

Abstract
Killer cell Ig-like receptors (KIRs) regulate the function of human natural killer and T cell subsets. A feature of the KIR locus is the clustering of homologous genes encoding for inhibitory and activating KIR. Inhibitory and activating KIR differ for ligand specificities and/or affinities. In particular, we show here with KIR tetramers that activating KIR2DS2 does not bind HLA-Cw3 molecules recognized by inhibitory KIR2DL2, despite 99% extracellular amino acid identity. We also report the 2.3-Å structure of KIR2DS2, which reveals subtle displacements of two residues (Tyr45 and Gln71) involved in the interaction of KIR2DL2 with HLA-Cw3. These results show that KIR molecules cannot tolerate any variability in their three-dimensional structure without altering their MHC class I recognition capacities. Therefore, the mode of recognition used by KIR largely differs from the conformational changes that characterize T cell receptor or NKG2D interaction with their respective ligands.
Keywords: KIR, crystal structure
Introduction
Eight genes encoding for inhibitory KIR (KIR-L) and six genes encoding for activating KIR (KIR-S) are clustered in the human chromosome 19q13.4 within the leukocyte receptor complex (1). KIR can harbor two (KIR2D) or three (KIR3D) extracellular C2-type Ig-like domains. The KIR-L recognition of HLA class I allotypes is determined by the amino acids belonging to the COOH-terminal portion of the MHC class I α1 helix (1). In particular, the recognition of inhibitory KIR2D is dictated to a large extent by the nature of the MHC class I amino acid present at position 80; KIR2DL1 (CD158a) recognizes HLA-C characterized by a Lys80 residue (HLA-Cw4 and related alleles), whereas KIR2DL2 (CD158b1) and KIR2DL3 (CD158b2) allelic forms recognize HLA-C with an Asn80 residue (HLA-Cw3 and related alleles).
In contrast to KIR-L, knowledge of the ligand specificity of KIR-S is limited. Using KIR-S/Fc constructs and a flow cytometry assay, only a weak interaction between KIR2DS1 (CD158h) and Lys80 HLA-C molecules has been reported, despite its homology to KIR2DL1 (2). The same approach failed to detect an interaction between KIR2DS2 and Asn80 HLA-C, although KIR2DS2 is highly homologous to KIR2DL2 and KIR2DL3 (3, 4). So far, only KIR-L three-dimensional structures have been resolved (KIR2DL1, KIR2DL2, and KIR2DL3; references 5–7), free or complexed to HLA-C ligands (KIR2DL2–HLA-Cw3, KIR2DL1–HLA-Cw4; references 8, 9). We report here the first crystal structure of an activating KIR, KIR2DS2 (CD158j), at 2.3-Å resolution.
Materials and Methods
Generation of KIR Tetrameric Complexes.
cDNA encoding the entire extracellular domain of KIR2DS2, KIR2DL1, and KIR2DL3 were amplified by PCR using Pwo DNA polymerase (Roche) and cloned into PLM-1 plasmid in frame with a sequence encoding a biotinylation signal. Soluble KIR proteins were produced as described previously (9), and biotinylated with biotin ligase BirA (Avidity). Soluble KIR tetramers were formed by incubating the biotinylated KIR with PE-coupled streptavidin (Molecular Probes) at a molar ratio of 4:1.
Binding Assay.
The HLA-A, -B, and -C negative mutant B lymphoblastoid cell line L.721.221 and its HLA-Cw3+ and -Cw4+ transfectants were provided by R. Biassoni (Center for Advanced Biotechnology, Genova, Italy). Cells were incubated for 1 h at 4°C with purified KIR tetramers. Fluorescence was analyzed by flow cytometry on a FACScan™ (Becton Dickinson). The ability of anti–MHC class I mAb to inhibit tetramer binding was assayed by preincubating cells with 6a4 mAb (provided by A. Moretta, School of Medicine, Genova University, Genova, Italy) for 30 min at 4°C.
Crystallization, Data Collection, Phase Determination, and Refinement.
A purified and renatured soluble form of truncated human KIR2DS2 (Arg6 to Thr200) was prepared according to Fan et al. (9), and crystallized at 20°C in a 2–4-μl hanging drop containing a 1:1 mixture of protein solution (13 mg/ml KIR2DS2 in 50 mM Tris, pH 8, and 50 mM NaCl) and reservoir solution (1.3–1.6 M ammonium sulfate, 50 mM sodium acetate, pH 4.6, and 20 mM ZnCl2). Diffraction data were recorded at 100 K on LN2 flash frozen crystals soaked previously in crystallization buffer containing 28% ethylene glycol (ID14 EH4; European Synchrotron Radiation Facility). The crystals diffract to 2.1-Å resolution and belong to the trigonal space group P3121, with unit-cell dimension of a = b = 97.535 Å and c = 54.375 Å with one KIR2DS2 molecule in the asymmetric unit (Table S1 available at http://www.jem.org/cgi/content/full/jem.20021624/DC1). Data were processed using DENZO (10) and CCP4 suite (11). The KIR2DS2 structure was determined by molecular replacement using AmoRe (12). The following KIR coordinates (8–3-Å data) were used as the initial model: KIR2DL1 (1nkr; reference 9), KIR2DL2 (2dl2, orthorhombic form, and 2dli, trigonal form; reference 6), and KIR2DL3 (1b6u; reference 7) solved free, and in complex with their respective HLA-C ligand, KIR2DL2 with HLA-Cw3 (1efx; reference 8) and KIR2DL1 with HLA-Cw4 (1im9; reference 9). The KIR2DL2 model, excluding the hinge region Ile101 to Glu105, yielded the clearest solution with a correlation coefficient of 61% and an R factor of 48%. After a rigid body refinement using CNS (13), the two domains of KIR2DS2 were easily identifiable. Different construction steps using Turbo-Frodo (14) were alternated with steps of simulated annealing and grouped B-factor refinement using the maximum likelihood method of CNS, including a bulk solvent correction and anisotropic B-factor scaling. The final model contains protein residues Pro8 to Thr200, 82 water molecules, one ethylene glycol molecule, and one sulfate ion. No electron density was visible for residues Arg6 and Lys7, which were apparently disordered in the crystals. The side chains of residues Glu35, His56, Asp57, His85, and His171 do not have any visible electron density in the 2fo-fc electron density maps contoured at 1σ and were substituted with alanine residues in the refinement steps. The final Rcryst is 22.1% and Rfree is 24.7% from 28 to 2.3-Å resolution (Table S1 available at http://www.jem.org/cgi/content/full/jem.20021624/DC1).
All KIR three-dimensional structures superpositions were performed using the RIGID and RIGID2 options of Turbo-Frodo (14), with a fixed cutoff of 8 Å for complete molecule superpositions and a cutoff of 4 Å for the superposition of the separated D1 and D2 KIR domains, respectively. Figures were realized using Molscript (15), Raster3D (16), and Turbo-Frodo (14).
Data Deposition.
Atomic coordinates for KIR2DS2 have been deposited with the Protein Data Bank, Brookhaven National Laboratory with the accession code 1m4k.
Online Supplemental Material.
The table describes the principal statistics of data collection and refinement of the KIR2DS2 crystals. Details are available at http://www.jem.org/cgi/content/full/jem.20021624/DC1.
Results and Discussion
KIR2DS2 Does Not Interact with HLA-Cw3 Molecules Recognized by KIR2DL3.
We investigated the binding specificities of KIR2DS2 using tetrameric probes known to present high avidity binding properties (17). KIR2DL3 and KIR2DL1 tetramers were also produced in parallel as controls. The integrity of the soluble recombinant biotinylated KIR monomers was ensured by surface plasmon resonance as well as by flow cytometry using anti-KIR mAb (GL183 and EB6) and streptavidin-coated beads. In addition, their monomeric status was verified by analytical and preparative Superdex gel filtration liquid chromatography. Moreover, monodispersity of the KIR2DS2 molecules was verified using dynamic light scattering. As expected, KIR2DL3 tetramers brightly bind HLA-Cw3+ transfectants of the HLA class I− lymphoblastoid cell line L721.221 (221.Cw3), but not parental or 221.Cw4 cells (Fig. 1) . Reciprocally, KIR2DL1 tetramers interact with 221.Cw4 cells, but not with parental or 221.Cw3 cells (Fig. 1). As a control, the binding of inhibitory KIR tetramers to their respective HLA-C ligands is inhibited by the 6a4 anti–HLA class I mAb (2). Despite the high avidity binding properties of these KIR tetramers, KIR2DS2 tetramers fail to bind 221.Cw3 and 221.Cw4 cells. In addition, no binding of KIR2DS2 tetramers was detected on all Asn80 HLA-C+ B lymphoblastoid cell lines tested, in contrast to the positive staining systematically obtained with KIR2DL3 tetramers (unpublished data). These data make it unlikely that the reported failure of KIR2DS2 to interact with HLA-Cw3 is the consequence of low avidity KIR2DS2/Fc probes used in the binding assay (3). Thus, our data formally demonstrate the intrinsic incapacity of KIR2DS2 to recognize Asn80 HLA-C molecules, at least with a binding mode similar to that used by inhibitory KIR2DL2/L3 molecules. The putative ligands for KIR2DS2 still remain to be identified, as KIR2DS2 is involved in susceptibility to autoimmune disorders such as rheumatoid vasculitis (18) and psoriatic arthritis (19).
Figure 1.

Binding of soluble KIR tetramers to HLA class I transfectants. Cells were incubated with indicated 20 µg/ml tetramers in the absence or presence of anti–MHC class I mAb 6a4 before flow cytometric analysis. Determination of cell-surface expression of HLA molecules was assessed using 6a4 mAb.
The Overall Structure of KIR2DS2.
The topology of the KIR2DS2 Ig-like domains is essentially identical to those of inhibitory KIR, with a β-sheet containing three antiparallel β-strands (A, B, and E) and a small β-strand A′, which makes a switch with the other β-sheet containing four antiparallel β-strands (C′CFG). A very short extra β-strand (D-strand, residues 54 and 55) adds a β-strand to the three-stranded β-sheets (ABE) of the D1 domain (8). Of note, there is no extra D β-strand in KIR2DS2 D2 domain as in KIR2DL1 and KIR2DL3, in contrast to KIR2DL2 (Fig. 2 A; references 5, 8).
Figure 2.
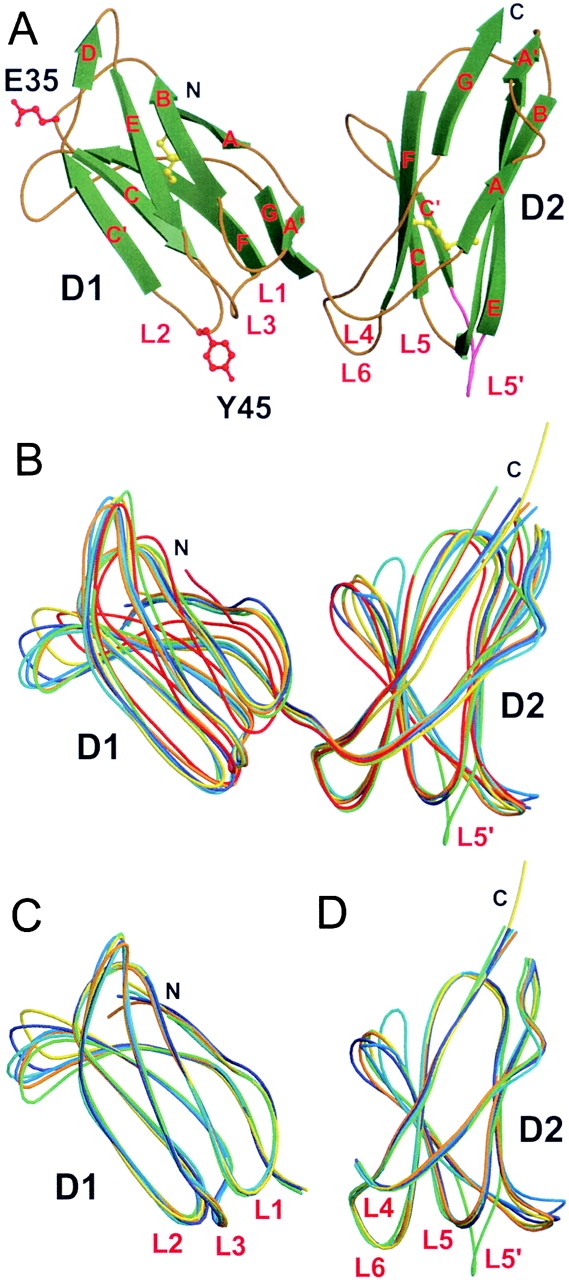
KIR2DS2 crystal structure and its comparison with other known KIR structures. (A) Overall structure of KIR2DS2 extracellular domains shown in molecular ribbon representation with the β-strands in green and the coils golden in color. The two amino acids different between KIR2DS2 and KIR2DL3 (within the sequence range 8–200), Tyr45(Y45) and Glu35(E35), are shown in red ball-and-stick representation. The L5′ loop between β-strands C′ and E of the D2 domain is represented in pink. The two cysteine disulfide bridges are shown in yellow ball-and-stick representation. Cα trace superpositions of (B) the complete KIR molecules (residues 8–200), (C) the isolated D1 domains (residues 8–102), and (D) the isolated D2 domains (residues 103–200) are represented as coils with different colors: KIR2DS2, green; KIR2DL1, red; KIR2DL2 (trigonal form), dark blue; KIR2DL2 (orthorhombic form), cyan; KIR2DL2 bound to HLA-Cw3 (KIR-A), orange; free KIR2DL2 (KIR-B) from the same KIR2DL2–HLA-Cw3 complex crystal, marine blue; and KIR2DL3, yellow.
Pairwise superpositions of the six KIR-L structures with the KIR2DS2 structure revealed (Fig. 2 B) that the root mean square deviations (rmsds) for 192 equivalent Cα atoms are as follows: 2.3 Å for KIR2DL1, 1.8 Å for KIR2DL2 (trigonal form), 2.0 Å for KIR2DL2 (orthorhombic form), 1.8 Å for the KIR2DL2-A molecule interacting with HLA-Cw3, 1.84 Å for the KIR2DL2-B molecule (the KIR2DL2 molecule free in the crystal complex), and 2.0 Å for KIR2DL3. One of the notable differences between KIR-L structures resides in the tertiary packing of their two extracellular domains. The value of the hinge angle between D1 and D2 domains is variable, demonstrating some degree of flexibility in the hinge loop region (L4) from residues 102 to 109 (8). The KIR2DS2 hinge angle value is 73°, higher than those of free KIR2DL1 (55°) and complexed KIR2DL1 (66°; reference 9), but lower than those reported for KIR2DL2 (81°) and KIR2DL3 (78°; reference 8). A consequence of this few degree variation in the D1–D2 hinge angle is shown in Fig. 2 B, where KIR2DS2 D1 and D2 domains do not align with superimposed KIR-L D1 and D2 domains. In addition, the total solvent accessible area buried at the D1–D2 interface of KIR2DS2 is only 830 Å2, lower than the values reported in KIR2DL2 (919 Å2), KIR2DL3 (1,050 Å2), and KIR2DL1 (1,076 Å2; reference 8). The residues found at the KIR2DS2 D1–D2 interface consist principally of those conserved in other KIR interdomain hydrophobic core. Yet, the D1–D2 interface of all inhibitory KIR molecules contains a conserved interdomain salt bridge Asp98-Arg149 that is not conserved in KIR2DS2 structure, where the NH1 and NH2 groups of the Arg149 side chain make hydrogen bonds to a SO4 ion and are distant from the Asp98 carbonyl groups from >8 Å (unpublished data). The superpositions of each separate domain of KIR2 molecules (Fig. 2 C, D1, and Fig. 2 D, D2) revealed conformational variabilities in the DE and FG loops of the D1 domain, the CC′ loop of the D2 domain, and in particular in the L5′ loop, a loop defined between β-strands C′E in D2 (Fig. 2 D). The rmsds for the D1 domains (93–95 equivalent Cα atoms within residues 8–102) of KIR2DS2 and the six other KIR-L molecules are as follows 1.27 Å for KIR2DL1, 0.96 Å for KIR2DL2 (trigonal form), 1.06 Å for KIR2DL2 (orthorhombic form), 0.86 Å for KIR2DL2-A, 0.72 Å for KIR2DL2-B, and 0.94 Å for KIR2DL3. The rmsds difference between the D2 domains (91–94 equivalent Cα atoms within residues 103–200) of KIR2DS2 and the other KIR-L molecules is 0.78 Å for KIR2DL1, 0.94 Å for KIR2DL2 (trigonal form), 1.2 Å for KIR2DL2 (orthorhombic form), 1.0 Å for KIR2DL2-A, 0.95 Å for KIR2DL2-B, and 1.0 Å for KIR2DL3. These rmsds values are similar to those obtained when the D1 and D2 domains were compared between all KIR-L structures (unpublished data).
The D2 Domain Flexible Loop C′E (L5′ Loop).
A distinctive feature of the KIR2DS2 structure is an almost 90° clockwise rotation of the L5′ loop (the Ser151 to Phe160 loop between the β-strands C′ and E of the D2 domain) along an axis joining Phe160 Cα and Ser151 Cα (Fig. 2, B and C). A 14-Å distance separates equivalent residues at position 157 in KIR2DS2 versus KIR2DL2. In KIR2DL2, the L5′ loop is poorly stabilized by only two hydrogen bonds: Thr159-Lys155 and between Gln161-Gly153 (unpublished data). In the case of KIR2DS2, the L5′ loop is highly stabilized by a network of hydrogen bonds that involves Thr159, Tyr137, Tyr134, Gly158, Lys155, Asp135, and buried water molecules (Fig. 3 ; unpublished data). In addition, a displacement of the Phe160 benzene ring side chain toward the external part of the loop shifts the position of the Ser133 side chain hydroxyl group to a new position that is 2.0 Å away from its strictly conserved position in KIR-L molecules. Interestingly, the differences in the position of the L5′ loop of KIR2DS2 do not result from amino acid differences with KIR2DL2 or KIR2DL3, because the sequence of the L5′ loop is strictly conserved. Instead, they may be reflecting an inherent loop flexibility made of the S151AGPKVNGTF160 peptide stretch rich in short amino acids. The reasons for the particular L5′ loop position of KIR2DS2 are unclear but likely involve KIR2DS2 crystal packing, as the L5′ COOH-terminal half portion interacts with the C′ β-strand of another KIR2DS2 monomer (unpublished data).
Figure 3.

Close-up stereo view of the intramolecular stabilization of KIR2DS2 L5′ loop. Amino acids involved in L5′ loop (pink) stabilization are labeled and represented as sticks with their atom colored according to their nature. β-strands, green; coils, golden; water, red balls; and hydrogen bonds, cyan dotted lines.
The L1–L6 Loops in KIR2DS2.
16 amino acids from six loops (L1–L6) in KIR2DL2 are involved in the interaction with HLA-Cw3 (8). In the D1 domain, four amino acids (Glu21, Lys44, Gln71, and Asp72) mediate four hydrogen bonds and one salt bridge (Glu21-Arg69). In the D2 domain, five amino acids (Glu106, Ser133, Asp135, Asp183, and Glu187) are involved in four hydrogen bonds and three salt bridges (Glu106-Arg151, Asp135-Arg145, and Asp183-Lys146; reference 8). Moreover, there are two water-mediated hydrogen bonds implicating residues Gln71, Glu187, and Ala8 of the GAV peptide presented by HLA-Cw3. In addition, hydrophobic contacts contribute to KIR2DL2 and HLA-Cw3 interaction, and involve 10 KIR2DL2 amino acids: Phe45, Met70, Gln71, Asp72, Leu104, Tyr105, Ser132, Phe181, Asp183, and to a lesser extent Ser184. There are only two amino acid differences between the amino acid sequences of the extracellular portion of KIR2DS2 and KIR2DL3, with the Tyr45Phe and Glu35Gln substitutions from KIR2DS2 to KIR2DL3, and four amino acid differences between KIR2DS2 and KIR2DL2 (Pro16Arg, Tyr45Phe, Arg148Cys, and Thr200Ile). Consistent with this high degree of homology, no significant modifications in the position of the following 12 key interface amino acids were detected when KIR2DS2, KIR2DL2, and KIR2DL3 structures were superimposed: Glu21, Met70, Asp72, Leu104, Tyr105, Glu106, Ser132, Asp135, Phe181, Asp183, Ser184, and Glu187 (Fig. 4, A–F) . As a consequence of these conserved positions, the failure of KIR2DS2 to bind HLA-Cw3 cannot be explained by a loss of any of the four salt bridges or by major alterations in the hydrophobic contacts involved in KIR2DL2–HLA-Cw3 interaction (unpublished data).
Figure 4.

Close-up view of the KIR amino acids involved in HLA-Cw3 recognition. The D1 and D2 domains of the inhibitory KIR2DL2 and KIR2DL3 molecules were superposed to the respective D1 and D2 domains of KIR2DS2. Distances between residues in KIR2DS2 and residues in a putative HLA-Cw3 ligand are extrapolated from these superpositions using Turbo-Frodo. Important interacting residues of HLA-Cw3 are represented as sticks in pink; residues from the GAV peptide are in red; and the residues from other KIR molecules are colored as in Fig. 2. Hydrogen bonds between KIR2DL2-A (orange) and HLA-Cw3 are indicated by cyan dotted lines (acceptor–donor distances between 2.5 and 3.4 Å), and important water molecules are indicated by red balls. Interacting residues in KIR2DL2/HLA-Cw3 are labeled from the (A) L1 loop, (B) L2 loop, (C) L3 loop, (D) L4 loop, (E) L5 loop, and (F) L6 loop.
However, the positions of four KIR amino acids (Lys44, Phe45, Gln71, and Ser133) appear altered between KIR2DS2 and KIR2DL2–HLA-Cw3 complex structures. Lys44 has a crucial role in the allotypic recognition of Asn80 HLA-C molecules by KIR2DL2 (1). In KIR2DS2, the position of Lys44 renders the hydrogen-bonded interaction with Asn80 on HLA-Cw3 unlikely (Fig. 4 B). However, the Lys44 position is highly variable in the other free KIR-L structures, probably because of the lack of stabilizing interactions. Position 45 is of great interest, as it is a natural substitution (Phe45 by Tyr45) in KIR2DS2 as compared with inhibitory KIR2DL2-L3. In the KIR2DS2 crystal structure, the Tyr45 benzene ring side chain is clearly visible in the final electronic density map, but its hydroxyl group is not well-defined in the structure. Nevertheless, the hydroxyl group position could be derived from its simple modelization. When the KIR2DS2 D2 domain is superimposed to the KIR2DL2–HLA-Cw3 structure, the tip of Tyr45 hydroxyl group is predicted to be distant from 2.7 Å with the Cβ and from 2.5 Å with the CG atoms of HLA-Cw3 Arg79 (Fig. 4 B). Although these distances are short, they do not support the possibility that Tyr45 in KIR2DS2 could impair HLA-Cw3 binding because of a lack of sufficient space to accommodate Tyr45 hydroxyl group and HLA-Cw3 α1 helix, as suggested previously (1). However, the hydrophobic contacts between Phe45 in KIR2DL2 and Arg75 as well as Val76 in HLA-Cw3 are predicted to be lost or highly perturbed for Tyr45 in KIR2DS2 because of the polar nature of the hydroxyl group present on the Tyr45 side chain (Fig. 4 B). Ser133 is involved in both KIR2DL2–HLA-Cw3 and KIR2DL1–HLA-Cw4 complexes (8, 9). In KIR2DL2, Ser133 mediates three hydrogen bonds with the Arg145 side chain in HLA-Cw3: two with the NH2 group and one with the NE atom (Fig. 4 E). In the KIR2DS2 structure, one of these hydrogen bonds is impaired because of the significant increase in the distance between the main chain Ser133 O atom and the NH2 group of Arg145 side chain (Fig. 4 E). This distance is evaluated at 3.9 Å for KIR2DS2, in contrast with 2.8 Å in the KIR2DL2–HLA-Cw3 crystal (Fig. 4 E). However, the displacement of Ser133 may likely be the consequence of the L5′ loop particular position (Fig. 3). Finally, Gln71 is also involved in KIR2DL2–HLA-Cw3 interface via two hydrogen bonds: one water-mediated (water 38) and one with the Ala8 GAV peptide main chain nitrogen atom. In the KIR2DS2 structure, the fully solvent accessible Gln71 side chain is positioned in a way that both hydrogen bonds are not possible (Fig. 4 C). Superpositions of KIR structures implies Gln71 OE1 in KIR2DS2 to be distant from 7.4 Å with the Ala8 N atom and from 5.6 Å with water 38, in contrast to 3.0 Å in both cases for Gln71 OE1 in KIR2DL2–HLA-Cw3 crystal (Fig. 4 C). However, because no stabilizing interactions of the Gln71 position are clearly revealed by the KIR2DS2 structure, we cannot rule out that Gln71 could take other possible rotamer positions. Nevertheless, of the seven possible Gln71 rotamers, the shortest distance measured between the Gln71 OE1 atom and the peptide Ala8 main chain N atom (4.26 Å) is longer than the 3.05-Å distance measured in the KIR2DL2–HLA-Cw3 complex, and not compatible with a Gln71–Ala8 interaction.
Conclusions.
Our report shows that differences between KIR2DS2 and KIR2DL2 structures are not major, but principally result from the presence of the hydroxyl group of Tyr45 and from the displacement of Gln71 side chain. Thus, we propose that Tyr45 and Gln71 are likely involved in the incapacity of KIR2DS2 to bind the HLA-Cw3–peptide complexes recognized by KIR2DL3. For Tyr45, our results are supported by the Phe45Tyr mutation in KIR2DL3 that abolishes its binding of to HLA-Cw3, and the reciprocal Tyr45Phe mutation in KIR2DS2 that restores to some extent the binding to HLA-Cw3 (3). For Gln71, the absence of other KIR2DS2 structures obtained from different crystal lattices is a limitation to the interpretation of its subtle side chain conformational modification. However, the predicted loss of the interaction between Gln71 (KIR2DS2) and Ala8 (peptide) is compatible with previous observations indicating that the loss of a single salt bridge or a single hydrogen bond destabilizes the electrostatic and hydrophobic interface between KIR and HLA-C ligands. Indeed, single replacements of KIR2DL2 residues (Lys44Met, Tyr105Ala, Asp135His, Asp183Ala, and to a lesser extent Glu106Ala) are responsible for a large decrease in HLA-Cw3 binding affinity (8). It is also possible that modifications observed at positions 44 and 133 contribute to some extent, alone or in combination, to the failure of KIR2DS2 to bind to HLA-Cw3. Consistent with the fast kinetics and the favorable binding entropy of KIR-L interaction with HLA-C molecules (20), our results thus emphasize the known low tolerance of KIR molecules to single mutations at the MHC binding site, and show, at the atomic resolution level, that KIR and HLA associate essentially as rigid bodies. This mode of KIR recognition strongly contrasts with the strategy used by TCR and NKG2D, where conformational changes in receptor binding loops are necessary for efficient ligand binding (21–23).
Acknowledgments
The authors thank E. Termine for excellent technical help.
This work was supported by institutional grants from INSERM, CNRS, the Ministère de l'Enseignement Supérieur et de la Recherche, and specific grants from the Ligue Nationale contre le Cancer (to E. Vivier [Equipe labellisée La Ligue] and X. Saulquin).
X. Saulquin and L.N. Gastinel contributed equally to this work.
The online version of this article contains supplemental material.
References
- 1.Vilches, C., and P. Parham. 2002. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu. Rev. Immunol. 20:217–251. [DOI] [PubMed] [Google Scholar]
- 2.Biassoni, R., A. Pessino, A. Malaspina, C. Cantoni, C. Bottino, S. Sivori, L. Moretta, and A. Moretta. 1997. Role of amino acid position 70 in the binding affinity of p50.1 and p58.1 receptors for HLA-Cw4 molecules. Eur. J. Immunol. 27:3095–3099. [DOI] [PubMed] [Google Scholar]
- 3.Winter, C.C., J.E. Gumperz, P. Parham, E.O. Long, and N. Wagtmann. 1998. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J. Immunol. 161:571–577. [PubMed] [Google Scholar]
- 4.Vales-Gomez, M., H.T. Reyburn, R.A. Erskine, and J. Strominger. 1998. Differential binding to HLA-C of p50-activating and p58-inhibitory natural killer cell receptors. Proc. Natl. Acad. Sci. USA. 95:14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan, Q.R., L. Mosyak, C.C. Winter, N. Wagtmann, E.O. Long, and D.C. Wiley. 1997. Structure of the inhibitory receptor for human natural killer cells resembles haematopoietic receptors. Nature. 389:96–100. [DOI] [PubMed] [Google Scholar]
- 6.Snyder, G.A., A.G. Brooks, and P.D. Sun. 1999. Crystal structure of the HLA-Cw3 allotype-specific killer cell inhibitory receptor KIR2DL2. Proc. Natl. Acad. Sci. USA. 96:3864–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maenaka, K., T. Juji, D.I. Stuart, and E.Y. Jones. 1999. Crystal structure of the human p58 killer cell inhibitory receptor (KIR2DL3) specific for HLA-Cw3-related MHC class I. Structure. 7:391–398. [DOI] [PubMed] [Google Scholar]
- 8.Boyington, J.C., S.A. Motyka, P. Schuck, A.G. Brooks, and P.D. Sun. 2000. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 405:537–543. [DOI] [PubMed] [Google Scholar]
- 9.Fan, Q.R., E.O. Long, and D.C. Wiley. 2001. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol. 2:452–460. [DOI] [PubMed] [Google Scholar]
- 10.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. [DOI] [PubMed] [Google Scholar]
- 11.CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta.Crystallogr. D. Biol. Crystallogr. 50:760–763. [DOI] [PubMed] [Google Scholar]
- 12.Navaza, J. 1994. Amore: an automated package for molecular replacement. Acta Crystallogr. 50:157–163. [Google Scholar]
- 13.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. DeLano, P. Gros, R.W. Grosse-Kunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 14.Roussel, A., and C. Cambillau. 1991. Turbo-Frodo. Silicon Graphics Geometry. P. Directory, ed., Silicon Graphics, Mountain View, CA.
- 15.Kraulis, P.J. 1991. Molscript: a program to produce both detailed and schematic plots of structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- 16.Merritt, E.A., and M. Murphy. 1994. Raster 3D V. 2.0: a program to produce detailed and schematic plots of structures. Acta. Crystallogr. 50:869–873. [DOI] [PubMed] [Google Scholar]
- 17.Altman, J.D., P.A.H. Moss, P.J.R. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]
- 18.Yen, J.H., B.E. Moore, T. Nakajima, D. Scholl, D.J. Schaid, C.M. Weyand, and J.J. Goronzy. 2001. Major histocompatibility complex class I–recognizing receptors are disease risk genes in rheumatoid arthritis. J. Exp. Med. 193:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin, M.P., G. Nelson, J.H. Lee, F. Pellett, X. Gao, J. Wade, M.J. Wilson, J. Trowsdale, D. Gladman, and M. Carrington. 2002. Cutting edge: susceptibility to psoriatic arthritis: influence of activating killer Ig-like receptor genes in the absence of specific HLA-C alleles. J. Immunol. 169:2818–2822. [DOI] [PubMed] [Google Scholar]
- 20.Maenaka, K., T. Juji, T. Nakayama, J.R. Wyer, G.F. Gao, T. Maenaka, N.R. Zaccai, A. Kikuchi, T. Yabe, K. Tokunaga, et al. 1999. Killer cell immunoglobulin receptors and T cell receptors bind peptide-major histocompatibility complex class I with distinct thermodynamic and kinetic properties. J. Biol. Chem. 274:28329–28334. [DOI] [PubMed] [Google Scholar]
- 21.Li, P., D.L. Morris, B.E. Willcox, A. Steinle, T. Spies, and R.K. Strong. 2001. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat. Immunol. 2:443–451. [DOI] [PubMed] [Google Scholar]
- 22.Radaev, S., B. Rostro, A.G. Brooks, M. Colonna, and P.D. Sun. 2001. Conformational plasticity revealed by the cocrystal structure of NKG2D and its class I MHC-like ligand ULBP3. Immunity. 15:1039–1049. [DOI] [PubMed] [Google Scholar]
- 23.Rudolph, M.G., and I.A. Wilson. 2002. The specificity of TCR/pMHC interaction. Curr. Opin. Immunol. 14:52–65. [DOI] [PubMed] [Google Scholar]