

Abstract
Signaling pathways involved in regulating T cell proliferation and survival are not well understood. Here we have investigated a possible role of the nuclear factor (NF)-κB pathway in regulating mature T cell function by using CD4+ T cells from p50−/− cRel−/− mice, which exhibit virtually no inducible κB site binding activity. Studies with these mice indicate an essential role of T cell receptor (TCR)-induced NF-κB in regulating interleukin (IL)-2 expression, cell cycle entry, and survival of T cells. Our results further indicate that NF-κB regulates TCR-induced expression of antiapoptotic Bcl-2 family members. Strikingly, retroviral transduction of CD4+ T cells with the NF-κB–inducing IκB kinase β showed that NF-κB activation is not only necessary but also sufficient for T cell survival. In contrast, our results indicate a lack of involvement of NF-κB in both IL-2 and Akt-induced survival pathways. In vivo, p50−/− cRel−/− mice showed impaired superantigen-induced T cell responses as well as decreased numbers of effector/memory and regulatory CD4+ T cells. These findings provide the first demonstration of a role for NF-κB proteins in regulating T cell function in vivo and establish a critically important function of NF-κB in TCR-induced regulation of survival.
Keywords: T lymphocytes, T cell receptor, cell death, NF-κB, transcription factor
Introduction
Activation of CD4+ Th cells is a critical step in initiating an adaptive immune response. Two signals, both of which are delivered by APCs, are required for activation-induced proliferation of Th cells: (a) engagement of the TCR by an antigen–MHC complex on APCs, and (b) engagement of costimulatory molecules, the best characterized of which is CD28, with the B7 family of proteins expressed by APCs. TCR and CD28-induced pathways synergize in stimulating T cell proliferation and in regulating expression of growth promoting cytokines, such as IL-2 (1–3). Significantly, T cell responses are also intimately dependent upon survival-promoting signals induced by TCR, CD28, and cytokine receptors.
Antigen encounter by T cells induces both proliferative and survival pathways, which drive T cell expansion and lead to the development of immunity. Conversely, after antigen is cleared, cessation of T cell–APC engagement can result in rapid induction of cell death (4, 5). This mode of T cell death, referred to as “passive” cell death, is thought to be crucial for terminating an immune response and maintaining T cell homeostasis (5). The best-defined survival pathways in T cells involve growth-promoting cyto-kines, such as IL-2, or costimulatory molecules, such as CD28, that appear to function by activating the antiapoptotic kinase, Akt (see below). TCR engagement itself also provides protection through a signaling pathway that has not yet been defined. In contrast, when T cells encounter high concentrations of antigens (e.g., autoantigens), TCR signals promote apoptosis either by triggering death receptors such as Fas or TNFR1 (4–6), or by activating the proapoptotic molecule Bim (7). This form of T cell death is referred to as “activation-induced” cell death and helps prevent autoimmunity. Thus, regulation of T cell responses is dependent upon both antiapoptotic and proapoptotic signaling pathways.
T cell activation is initiated by protein tyrosine kinases, which induce the activation of multiple signaling pathways (8). Several transcription factors have been shown to be activated by TCR, CD28, and cytokine receptor engagement, including those belonging to the Ets, NFAT, AP-1, nuclear factor (NF)*-κB, and signal transducer and activator of transcription families (9). However, the specific roles played by these factors in regulating T cell activation and survival is only beginning to be understood. Gene targeting studies have recently shed some light on the function of these transcription factors in T cells. Studies of mature T cells singly or doubly deficient in NFAT proteins (10) have primarily revealed defects in T cell differentiation rather than activation or survival (11–13). c-Jun−/− T cells also show no proliferative defects (14) whereas the role of other AP-1 family proteins in T cells is still unclear. Recent studies have indicated that the PI3K/Akt pathway is a key survival mediator of both CD28 and IL-2R (15–18). However, it is not known how the Akt protein kinase blocks apoptosis in T cells, although NF-κB is thought to be one of the targets of Akt (18–21).
The NF-κB transcription factors are key regulators of inflammatory and immune response genes (22). NF-κB activation is controlled by signal-dependent phosphorylation of NF-κB–associating IκB inhibitory proteins by IκB kinases (IKKα and IKKβ; reference 23). Phosphorylated IκB proteins are rapidly degraded, allowing translocation of NF-κB to the nucleus. Recent studies have shown an important role for NF-κB proteins in regulating cell death in many different cell types (22), including B cells (24–26) and dendritic cells (DCs; reference 27). In T cells, NF-κB is activated by both TCR and CD28 engagement and comprises of p50+RelA and p50+cRel heterodimers and p50 homodimers (28). However, in vitro proliferative responses in p50−/−, RelA−/−, and cRel−/− CD4+ T cells are only moderately reduced compared with WT T cells (28). In contrast, proliferation of p50−/− cRel−/− doubly deficient T cells is drastically reduced, indicating important but redundant functions for p50 and cRel in T cells (28). NF-κB function in T cells has also been studied in transgenic (Tg) mice expressing a degradation-resistant form of the IκBα inhibitor in T cells (29–31). Studies of IκB-Tg T cells also showed impaired in vitro proliferative responses (29). Gene targeting studies of PKCθ and Bcl-10 have revealed both impaired NF-κB activation and T cell proliferative responses in vitro (32, 33). However, impaired proliferation may result from defects in cell cycle regulation and/or decreased survival of activated T cells. Studies to date have not discriminated between these different possible functions for NF-κB in T cells. Significantly, the possible function of NF-κB in regulating TCR, CD28, and cytokine receptor–induced pathways has not been determined. Finally, in vitro studies of T cell function may not accurately reflect in vivo events. At present, the in vivo function of NF-κB in T cells remains to be addressed.
Here we have examined the function of NF-κB in regulating mature T cell signaling pathways by using p50−/− cRel−/− CD4+ T cells, which have virtually no inducible NF-κB activity. We show that NF-κB plays essential roles in regulating TCR-induced cell cycle entry and survival both in vitro and in vivo. In contrast, NF-κB is not necessary for survival pathways induced by Akt or IL-2. Our findings define NF-κB as a key participant in the TCR-induced survival pathway that is not only essential but also sufficient for maintaining T cell survival.
Materials and Methods
Isolation, Activation, and Cell Division Analysis of CD4+ T Cells
p50−/− cRel−/− mice were generated by crossing p50−/− mice (34) with cRel−/− mice (provided by S. Gerondakis, The Walter and Eliza Hall Institute of Medical Research, The Royal Melbourne Hospital, Victoria, Australia; reference 35). OTII×IL-2−/− mice were generated by crossing OT-II mice (36) with IL-2−/− mice (37). All mice were used between 2 to 4 mo after birth. CD4+ T cells were isolated from mouse spleens using CD4+ Dynabeads (Dynal) according to the manufacturer's instructions. Isolated T cells were cultured in T cell medium (RPMI containing 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM Hepes, and 0.1 mM 2-mercaptoethanol; GIBCO BRL). T cells were activated by culture in the presence of 1 μg/ml plate-bound αCD3 and 1 μg/ml αCD28 (BD Biosciences) monoclonal antibodies. In some experiments, 20 ng/ml murine IL-2 (R&D Systems) was also added to T cell culture media. For proliferation analysis, naive CD4+ T cells were incubated in T cell media containing 1 μM 5-chloromethylfluorescein diacetate (Molecular Probes) at 37°C for 30 min, washed twice, and cultured under different activation conditions for 1–3 d before FACS® analysis. 5-chloromethylfluorescein diacetate breaks into carboxyfluorescein diacetate succinimidyl ester (CFSE) in cells and then the latter covalently associates with cellular proteins. When a cell divides, CFSE gets evenly distributed in two cells. Each cell division causes a “left shift” in the FACS® histograph of ∼0.3 log scale units. CFSE analysis was performed on living cells after gating on them based on their forward and side scatter characteristics.
FACS®, Electrophoretic Mobility Shift Assay (EMSA), RT-PCR, and Northern Analysis
PE-αCD4, PerCP-αCD4, FITC-αCD8, PE-αB220, FITC-αCD3, FITC-αCD25, PE-αCD25, FITC-αTCRVβ6, FITC-αTCRVβ8, FITC-αCD44, and PE-αCD62L antibodies were purchased from BD Biosciences. FACS® analysis was performed on a FACSCalibur® cytometer (Becton Dickinson). EMSAs were performed as previously described (28). The hairpin oligonucleotide probes used were GAGAGGGGATTCCCCGATTACCTTTCGGGGAATCCCCTCT (H2 site) and GAGAGGGGAATCTCCCATTAGCTTTGGGAGATTCCCCTCT (IL-2Rα site). Total RNA of T cells stimulated under different conditions were extracted using TRI reagent (Molecular Research Center) as recommended by the manufacturer. RT was performed using PRO-Star RT-PCR First Strand Kit from Stratagene according to the manufacturer's instructions. The primers used in the experiments were: IL-2: 5′ primer AACAGCGCACCCACTTCAA, 3′ primer TTGAGATGATGCTTTGACA; c-Myc: 5′ primer CGACGATGCCCCTCAACGTG, 3′ primer CGAGTTAGGTCAGTTTATGC; Bcl2: 5′ primer GCTTCTTTTCGGGGAAGGAT, 3′ primer AAGCCCAGACTCATTCAACC; Bcl-XL: 5′ primer AAG CAA GCG CTG AGA GAG GCA, 3′ primer ACA GTC ATG CCC GTC AGG AAC; and β actin: 5′ primer ATGGATGACGATATCGCT, 3′ primer ATGAGGTAGTCTGTCAGGT. Northern blotting was performed as previously described (38), using specific radiolabeled cDNA probes for Bcl2, Bcl-XL, and β actin genes.
Staphylococcal Enterotoxin B (SEB) Challenge Experiments
WT and p50−/− cRel−/− mice were injected intravenously with 50 μg SEB (Toxin Technology). Mice were killed before injection (day 0) and 3 and 6 d after injection. Splenocytes were double stained with CD4 and TCR-Vβ8 or TCR-Vβ6 antibodies to determine the percentage of Vβ8+ or Vβ6+ T cells in total CD4+ T cell population.
Retroviral Infection of CD4+ T Cells and Infected Cell Survival Assays
The constitutively active IKKβ mutant, IKKβ(EM), which contains both EE and M10 mutations, was provided by M. Delhase and M. Karin (University of California San Diego, San Diego, CA; reference 39). It was subcloned into murine stem cell virus internal ribosome entry site green fluorescent protein (GFP; MIG) retroviral expression vector (16). MIG-Bcl2 and MIG-MyrAKT constructs have been described (15, 16). Retroviruses were produced by transfecting BOSC 293T cells (40) with retroviral plasmid DNA and the pCL-Eco packaging plasmid.
During infection, WT and p50−/− cRel−/− T cells were activated with 1 μg/ml αCD3 and 1 μg/ml αCD28 in the presence of 20 ng/ml IL-2. OTII T cells were activated with 1 μg/ml OVA peptide in the presence of 10 ng/ml IL-2. CD4+ T cells were spin infected at 2,500 rpm for 1 h at 30°C on days 1 and 2 of T cell activation. Viable cells were harvested on day 4 by Ficoll (Amersham Biosciences) gradient centrifugation. Because all retroviral constructs based on MIG vector contain an internal ribosome entry site GFP cassette, infected cells were distinguished from uninfected cells by GFP expression using FACS®. T cell survival assays were performed as follows: after infection, viable T cells were obtained by Ficoll spin. Then, the percentage of GFP+ cells was determined by FACS®. This was the starting time point T0 (day 0). These T cells were plated in T cell medium without αCD3 antibody. At different time points Tn (days 1, 2, and 3), cells were stained with 2.5 μg/ml propidium iodide (PI) for 10 min before FACS® analysis. Viable infected T cells were represented by the GFP+ PI− population. The survival rate of the infected T cells was calculated according to this formula: (the percentage of GFP+ PI− cells at Tn)/(the percentage of GFP+ PI− cells at T0) × 100.
Cell Death Assays
DNA Content Staining.
DNA content staining was performed as previously described (28). In brief, T cells were fixed at 4°C in 70% ethanol for 24 h and stained with a PI staining solution (PBS containing 50 μg/ml PI, 100 U/ml RNase A, and 1 mg/ml glucose) for 2 h at room temperature before FACS® analysis. Apoptosis was determined by quantification of the sub-G0 population.
Passive Cell Death Determination.
After 3 d of activation, CD4+ T cells were harvested by Ficoll (Amersham Biosciences) density gradient centrifugation. Typically, T cells obtained by this method contained <5% dead cells. These T cells were plated in T cell medium, with or without 20 ng/ml IL-2 for 1 or 2 d before being assayed for apoptosis by DNA content staining and quantification of the sub-G0 population. In certain experiments, SYTOX was used to stain the apoptotic cell population. SYTOX (Molecular Probes) is a fluorescent dye that emits a strong green fluorescence after binding to DNA. Cells were double stained with PE-αCD25 and SYTOX (2 nM in PBS) before FACS® analysis. Apoptosis rate was calculated using this formula: (percentage of CD25+ SYTOX+ cells)/(percentage of total CD25+ cells) × 100.
Cell Death of Retrovirus-infected T Cells.
This is described above in the section on retrovirus infection.
Results
Normal T Cell Development in the Absence of p50 and cRel.
Amongst known NF-κB subunits, p50, cRel, and RelA comprise a large proportion of NF-κB activity in mature T lymphocytes (28). Although T cell development is normal in p50−/−, cRel−/−, or RelA−/− mice, doubly deficient p50−/− RelA−/− and IKKβ−/− mice lack lymphocytes, probably because of impaired survival of common lymphocyte precursors (34, 35, 41–44). Because p50+RelA and p50+cRel heterodimers comprise two major NF-κB complexes in many cell types, including T cells, we determined the consequence of the absence of p50 and cRel on lymphocyte development. p50−/− and cRel−/− mice (34, 35) were interbred to obtain p50−/− cRel−/− mice, which were fertile and apparently healthy. As shown in Fig. 1 A, no differences in the numbers of double-positive and CD4 and CD8 single-positive thymocytes were noticed between WT and p50−/− cRel−/− mice. Mature T and B cells were also present in normal numbers in the spleens of p50−/− cRel−/− mice, as were CD4+ and CD8+ T cells (Fig. 1 B). We have also found that DC development proceeds normally in these mice (27). Expression of the CD3 component of the TCR was indistinguishable between WT and p50−/− cRel−/− CD4+ T cells (Fig. 1 C).
Figure 1.

Combined absence of p50 and cRel NF-κB subunits does not affect T cell development. (A) FACS® analysis of CD4 and CD8 expression in WT and p50−/− cRel−/− thymocytes. (B) FACS® analysis of B220, CD3, CD4, and CD8 expression in WT and p50−/− cRel−/− splenocytes. (C) CD3 expression level in WT and p50−/− cRel−/− CD4+ T cells. FACS® was performed by gating on CD4+ cells. (D) EMSAs were performed with nuclear extracts from naive CD4+ T cells either untreated or activated with 1 μg/ml plate-bound αCD3 or 1 μg/ml plate-bound αCD3 plus 1 μg/ml αCD28 for 6 h. NF-κB binding sites used were from H-2Kband IL-Rα (CD25). Complexes 1 and 2 are described in the text. (E) EMSAs were performed with nuclear extracts from naive CD4+ T cells activated with 1 μg/ml plate-bound αCD3 plus 1 μg/ml αCD28 for 6 h. NF-κB binding site was from H-2Kb. The addition of antibodies to RelA and p100/p52 is indicated.
To determine the specific consequence of engagement of TCR and CD28 on NF-κB activation in WT and p50−/− cRel−/− CD4+ T cells, we used plate-bound anti-CD3 (αCD3) and anti-CD28 (αCD28) monoclonal antibodies. EMSA analysis showed that in WT T cells, nuclear NF-κB activity binding with the high affinity H-2 or IL-2Rα (CD25) κB sites was significantly enhanced after a 6-h stimulation with αCD3 or αCD3+αCD28 (Fig. 1 D). The two indicated complexes have previously been characterized as heterodimers of p50 with RelA and cRel (complex 1) or homodimers of p50 (complex 2; reference 28). As expected from previous studies, αCD3-induced NF-κB activity could be enhanced in the presence of αCD28 (Fig. 1 D, bottom). Strikingly, both constitutive and αCD3-inducible κB site binding activities were virtually abolished in p50−/− cRel−/− T cells (Fig. 1 D). Furthermore, CD3+CD28 engagement did not enhance κB site binding activity in p50−/− cRel−/− T cells. Similar results were also obtained when T cells were used after 3 d of activation (unpublished data). These results demonstrate that p50+cRel comprise a major proportion of NF-κB activity in mature CD4+ T cells, as they do in mature B cells and DCs (27, 35, 45).
We then tested for the presence of RelA and p52, two NF-κB subunits that may substitute for p50 and cRel in p50−/− cRel−/− T cells. As previously shown (28) and in Fig. 1 E, the κB site binding activity in αCD3+αCD28–stimulated WT T cells is reduced by a RelA Ab (complex 1) and a p100/p52 Ab (complex 2). In contrast, the RelA Ab did not affect p50−/− cRel−/− T cell DNA binding activity although reactivity was evident with the p100/p52 Ab (complex 3). Importantly, RelA protein levels were no different between WT and p50−/− cRel−/− T cells (unpublished data). Thus, lack of RelA DNA binding in p50−/− cRel−/− T cells is not due to absence of RelA protein. Although it is unclear why RelA DNA binding is not detected in p50−/− cRel−/− T cells, a likely explanation might be that in the absence of p50 and cRel, RelA remains monomeric and/or forms homodimers. Although monomeric RelA cannot bind DNA, previous studies have shown that RelA homodimers poorly bind DNA compared with heterodimers with p50 (46), which may also explain why RelA is typically present in heterodimeric complexes with these proteins. Thus, the absence of p50 and cRel also diminishes the formation of RelA DNA binding complexes, resulting in further reduction in κB site binding activity. Overall, these results indicate that the normal development and greatly reduced κB site binding activity in p50−/− cRel−/− CD4+ T cells make them an excellent tool for studying NF-κB function in mature T cells.
Impaired Cell Cycle Entry after TCR Stimulation of p50−/− cRel−/− T Cells In Vitro.
Using p50−/− cRel−/− cells, we first determined the role of NF-κB in cell cycle regulation of CD4+ T cells. To this end, WT and p50−/− cRel−/− cells were stimulated with αCD3 for 2 d, after which DNA content per cell was quantified to simultaneously determine potential defects in regulation of cell cycle and cell survival. αCD3 treatment of WT T cells induced significant cell cycle entry, evidenced by S and G2/M phase cells (Fig. 2 A). In contrast, very few S and G2/M phase cells were detected after αCD3 treatment of p50−/− cRel−/− T cells. Significantly, a large proportion of p50−/− cRel−/− cells underwent apoptosis as evidenced by the sub-G0 population (Fig. 2 A). However, costimulation of p50−/− cRel−/− cells with αCD28 significantly enhanced cell cycle entry and reduced the number of apoptotic cells (Fig. 2 A), suggesting a potentially NF-κB–independent role for CD28 in regulating these processes. In contrast, the addition of exogenous IL-2 did not significantly affect T cell survival or cell cycle entry (Fig. 2 A). Next, we determined whether impaired cell cycle entry and survival of p50−/− cRel−/− cells was due to impaired expression of genes involved in the regulation of these processes. A 6-h treatment of WT T cells with αCD3 or αCD3+αCD28 increased the expression of mRNAs for IL-2, c-Myc, Bcl-2, and Bcl-xL (Fig. 2 B). On the other hand, similar treatment of p50−/− cRel−/− cells showed significantly impaired induction of IL-2 and Bcl-xL whereas induction of c-Myc and Bcl-2 was also moderately reduced. Thus, defects in cell cycle entry and/or survival of p50−/− cRel−/− T cells might be due to impaired induction of expression of these key genes after TCR engagement. Although our results do not demonstrate that NF-κB proteins directly regulate these genes, previous studies have identified κB sites in control regions of these genes (e.g., Bcl-xL; references 47 and 48), suggesting a possible direct role.
Figure 2.

Impaired cell cycle entry and survival after TCR stimulation of p50−/− cRel−/− T cells. (A) WT and p50−/− cRel−/− CD4+ T cells were activated with plate-bound αCD3, αCD3+IL-2, αCD3+αCD28, or αCD3+ αCD28+IL-2 for 2 d before DNA content staining and FACS® were performed. The percentages show the sub-G0 population, which represents apoptotic cells and cells in different phases of the cell cycle. Typical results of several independent experiments are shown. (B) RT-PCR was performed to determine expression of IL-2, c-Myc, Bcl2, and Bcl-XL in WT and p50−/− cRel−/− CD4+ T cells after 6 h of activation.
These results indicate potential defects in both cell cycle entry and survival of p50−/− cRel−/− T cells. To specifically determine whether p50−/− cRel−/− T cells exhibit a defect in cell cycle control, both WT and p50−/− cRel−/− cells were CFSE labeled and stimulated for 1–3 d with αCD3. At the end of stimulation, dead cells were gated out during FACS® and the level of CFSE was determined specifically in the viable cell population. The level of CFSE was used to determine the number of cell divisions that viable WT and p50−/− cRel−/− T cells underwent after stimulation. As shown in Fig. 3 A, either a 2- or 3-d αCD3 treatment led to multiple cell divisions in WT T cells whereas the vast majority of p50−/− cRel−/− did not undergo cell division. As shown above, IL-2 expression was significantly reduced in p50−/− cRel−/− cells. Therefore, we tested whether the addition of exogenous IL-2 could allow p50−/− cRel−/− T cell division. A 3-d treatment with αCD3+IL-2 moderately increased the number of dividing p50−/− cRel−/− cells (Fig. 3 B), but this number remained significantly lower than WT cells treated with αCD3 alone or with αCD3+IL-2. In contrast, CD28 costimulation was able to significantly increase the number of p50−/− cRel−/− cells that underwent cell division (consistent with the results shown in Fig. 2 A). As shown in Fig. 3 B, bottom right, a majority of WT cells stimulated with αCD3+CD28+IL-2 divided three to four times over a 3-d period. In contrast, the number of p50−/− cRel−/− cells that underwent more than two cell divisions when stimulated in the same manner was significantly reduced. Furthermore, most p50−/− cRel−/− cells failed to undergo even a single cell division. Significantly, p50−/− cRel−/− T cells stimulated with αCD3+CD28+IL-2 for 3 d expressed high levels of the IL-2Rα chain (CD25; Fig. 3 C), although less than in similarly treated WT cells. These results indicate that impaired cell cycle entry of p50−/− cRel−/− T cells is not due to lack of responsiveness to stimulation. Instead, p50−/− cRel−/− cells do undergo certain activation events, but fail to cycle. Therefore, these results demonstrate a crucial role for NF-κB proteins in regulating TCR-induced cell division. In addition, our results suggest that defects in cell division are not due to impaired IL-2 or IL-2R expression.
Figure 3.

Impaired cell division of p50−/− cRel−/− CD4+ T cells after activation. (A and B) WT and p50−/− cRel−/− CD4+ T cells were CFSE labeled and activated under different conditions as shown for 1–3 d. FACS® was performed on viable cells by gating on the forward and side scatter characteristics. As shown in the bottom right of B, peaks/shoulders represent the number of times cells underwent division. The percentage indicates cell population that has divided at least once. (C) FACS® analysis of CD25 (IL-2Rα) expression on WT and p50−/− cRel−/− CD4+ T cells after 3 d of activation by plate-bound αCD3+αCD28 and IL-2.
Increased Cell Death in Activated p50−/− cRel−/− T Cells Can Be Rescued by Bcl-2.
The results shown above also indicate that the activation of p50−/− cRel−/− T cells render them more susceptible to cell death than WT T cells (Fig. 2 A). To test this possibility and the potential mechanisms involved, WT and p50−/− cRel−/− cells were stimulated with αCD3+αCD28 for 3 d, after which viable cells were isolated on a Ficoll gradient. These cells were then double labeled with αCD25 to detect activated T cells and the DNA dye SYTOX to detect apoptotic cells. This method was used to determine the percentage of activated T cells undergoing apoptosis (i.e., CD25+ SYTOX+) out of the total number of CD25+ cells (CD25+ SYTOX− and CD25+ SYTOX+). As shown in Fig. 4 A, immediately after activation (day 0), only 5% WT and p50−/− cRel−/− Ficoll-isolated cells were apoptotic. In the absence of stimulation, activated T cells rapidly undergo cell death. After 1 d in culture without stimulation, ∼25% activated CD25+ WT T cells underwent cell death. In contrast, 75% of CD25+ p50−/− cRel−/− cells underwent cell death over the same period. Thus, activated p50−/− cRel−/− cells are significantly more susceptible to cell death than WT cells. Therefore, these results demonstrate that NF-κB proteins play an essential role in regulating not only cell division (Fig. 3), but also survival of activated T cells.
Figure 4.
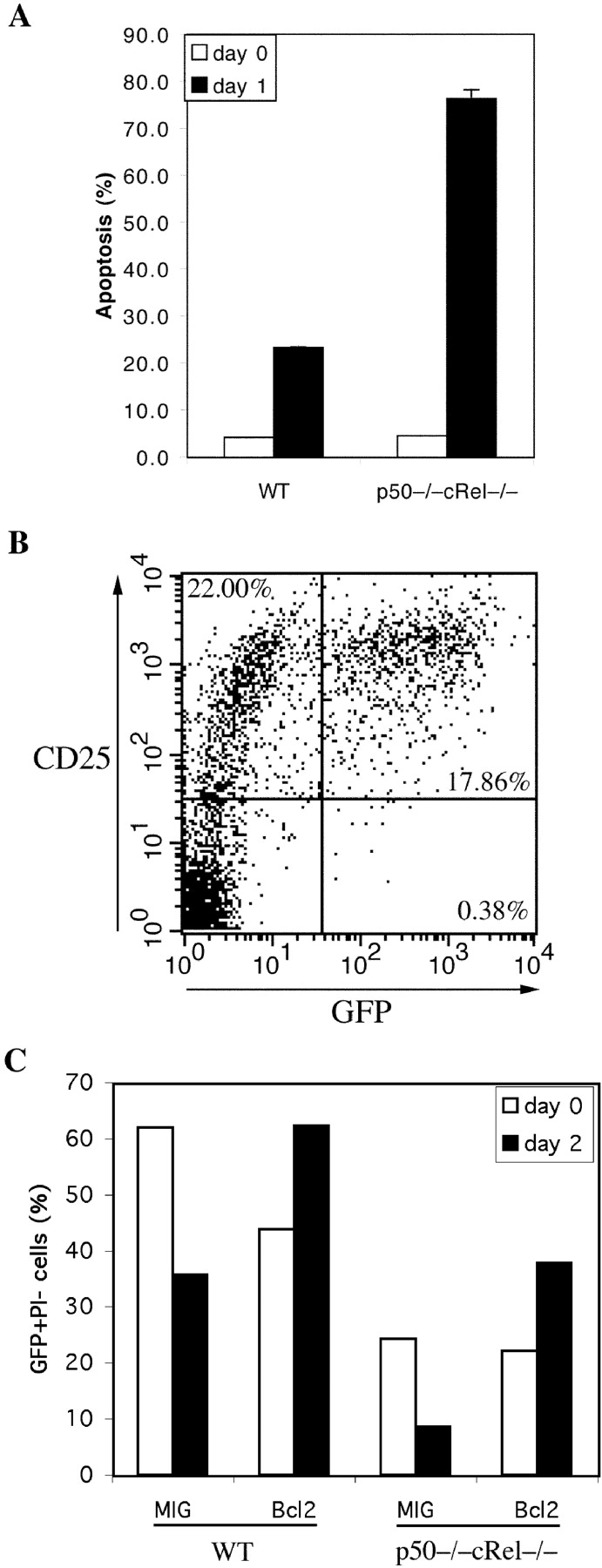
High susceptibility of activated p50−/− cRel−/− T cells to cell death can be rescued by Bcl-2. (A) WT and p50−/− cRel−/− CD4+ T cells were activated by plate-coated αCD3+αCD28 for 3 d after which dead cells were removed on a Ficoll gradient. These cells were either stained with PE-αCD25 and SYTOX immediately (day 0) or cultured in T cell medium without αCD3 or IL-2 for 24 h before PE-αCD25 and SYTOX staining (day 1). Apoptosis rate represent the percentage of CD25+ SYTOX+ cells (apoptotic) in the total CD25+ population. (B) p50−/− cRel−/− CD4+ T cells were infected with MIG retrovirus during a 4-d stimulation in the presence of αCD3+ αCD28. On day 4, viable cells were stained by αCD25 before FACS® analysis. (C) MIG and Bcl2 retroviral-infected WT and p50−/− cRel−/− CD4+ T cells were obtained as described in B, and either stained with PI and analyzed by FACS® immediately (day 0) or cultured in T cell medium without αCD3 or IL-2 for 2 d before PI staining and FACS® (day 2). The viable infected cells were GFP+ PI−. The percentage was calculated based on the percentage of GFP+ PI− cells in the total cell population. The increase in the percentage of Bcl-2–infected cells after 2 d in this experiment is likely due to the decrease in the number of uninfected cells because of cell death, rather than an increase in the absolute number of Bcl-2–infected cells.
As shown in Fig. 2 B, expression of Bcl-2 was moderately reduced whereas expression of Bcl-xL was dramatically impaired in p50−/− cRel−/− T cells after TCR engagement. Importantly, it has been shown that Bcl-2 and Bcl-xL inhibit cell death by biochemically similar mechanisms (49). Therefore, we wanted to determine whether impaired survival of activated p50−/− cRel−/− cells was due to decreased expression of these Bcl-2 family members after TCR engagement. To this end, we first infected p50−/− cRel−/− T cells with a GFP-expressing polycistronic retrovirus (MIG; reference 16). All GFP+ p50−/− cRel−/− cells were also CD25+ because oncoretroviruses can only infect proliferating cells (Fig. 4 B; reference 50). WT and p50−/− cRel−/− T cells were then infected with MIG or MIG-Bcl-2 and incubated for 2 d without stimulation, after which the percentage of viable infected cells (GFP+ PI−) cells was determined. Although, as expected, WT T cells underwent less death than p50−/− cRel−/− T cells, their survival was enhanced by Bcl-2 expression (Fig. 4 C). Of ∼24% p50−/− cRel−/− T cells infected with MIG-GFP, only 8% survived after 2 d in culture (Fig. 4 C). In contrast, survival of Bcl-2 retrovirus-infected p50−/− cRel−/− T cells was dramatically enhanced compared with MIG-infected T cells. Together with the above findings, these results suggest that increased susceptibility of p50−/− cRel−/− cells to apoptosis is due to impaired TCR-induced expression of Bcl-2 family members.
IL-2-induced Survival Pathway Does Not Require p50+ cRel.
T cell proliferation and survival is dependent on TCR+CD28– and cytokine- (e.g., IL-2) driven pathways. During early stages of activation, before significant generation of antiapoptotic cytokines and/or expression of cytokine receptors, TCR-driven pathways predominate. As described above, increased susceptibility of p50−/− cRel−/− cells to apoptosis is likely due to impaired TCR-induced expression of Bcl-2 family members. In addition, impaired IL-2 expression may also contribute to increased susceptibility of p50−/− cRel−/− T cells to apoptosis (discussed later). Although the addition of IL-2 did not prevent p50−/− cRel−/− or WT T cell death during initial stages of TCR-induced activation (Fig. 2 A), this could be due to lack of sufficient IL-2Rα expression. Here we have investigated a possible biochemical requirement for NF-κB in the IL-2–induced survival pathway in T cells activated with αCD3+αCD28 for 3 d. As shown above, expression of IL-2Rα is significantly enhanced in both WT and p50−/− cRel−/− T cells activated with αCD3+αCD28 for 3 d (Fig. 3 C and see below). These activated T cells were further cultured in the presence or absence of IL-2 for 1 or 2 d. As shown in Fig. 5 , survival of both activated WT cells and p50−/− cRel−/− was dramatically enhanced by IL-2. Thus, although IL-2Rα expression is a little lower in p50−/− cRel−/− T cells (Fig. 3 C), it is sufficient for potent enhancement of survival by IL-2. However, this does not preclude that other aspects of IL-2 function are also unaffected in p50−/− cRel−/− T cells. Consistent with the above results, we have found that Bcl-2 family members, previously shown to be induced by IL-2 (16, 51), were also induced by IL-2 in p50−/− cRel−/− T cells (unpublished data). Therefore, these results indicate an essential requirement for NF-κB in the TCR-induced but not IL-2–induced survival pathway.
Figure 5.

IL-2–induced survival pathway does not require p50+cRel. αCD3+αCD28–activated WT and p50−/− cRel−/− CD4+ T cells were cultured in T cell medium alone or in the presence of 20 ng/ml IL-2 for 1 or 2 d. Cell death was determined by DNA content staining and sub-G0 quantification.
Constitutive NF-κB Activation Is Sufficient to Promote Activated T Cell Survival.
Our results indicate that NF-κB activation by TCR engagement is necessary for T cell survival. Next, we determined whether NF-κB is sufficient to promote activated T cell survival in the absence of stimulation. We did this by using a complementation approach involving retroviral transduction of WT T cells with a constitutively active mutant (EM, refer to Materials and Methods) of the NF-κB–activating IκB kinase β (CA-IKKβ; reference 39). Nuclear NF-κB activity was readily detected in WT CD4+ T cells immediately after activation. However, after a 12-h incubation without αCD3, αCD28, nor IL-2, NF-κB complex 1 completely disappeared whereas complex 2 was also reduced (Fig. 6 A). As shown in Fig. 6 B, MIG/GFP-infected T cells also showed almost complete loss of NF-κB activity after 12 h (infection efficiencies in this experiment and others described later varied from 30 to 40%). In contrast, CA-IKKβ–expressing T cells were able to maintain high NF-κB levels after 12 h.
Figure 6.

NF-κB activation is sufficient to promote T cell survival. (A) αCD3+αCD28+IL-2–activated WT CD4+ T cells were either used to make nuclear extract immediately or cultured in T cell medium without stimulation for 12 h before nuclear extract was made. EMSA was performed with an H2 site probe. The two complexes are described in the text. (B) MIG and CA-IKKβ retrovirus-infected WT CD4+ T cells were used immediately (0 h) or cultured in T cell medium without stimulation for 12 h. Nuclear extracts were made and EMSA was performed with the H2 site probe. (C and D) MIG, CA-IKKβ, and Bcl2 retrovirus-infected WT T cells were cultured in T cell medium without stimulation for 0, 1, 2, or 3 d, after which cells were stained with PI and analyzed by FACS®. FACS® data on days 0 and 3 are shown in C. The percentage indicates the proportion of GFP+ PI− cells in total cell population. The percentage survival of infected T cells from days 0 to 3 are shown in D. (E) MIG, CA-IKKβ, and Bcl2 retroviral-infected WT T cells were cultured in T cell medium without αCD3 or IL-2 for 12 h. RNA was extracted before and after culturing. Bcl2, Bcl-XL, and β actin expression was examined by Northern blotting. 1, endogenously expressed Bcl2; 2, ectopically expressed Bcl2. (F) MIG, CA-IKKβ retrovirus-infected OT-II×IL-2+/+ and OT-II×IL-2−/− T cells were cultured in T cell medium without stimulation for 0–3 d. Survival rate of the infected cells was determined by PI staining and FACS® analysis as in C and D.
We then tested the effect of continued presence of NF-κB on T cell survival. As shown above, cessation of TCR engagement leads to the induction of activated T cell death. After 3 d without stimulation, the number of MIG-infected T cells was greatly reduced (Fig. 6, C and D). In striking contrast, IKKβ-infected T cells showed no loss of viability (Fig. 6, C and D). As expected, retrovirus-mediated expression of Bcl-2 also substantially enhanced survival compared with MIG-infected T cells (Fig. 6, C and D). Significantly, only survival of IKKβ-infected but not uninfected T cells was enhanced (Fig. 6 C). These results indicate that the survival-promoting effect of IKKβ is likely mediated by a cell-intrinsic mechanism rather than through enhancement of cytokine production (e.g., IL-2), which would also be expected to affect survival of uninfected cells (also see below). These results suggest that the maintenance of NF-κB activity is sufficient for promoting T cell survival, likely by a cell-intrinsic mechanism.
Next, we determined whether enhanced survival of IKKβ-expressing T cells was due to an increase in the expression of Bcl-2 family members. Both Bcl-2 and Bcl-xL mRNAs were readily detected in T cells immediately after activation (Fig. 6 E). However, a 12-h incubation of MIG-infected cells led to a significant reduction in expression of both Bcl-2 and Bcl-xL. In contrast, IKKβ expression was sufficient to maintain expression of both genes (Fig. 6 E). Thus, NF-κB activity is sufficient for maintaining expression of these key antiapoptotic genes. Interestingly, although Bcl-2 and Bcl-xL mRNAs levels were greatly reduced by 12 h, significant reduction in Bcl-2 and Bcl-xL protein levels were only seen after 2–3 d (unpublished data). Thus, the loss of Bcl-2 and Bcl-xL protein levels better correlate with the kinetics of T cell death (Fig. 6, C and D). We also tested whether activated T cell death induced after antigen deprivation could be inhibited by IKKβ. To this end, OVA-specific OT-II T cells (36) were infected with MIG or IKKβ during a 3-d stimulation with antigen. Significantly, cell death induced after antigen deprivation was also inhibited by IKKβ (Fig. 6 F). Because IL-2 is an NF-κB target gene, we used OT-II T cells to determine whether IL-2 was required for IKKβ-induced enhancement of survival. As shown in Fig. 6 F, survival of OT-II IL-2−/− could also be enhanced by IKKβ. Together, these results lead us to two key conclusions: (a) NF-κB activation is sufficient to promote T cell survival by an IL-2–independent and likely cell-intrinsic mechanism, and (b) down-regulation of NF-κB after termination of TCR engagement may play a crucial role in inducing activated T cell death.
Lack of Involvement of p50+cRel in Akt-induced T Cell Survival.
Similar to other cell types, Akt has been shown to play an important role in regulating the survival of T cells. NF-κB is thought to be one of the key mediators of Akt-induced inhibition of cell death (18, 21). Furthermore, Akt-induced NF-κB activation has been shown to require IKKβ (20). Using p50−/− cRel−/− T cells, we also investigated a possible role of NF-κB in Akt-mediated control of T cell survival. Infection of WT T cells with constitutively active (myr mutant) Akt (15) or CA-IKKβ–encoding retrovirus substantially inhibited cell death (Fig. 7) . On the other hand, IKKβ expression in p50−/− cRel−/− T cells did not inhibit cell death (Fig. 7), further demonstrating the absence of functional NF-κB activity in these cells. In striking contrast, Akt expression significantly inhibited cell death of p50−/− cRel−/− cells (Fig. 7). Therefore, these surprising results suggest a lack of requirement for NF-κB in regulating the Akt-induced survival pathway in T cells (see Discussion). Thus, NF-κB and Akt-induced pathways may independently regulate survival of T cells.
Figure 7.

Lack of involvement of p50+cRel in Akt-induced T cell survival. MIG, myr-AKT, and CA-IKKβ retrovirus-infected WT and p50−/− cRel−/− CD4+ T cells were cultured in T cell medium without αCD3 or IL-2 for 1 or 2 d. Survival rate of the infected cells was determined by PI staining and FACS® analysis as in Fig. 6, C and D.
Impaired Antigen Responsiveness In Vivo and Decreased Proportion of Effector/Memory and Regulatory T Cells in p50−/− cRel−/− Mice.
Very little is known about the function of NF-κB proteins in regulating T cell responses in vivo. Although the results described above indicate an important role for NF-κB in regulating T cell function in vitro, in vivo regulatory mechanisms can often be different. To test the in vivo role of NF-κB in T cell responses, mice were challenged with the superantigen SEB, a widely used reagent to study antigen-induced responses in vivo (52, 53). SEB specifically binds to TCRs that contain the Vβ8 element and drive their expansion (52). SEB-mediated T cell responses require TCR expression on T cells and MHC II expression on APC, notably DCs. Significantly, p50−/− cRel−/− mice have normal DC development and MHC II expression on spleen DCs (unpublished data and reference 27). Both WT and p50−/− cRel−/− mice were injected intravenously with SEB and the percentage of CD4+ Vβ8+ T cells in total CD4+ T cells was determined at days 0 (uninjected), 3, and 6. In addition, CD4+ Vβ6+ T cell numbers were also determined at the same periods as negative controls. SEB does not bind to TCRs containing this Vβ element. As shown in Fig. 8 A, in WT mice, the CD4+ Vβ8+ T cell population increased from ∼24 to ∼32% 3 d after SEB injection and then dropped to ∼22% on day 6. The kinetics of these changes in cell numbers is consistent with previous reports (53). The initial increase in Vβ8+ T cells results from a proliferative response (day 3) whereas the decrease is due to cell death (day 6; reference 53). The CD4+ Vβ6+ T cell population remained unchanged during the same period, demonstrating the specificity of the SEB response for Vβ8+ T cells. In contrast to their WT counterparts, CD4+ Vβ8+ T cell populations in p50−/− cRel−/− mice showed no increase after SEB injection. Instead, over a 6-d period, Vβ8+ T cells dropped from ∼23 to ∼16% (Fig. 8 A). The CD4+ Vβ6+ T cells in these mice were not altered during the same period. These results support our in vitro findings and suggest that impaired SEB responsiveness of p50−/− cRel−/− T cells in vivo is due to defects in proliferation and survival.
Figure 8.
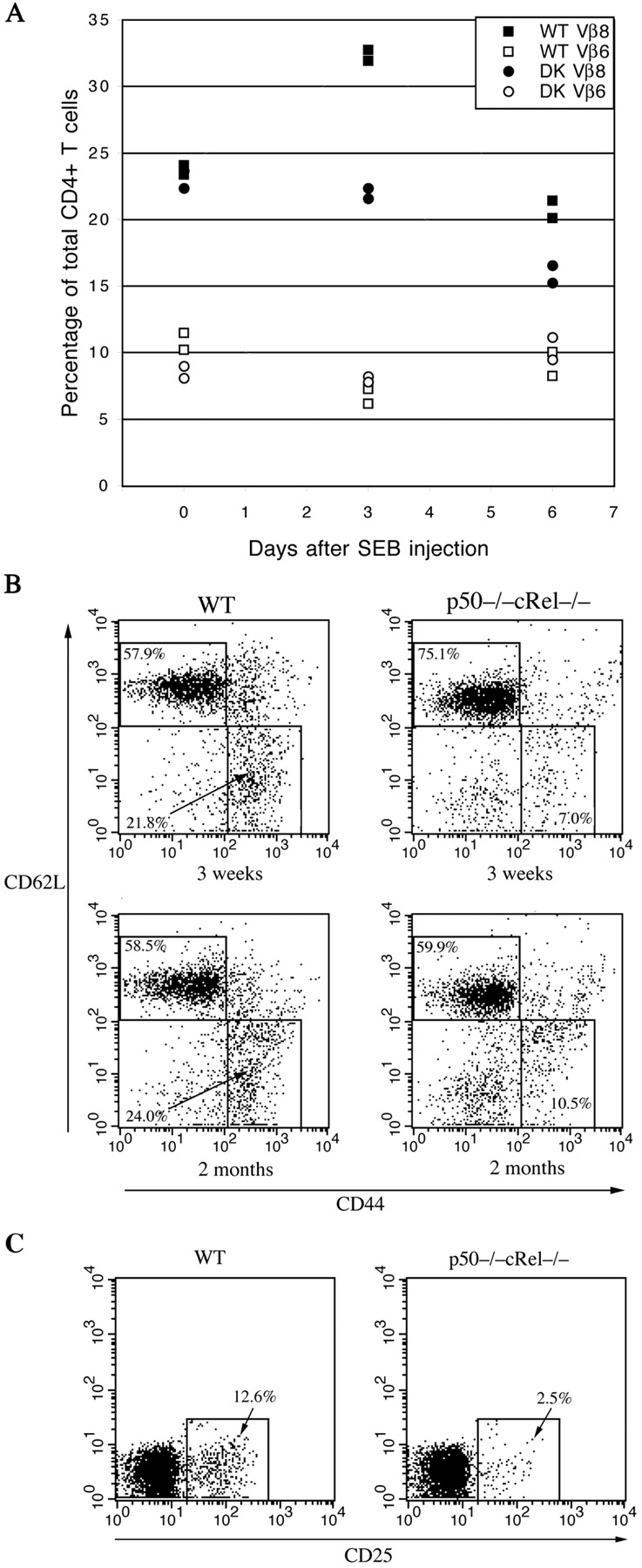
Impaired antigen-induced responses and effector/memory and regulatory T cell generation in WT and p50−/− cRel−/− mice. (A) WT and p50−/− cRel−/− mice were either uninjected (day 0) or injected with SEB. Vβ8+ and Vβ6+ T cell populations were determined 3 and 6 d after injection. Two mice of each genotype were used per condition. (B) Naive and memory T cell populations were determined using 3- and 8-wk-old WT and p50−/− cRel−/− mice. Splenocytes from these mice were stained with CD4, CD44, and CD62L antibodies. FACS® analysis was performed on gated CD4+ T cells. Naive T cells were CD44low CD62L+ and memory T cells were CD44high CD62L−. (C) Splenocytes from 2-mo-old WT and p50−/− cRel−/− mice were stained with CD4 and CD25. FACS® analysis was performed on gated CD4+ T cells.
It is generally believed that memory T cells are derived from a small subset of effector T cells activated during a primary immune response (54, 55). The proportion of memory T cells in unchallenged mice increases with age, likely through interactions with environmental antigens. Because p50−/− cRel−/− T cells showed impaired proliferation and survival after activation in vivo, we investigated whether memory T cell development was affected in these mice. Spleen cells from 3- and 8-wk-old WT and p50−/− cRel−/− mice were stained with CD4, CD44, and CD62L antibodies and FACS® analysis was performed on gated CD4+ cells. As shown in Fig. 8 B, naive T cells were identified as the CD44low CD62L+ population and memory T cells as the CD44high CD62L− population (55). Strikingly, 3-wk-old WT mice had approximately threefold more effector/memory T cells than p50−/− cRel−/− mice (21.8 vs. 7%) whereas in 8-wk-old mice the difference was ∼2.5-fold (24 vs. 10.5%). Thus, the proportion of memory T cells is significantly reduced in p50−/− cRel−/− mice. We also tested regulatory T cell (CD4+ CD25+) populations in WT and p50−/− cRel−/− mice. In 2-mo-old p50−/− cRel−/− mice, CD4+ CD25+ T cells were reduced fivefold compared with WT mice (Fig. 8 C). Significantly, the differentiation of naive T cells into effector, memory, and regulatory T cells requires initial antigen-induced activation. Therefore, the decreased numbers of all three populations in p50−/− cRel−/− mice are consistent with our results showing impaired in vitro and in vivo proliferation and survival of p50−/− cRel−/− T cells. Together, these results show for the first time an important role for NF-κB in regulating mature T cell function in vivo.
Discussion
Multiple signaling pathways are induced during mature T cell activation that together regulate proliferation, survival, and cytokine production. However, the precise role of specific signaling molecules and transcription factors in regulating these pathways is not well understood. To this end, we have studied the role of the NF-κB transcription factor in mature T cells by using doubly deficient p50−/− cRel−/− T cells, which exhibit virtually no TCR-inducible κB site binding activity. Our results demonstrate an essential role for NF-κB in regulating cell cycle entry and survival of activated T cells in vitro and in vivo. These results indicate that of the many transcription factors considered important in T cells, NF-κB might be among the most crucial. The findings reported here help us understand many key aspects of regulatory mechanisms involved in T cell function.
Regulation of T Cell Survival by NF-κB.
A primary goal of this study was to elucidate NF-κB–dependent mechanisms involved in regulating T cell survival. We have found, using both in vitro and in vivo approaches, that NF-κB activation after TCR engagement plays a crucial role in regulating T cell survival. Significantly, our results indicate that NF-κB activation is not only necessary but also sufficient for T cell survival. We have also found that NF-κB plays an essential role in TCR-induced regulation of Bcl-2 and Bcl-xL gene expression. Significantly, the high susceptibility of activated p50−/− cRel−/− T cells to apoptosis was inhibited by retroviral expression of Bcl-2, suggesting that NF-κB prevents cell death by regulating Bcl-2 family member expression. These findings establish a critically important function of NF-κB in TCR-induced regulation of survival. Our results also demonstrate that the combined absence of p50 and cRel subunits is required for significant impairment of mature T cell function. Although cRel−/− T cells were thought to have greatly impaired proliferation (35), we have previously found relatively intact responsiveness in these and p50−/− T cells (28). Thus, our findings with p50−/− cRel−/− T cells indicate redundant functions for these two proteins in mature T cells.
p50−/− cRel−/− T cells show impaired IL-2 expression after activation. Thus, the antiapoptotic function of NF-κB may involve both TCR induction of expression of Bcl-2 family members and IL-2. We believe both these NF-κB–dependent mechanisms are important, but at different stages. During the initial TCR-dependent phase of activation (1–2 d), i.e., before there is significant generation of antiapoptotic cytokines and/or expression of cytokine receptors, NF-κB directly induces expression of Bcl-2 family members and enhances activated T cell survival (Fig. 2). The ability of NF-κB to enhance survival in a cytokine- (e.g., IL-2) independent manner is further evidenced by IKKβ transduction studies with WT and IL-2−/− T cells (Fig. 6). At this early stage, T cell survival might be regulated by cooperative interactions between NF-κB and other TCR+CD28–induced pathways. During later stages of T cell activation (3–4 d), cytokine-driven responses likely predominate. At this stage, NF-κB may also enhance survival through generation of antiapoptotic cytokines, such as IL-2. Interestingly, we have shown a lack of involvement of NF-κB in the IL-2–induced survival pathway. Thus, although NF-κB is important for IL-2 expression, it is not a mediator of IL-2–induced survival, which also depends on Bcl-2 family members. Thus, T cell survival can be controlled independently or through cooperative regulation of Bcl-2 family expression by TCR+CD28 and cytokine-induced pathways. It was recently shown that T cell activation and generation of memory/effector T cells can still occur in γc-deficient mice (56), which are nonresponsive to many cytokines important for T cell function including IL-2, IL-4, IL-7, and IL-15. It is interesting to speculate whether immune responses in these mice are mediated largely by TCR-induced NF-κB activation.
A key question is why TCR engagement induces an antiapoptotic pathway in the first place. One reason might be to block simultaneously induced apoptotic pathways for T cell expansion to occur. It has recently been shown that TCR signals lead to activation of Bim, a proapoptotic member of the Bcl-2 family (7, 57). TCR signals also lead to generation of reactive oxygen species in activated T cells, which contribute to induction of apoptosis (58). This would be consistent with the function of Bcl-2 proteins, which are known to inhibit apoptosis induced by reactive oxygen species (59, 60). However, an additional function of antiapoptotic pathways may also allow efficient removal of activated T cells once TCR stimulation ends. Because continued antiapoptotic pathway activation will depend on TCR signaling, this would allow rapid elimination of T cells when TCR engagement ends. Such cell death may occur as a result of down-regulation of one or multiple antiapoptotic signaling pathways. We have shown here that nuclear NF-κB levels rapidly decline in WT T cells after cessation of TCR stimulation, an event that was concomitant with induction of cell death. However, T cells complemented with a retrovirus-encoding CA-IKKβ maintained nuclear NF-κB levels and survived in the absence of stimulation. Thus, NF-κB activation is not only sufficient to promote T cell survival but down-regulation of NF-κB can also provide a mechanism for induction of cell death. Although IKKβ expression apparently enhances survival in a cell-intrinsic manner by inducing Bcl-2 family member expression, it is likely that down-regulation of NF-κB after termination of TCR engagement leads to apoptosis both through decreased expression of Bcl-2 proteins and cytokines such as IL-2.
The PKB/Akt kinase is a key mediator of T cell survival pathways and has previously been shown to function through NF-κB (19–21, 61). However, our results show that Akt but not IKKβ is fully capable of enhancing p50−/− cRel−/− T cell survival. In addition, we have found that the PI3K inhibitor LY294002, which inhibits Akt activation, does not inhibit αCD3+αCD28–induced NF-κB activation in WT T cells (unpublished data). Furthermore, IL-2 induces Akt but not NF-κB activation in T cells (unpublished data). Together, these different findings strongly suggest that NF-κB may not be a mediator of the Akt-induced survival pathway. Therefore, these results indicate the existence of two parallel and potentially independent survival pathways in T cells. They may also help explain how IL-2, which activates Akt, enhances the survival of p50−/− cRel−/− T cells. In addition, Akt is also thought to be a key mediator of CD28-induced responses. However, CD28-induced responses are generally evident only in the presence of TCR engagement. Because TCR-induced NF-κB itself is so crucial for survival, it is difficult to determine the specific role of NF-κB in the CD28 survival pathway using p50−/− cRel−/− T cells (the role NF-κB in CD28-induced proliferation is discussed below). The potentially NF-κB–independent nature of Akt prosurvival function suggests the existence of alternate Akt-induced mechanisms for regulating Bcl-2 family member expression. Although the identity of such transcription factors is presently unknown, they may include members of signal transducer and activator of transcription, Ets, or AP-1 families. Interestingly, we have also found that activated p50−/− cRel−/− T cells, similar to T cells deficient in p50−/−, RelA−/−, or cRel−/− (28), are no more susceptible to Fas-induced killing than WT T cells (unpublished data). These results indicate that a distinct survival pathway might be required for regulating Fas killing and further underscore a specific function for NF-κB in regulating TCR-induced survival.
Immunological adjuvants were shown to induce expression of the IκB family member Bcl-3 in T cells, resulting in enhanced T cell survival (62). These results, together with our findings, suggest that Bcl-3 likely enhances transcriptional functions of NF-κB in T cells. One interesting possibility might be that Bcl-3 promotes T cell survival by maintaining NF-κB nuclear activity in the absence of TCR engagement. Gene targeting studies of PKCθ and Bcl-10 have revealed an essential role for these proteins in NF-κB activation and in regulating T cell proliferative responses (32, 33). Based on our results with p50−/− cRel−/− T cells, it is possible that impaired proliferation of T cells deficient in PKCθ or Bcl-10 might be due to defects in both cell division and cell survival.
Control of T Cell Proliferation and Effector/Memory and Regulatory T Cell Generation by NF-κB.
We have found that in addition to regulating survival, NF-κB proteins also regulate CD4+ T cell proliferation. TCR-induced cell cycle entry of p50−/− cRel−/− T cells is both significantly reduced and delayed compared with WT T cells. Previous studies have implicated NF-κB as a key component of the CD28 costimulatory pathway (63, 64). However, our results indicate that CD28 could significantly increase proliferation of p50−/− cRel−/− T cells, suggesting that NF-κB control of proliferation might be more specific for the TCR pathway. One possibility is that CD28-induced enhancement of p50−/− cRel−/− T cell proliferation is through Akt-induced pathways. In addition to NF-κB, CD28 also induces activation of the AP-1 family of transcription factors. Thus, AP-1 factors may play a more crucial role in the CD28 pathway than NF-κB and CD28-induced AP-1 activity may synergize with TCR-induced NF-κB and NFAT pathways in regulating T cell proliferation, survival, and other functions. Interestingly, PKCθ−/− T cells are deficient in both NF-κB and AP-1 activation (33). Comparative analysis of T cell function in PKCθ−/− and p50−/− cRel−/− mice may therefore help us better understand the specific role played by AP-1 in regulating T cell proliferation.
IL-2 expression was significantly reduced in p50−/− cRel−/− cells, but the addition of exogenous IL-2 failed to rescue proliferation defects. p50−/− cRel−/− cells, however, express IL-2R and are protected from cell death by IL-2. Thus, impaired proliferative responses cannot simply be due to reduced IL-2 or IL-2R expression, but instead may result from impaired TCR induction of genes involved in cell cycle control. The identity of such NF-κB–regulated genes has yet to be determined. One of the important findings reported here is the decreased number of effector/memory and regulatory T cells in p50−/− cRel−/− mice. Because differentiation of naive T cells into effector, memory, and regulatory T cells requires antigen-induced activation, these results provide further evidence for the crucial role of NF-κB in regulating activation-induced proliferation and survival of T cells. However, our results also indicate that a certain proportion of effector/memory T cells are generated in p50−/− cRel−/− mice, which appear to increase with age. One possibility is that decreased effector/memory T cell in p50−/− cRel−/− mice are a consequence of impaired proliferation and/or survival during a primary response. However, the small number of effector/memory T cells that do form in p50−/− cRel−/− mice may undergo expansion by cytokine-driven homeostatic mechanisms, which might not require NF-κB.
NF-κB Function in T Cell Development.
Consistent with our findings, previous studies of IκB-Tg mice have also shown impaired proliferative responses and IL-2 expression, but unlike p50−/− cRel−/− mice, IκB-Tg mice also showed impaired thymocyte development and reduction in numbers of peripheral T cells (29–31). The effect was especially pronounced for the CD8 lineage (29–31). As recently shown, impaired thymocyte development likely reflects a requirement for NF-κB in regulating survival of developing thymocytes (65). Unlike mature T cells (Fig. 1 E), p50−/− cRel−/− thymocytes still exhibit low levels of RelA (unpublished data). Thus, RelA may play an important role in the development of thymocytes in the absence of p50 and cRel. Notably, a proapoptotic role for NF-κB in thymocytes has also been proposed (30, 66). Thus, although the findings presented here demonstrate a crucial role for NF-κB in mature T cells, the precise role played by NF-κB in thymocytes remains to be determined.
Acknowledgments
We thank Drs. Paul Rothman (Columbia University) and Timothy Finco (Agnes Scott College) for helpful comments on this manuscript.
This work was supported in part by National Institutes of Health grant CA74892-05A2 to A.A. Beg and a Career Development Award from the Juvenile Diabetes Research Foundation to L.V. Parijs.
Footnotes
Abbreviations used in this paper: CFSE, carboxyfluorescein diacetate succinimidyl ester; DC, dendritic cell; EMSA, electrophoretic mobility shift assay; GFP, green fluorescent protein; NF, nuclear factor; PI, propidium iodide; SEB, staphylococcal enterotoxin B; Tg, transgenic.
References
- 1.Kane, L.P., J. Lin, and A. Weiss. 2000. Signal transduction by the TCR for antigen. Curr. Opin. Immunol. 12:242–249. [DOI] [PubMed] [Google Scholar]
- 2.Frauwirth, K.A., and C.B. Thompson. 2002. Activation and inhibition of lymphocytes by costimulation. J. Clin. Invest. 109:295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins, M.K., A. Khoruts, E. Ingulli, D.L. Mueller, S.J. McSorley, R.L. Reinhardt, A. Itano, and K.A. Pape. 2001. In vivo activation of antigen-specific CD4 T cells. Annu. Rev. Immunol. 19:23–45. [DOI] [PubMed] [Google Scholar]
- 4.Van Parijs, L., and A.K. Abbas. 1998. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 280:243–248. [DOI] [PubMed] [Google Scholar]
- 5.Lenardo, M., K.M. Chan, F. Hornung, H. McFarland, R. Siegel, J. Wang, and L. Zheng. 1999. Mature T lymphocyte apoptosis-immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 17:221–253. [DOI] [PubMed] [Google Scholar]
- 6.Siegel, R.M., F.K. Chan, H.J. Chun, and M.J. Lenardo. 2000. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat. Immunol. 1:469–474. [DOI] [PubMed] [Google Scholar]
- 7.Bouillet, P., J.F. Purton, D.I. Godfrey, L.C. Zhang, L. Coultas, H. Puthalakath, M. Pellegrini, S. Cory, J.M. Adams, and A. Strasser. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 415:922–926. [DOI] [PubMed] [Google Scholar]
- 8.Samelson, L.E. 2002. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20:371–394. [DOI] [PubMed] [Google Scholar]
- 9.Kuo, C.T., and J.M. Leiden. 1999. Transcriptional regulation of T lymphocyte development and function. Annu. Rev. Immunol. 17:149–187. [DOI] [PubMed] [Google Scholar]
- 10.Rao, A., C. Luo, and P.G. Hogan. 1997. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15:707–747. [DOI] [PubMed] [Google Scholar]
- 11.Peng, S.L., A.J. Gerth, A.M. Ranger, and L.H. Glimcher. 2001. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 14:13–20. [DOI] [PubMed] [Google Scholar]
- 12.Rengarajan, J., B. Tang, and L.H. Glimcher. 2002. NFATc2 and NFATc3 regulate T(H)2 differentiation and modulate TCR-responsiveness of naive T(H)cells. Nat. Immunol. 3:48–54. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida, H., H. Nishina, H. Takimoto, L.E. Marengere, A.C. Wakeham, D. Bouchard, Y.Y. Kong, T. Ohteki, A. Shahinian, M. Bachmann, et al. 1998. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity. 8:115–124. [DOI] [PubMed] [Google Scholar]
- 14.Chen, J., V. Stewart, G. Spyrou, F. Hilberg, E.F. Wagner, and F.W. Alt. 1994. Generation of normal T and B lymphocytes by c-jun deficient embryonic stem cells. Immunity. 1:65–72. [DOI] [PubMed] [Google Scholar]
- 15.Kelly, E., A. Won, Y. Refaeli, and L. Van Parijs. 2002. IL-2 and related cytokines can promote T cell survival by activating AKT. J. Immunol. 168:597–603. [DOI] [PubMed] [Google Scholar]
- 16.Van Parijs, L., Y. Refaeli, J.D. Lord, B.H. Nelson, A.K. Abbas, and D. Baltimore. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 11:281–288. [DOI] [PubMed] [Google Scholar]
- 17.Burr, J.S., N.D. Savage, G.E. Messah, S.L. Kimzey, A.S. Shaw, R.H. Arch, and J.M. Green. 2001. Cutting edge: distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-XL. J. Immunol. 166:5331–5335. [DOI] [PubMed] [Google Scholar]
- 18.Jones, R.G., M. Parsons, M. Bonnard, V.S. Chan, W.C. Yeh, J.R. Woodgett, and P.S. Ohashi. 2000. Protein kinase B regulates T lymphocyte survival, nuclear factor κB activation, and Bcl-X(L) levels in vivo. J. Exp. Med. 191:1721–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kane, L.P., V.S. Shapiro, D. Stokoe, and A. Weiss. 1999. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 9:601–604. [DOI] [PubMed] [Google Scholar]
- 20.Madrid, L.V., M.W. Mayo, J.Y. Reuther, and A.S. Baldwin, Jr. 2001. Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 276:18934–18940. [DOI] [PubMed] [Google Scholar]
- 21.Romashkova, J.A., and S.S. Makarov. 1999. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 401:86–90. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh, S., M.J. May, and E.B. Kopp. 1998. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260. [DOI] [PubMed] [Google Scholar]
- 23.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18:621–663. [DOI] [PubMed] [Google Scholar]
- 24.Grumont, R.J., I.J. Rourke, and S. Gerondakis. 1999. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 13:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pohl, T., R. Gugasyan, R.J. Grumont, A. Strasser, D. Metcalf, D. Tarlinton, W. Sha, D. Baltimore, and S. Gerondakis. 2002. The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc. Natl. Acad. Sci. USA. 99:4514–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owyang, A.M., J.R. Tumang, B.R. Schram, C.Y. Hsia, T.W. Behrens, T.L. Rothstein, and H.C. Liou. 2001. c-Rel is required for the protection of B cells from antigen receptor-mediated, but not Fas-mediated, apoptosis. J. Immunol. 167:4948–4956. [DOI] [PubMed] [Google Scholar]
- 27.Ouaaz, F., J. Arron, Y. Zheng, Y. Choi, and A.A. Beg. 2002. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 16:257–270. [DOI] [PubMed] [Google Scholar]
- 28.Zheng, Y., F. Ouaaz, P. Bruzzo, V. Singh, S. Gerondakis, and A.A. Beg. 2001. Nf-kappab rela (p65) is essential for tnf-alpha-induced fas expression but dispensable for both tcr-induced expression and activation-induced cell death. J. Immunol. 166:4949–4957. [DOI] [PubMed] [Google Scholar]
- 29.Boothby, M.R., A.L. Mora, D.C. Scherer, J.A. Brockman, and D.W. Ballard. 1997. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-κB. J. Exp. Med. 185:1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hettmann, T., J. DiDonato, M. Karin, and J.M. Leiden. 1999. An essential role for nuclear factor κB in promoting double positive thymocyte apoptosis. J. Exp. Med. 189:145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esslinger, C.W., A. Wilson, B. Sordat, F. Beermann, and C.V. Jongeneel. 1997. Abnormal T lymphocyte development induced by targeted overexpression of IkappaB alpha. J. Immunol. 158:5075–5078. [PubMed] [Google Scholar]
- 32.Ruland, J., G.S. Duncan, A. Elia, I. del Barco Barrantes, L. Nguyen, S. Plyte, D.G. Millar, D. Bouchard, A. Wakeham, P.S. Ohashi, et al. 2001. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell. 104:33–42. [DOI] [PubMed] [Google Scholar]
- 33.Sun, Z., C.W. Arendt, W. Ellmeier, E.M. Schaeffer, M.J. Sunshine, L. Gandhi, J. Annes, D. Petrzilka, A. Kupfer, P.L. Schwartzberg, et al. 2000. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 404:402–407. [DOI] [PubMed] [Google Scholar]
- 34.Sha, W.C., H.C. Liou, E.I. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 80:321–330. [DOI] [PubMed] [Google Scholar]
- 35.Kontgen, F., R.J. Grumont, A. Strasser, D. Metcalf, R. Li, D. Tarlinton, and S. Gerondakis. 1995. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 9:1965–1977. [DOI] [PubMed] [Google Scholar]
- 36.Barnden, M.J., J. Allison, W.R. Heath, and F.R. Carbone. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40. [DOI] [PubMed] [Google Scholar]
- 37.Schorle, H., T. Holtschke, T. Hunig, A. Schimpl, and I. Horak. 1991. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 352:621–624. [DOI] [PubMed] [Google Scholar]
- 38.Ouaaz, F., M. Li, and A.A. Beg. 1999. A critical role for the RelA subunit of nuclear factor κB in regulation of multiple immune-response genes and in Fas-induced cell death. J. Exp. Med. 189:999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delhase, M., M. Hayakawa, Y. Chen, and M. Karin. 1999. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 284:309–313. [DOI] [PubMed] [Google Scholar]
- 40.Pear, W.S., G.P. Nolan, M.L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 90:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beg, A.A., W.C. Sha, R.T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- 42.Doi, T.S., T. Takahashi, O. Taguchi, T. Azuma, and Y. Obata. 1997. NF-κ B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J. Exp. Med. 185:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horwitz, B.H., M.L. Scott, S.R. Cherry, R.T. Bronson, and D. Baltimore. 1997. Failure of lymphopoiesis after adoptive transfer of NF-kappaB-deficient fetal liver cells. Immunity. 6:765–772. [DOI] [PubMed] [Google Scholar]
- 44.Senftleben, U., Z.W. Li, V. Baud, and M. Karin. 2001. IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity. 14:217–230. [DOI] [PubMed] [Google Scholar]
- 45.Liou, H.-C., W.C. Sha, M.L. Scott, and D. Baltimore. 1994. Sequential induction of NF-κB/Rel family proteins during B-cell terminal differentiation. Mol. Cell. Biol. 14:5349–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nolan, G.P., S. Ghosh, H.-C. Liou, P. Tempst, and D. Baltimore. 1991. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell. 64:961–969. [DOI] [PubMed] [Google Scholar]
- 47.Khoshnan, A., C. Tindell, I. Laux, D. Bae, B. Bennett, and A. Nel. 2000. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J. Immunol. 165:1743–1754. [DOI] [PubMed] [Google Scholar]
- 48.Chen, C., L.C. Edelstein, and C. Gelinas. 2000. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell. Biol. 20:2687–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chao, D.T., G.P. Linette, L.H. Boise, L.S. White, C.B. Thompson, and S.J. Korsmeyer. 1995. Bcl-XL and Bcl-2 repress a common pathway of cell death. J. Exp. Med. 182:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lois, C., Y. Refaeli, X.F. Qin, and L. Van Parijs. 2001. Retroviruses as tools to study the immune system. Curr. Opin. Immunol. 13:496–504. [DOI] [PubMed] [Google Scholar]
- 51.Boise, L.H., A.J. Minn, P.J. Noel, C.H. June, M.A. Accavitti, T. Lindsten, and C.B. Thompson. 1995. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 3:87–98. [DOI] [PubMed] [Google Scholar]
- 52.Marrack, P., and J. Kappler. 1990. The staphylococcal enterotoxins and their relatives. Science. 248:705–711. [DOI] [PubMed] [Google Scholar]
- 53.Kawabe, Y., and A. Ochi. 1991. Programmed cell death and extrathymic reduction of Vbeta8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature. 349:245–248. [DOI] [PubMed] [Google Scholar]
- 54.Sprent, J., and D.F. Tough. 2001. T cell death and memory. Science. 293:245–248. [DOI] [PubMed] [Google Scholar]
- 55.Sprent, J., and C.D. Surh. 2002. T cell memory. Annu. Rev. Immunol. 20:551–579. [DOI] [PubMed] [Google Scholar]
- 56.Lantz, O., I. Grandjean, P. Matzinger, and J.P. Di Santo. 2000. Gamma chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 1:54–58. [DOI] [PubMed] [Google Scholar]
- 57.Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- 58.Hildeman, D.A., T. Mitchell, T.K. Teague, P. Henson, B.J. Day, J. Kappler, and P.C. Marrack. 1999. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 10:735–744. [DOI] [PubMed] [Google Scholar]
- 59.Hockenbery, D.M., Z.N. Oltvai, X.M. Yin, C.L. Milliman, and S.J. Korsmeyer. 1993. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 75:241–251. [DOI] [PubMed] [Google Scholar]
- 60.Gottlieb, E., M.G. Vander Heiden, and C.B. Thompson. 2000. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 20:5680–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kane, L.P., P.G. Andres, K.C. Howland, A.K. Abbas, and A. Weiss. 2001. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat. Immunol. 2:37–44. [DOI] [PubMed] [Google Scholar]
- 62.Mitchell, T.C., D. Hildeman, R.M. Kedl, T.K. Teague, B.C. Schaefer, J. White, Y. Zhu, J. Kappler, and P. Marrack. 2001. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat. Immunol. 2:397–402. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh, P., T.H. Tan, N.R. Rice, A. Sica, and H.A. Young. 1993. The interleukin 2 CD28-responsive complex contains at least three members of the NF kappa B family: c-Rel, p50, and p65. Proc. Natl. Acad. Sci. USA. 90:1696–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maggirwar, S.B., E.W. Harhaj, and S.C. Sun. 1997. Regulation of the interleukin-2 CD28-responsive element by NF-ATp and various NF-kappaB/Rel transcription factors. Mol. Cell. Biol. 17:2605–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voll, R.E., E. Jimi, R.J. Phillips, D.F. Barber, M. Rincon, A.C. Hayday, R.A. Flavell, and S. Ghosh. 2000. NF-kappa B activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity. 13:677–689. [DOI] [PubMed] [Google Scholar]
- 66.Kim, D., M. Xu, L. Nie, X.C. Peng, E. Jimi, R.E. Voll, T. Nguyen, S. Ghosh, and X.H. Sun. 2002. Helix-loop-helix proteins regulate pre-TCR and TCR signaling through modulation of Rel/NF-kappaB activities. Immunity. 16:9–21. [DOI] [PubMed] [Google Scholar]