

Abstract
Dengue virus is a single-stranded, enveloped RNA virus that productively infects human dendritic cells (DCs) primarily at the immature stage of their differentiation. We now find that all four serotypes of dengue use DC-SIGN (CD209), a C-type lectin, to infect dendritic cells. THP-1 cells become susceptible to dengue infection after transfection of DC-specific ICAM-3 grabbing nonintegrin (DC-SIGN), or its homologue L-SIGN, whereas the infection of dendritic cells is blocked by anti–DC-SIGN antibodies and not by antibodies to other molecules on these cells. Viruses produced by dendritic cells are infectious for DC-SIGN– and L-SIGN–bearing THP-1 cells and other permissive cell lines. Therefore, DC-SIGN may be considered as a new target for designing therapies that block dengue infection.
Keywords: receptor, flavivirus, lectin, antigen-presenting cells, virus receptor
Introduction
Dengue virus (DV)* is the most common human arbovirus infection worldwide. It is an emerging and volatile public health concern. DV is composed of four antigenically distinct serotypes: DV 1, 2, 3, and 4 (1). All serotypes cause human disease; viremia is detected early, in essentially all DV cases at the onset of symptoms (2). Although most infections are generally mild, a complicated DV infection can result in dengue hemorrhagic fever and dengue shock syndrome. These life-threatening complications usually occur after a second DV infection with a heterologous strain (3). Epidemiologic evidence indicates that the immune-mediated enhancement of infection in the presence of a waning heterologous antibody is the underlying mechanism (4, 5). This process is referred to as antibody-dependent enhancement and was first reported over 30 yr ago (6, 7). The phenomenon of antibody-dependent enhancement notwithstanding, the fact remains that no long-term cross protection is conferred by any dengue serotype (8). This makes the prospects of vaccine design demanding and creates an impetus for new types of therapy.
The design of strategies to counter DV infection requires information on cellular sites and mechanisms of infection. However, limited data are available to establish the major sites of DV replication in vivo. In natural infection, DV is deposited by the mosquito vector into the skin during a blood meal. Recent studies (9–12) indicate that immature dendritic cells (DCs), which are normal residents of the skin, support infection with DV, and that this infection is not altered by a DV-enhancing immune serum (10). Cellular receptors for DV also are not well-defined. Heparan sulfates (13), LPS/CD14-associated binding proteins (14), and other glycoproteins (15, 16) have been proposed as cellular receptors for DV, but these would not explain our prior findings (9) that DCs and not monocytes or macrophages are preferentially infected with DV.
Certain subsets of DCs, especially those susceptible to infection with DV in culture (9–12), express DC-specific intracellular adhesion molecule (ICAM) 3–grabbing nonintegrin (DC-SIGN; reference 17). This C-type lectin allows direct infection of DCs with Ebola virus (18, 19), human cytomegalovirus (20), Leishmania pifanoi amastigotes (21), and Mycobacterium tuberculosis (22–24). DC-SIGN also accounts, in large part, for the capacity of DCs to capture and retain HIV-type 1 (HIV-1) for the infection of T cells (17, 25–28). Using primary human DCs naturally expressing DC-SIGN, as well as cell lines transfected with DC-SIGN and its homologue liver/lymph node–specific ICAM-3–grabbing nonintegrin (L-SIGN), we show extensive infection with DV as a direct result of expression of these lectins. Because DC-SIGN–mediated DV entry allows for productive infection, releasing infectious virions capable of transmitting DV infection to susceptible cells, these results make DC-SIGN a logical new candidate for interrupting DV infection in humans.
Materials and Methods
Viral Stocks and Infection.
All viral stocks and cell lines were mycoplasma-free. The dengue-2 strain, S16803, was grown in an African green monkey Vero cell line (American Type Culture Collection), and cell-free supernatants with a titer of 106–107 PFU/ml were used as virus stocks. In some experiments, DV 1, 3, and 4 from two sources were also used. The isolates were either nonattenuated (not passaged in primary dog kidney cells) and grown in the lab in C6/36 mosquito cells and Vero cells, or the viruses were low passage clinical isolates from Bandung, Indonesia and grown in C6/36 mosquito cells after isolation. For DV infection, the cells were exposed to DV for 2 h with a multiplicity of infection (MOI) of 0.02–1. The exposed cells were washed with a complete medium (cRPMI consisting of RPMI 1640 [Quality Biologicals] supplemented with 10% heat-inactivated FCS [PAA Laboratories, Inc.], 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin [Quality Biologicals]) at least twice to remove excess virus. For antibody-blocking studies, we tested azide-free anti–human-DC-SIGN1 mAb (clone 120507), anti–DC-SIGN2 (clone 120612), anti–L-SIGN (clone 120604; R&D Systems), anti-CD11a (lymphocyte function–associated antigen 1, LFA-1), anti-CD58 (LFA-3), anti-CD74 (invariant chain), and matched isotype controls (Becton Dickinson). The test cells were pretreated with 2–20 µg/ml of the indicated mAb for 60 min at 37°C before exposure to DV.
Monocyte-derived DCs.
PBMCs were cultured as described previously (29) with some modifications. In brief, leukapheresis blood from healthy donors was layered over Ficoll-Hypaque and centrifuged to isolate the mononuclear cells, which were adhered to Petri dishes for 60 min at 37°C. After six to eight washes with complete media, the adherent cells were cultured in 10 ml cRPMI with 800 U/ml rhuGM–CSF (Immunex) and 1,000 U/ml rhuIL-4 (R&D Systems). Cytokines were added every other day. To produce mature DCs, 20% (vol/vol) monocyte-conditioned media (MCM) was added to the cells for an additional 2 d. MCM was prepared as described previously (29). The appropriate phenotype of immature and mature DCs was confirmed by cytofluorometry before each experiment. Specifically, immature and mature DCs lacked CD3, CD14, CD20, and CD56, but expressed high levels of MHC class I, class II, and CD1a; only mature cells expressed high levels of CD25, CD83, and CD86 as described previously (30).
THP-1 Human Monocytic Cell Lines.
We were provided with DC-SIGN–transfected THP-1 (THP DC-SIGN), THP DC-SIGN Δ35 (fully truncated cytoplasmic tail), and THP-1 cells by Dr. D. Littman (New York University School of Medicine, New York, NY; reference 27) and L-SIGN/DC-SIGNR expressing THP (THP L-SIGN) by Dr. V. Kewal Ramani (National Cancer Institute, Frederick, MD; reference 25). All THP-1 cells were grown in complete media.
Monitoring Infection with DV.
The DV or mock-infected cells were cytospun onto slides, air-dried, and fixed with 4% paraformaldehyde. Specific anti–DV mAbs, 2H2, or 3H5 (anti–DV envelope complex) or anti–DC-SIGN (120507) were applied after permeabilization with 1% saponin. After several washes, the appropriate secondary antibodies were added; i.e., for immunofluorescence, directly conjugated goat anti–mouse Alexa Fluor 546 or 488 (Molecular Probes) or for immunocytochemistry, biotinylated horse anti–mouse followed by Vectastain ABC alkaline phosphatase or peroxidase kits (Vector Labs). Nuclei were labeled with DAPI (4′, 6′-diamidino-2-phenylindole · 2HCl; Sigma-Aldrich) for immunofluorescence. Matched isotype control antibodies were used in both mock-infected and DV–exposed cells to assess background staining. The slides were mounted and analyzed using either standard light microscopy or a deconvolution microscope (AX-70 laser scanning microscope; Olympus). To quantify the infection virus, a Vero cell plaque assay was performed as described previously (31). Six 10-fold serial dilutions (1:10–1:106) were made for each supernatant sample and inoculated into duplicate wells of six-well tissue culture plates containing confluent Vero cell monolayers. After virus adsorption for 1 h, the Vero monolayers were overlaid with complete MEM media (Cellgro) containing agarose (Invitrogen) to restrict dissemination of progeny virions. The cells were incubated for 5 d at 37°C in a 5%-CO2 incubator and overlaid with the vital stain, neutral red (Sigma-Aldrich). Plaques were counted by visual inspection at 24 and 48 h after neutral red overlay to determine the number of plaque-forming units of DV per milliliter of supernatant.
Flow Cytometry.
To characterize DCs, a FACS® Calibur (Becton Dickinson) was used to monitor surface staining with a panel of PE-conjugated mAbs to HLA-DR, CD80, CD86, CD3, CD14, CD20, CD25 (Becton Dickinson), CD83 (Immunotech), CD11c, CD1a (BD Biosciences), and matched isotype controls. To detect intracellular DV antigens, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% saponin, stained with FITC-conjugated–2H2 (anti–DV envelope complex mAb) and/or FITC-conjugated–7E11 (anti–DV-NS1 mAb, first nonstructural protein), with antibodies provided by R. Putnak (Walter Reed Army Institute of Research, Washington, DC).
Statistical Analysis.
This was performed with StatView 5 (SAS Institute).
Results
Cell-surface DC-SIGN Expression Correlates with DV Infection Rates.
We used flow cytometry to follow the expression of DC-SIGN and infection with DV. We suspected that DC-SIGN (CD209) might serve as a DV receptor. For example, prior work showed that immature DCs were much more susceptible to DV infection than their mature counterparts (5–10-fold higher levels; reference 9), and we found that the expression of DC-SIGN mean fluorescence intensity (MFI) was threefold higher on immature DCs (MFI = 175 ± 49, n = 4) compared with mature DCs (MFI = 60 ± 3, n = 3; Fig. 1 A) in agreement with other reports (18, 27, 32, 33). To study the role of DC-SIGN in DV entry, we used a DC-SIGN–transfected THP-1 (THP DC-SIGN) cell line, and the corresponding nontransfected THP cells as controls (27). We confirmed high levels of DC-SIGN on the transfected cells and no DC-SIGN expression on the nontransfected cells (Fig. 1 A). When these different cell types were exposed to DV (MOI = 1) and evaluated for infection 48 h later using a DV-specific monoclonal antibody (2H2) that binds to a dengue envelope complex protein, only the DCs and the THP DC-SIGN transfectants were infected (Fig. 1 B). In multiple experiments, the amount of infection of each cell type was directly correlated (r = 0.89) with the level of DC-SIGN expression (Fig. 1 C). These data implicate DC-SIGN in DV infection.
Figure 1.

Cytofluorometry of DV infection of DC-SIGN–expressing cells. (A) The MFI of DC-SIGN surface expression on DC and THP-1 cells is shown (left). The bars represent means (± SEM) of at least three independent experiments. The top right histogram shows the relative DC-SIGN expression (MFI) on a representative donor's immature (shaded) and MCM-matured DCs (heavy line). The bottom right histogram shows relative DC-SIGN on the surface of THP DC-SIGN (shaded) and THP-1 cells (heavy line). Matched isotype controls are represented by the light dashed line. (B) The mean (± SEM) percentage of DV infection of immature and mature DC (left) and THP DC-SIGN and THP-1 (right) after intracellular staining for dengue envelope antigen (2H2) in at least three independent experiments. (C) Positive correlation between DC-SIGN expression (MFI) on the x axis and percent DV infection as determined by 2H2 binding on the y axis on both DC and THP cells (n = 16).
THP DC-SIGN Can Be Infected with All Four Serotypes of DV.
We extended the study of DV infection to two different DV-specific monoclonal antibodies, the previously mentioned 2H2 anti–envelope complex antibody and the 7E11 antibody, to a nonstructural protein 1 (NS1) that is expressed during viral replication but is not a component of the virion. Infection, as assessed by the binding of monoclonal antibodies, was proportional to the dose of added virus (Fig. 2 A). The 2H2 binding typically was slightly higher than the 7E11 binding and again, no infection was detected in the nontransfected THP with either antibody. Kinetic studies with the DEN-2 virus (MOI = 0.2) were used to determine the peak time for detection of infection (Fig. 2 B). The optimal time point was at 48 h after viral exposure, which is consistent with previous studies in primary DCs (9, 11, 12). All studies used this 48-h time point, unless otherwise noted. All four DV serotypes were able to infect THP DC-SIGN transfectants, as previously shown in immature DCs (Fig. 2 C; reference 9). Of note, both low passage clinical isolates (two passages) and more lab-adapted isolates were able to infect at similar levels. When DV infection was evaluated in THP cells transfected with L-SIGN, a close relative of DC-SIGN (25), high levels of infection were again observed with all four serotypes (unpublished data). To inspect DV infection at the cellular level, we looked for DV antigens (2H2; red) using immunofluorescence and confocal microscopy in DC-SIGN–bearing cells (Fig. 2 D, green) at early and late time points, 2 and 24 h, respectively (Fig. 2 D). Newly produced virions accumulated in both DCs and THP transfectants at the later time point (Fig. 2 D, bottom), which is consistent with prior EM observations that replicating DV (at 24–48 h) is found in the endoplasmic reticulum of DCs (9, 12). The presence of the replicating virus within the DC-SIGN–bearing cells was confirmed by staining viral antigens with 2H2, using immunohistochemistry techniques. Abundant viral antigen was noted in the cytoplasm (Fig. 2 E, red) in DC-SIGN–bearing cells (Fig. 2 E, blue) at 48 h (but not at 2 h) for both immature DC and THP DC-SIGN (Fig. 2 E) and THP L-SIGN (unpublished data). No viral antigens were detected in non DC-SIGN–bearing cells (unpublished data). Therefore, transfection of THP cells with DC-SIGN and L-SIGN renders the cells permissive to infection with all DV serotypes.
Figure 2.
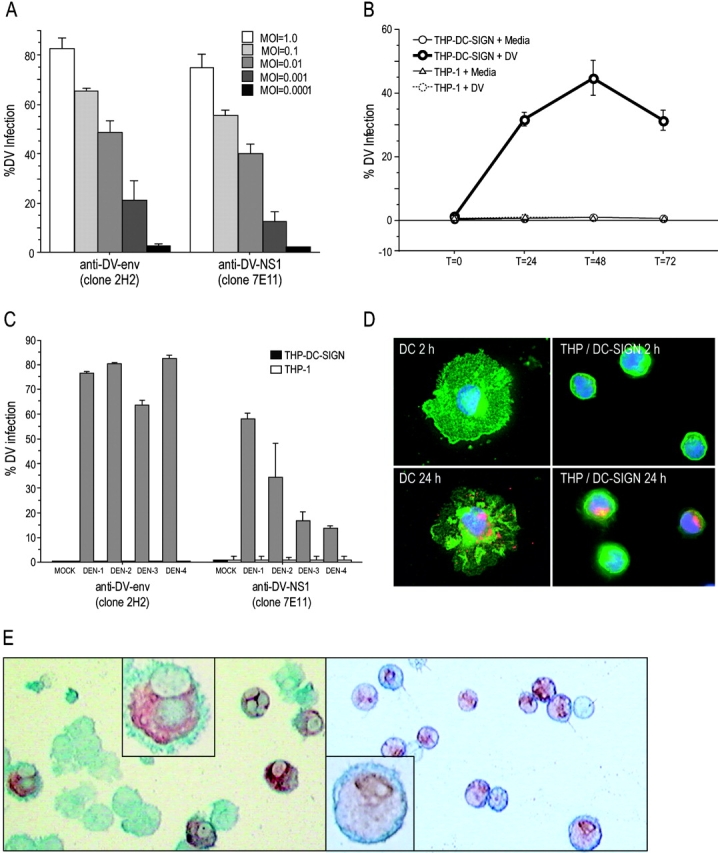
Dose and time dependence of DV infection in THP DC-SIGN cells. (A) Titration of DV2 infection in THP DC-SIGN and THP-1 2 d after infection. The infected cells were stained for DV envelope antigen (clone 2H2) and the NS1 (clone 7E11) and infection was determined by calculating the percentage of fluorescence-positive cells. (B) Kinetics of DV2 infection in THP DC-SIGN and THP-1. The infected cells were harvested at t = 0, 24, 48, and 72 h and stained with 2H2 and 7E11. (C) DV infection rates of all four DV serotypes in THP DC-SIGN and THP-1, using DV 1, 2, 3, and 4. Data are averages of two independent experiments. (D) Representative immunofluorescence experiment at two time points (2 h, top, and 24 h, bottom) showing bound DV envelope complex antigens (red) in DC-SIGN (green)–bearing DC (left) and THP-DC-SIGN (right) cells. Nuclei were labeled with DAPI stain. (E) Representative immunohistochemistry experiment showing bound intracellular DV envelope complex antigens (dark red) in DC-SIGN–bearing cells (blue). The left (immature DC) and right panels (THP DC-SIGN) show cells infected with DV. Original magnifications at 200 (insets, 600).
Anti–DC-SIGN Monoclonal Antibodies Block DV Infection in Dendritic Cells and THP DC-SIGN Cells.
To further evaluate DC-SIGN as a potential DV receptor, we used two different anti–DC-SIGN antibodies (clones 120612 and 120507). Preliminary antibody titration experiments were performed, which indicated that a range of 2–20 µg/ml antibody was sufficient to reduce infection of the DC (unpublished data). All subsequent blocking experiments were conducted using 2 µg/ml anti–DC-SIGN antibody and matched isotype control antibodies including anti-CD11a. In multiple experiments, both DC-SIGN antibodies independently and significantly blocked DV infection of either immature or mature DCs at 2 µg/ml (Fig. 3 A). We used similar experimental conditions for blocking DV infection of THP cells (Fig. 3 B). We added two other relevant antibodies that react strongly with the cells: anti-CD58 (LFA-3) and anti-CD74 (invariant chain). The levels of DV infection were unaltered in the presence of antibodies to CD58, CD74, or CD11a. Interestingly, we found that the 120507 clone was less effective at blocking DV infection (reduced by 50%) when compared with the “cross-reactive” 120612 clone (reduced by 90%) in the THP DC-SIGN cells. The THP L-SIGN cells were readily blocked using either the cross-reactive clone 120612 (>75% reduction) or the L-SIGN–specific clone 120604 (95% reduction; Fig. 3 C). Thus, DV infection of DCs, DC-SIGN, or L-SIGN–transfected cells can be significantly blocked by antibody-selective targeting of an epitope shared by DC-SIGN and L-SIGN.
Figure 3.

Blocking studies of DV infection in DCs and THP-1. (A) Comparison of percent DV2 infection rate of immature (left) and mature DCs (right) in the presence and absence of anti–DC-SIGN mAbs (clones 120507-specific or 120612–cross-reactive), anti-CD11a, or an irrelevant matched isotype control. (B) Comparison of percent DV2 infection rate of THP DC-SIGN and THP-1 in the presence and absence of specific anti–DC-SIGN mAbs, anti-CD11a, anti-CD58, anti-CD74, or an irrelevant matched isotype control. Data are means (± SEM) of four independent experiments. (C) A representative blocking experiment (one of two) in the THP L-SIGN cells in the presence and absence of specific anti–DC-SIGN or L-SIGN mAbs (120604), anti-CD11a, or an irrelevant matched isotype control.
The Cytoplasmic Domain of DC-SIGN Enhances but Is Not Essential for Infection.
To gain information on the mechanism of DC-SIGN function, we considered a THP DC-SIGN cell line in which the DC-SIGN molecule lacked its cytoplasmic domain (THP DC-SIGN Δ35). The cytosolic tail of DC-SIGN mediates endocytosis and is important for sequestering and transmitting HIV-1 when DCs are exposed to low doses of virus (27). DC-SIGN expression levels on the wild-type and THP DC-SIGN Δ35 cells were comparable (300–500 MFI). We found a significant reduction in viral entry in the THP DC-SIGN Δ35 (75% reduction) after DV exposure (Fig. 4) . Nevertheless, the residual DV infection could be completely abrogated in the presence of the anti–DC-SIGN antibody (clone 120612). This residual infection might have resulted from the constitutive entry of the truncated DC-SIGN receptor during bulk phase endocytosis, which is active in the cells we studied.
Figure 4.

Internalization of DV. Comparison of DV infection of THP DC-SIGN, THP DC-SIGN Δ35 (fully truncated cytoplasmic domain DC-SIGN), and THP-1 in the presence of specific anti–DC-SIGN antibody (clone 120612) or a matched isotype control. Data are the mean (± SEM) of three independent experiments.
Spreading Infection of DCs Is Inhibited by Anti–DC-SIGN Antibodies.
To determine if DV infection of DCs produced infectious virions, we performed a standard DV plaque assay (31) in susceptible Vero cells. We infected DCs or THP DC-SIGN/THP L-SIGN cells for 2 h in the absence or presence of blocking anti–DC-SIGN mAbs, washed, cultured the cells for 48 h to allow for production of infectious virus, and checked the 48-h supernatants for infection of Vero cells. The data showed that supernatants from the DC-SIGN–bearing cells induced plaque formation and transmitted DV infection; furthermore, the “trans” DV infection was significantly reduced (>1 log drop [PFU/ml]) when the initial infection of DCs had been performed in the presence of anti–DC-SIGN antibodies (Fig. 5) .
Figure 5.

Plaque assays of cell-free culture supernatants show infectivity. Supernatants transmit DV infection from DV-infected DCs (left) and THP DC-SIGN cells (right) to Vero cells. DCs or THPs were pretreated with an anti–DC-SIGN mAb (clone 120612) or a matched isotype control and exposed to DV for 2 h. Virus and mAb were washed away, and the supernatants were collected 48 h later. Plaque assays were used to determine PFUs in the collected supernatants. Data are the averages of two independent experiments.
Discussion
This paper identifies DC-SIGN and L-SIGN as mediators of DV infection. DC-SIGN is expressed primarily by a subset of DCs, whereas L-SIGN is expressed by certain endothelial cells, especially sinusoidal endothelium in liver and lymphoid organs and placental capillaries. We suggest that DC-SIGN allows immature DCs to capture and replicate DV after transmission from the mosquito vector. It is not clear if standard targets for DV infection, e.g., monkey Vero cells and C6/36 mosquito cells, express a functionally equivalent molecule to DC-SIGN or if they use an entirely distinct pathway of infection.
Although DC-SIGN binds at least two normal cellular adhesion molecules, ICAM-3–CD50 (32) and ICAM-2–CD102 (34), much of the research on this C-type lectin has involved its interaction with pathogens. DC-SIGN is proving to be a receptor used by DCs to capture, replicate, and/or transmit many pathogens, at least in culture. DC-SIGN is known to play a critical role in tissue culture by enhancing HIV transmission from DC to T cells (17, 27, 35). Recently, the Ebola virus (18, 19), cytomegalovirus (20), L. pifanoi amastigotes (21), and M. tuberculosis (22–24) were shown to use DC-SIGN to gain entry into DCs. Not only are large numbers of DCs and transfected THP-1 cells infected quickly with DV, but these cells yield a virus that is infectious for other cells. Thus, very low MOIs are sufficient to bring about a productive infection. In HIV-1 infection, direct infection of DCs is actually minimal at a time that DC-SIGN is allowing the DCs to sequester and transmit HIV-1 to T cells. With cytomegalovirus, like DV, the DCs themselves are infected (cis) and enhance infection of a permissive cell type in trans (20); in Ebola, L. pifanoi, and M. tuberculosis, it remains to be determined if DC-SIGN promotes spreading infection. Therefore, DC-SIGN likely recognizes “pathogen-associated microbial patterns.” One of the consequences of the DC-SIGN–pathogen interaction may be to enhance infection or pathogenesis by allowing the pathogen to enter the endocytic system. Once inside the endocytic pathway, the pathogen either replicates or fuses with the vacuole membrane to enter the cytoplasm. The latter is proposed for a pathogen like DV, where an acidic environment is likely to be required for the fusion of the DV envelope glycoproteins with the host cell membrane (36).
We used specific monoclonal Abs to block infection of DCs and THP DC-SIGN/L-SIGN to prove that viral entry depends specifically on these lectins. Control antibodies that reacted strongly with DCs and THPs (CD11a, 58, and 74) did not block infection. The two receptors, DC-SIGN and L-SIGN/DC-SIGNR, exhibit 77% amino acid identity and likely share ligands (25). We used two anti–DC-SIGN clones: DC-SIGN–specific (120507) and another cross-reactive clone (120612) that binds to shared epitopes on DC-SIGN and L-SIGN/DC-SIGNR. The antibodies all blocked infection in primary DCs and THP DC-SIGN, but the latter were most efficiently blocked with the cross-reactive monoclonal antibody. This suggests that a critical epitope for DV infection involves an epitope shared by DC-SIGN and L-SIGN. However, this does not exclude other possibilities such as antibody-induced steric hindrance of a nearby DV epitope or antibody-induced conformational changes at a distant epitope.
In an extension of the work presented here, we performed neutralization assays with sera from DV-infected patients and monoclonal antibodies to the DV envelope. Both the patients' sera and monoclonal antibody independently blocked infection of immature DC and THP DC-SIGN cells in a dose-dependent manner (unpublished data). It is possible that antibodies to DV have two contrasting functions. The valuable one, in terms of vaccine design, would be to block primary infection at the level of the DV envelope interacting with DC-SIGN receptors on DCs (and possibly L-SIGN on endothelial cells). The other contrasting function would be for an antibody to enhance infection via macrophages, which express Fc receptors but lack DC-SIGN. Prior work showed that antibody-mediated enhancement of DV infection, using select “enhancing” sera obtained in the waning stages of heterotypic DV infection, is not observed in DCs (10). The presence of DC-SIGN may override the need for other binding mechanisms to enhance infection in DCs.
Highly conserved, relevant epitopes of DV involved in cellular infection need to be identified to advance the development of DV vaccines and therapies. Current DV vaccine strategies need to incorporate all four serotypes due to the potential risk of DV hemorrhagic fever and shock syndromes. Recent public health concerns also extend to the potential use of the viruses that cause hemorrhagic fevers as weaponzied biological agents (37, 38). The possibility of limiting productive infection of important target cells at the level of virus entry, which for DV includes the DC-SIGN and L-SIGN receptors, may be an option worthy of consideration for translation from the laboratory to the clinic.
Acknowledgments
The authors thank S. Widjaja and R. Putvatana for technical assistance and M. Desouza for manuscript review.
B. Tassaneetrithep is a Medical Scholar Program Student (Ph.D-MD), Mahidol University, Thailand. National Institutes of Health grants AI40045 and AI40874 to R.M. Steinman. This work was supported in part by the cooperative agreement DAMD17-98-2-8007, between the U.S. Army Medical Research and Materiel Command, the Henry M. Jackson Foundation for the Advancement of Military Medicine, and the Military Infectious Disease Research Program. The views and opinions expressed herein are those of the authors and do not purport to reflect the official policy or position of the Department of Defense.
R.M. Steinman, S. Schlesinger, and M.A. Marovich all contributed equally to this work.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; DC-SIGN, DC-specific ICAM-3 grabbing nonintegrin; DV, dengue virus; HIV-1, HIV-type 1; ICAM, intracellular adhesion molecule; L-SIGN, liver/lymph node–specific ICAM-3–grabbing nonintegrin; MCM, monocyte-conditioned media; MFI, mean fluorescence intensity; MOI, multiplicity of infection; NS1, nonstructural protein 1; THP DC-SIGN, DC-SIGN–transfected THP.
References
- 1.Monath, T.P., and F.X. Heinz. 1996. Flaviviruses. In Fields Virology. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven, Philadelphia. 961–1034.
- 2.Gubler, D.J., S. Nalim, R. Tan, H. Saipan, and J. Sulianti Saroso. 1979. Variation in susceptibility to oral infection with dengue viruses among geographic strains of Aedes aegypti. Am. J. Trop. Med. Hyg. 28:1045–1052. [DOI] [PubMed] [Google Scholar]
- 3.Halstead, S.B. 1997. Epidemiology of dengue and dengue hemorrhagic fever. In Dengue and Dengue Hemorrhagic Fever. D.J. Gubler and G. Kuno, editors. Cab International, London. 23–44.
- 4.Burke, D.S., A. Nisalak, D.E. Johnson, and R.M. Scott. 1988. A prospective study of dengue infections in Bangkok. Am. J. Trop. Med. Hyg. 38:172–180. [DOI] [PubMed] [Google Scholar]
- 5.Kliks, S.C., A. Nisalak, W.E. Brandt, L. Wahl, and D.S. Burke. 1989. Antibody-dependent enhancement of dengue virus growth in human monocytes as a risk factor for dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 40:444–451. [DOI] [PubMed] [Google Scholar]
- 6.Halstead, S.B., E.J. O'Rourke, and A.C. Allison. 1977. Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J. Exp. Med. 146:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halstead, S.B., and E.J. O'Rourke. 1977. Antibody-enhanced dengue virus infection in primate leukocytes. Nature. 265:739–741. [DOI] [PubMed] [Google Scholar]
- 8.Innis, B. 1995. Dengue and dengue haemorrhagic fever. Exotic Viral Infections. J. Porterfield, editor. Chapman and Hall, London. 103–146.
- 9.Wu, S.J., G. Grouard-Vogel, W. Sun, J.R. Mascola, E. Brachtel, R. Putvatana, M.K. Louder, L. Filgueira, M.A. Marovich, H.K. Wong, et al. 2000. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 6:816–820. [DOI] [PubMed] [Google Scholar]
- 10.Marovich, M., G. Grouard-Vogel, M. Louder, M. Eller, W. Sun, S.J. Wu, R. Putvatana, G. Murphy, B. Tassaneetrithep, T. Burgess, et al. 2001. Human dendritic cells as targets of dengue virus infection. J. Investig. Dermatol. Symp. Proc. 6:219–224. [DOI] [PubMed] [Google Scholar]
- 11.Libraty, D.H., S. Pichyangkul, C. Ajariyakhajorn, T.P. Endy, and F.A. Ennis. 2001. Human dendritic cells are activated by dengue virus infection: enhancement by gamma interferon and implications for disease pathogenesis. J. Virol. 75:3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho, L.J., J.J. Wang, M.F. Shaio, C.L. Kao, D.M. Chang, S.W. Han, and J.H. Lai. 2001. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J. Immunol. 166:1499–1506. [DOI] [PubMed] [Google Scholar]
- 13.Chen, Y., T. Maguire, R.E. Hileman, J.R. Fromm, J.D. Esko, R.J. Linhardt, and R.M. Marks. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866–871. [DOI] [PubMed] [Google Scholar]
- 14.Chen, Y.C., S.Y. Wang, and C.C. King. 1999. Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/macrophages by blockade of virus entry via a CD14-dependent mechanism. J. Virol. 73:2650–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marianneau, P., F. Megret, R. Olivier, D.M. Morens, and V. Deubel. 1996. Dengue 1 virus binding to human hepatoma HepG2 and simian Vero cell surfaces differs. J. Gen. Virol. 77:2547–2554. [DOI] [PubMed] [Google Scholar]
- 16.Hung, S.L., P.L. Lee, H.W. Chen, L.K. Chen, C.L. Kao, and C.C. King. 1999. Analysis of the steps involved in dengue virus entry into host cells. Virology. 257:156–167. [DOI] [PubMed] [Google Scholar]
- 17.Geijtenbeek, T.B., D.S. Kwon, R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, J. Middel, I.L. Cornelissen, H.S. Nottet, V.N. KewalRamani, D.R. Littman, et al. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 100:587–597. [DOI] [PubMed] [Google Scholar]
- 18.Alvarez, C.P., F. Lasala, J. Carrillo, O. Muniz, A.L. Corbi, and R. Delgado. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76:6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baribaud, F., S. Pohlmann, G. Leslie, F. Mortari, and R.W. Doms. 2002. Quantitative expression and virus transmission analysis of DC-SIGN on monocyte-derived dendritic cells. J. Virol. 76:9135–9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halary, F., A. Amara, H. Lortat-Jacob, M. Messerle, T. Delaunay, C. Houles, F. Fieschi, F. Arenzana-Seisdedos, J.F. Moreau, and J. Dechanet-Merville. 2002. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 17:653–664. [DOI] [PubMed] [Google Scholar]
- 21.Colmenares, M., A. Puig-Kroger, O. Muniz Pello, A.L. Corbi, and L. Rivas. 2002. Dendritic-cell specific ICAM-3 grabbing nonintegrin (DC-SIGN, CD209), a C-type surface lectin in human dendritic cells, is a receptor for Leishmania amastigotes. J. Biol. Chem. 277:36766–36769. [DOI] [PubMed] [Google Scholar]
- 22.Maeda, N., J. Nigou, J.L. Herrmann, M. Jackson, A. Amara, P.H. Lagrange, G. Puzo, B. Gicquel, and O. Neyrolles. 2003. The cell surface receptor DC-SIGN discriminates between Mycobacterium species through selective recognition of the mannose caps on lipoarabinomannan. J. Biol. Chem. 278:5513–5516. [DOI] [PubMed] [Google Scholar]
- 23.Tailleux, L., O. Schwartz, J.-L. Herrmann, E. Pivert, M. Jackson, A. Amara, L. Legres, D. Dreher, L.P. Nicod, J.C. Gluckman, et al. 2003. DC-SIGN is the major mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 197:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geijtenbeek, T.B.H., S.J. van Vliet, E.A. Koppel, M. Sanchez-Hernandez, C.M.J.E. Vandenbroucke-Grauls, B. Appelmelk, and Y. Van Kooyk. 2003. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bashirova, A.A., T.B. Geijtenbeek, G.C. van Duijnhoven, S.J. van Vliet, J.B. Eilering, M.P. Martin, L. Wu, T.D. Martin, N. Viebig, P.A. Knolle, et al. 2001. A dendritic cell–specific intercellular adhesion molecule 3–grabbing nonintegrin (DC-SIGN)–related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Exp. Med. 193:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pohlmann, S., E.J. Soilleux, F. Baribaud, G.J. Leslie, L.S. Morris, J. Trowsdale, B. Lee, N. Coleman, and R.W. Doms. 2001. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc. Natl. Acad. Sci. USA. 98:2670–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon, D.S., G. Gregorio, N. Bitton, W.A. Hendrickson, and D.R. Littman. 2002. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 16:135–144. [DOI] [PubMed] [Google Scholar]
- 28.Trumpfheller, C., C.G. Parke, J. Finke, R.M. Steinman, and A. Granelli-Piperno. 2003. Cell type dependent retention and transmission of HIV-1 by DC-SIGN. Int. Immunol. 15:289–298. [DOI] [PubMed] [Google Scholar]
- 29.Thurner, B., C. Roder, D. Dieckmann, M. Heuer, M. Kruse, A. Glaser, P. Keikavoussi, E. Kampgen, A. Bender, and G. Schuler. 1999. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J. Immunol. Methods. 223:1–15 (published erratum appears in J. Immunol. Methods. 1999. 224:211). [DOI] [PubMed] [Google Scholar]
- 30.Marovich, M.A., J.R. Mascola, M.A. Eller, M.K. Louder, P.A. Caudrelier, R. El-Habib, S. Ratto-Kim, J.H. Cox, J.R. Currier, B.L. Levine, et al. 2002. Preparation of clinical-grade recombinant canarypox-human immunodeficiency virus vaccine-loaded human dendritic cells. J. Infect. Dis. 186:1242–1252. [DOI] [PubMed] [Google Scholar]
- 31.Eckels, K.H., W.E. Brandt, V.R. Harrison, J.M. McCown, and P.K. Russell. 1976. Isolation of a temperature-sensitive dengue-2 virus under conditions suitable for vaccine development. Infect. Immun. 14:1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geijtenbeek, T.B., R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, G.J. Adema, Y. van Kooyk, and C.G. Figdor. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 100:575–585. [DOI] [PubMed] [Google Scholar]
- 33.Sanders, R.W., E.C. De Jong, C.E. Baldwin, J.H. Schuitemaker, M.L. Kapsenberg, and B. Berkhout. 2002. Differential transmission of human immunodeficiency virus type 1 by distinct subsets of effector dendritic cells. J. Virol. 76:7812–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geijtenbeek, T.B., D.J. Krooshoop, D.A. Bleijs, S.J. van Vliet, G.C. van Duijnhoven, V. Grabovsky, R. Alon, C.G. Figdor, and Y. van Kooyk. 2000. DC-SIGN-ICAM-2 interaction mediates dendritic cell trafficking. Nat. Immunol. 1:353–357. [DOI] [PubMed] [Google Scholar]
- 35.Lee, B., G. Leslie, E. Soilleux, U. O'Doherty, S. Baik, E. Levroney, K. Flummerfelt, W. Swiggard, N. Coleman, M. Malim, and R.W. Doms. 2001. cis expression of DC-SIGN allows for more efficient entry of human and simian immunodeficiency viruses via CD4 and a coreceptor. J. Virol. 75:12028–12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn, R.J., W. Zhang, M.G. Rossmann, S.V. Pletnev, J. Corver, E. Lenches, C.T. Jones, S. Mukhopadhyay, P.R. Chipman, E.G. Strauss, et al. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 108:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borio, L., T. Inglesby, C.J. Peters, A.L. Schmaljohn, J.M. Hughes, P.B. Jahrling, T. Ksiazek, K.M. Johnson, A. Meyerhoff, T. O'Toole, et al. 2002. Hemorrhagic fever viruses as biological weapons: medical and public health management. JAMA. 287:2391–2405. [DOI] [PubMed] [Google Scholar]
- 38.Gubler, D.J. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social, and economic problem in the 21st century. Trends Microbiol. 10:100–103. [DOI] [PubMed] [Google Scholar]