, NF-κB, and GATA-3 translocation and, of course, proliferation and cell cycle progression (13, 14, 51) in the absence of TCRζ or Zap70 hyperphosphorylation. Studies of the effects of conventional mAbs or B7 transfected cells have previously shown that the modification of grb2, p62dok, vav, and slp76 (10, 52, 53) can all occur in the absence of TCR ligation. Superagonistic stimulation in T cell lines nevertheless requires the presence of the TCR at the cell-surface (unpublished data). Therefore, we favor the view that the constitutive, low level triggering of the TCR enhances the recruitment of key intermediates at the point where the signal 1 and signal 2 pathways converge or, alternatively, provides a sufficient amount of preactivated CD28 molecules to be addressed by superagonist stimulation. Importantly, the activation of resting primary T cells with superagonistic mAbs proceeds without a detectable increase above the basal constitutive phosphorylation of ZAP-70 (Fig. 7; reference 13) or the TCRζ chain (Fig. 7) excluding indirect or cross-reactive stimulation of the TCR complex. The effects of cross-linking by the superagonistic mAbs we observe may therefore be mimicking the very early events after TCR triggering and synapse formation.
The oligomerization of the CD28 homodimer via the laterally exposed C′′D loop releases the full signaling potential of this key regulator of human immune responses. This makes superagonistic CD28 mAbs unique polyclonal T cell activators with the potential to exploit the mitogenic, anti-apoptotic, and antiinflammatory effects associated with CD28-mediated T cell activation, and hence promising drugs for immunotherapy. A more complete understanding of the mechanism(s) of superagonistic signaling awaits the determination of the structure of CD28.
Acknowledgments
We gratefully acknowledge D. Lindemann for retroviral vectors, T. Herrmann for the TCR-transfected 58 T cell line, S. Wagner for help in cloning mouse CD28, P. Zigan for lymphocyte preparation, and T. Herrmann and A. Schimpl for critically reading the manuscript.
Funding was provided by a joint grant from TeGenero ImmunoTherapeutics AG and the Bayerisches Technologie-Förderprogramm (BayTP), and by the Deutsche Forschungsgemeinschaft through SFB 465. F. Lühder was supported by the Bundesministerium für Bildung und Forschung (project 01 KX 9820E) and by the Bayerisches Staatsministerium für Wissenschaft, Forschung und Kunst (Bayerischer Habilitationsförderpreis 2000).
Footnotes
Abbreviations used in this paper: aa, amino acid(s); c-SMAC, central supramolecular activation cluster; EPO, erythropoietin; NF, nuclear factor.
References
- 1.Schwartz, R.H. 1990. A cell culture model for T lymphocyte clonal anergy. Science. 248:1349–1356. [DOI] [PubMed] [Google Scholar]
- 2.Sharpe, A.H., and G.J. Freeman. 2002. The B7-CD28 superfamily. Nat. Rev. Immunol. 2:116–126. [DOI] [PubMed] [Google Scholar]
- 3.Rudd, C.E. 1996. Upstream-downstream: CD28 cosignaling pathways and T cell function. Immunity. 4:527–534. [DOI] [PubMed] [Google Scholar]
- 4.Viola, A., S. Schroeder, Y. Sakakibara, and A. Lanzavecchia. 1999. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 283:680–682. [DOI] [PubMed] [Google Scholar]
- 5.Wulfing, C., and M.M. Davis. 1998. A receptor/cytoskeletal movement triggered by costimulation during T cell activation. Science. 282:2266–2269. [DOI] [PubMed] [Google Scholar]
- 6.Bromley, S.K., A. Iaboni, S.J. Davis, A. Whitty, J.M. Green, A.S. Shaw, A. Weiss, and M.L. Dustin. 2001. The immunological synapse and CD28-CD80 interactions. Nat. Immunol. 2:1159–1166. [DOI] [PubMed] [Google Scholar]
- 7.Grakoui, A., S.K. Bromley, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 8.Monks, C.R., B.A. Freiberg, H. Kupfer, N. Sciaky, and A. Kupfer. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 395:82–86. [DOI] [PubMed] [Google Scholar]
- 9.Tuosto, L., and O. Acuto. 1998. CD28 affects the earliest signaling events generated by TCR engagement. Eur. J. Immunol. 28:2131–2142. [DOI] [PubMed] [Google Scholar]
- 10.Michel, F., G. Attal-Bonnefoy, G. Mangino, S. Mise-Omata, and O. Acuto. 2001. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity. 15:935–945. [DOI] [PubMed] [Google Scholar]
- 11.Lee, K.H., A.D. Holdorf, M.L. Dustin, A.C. Chan, P.M. Allen, and A.S. Shaw. 2002. T cell receptor signaling precedes immunological synapse formation. Science. 295:1539–1542. [DOI] [PubMed] [Google Scholar]
- 12.Tacke, M., G. Hanke, T. Hanke, and T. Hünig. 1997. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur. J. Immunol. 27:239–247. [DOI] [PubMed] [Google Scholar]
- 13.Bischof, A., T. Hara, C.H. Lin, A.D. Beyers, and T. Hünig. 2000. Autonomous induction of proliferation, JNK and NF-kappaB activation in primary resting T cells by mobilized CD28. Eur. J. Immunol. 30:876–882. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Palmero, M., T. Hara, A. Thumbs, and T. Hünig. 1999. Triggering of T cell proliferation through CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur. J. Immunol. 29:3914–3924. [DOI] [PubMed] [Google Scholar]
- 15.Tacke, M., G.J. Clark, M.J. Dallman, and T. Hünig. 1995. Cellular distribution and costimulatory function of rat CD28. Regulated expression during thymocyte maturation and induction of cyclosporin A sensitivity of costimulated T cell responses by phorbol ester. J. Immunol. 154:5121–5127. [PubMed] [Google Scholar]
- 16.Gross, J.A., E. Callas, and J.P. Allison. 1992. Identification and distribution of the costimulatory receptor CD28 in the mouse. J. Immunol. 149:380–388. [PubMed] [Google Scholar]
- 17.Dennehy, K.M., R. Broszeit, D. Garnett, G.A. Durrheim, L.L. Spruyt, and A.D. Beyers. 1997. Thymocyte activation induces the association of phosphatidylinositol 3-kinase and pp120 with CD5. Eur. J. Immunol. 27:679–686. [DOI] [PubMed] [Google Scholar]
- 18.Hünig, T., H.J. Wallny, J.K. Hartley, A. Lawetzky, and G. Tiefenthaler. 1989. A monoclonal antibody to a constant determinant of the rat T cell antigen receptor that induces T cell activation. Differential reactivity with subsets of immature and mature T lymphocytes. J. Exp. Med. 169:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark, G.J., and M.J. Dallman. 1992. Identification of a cDNA encoding the rat CD28 homologue. Immunogenetics. 35:54–57. [DOI] [PubMed] [Google Scholar]
- 20.Kuss, A.W., M. Knodel, F. Berberich-Siebelt, D. Lindemann, A. Schimpl, and I. Berberich. 1999. A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur. J. Immunol. 29:3077–3088. [DOI] [PubMed] [Google Scholar]
- 21.Letourneur, F., and B. Malissen. 1989. Derivation of a T cell hybridoma variant deprived of functional T cell receptor alpha and beta chain transcripts reveals a nonfunctional alpha-mRNA of BW5147 origin. Eur. J. Immunol. 19:2269–2274. [DOI] [PubMed] [Google Scholar]
- 22.Pietschmann, T., M. Heinkelein, M. Heldmann, H. Zentgraf, A. Rethwilm, and D. Lindemann. 1999. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 73:2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soneoka, Y., P.M. Cannon, E.E. Ramsdale, J.C. Griffiths, G. Romano, S.M. Kingsman, and A.J. Kingsman. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 23:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostrov, D.A., W. Shi, J.C. Schwartz, S.C. Almo, and S.G. Nathenson. 2000. Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science. 290:816–819. [DOI] [PubMed] [Google Scholar]
- 25.Metzler, W.J., J. Bajorath, W. Fenderson, S.Y. Shaw, K.L. Constantine, J. Naemura, G. Leytze, R.J. Peach, T.B. Lavoie, L. Mueller, et al. 1997. Solution structure of human CTLA-4 and delineation of a CD80/CD86 binding site conserved in CD28. Nat. Struct. Biol. 4:527–531. [DOI] [PubMed] [Google Scholar]
- 26.Jones, T.A., J.Y. Zou, S.W. Cowan, and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 27.Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15:132–134, 112–113 [DOI] [PubMed] [Google Scholar]
- 28.Schreiber, E., P. Matthias, M.M. Muller, and W. Schaffner. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peach, R.J., J. Bajorath, W. Brady, G. Leytze, J. Greene, J. Naemura, and P.S. Linsley. 1994. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J. Exp. Med. 180:2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webb, M., D.W. Mason, and A.F. Williams. 1979. Inhibition of mixed lymphocyte response by monoclonal antibody specific for a rat T lymphocyte subset. Nature. 282:841–843. [DOI] [PubMed] [Google Scholar]
- 31.Cobbold, S.P., A. Jayasuriya, A. Nash, T.D. Prospero, and H. Waldmann. 1984. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 312:548–551. [DOI] [PubMed] [Google Scholar]
- 32.Waldmann, H. 1989. Manipulation of T-cell responses with monoclonal antibodies. Annu. Rev. Immunol. 7:407–444. [DOI] [PubMed] [Google Scholar]
- 33.Kropshofer, H., S. Spindeldreher, T.A. Rohn, N. Platania, C. Grygar, N. Daniel, A. Wolpl, H. Langen, V. Horejsi, and A.B. Vogt. 2002. Tetraspan microdomains distinct from lipid rafts enrich select peptide-MHC class II complexes. Nat. Immunol. 3:61–68. [DOI] [PubMed] [Google Scholar]
- 34.Kappler, J., R. Kubo, K. Haskins, J. White, and P. Marrack. 1983. The mouse T cell receptor: comparison of MHC-restricted receptors on two T cell hybridomas. Cell. 34:727–737. [DOI] [PubMed] [Google Scholar]
- 35.Kaye, J., S. Porcelli, J. Tite, B. Jones, and C.A. Janeway, Jr. 1983. Both a monoclonal antibody and antisera specific for determinants unique to individual cloned helper T cell lines can substitute for antigen and antigen-presenting cells in the activation of T cells. J. Exp. Med. 158:836–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meuer, S.C., R.E. Hussey, M. Fabbi, D. Fox, O. Acuto, K.A. Fitzgerald, J.C. Hodgdon, J.P. Protentis, S.F. Schlossman, and E.L. Reinherz. 1984. An alternative pathway of T-cell activation: a functional role for the 50 kd T11 sheep erythrocyte receptor protein. Cell. 36:897–906. [DOI] [PubMed] [Google Scholar]
- 37.Li, J., A. Smolyar, R. Sunder-Plassmann, and E.L. Reinherz. 1996. Ligand-induced conformational change within the CD2 ectodomain accompanies receptor clustering: implication for molecular lattice formation. J. Mol. Biol. 263:209–226. [DOI] [PubMed] [Google Scholar]
- 38.Ikemizu, S., R.J. Gilbert, J.A. Fennelly, A.V. Collins, K. Harlos, E.Y. Jones, D.I. Stuart, and S.J. Davis. 2000. Structure and dimerization of a soluble form of B7-1. Immunity. 12:51–60. [DOI] [PubMed] [Google Scholar]
- 39.Collins, A., D. Brodie, R. Gilbert, A. Iaboni, R. Manso-Sancho, B. Walse, D. Stuart, P. van der Merwe, and S. Davis. 2002. The interaction properties of costimulatory molecules revisited. Immunity. 17:201–210. [DOI] [PubMed] [Google Scholar]
- 40.Yoon, S.T., U. Dianzani, K. Bottomly, and C.A. Janeway, Jr. 1994. Both high and low avidity antibodies to the T cell receptor can have agonist or antagonist activity. Immunity. 1:563–569. [DOI] [PubMed] [Google Scholar]
- 41.Livnah, O., D.L. Johnson, E.A. Stura, F.X. Farrell, F.P. Barbone, Y. You, K.D. Liu, M.A. Goldsmith, W. He, C.D. Krause, et al. 1998. An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat. Struct. Biol. 5:993–1004. [DOI] [PubMed] [Google Scholar]
- 42.Garcia, K.C., L. Teyton, and I.A. Wilson. 1999. Structural basis of T cell recognition. Annu. Rev. Immunol. 17:369–397. [DOI] [PubMed] [Google Scholar]
- 43.Aivazian, D., and L.J. Stern. 2000. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 7:1023–1026. [DOI] [PubMed] [Google Scholar]
- 44.Gil, D., W.W. Schamel, M. Montoya, F. Sanchez-Madrid, and B. Alarcon. 2002. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 109:901–912. [DOI] [PubMed] [Google Scholar]
- 45.Beglova, N., S.C. Blacklow, J. Takagi, and T.A. Springer. 2002. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9:282–287. [DOI] [PubMed] [Google Scholar]
- 46.Ogiso, H., R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J.H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu, et al. 2002. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 110:775–787. [DOI] [PubMed] [Google Scholar]
- 47.Garrett, T.P., N.M. McKern, M. Lou, T.C. Elleman, T.E. Adams, G.O. Lovrecz, H.J. Zhu, F. Walker, M.J. Frenkel, P.A. Hoyne, et al. 2002. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 110:763–773. [DOI] [PubMed] [Google Scholar]
- 48.Livnah, O., E.A. Stura, S.A. Middleton, D.L. Johnson, L.K. Jolliffe, and I.A. Wilson. 1999. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 283:987–990. [DOI] [PubMed] [Google Scholar]
- 49.van der Merwe, P.A., D.L. Bodian, S. Daenke, P. Linsley, and S.J. Davis. 1997. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 185:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stamper, C.C., Y. Zhang, J.F. Tobin, D.V. Erbe, S. Ikemizu, S.J. Davis, M.L. Stahl, J. Seehra, W.S. Somers, at al. 2001. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 410:608–611. [DOI] [PubMed] [Google Scholar]
- 51.Siefken, R., S. Klein-Hessling, E. Serfling, R. Kurrle, and R. Schwinzer. 1998. A CD28-associated signaling pathway leading to cytokine gene transcription and T cell proliferation without TCR engagement. J. Immunol. 161:1645–1651. [PubMed] [Google Scholar]
- 52.Raab, M., S. Pfister, and C.E. Rudd. 2001. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity. 15:921–933. [DOI] [PubMed] [Google Scholar]
- 53.Nunes, J.A., A. Truneh, D. Olive, and D.A. Cantrell. 1996. Signal transduction by CD28 costimulatory receptor on T cells. B7-1 and B7-2 regulation of tyrosine kinase adaptor molecules. J. Biol. Chem. 271:1591–1598. [DOI] [PubMed] [Google Scholar]