

Abstract
The chemokines CXCL9/Mig, CXCL10/IP-10, and CXCL11/I-TAC regulate lymphocyte chemotaxis, mediate vascular pericyte proliferation, and act as angiostatic agents, thus inhibiting tumor growth. These multiple activities are apparently mediated by a unique G protein–coupled receptor, termed CXCR3. The chemokine CXCL4/PF4 shares several activities with CXCL9, CXCL10, and CXCL11, including a powerful angiostatic effect, but its specific receptor is still unknown. Here, we describe a distinct, previously unrecognized receptor named CXCR3-B, derived from an alternative splicing of the CXCR3 gene that mediates the angiostatic activity of CXCR3 ligands and also acts as functional receptor for CXCL4.
Human microvascular endothelial cell line-1 (HMEC-1), transfected with either the known CXCR3 (renamed CXCR3-A) or CXCR3-B, bound CXCL9, CXCL10, and CXCL11, whereas CXCL4 showed high affinity only for CXCR3-B. Overexpression of CXCR3-A induced an increase of survival, whereas overexpression of CXCR3-B dramatically reduced DNA synthesis and up-regulated apoptotic HMEC-1 death through activation of distinct signal transduction pathways. Remarkably, primary cultures of human microvascular endothelial cells, whose growth is inhibited by CXCL9, CXCL10, CXCL11, and CXCL4, expressed CXCR3-B, but not CXCR3-A. Finally, monoclonal antibodies raised to selectively recognize CXCR3-B reacted with endothelial cells from neoplastic tissues, providing evidence that CXCR3-B is also expressed in vivo and may account for the angiostatic effects of CXC chemokines.
Keywords: CXCR3-B, CXCR3-A, angiogenesis, chemokines, chemokines receptor
Introduction
Chemokines are a family of small, structurally related molecules that regulate cell trafficking of various subsets of leukocytes (1–2). Moreover, chemokines regulate embryonic development, pathophysiology of the central nervous system, wound healing repair, control angiogenesis, and tumor growth and spread (1–4). The nomenclature for chemokines is based on the configuration of a conserved amino-proximal cysteine-containing motif. Based on this system, there are currently four branches of the chemokine family: CXC, CC, CX3C, and C (where X is any amino acid). CXC chemokines are characteristically heparin-binding proteins and exhibit the selective property to promote or inhibit angiogenesis (4–6). The angiostatic members of the CXC chemokine family include IP-10/CXCL10, MIG/CXCL9, I-TAC/CXCL11, and platelet factor 4/CXCL4 (4–6). These chemokines induce injury to established tumor-associated vasculature and promote extensive tumor necrosis (5–10) and thus have been proposed as useful therapeutic agents in cancer (7).
The angiostatic effects of CXCL9, CXCL10, and CXCL11 on human microvascular endothelial cells (HMVEC)* are mediated by the CXC chemokine receptor 3 (CXCR3; 4–6, 11–13), whereas the receptor for CXCL4 has not yet been identified (4, 6). Several studies have pointed out that CXCL10 and CXCL4 share a great number of activities, such as inhibition of chemotaxis and proliferation of endothelial cells (14, 15), or inhibition of hematopoiesis (16). However, the interaction of CXCL9, CXCL10, and CXCL11 with CXCR3 also results in the chemotaxis of activated Th1 cells (17, 18), NK cells (19), macrophages (20), and dendritic cells (21), whereas CXCL4 does not exert chemotactic activity on the same cell types. Furthermore, CXCL9 and CXCL10 have been implicated in the pathogenesis of proliferative glomerulonephritis, a common renal disease characterized by glomerular hypercellularity, because they induce increased survival and growth of human mesangial cells (HMC) through their receptor CXCR3 (22, 23). Recently, we and others found that CXCL9 and CXCL10 induce chemotaxis and proliferation (22, 23) of HMC through pertussis toxin–sensitive mechanisms (24). Thus, the apparently opposite effects of CXCL9, CXCL10, and CXCL11 on HMVEC and on HMC allow the hypothesis of the existence of cell-specific signal transduction pathways and/or distinct CXCR3 receptor variants.
In this study, we have identified a novel mRNA generated by an alternative splice site within the intron of the CXCR3 gene by using rapid amplification of cDNA ends (RACE) and Northern blot analysis. Real-time quantitative RT-PCR and transfection of the human microvascular endothelial cell line-1 (HMEC-1) provided evidence for the existence of a previously unrecognized receptor, named CXCR3-B, which mediated the inhibitory activity of CXCL9, CXCL10, and CXCL11 on the growth of HMVEC. Binding assay and signal transduction analysis demonstrated that CXCR3-B also acts as a functional receptor for CXCL4, thus explaining the large overlap between the biological activities of CXCL10 and CXCL4. By contrast, the classic CXCR3 receptor, renamed as CXCR3-A, mediated the proliferation of HMC in response to CXCL9, CXCL10, and CXCL11, and was responsible for both increased survival and angiogenic properties of transfected HMEC-1 cells, but did not react with CXCL4. Finally, mAbs selectively developed against CXCR3-B, reacted with endothelial cells of different human tumor tissues, but poorly or not with those of their normal counterparts, consistently with the previously described selective activity of both CXCL4 and CXCL10 on the proliferation of endothelial cells.
Materials and Methods
Reagents.
Recombinant human chemokines were obtained from R&D Systems (Minneapolis, MN). Pertussin toxin (PTX) was obtained from Calbiochem.
RACE.
5′ and 3′ RACE experiments were performed on 5 μg total RNA obtained from primary cultures of HMVEC or human thymus using the RLM-RACE kit (GeneRacer; Invitrogen), according to the manufacturer's instructions. Gene-specific primers used in 5′-RACE were 5′-ACTCTGAGCAGCTCCTCCTATAACT-3′ and nested 5′-CGCCAGTCTTCCAGGGCCGTACTT-3′. Gene-specific primers used in 3′ RACE were 5′-CCTGGAAGACTGGCGGGGACAGTTA-3′ and nested 5′-GAGTTCCTGCCAGGCCTTTACAC-3′. The RACE products were cloned into pGEM-T easy vector (Promega) and sequenced.
Northern Blot Analysis.
Human tissue Northern blots prepared with 2 μg poly-A mRNA of each sample (CLONTECH Laboratories, Inc.) were treated as previously described (25).
Real-Time Quantitative RT-PCR (TaqMan™).
Total RNA was extracted (26) and treated with DNase I (QIAGEN) to eliminate possible genomic DNA contamination. TaqMan RT-PCR was performed as previously described (26).
The following primers and probes were used: CXCR3-A: FAM probe, 5′-TGAGTGACCACCAAGTGCTAAATGACGC-3′; forward 5′-ACCCAGCAGCCAGAGCACC-3′; reverse 5′-TCATAGGAAGAGCTGAAGTTCTCCA-3′. CXCR3-B: VIC probe, 5′-CCCGTTCCCGCCCTCACAGG-3′; forward 5′-TGCCAGGCCTTTACACAGC-3′; reverse 5′-TCGGCGTCATTTAGCACTTG-3′. The two sets of primers displayed similar amplification efficiency and selective specificity, as tested on a plasmid encoding the CXCR3-A or CXCR3-B cDNA sequence, respectively. mRNA levels were quantitated by comparing experimental levels to standard curves generated using serial dilutions of the same amount of the plasmids. CXCL11, CXCL10, p21, p53, and GAPDH quantitation was performed using predeveloped TaqMan assay reagents target kits (Applied Biosystems). The CXCL9 primers and probes were: FAM probe, 5′-AAGGGTCGCTGTTCCTGCATCAGC-3′; forward 5′-TGCAAGGAACCCCAGTAGTGA-3′; reverse 5′-GGTGGATAGTCCCTTGGTTGG-3′. GAPDH was used for normalization.
Generation of HMEC-1 and HEK-293EBNA Stable Transfectants for CXCR3-A and CXCR3-B.
Full-length CXCR3-A and CXCR3-B cDNAs were obtained by RT-PCR performed on total mRNA derived from human thymus and HMVEC, respectively, cloned in expression vectors carrying the same promoter and polyadenylation signal, pCEP4 (27) and pTarget (Promega), respectively, sequenced, and transfected into HMEC-1 (28) and HEK-293EBNA (Invitrogen) by electroporation. Stably transfected cells were selected using MCDB 131 or DMEM media containing 300 μg/ml hygromycin or 500 μg/ml G-418, respectively. CXCR3-A and CXCR3-B expression was revealed by FACS® analysis with CXCR3-specific 1C6 (BD Biosciences) and 49801.111 (R&D Systems) mAbs, as previously described (11).
Binding Assays.
Iodination of human recombinant CXCL4 was performed using the chloramine-T method (29). The specific activity of the 125I-CXCL4 was 360.2 μCi/μg. Binding assays were performed as previously described (30). In brief, cells were plated in 24-well culture plates and incubated overnight in standard medium. For the binding assay, cells were washed twice with washing buffer (0.5 M NaCl, 50 mM Hepes, 1mM CaCl2, 5mM MgCl2, and 1% BSA, pH 7.2), once with binding buffer (washing buffer without NaCl), and then incubated in duplicate with a constant concentration (18 pM) of 125I-labeled CXCL10 (Amersham Biosciences) or 125I-labeled CXCL4 in the presence of increasing concentrations of unlabeled chemokines. Incubations took place in 200 μl binding buffer. After incubation at room temperature for 2 h, binding buffer was aspirated and cells were washed once in PBS (without Ca and Mg) and then lysed in 0.5 ml 1 N NaOH. Radioactivity was determined using a gamma counter. Data were analyzed using the computer program ALLFIT (31). These salt conditions have been optimized for chemokine binding and minimize the potential for chemo-kine self-association (32).
Flow Cytometric Analysis of Calcium Mobilization.
106 cells were loaded with 5 mM Indo-1 acetyloxy methyl ester for 45 min at 37°C, washed, and then incubated for an additional 45 min at 37°C. Variation in the intracellular calcium concentration [Ca++]i was measured by evaluating the 525/405-nm emission ratio of unstimulated (baseline), chemokine-, or ionomycin- (cell loading control) stimulated Indo-1–loaded cells.
Cell Cultures.
HMEC-1, HMC, and ACHN cells were cultured as previously described (28, 25, 33).
cAMP Detection Immunoassay.
cAMP levels were assessed by using a commercially available colorimetric kit (Calbiochem) on 40,000 cells/well.
Cell Proliferation Assay.
[3H]thymidine incorporation was assessed as previously described (11). To perform blocking experiments, a neutralizing anti–human CXCL11/I-TAC mAb (clone 87328) was obtained from R&D Systems.
Terminal Deoxynucleotidyl Transferase-mediated dUTP Nick End Labeling (TUNEL) Assay.
HMEC-1 transfectants were stained using the TUNEL method (Apoptag® kit, Oncor®; reference 34), and the number of labeled nuclei was assessed by cell counting.
Flow Cytometric Apoptosis Assays.
HMEC-1 transfectants were processed for FACS® analysis as previously described (11). The percentage of cells with a sub-G1 DNA content was taken as a measure of apoptosis cell rate.
Matrigel Morphogenetic Assay.
HMEC-1 transfectants (2 × 104/well) were plated onto 0.2 ml Matrigel (Becton Dickinson) and observed after a 48-h culture.
Chemotactic Assay.
Chemotaxis on HMEC-1 transfectants (5 × 104/well) was performed as previously described (24).
mAb Production.
CXCR3-B–specific mAbs were generated at Primm by immunizing BALB/c mice with 100 μg of a 51-mer synthetic peptide corresponding to the first 51 NH2-terminal amino acids of CXCR3-B (MELRKYGPGRLAGTVIGGAAQSKSQTKSDSITKEFLPGLYTAPSSPFPPSQ). This peptide was synthesized and coupled to ovalbumin. mAbs reactive with the peptide used for immunization were screened by ELISA of ibridomas supernatants. CXCR3-B specificity of the obtained mAbs was checked using untransfected and CXCR3-A– or CXCR3-B–transfected HMEC-1 cells and immunostaining. The two main mAbs used in this study (PL1 and PL2) had an IgG1 isotype.
Immunofluorescence and Immunohistochemistry.
Immunohistochemistry was performed as previously described (11). Anti–CXCR3-B mAbs purified ascites fluid of clones PL1 and PL2 were used at a final dilution of 3 μg/ml and 20 μg/ml, respectively, and the Vectastain ABC kit (Vector Laboratories) was used as a detection system. Pre-adsorption test was performed by preincubating the mAbs overnight at 4°C with the specific peptide used to immunize mice (2 mg). CXCR3-B and von Willebrand factor (anti-vWf Ab, 1:8,000; DakoCytomation) were colocalized on the same sections by double label immunohistochemistry (11).
Tumor Tissues.
Tumor specimens were obtained from 10 subjects suffering from nonsmall cell lung cancer and from 7 subjects suffering from renal cell carcinoma and their normal counterparts, who underwent surgical excision, in accordance with the Regional Ethical Committee on human experimentation.
Results
Identification, Molecular Characterization, and Tissue Distribution of a Novel CXCR3 Variant (CXCR3-B).
Translation of the CXCR3 gene with an automated computer program (ExPASy, available at http://ca.expasy.org) to search for novel sequence variants, identified the possible existence of a second mRNA. The predicted 1,248-bp open reading frame of this mRNA was initiated from an alternate in-frame ATG start codon found within the intron sequence and 151-bp upstream of the known AG acceptor site used for intron splicing (Fig. 1 a). Translation of CXCR3-B mRNA generated a 416–amino acid receptor containing a longer NH2-terminal extracellular domain different from the CXCR3-A sequence in the first 52 amino acid residues, whereas the remaining protein was identical (Fig. 1 a). To provide evidence for the existence of, and to better characterize, this new mRNA, a rapid amplification of complementary cDNA 5′ and 3′ ends (RACE) on total mRNA obtained from either primary cultures of HMVEC or human thymus was performed by using primers selectively hybridizing the final portion of the intron (Fig. 1, a and b). This new mRNA, named CXCR3-B, showed a large overlap with the known CXCR3 sequence, but a novel 5′ end resulting from an alternative splicing between the same donor site used by the known CXCR3 sequence (renamed CXCR3-A) and a novel acceptor site localized 233 bp upstream of the known AG acceptor site (Fig. 1, a–c). The size of CXCR3-B, including the 5′ UTR region, was 1,860 bp (sequence data are available from GenBank/EMBL/DDBJ under accession no. AF469635). Furthermore, screening of the ResGene Clone Collection database identified a clone (ID 19600412004045), obtained from a cDNA leukocyte library (NFLHAL1), showing complete homology with the first 567 bp of the CXCR3-B sequence, the last 269 bp of this clone overlapping the sequence common to both receptors. The expression of this new putative mRNA was assessed in different tissues by using Northern blot analysis. To this end, a 182-bp probe (Fig. 1 a, yellow) selectively hybridizing with the CXCR3-B mRNA was used, which recognized an mRNA of ∼1.8 kb, mainly expressed in the heart, kidney, liver, and skeletal muscle (Fig. 1 d, top). By using a 401-bp cDNA probe (Fig. 1 a, light green) encompassing a region shared by both receptors, two distinct mRNAs of ∼1.8 (CXCR3-B) and 1.6 kb (CXCR3-A) were detected in the heart, kidney, liver, and skeletal muscle, with CXCR3-A being consistently prevalent (Fig. 1 d, bottom). Only CXCR3-A mRNA was observed in the placenta (Fig. 1 d, bottom).
Figure 1.

Identification, molecular characterization, and tissue distribution of CXCR3-B. (a) Structure of the human CXCR3 gene. Upper case, the exons; lower case, the intron; arrow, the CXCR3-B transcription start; underline, the CXCR3-B translation start site (ATG); light shading, the donor (AG/gt, common for both CXCR3-A and CXCR3-B) and acceptor (ag/GG, CXCR3-B; AG/GT, CXCR3-A) splice sites. The nested primers used for 5′ and 3′ RACE are boxed and the direction is pointed by the arrow. The 182-bp probe (recognizing CXCR3-B) and the 401-bp probe (recognizing both CXCR3-A and CXCR3-B) used for Northern blot analysis are evidenced in yellow and light green, respectively. (b) RACE performed on primary cultures of HMVEC. Lane 1, DNA ladder Marker VIII; lane 2, 5′ RACE product; lane 3, 3′ RACE product; lane 4, DNA ladder 1 Kb plus. (c) Schematic representation illustrating the generation of CXCR3-A and CXCR3-B mRNA through alternative splicing. (d) Multiple tissue Northern blot hybridization with the 182-bp probe (top) or the 401-bp probe (bottom). These sequence data are available from GenBank/EMBL/DDBJ under accession no. AF469635.
CXCR3-B Binds CXCL10, CXCL11, CXCL9, and CXCL4.
To better investigate whether CXCR3-B and CXCR3-A had different properties, stable CXCR3-A or CXCR3-B transfectants were generated by using the HMEC-1 cell line. CXCR3-A transfectants exhibited high surface receptor expression (Fig. 2 a), whereas surface receptor expression in CXCR3-B transfectants was usually consistently lower (Fig. 2 b). One clone of CXCR3-A– and one of CXCR3-B–transfected cells, which exhibited comparable levels of surface receptor, were assessed for their ability to bind CXCL9, CXCL10, and CXCL11. All CXCR3 ligands displaced binding of 125I-CXCL10 from CXCR3-A with a higher affinity than from CXCR3-B. CXCL10 displaced 125I-CXCL10 bound to CXCR3-A transfectants with an IC50 of 0.35 ± 0.1 nM, CXCL11 with an IC50 of 0.41 ± 0.14 nM, and CXCL9 with an IC50 of 33 ± 24 nM (Fig. 2 c). The highest affinity binding for CXCR3-B was displayed by CXCL10 (IC50 = 6.9 ± 2.1 nM), whereas CXCL11 displaced radiolabeled CXCL10 bound to CXCR3-B transfectants with an IC50 of 32 ± 10.2 nM and CXCL9 with an IC50 of 133 ± 46.5 nM (Fig. 2 d). The binding of 125I-CXCL10 to either CXCR3-A or CXCR3-B transfectants was completely inhibited by the 49801.111 mAb used at concentrations of 10 or 1 μg/ml, respectively, demonstrating that this mAb was able to neutralize the functional activity of both CXCR3 receptor variants (unpublished data). HMEC-1 cells transfected with the empty vector (mock) did not bind 125I-CXCL10.
Figure 2.
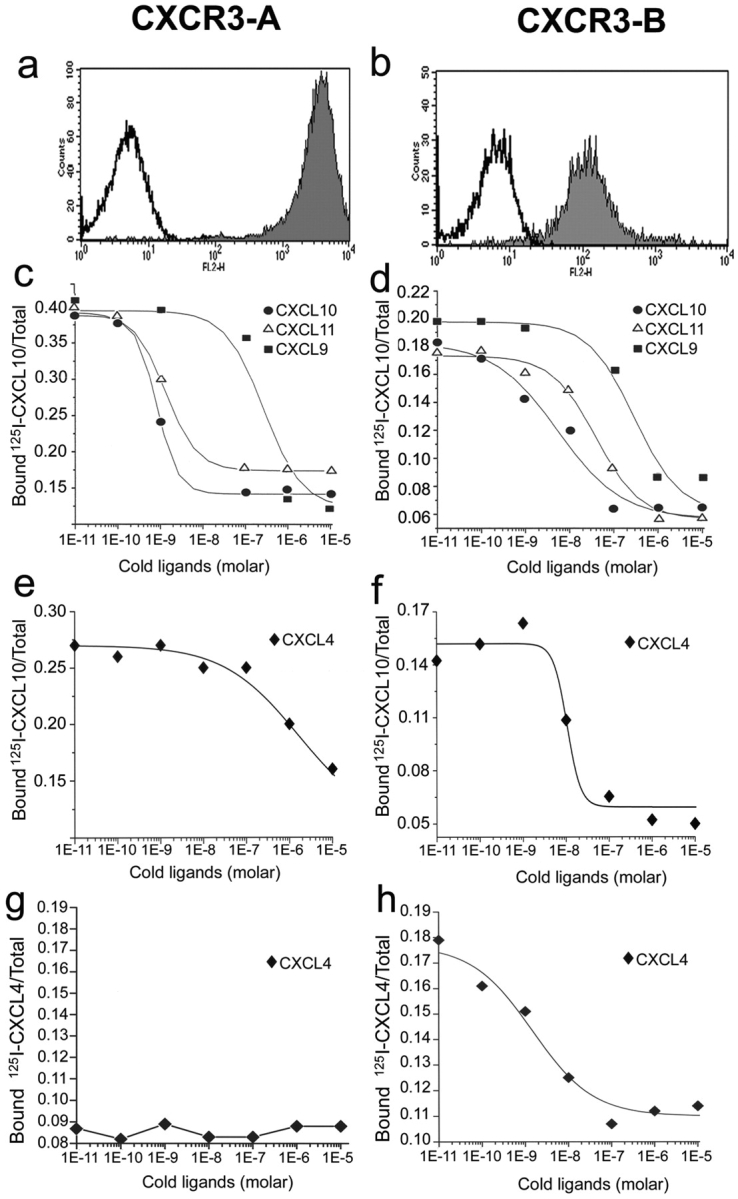
CXCR3-A and CXCR3-B expression by HMEC-1 transfectants and their binding affinity to cognate ligands. (a) Flow cytometric analysis of surface CXCR3-A. (b) Flow cytometric analysis of surface CXCR3-B. (c) CXCL10, CXCL9, and CXCL11 displacement of 125I-labeled CXCL10 bound to CXCR3-A transfectants. (d) CXCL10, CXCL9, and CXCL11 displacement of 125I-labeled CXCL10 bound to CXCR3-B transfectants. (e) Lack of significant displacement by cold CXCL4 of 125I-labeled CXCL10 bound to CXCR3-A transfectants. (f) CXCL4 displacement of 125I-labeled CXCL10 bound to CXCR3-B transfectants. (g) Lack of significant binding of CXCL4 to CXCR3-A transfectants. (h) High affinity binding of CXCL4 to CXCR3-B transfectants. Figures show mean displacement curves generated by analysis with the Allfit software obtained in eight separate experiments.
Because CXCR3-B apparently acted as a novel chemokine receptor, the binding to CXCR3-B transfectants of many chemokines was assessed. CCL1/I-309, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, CCL11/eotaxin, CCL17/TARC, CCL19/ELC, CCL21/SLC, CCL22/MDC, CCL25/TECK, CXCL1/GRO-α, CXCL2/GRO-β, CXCL3/GRO-γ, CXCL5/ENA-78, CXCL8/IL-8, and CXCL12/SDF-1 did not compete for radiolabeled CXCL10 binding on CXCR3-B transfectants (unpublished data). However, CXCL4, which shares with CXCL10, CXCL9, and CXCL11 several activities including a powerful angiostatic effect, displaced the binding of 125I-CXCL10 to CXCR3-B transfectants under the same conditions with an IC50 of 7.46 ± 3.05 nM (Fig. 2 f). By contrast, very poor displacement of 125I-CXCL10 binding to CXCR3-A transfectants (IC50 = 448 ± 152 nM; Fig. 2 e) was observed. To provide direct evidence that CXCL4 binds to CXCR3-B and to measure its affinity, binding assays with 125I-CXCL4 were performed. Radiolabeled CXCL4 bound specifically to CXCR3-B transfectants (Fig. 2 h), but neither to mock (unpublished data) nor to CXCR3-A transfectants (Fig. 2 g). More importantly, the binding of radiolabeled CXCL4 to CXCR3-B transfectants was displaceable by increasing concentrations of unlabeled CXCL4, with an IC50 of 1.85 ± 0.9 nM (Fig. 2 h). The binding of 125I-CXCL4 to CXCR3-B transfectants was completely inhibited by the addition of anti-CXCR3 mAb 49801.111 used at the concentration of 20 μg/ml (unpublished data). Because CXCL4 exhibited high affinity and selectivity for CXCR3-B, whereas CXCL10, CXCL11, and CXCL9 bound CXCR3-A with higher affinity, we suggest that CXCL4 is the dominant ligand for CXCR3-B.
Selective Overexpression of CXCR3-A or CXCR3-B Has Opposite Effects on Both Growth and Angiogenic Activity of Endothelial Cells.
To further investigate the functions of CXCR3-B, 29 clones of CXCR3-B, and 26 clones of CXCR3-A, transfectants were screened for cell viability, proliferation, apoptosis, and ability to induce vessel formation in vitro. All clones expressing high CXCR3-B levels on their surface rapidly died within two or three passages. Only six clones expressing low CXCR3-B levels survived for 8–10 passages. However, all exhibited a lower proliferation rate than the respective mock transfectants, as demonstrated by either assessing [3H]thymidine incorporation on day 3 (2,623 ± 234 vs. 10,748 ± 1,820 cpm/well; P < 0.001) or counting the numbers of viable cells in 60-mm wells on day 7 (370,000 ± 20,000 vs. 1,570,000 ± 250,000; P < 0.001). Low proliferation of CXCR3-B clones was related to a high degree of apoptotic cell death as shown by either FACS® analysis of sub-G1 DNA content (Fig. 3 a) or detection of DNA fragmentation with TUNEL immunostaining (Fig. 3 a, inset). Furthermore, these clones were unable to induce tube-like vessel formation in the Matrigel assay in vitro (Fig. 3 b) and did not reach confluence in culture, suggesting the presence of contact inhibition. Of note, however, CXCR3-B transfection of HEK293-EBNA cells resulted in little, if any, inhibitory effect on their proliferation (unpublished data), suggesting that CXCR3-B expression does not have the same effects on any cell type. By contrast, all 26 clones exhibiting high levels of CXCR3-A displayed higher [3H]thymidine incorporation on day 3 (36,323 ± 8,234 vs. 9,932 ± 2,287 cpm/well; P < 0.001), higher numbers of viable cells in 60-mm wells on day 7 (2,450,000 ± 150,000 vs. 1,250,000 ± 250,000; P < 0.001), lower degree of apoptotic cell death (Fig. 3 c), and higher capability to induce tube-like vessel formation in the Matrigel assay (Fig. 3 d) than the respective mock transfectants. Moreover, although CXCR3-A transfectants showed chemotactic response to CXCL10, CXCL9, CXCL11, CXCL8, CXCL12, or basic fibroblast growth factor, CXCR3-B transfectants did not exhibit chemotactic response to any of these factors (unpublished data), suggesting inhibition by CXCR3-B expression even on endothelial cell chemotaxis.
Figure 3.
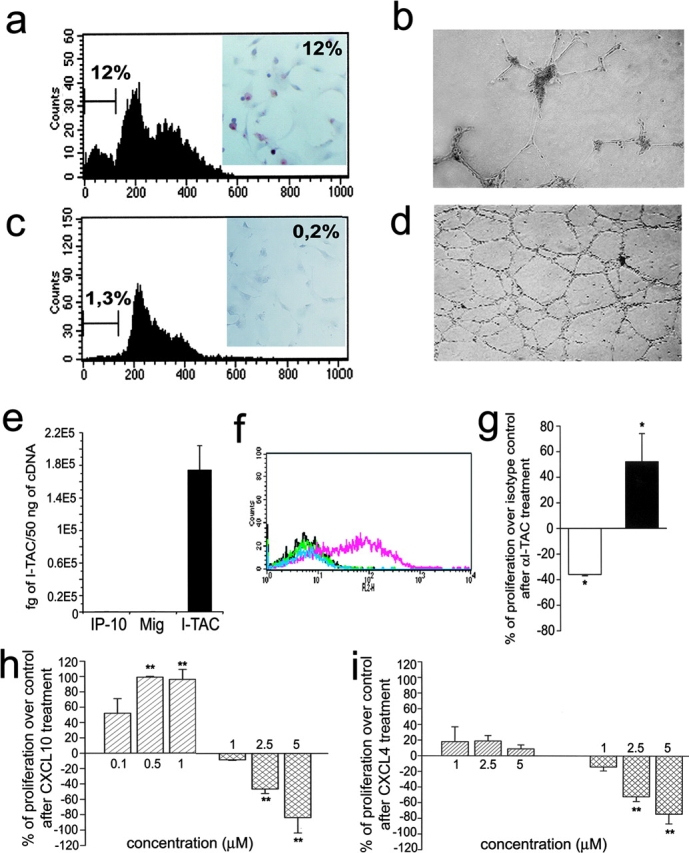
Functional properties of CXCR3-A- and CXCR3-B-transfected cells. (a) Apoptosis in CXCR3-B transfectants as assessed by detection with flow cytometry of the percentage of cells with a sub-G1 DNA content or of cells showing DNA fragmentation by using the TUNEL technique (inset). Results are from one representative of four experiments. (b) In vitro vessel formation capability of CXCR3-B transfectants as assessed by the Matrigel assay. Data are from one representative of four independent experiments. (c) Apoptosis in CXCR3-A–transfected cells (for details see Fig. 4 a). (d) In vitro vessel formation capability of CXCR3-A transfectants (for details see Fig. 4 b). (e) Detection of CXCL11 mRNA expression by HMEC-1 as assessed by real-time quantitative RT-PCR. Columns represent mean values (±SD) of four separate experiments. (f) Flow cytometric analysis on HMEC-1 of CXCL9, CXCL10, and CXCL11 protein. Black line, isotype control mAb; green line, CXCL10; light blue, CXCL9; pink line, CXCL11. Data are from one representative of five independent experiments. (g) Opposite effects of neutralizing anti-CXCL11 mAb on the proliferation of CXCR3-A (open bar) and CXCR3-B (solid bar) transfectants. Results represent mean values (±SD) of the percentage of proliferation over isotype controls obtained in four separate experiments. (h) Opposite effects of CXCL10 on the proliferation of CXCR3-A (left bars) and CXCR3-B (right bars) transfectants. (i) Effects of CXCL4 on the proliferation of CXCR3-A (left bars) and CXCR3-B (right bars) transfectants. Results represent mean values (±SD) of the percentage of proliferation over controls (unstimulated cells) from four separate experiments *, P < 0.05; **, P < 0.001.
The differences in cell growth, viability, and angiogenic capability between CXCR3-A or CXCR3–B transfectants, allowed us to hypothesize the constitutive production by these cells of one or more CXCR3 ligand. By using real-time quantitative RT-PCR and FACS® analysis, high mRNA and protein levels for CXCL11 were indeed observed in all transfectants, including mock (Fig. 3, e and f). Accordingly, the addition in culture of an anti-CXCL11 mAb reduced the spontaneous proliferation of CXCR3-A transfectants, but enhanced the proliferation of CXCR3-B–transfected cells (Fig. 3 g). However, despite the high constitutive expression of CXCL11 on their surface, both CXCR3-A and CXCR3-B transfectants still responded to CXCL10 with increased or decreased proliferation, respectively (Fig. 3 h). Notably, in agreement with the higher affinity of CXCR3-A than CXCR3-B for CXCL10, the inhibition of proliferation on CXCR3-B transfectants could be observed at 2.5–5 μM, whereas proliferative effects on CXCR3-A transfected cells peaked at 500-nM concentrations (Fig. 3 h). Inhibitory effects on the proliferation of CXCR3-B transfectants quite similar to those induced by CXCL10 were also induced by CXCL4, whereas even at higher concentrations the latter chemokine exerted poor, if any, stimulatory effects on the proliferation of CXCR3-A transfectants (Fig. 3 i).
CXCR3-A and CXCR3-B Activate Distinct Signal Transduction Pathways.
Chemotactic activity of chemokine receptors is due to their coupling to heterotrimeric G protein Gi, which mediates intracellular Ca++ flux and can be inactivated by pretreatment with PTX. However, differently from CXCR3-A, CXCR3-B was apparently unable to mediate chemotactic activity. To investigate whether CXCR3-B was also coupled to Gi protein, CXCR3-A and CXCR3-B transfectants were incubated for 24–72 h with 1 μg/ml PTX and thymidine incorporation was assessed. As expected, PTX strongly reduced proliferation of CXCR3-A transfectants in basal conditions as well as after treatment with 500 nM CXCL10 (Fig. 4 a), whereas it did not affect the proliferation of CXCR3-B transfectants either in basal conditions nor after treatment with 4 μM CXCL10 (Fig. 4 b). Effects were also not observed in mock nor in untransfected cells (unpublished data). The signal transduction pathways activated by CXCR3-A or CXCR3-B were then investigated. CXCR3-A transfectants exhibited a rapid, dose-dependent [Ca++]i flux in response to CXCL9, CXCL10, and CXCL11, but poor if any to CXCL4 (Fig. 4 c). By contrast, CXCR3-B transfectants did not mediate calcium flux in response to the four chemokines over a dose ranging from 10 pM to 1 μM (unpublished data).
Figure 4.

Activation of distinct signal transduction pathways in CXCR3-A and CXCR3-B transfectants. (a) Increased proliferative activity in CXCR3-A transfectants is strongly reduced by PTX both in basal conditions and after treatment with 500 nM CXCL10. (b) PTX has no effect on the proliferation of CXCR3-B transfectants both in basal conditions and after treatment with 4 μM CXCL10. Cells were incubated for 60 h with 1 μg/ml PTX and thymidine incorporation was assessed in the last 12 h. Columns represent mean values (±SD) of three separate experiments. *, P < 0.05. (c) Induction by CXCL10, CXCL9, and CXCL11 (1 μM), but not by CXCL4 (1 μM), of Ca++ mobilization in CXCR3-A transfectants. Results are from one representative of four experiments. (d) Effect of CXCL10, CXCL9, CXCL11, and CXCL4 (2 μM) on basal cAMP production and of CXCL10 and CXCL4 (2 μM) on forskolin (1 μM)-stimulated cAMP production in CXCR3-B transfectants. Columns represent mean values (±SD) of six separate experiments. *, P < 0.05; **, P < 0.001. (e) Absence of p21Cip1/Waf1 and p53 regulation by CXCL4 in CXCR3-A transfectants as assessed by real-time quantitative RT-PCR. (f) Up-regulation in CXCR3-B transfectants of p21Cip1/Waf1, but not of p53, mRNA levels by increasing concentrations of CXCL4 as assessed by real-time quantitative RT-PCR.
To identify the alternative pathway involved in CXCR3-B–mediated signal transduction, cAMP production in CXCR3-B transfectants was assessed. Basal cAMP levels were highly increased in comparison with mock (1.9 ± 0.47 vs. 0.69 ± 0.09 pmol/ml; P < 0.05), suggesting chronic activation of adenylyl cyclase activity due to a receptor-mediated mechanism in CXCR3-B transfectants. However, despite their high constitutive cAMP levels, the addition of 1–5 μM CXCL10, CXCL9, CXCL11, or CXCL4 significantly increased intracellular cAMP levels in CXCR3-B transfectants and also potentiated forskolin-induced cAMP levels (Fig. 4 d) with a peak response at 2 h. The increase of cAMP levels induced by CXCL10, CXCL9, CXCL11, or CXCL4 in CXCR3-B transfectants was PTX insensitive (unpublished data). Of note, the same chemokines did not induce any significant increase of cAMP levels in either mock cells nor CXCR3-A transfectants (unpublished data).
The effects of CXCL10 and CXCL4 on the expression of cell cycle regulatory molecules, such as p21CIP1/WAF1 and p53, was then investigated. Basal levels of p53 mRNA were comparable in CXCR3-A and CXCR3-B transfectants, whereas p21 mRNA levels were at least 10 times higher in CXCR3-B compared with CXCR3-A transfectants (Fig. 4, e and f). CXCL4 had virtually no effect on both p21 and p53 mRNA expression by CXCR3-A–transfected cells. By contrast, CXCL4 significantly up-regulated p21 mRNA transcription in CXCR3-B transfectants (Fig. 4 c), but had no effect on the transcription of p53 mRNA (Fig. 4 f). Quite similar effects were induced by the same concentrations of CXCL10 (unpublished data).
Cell Type–specific Transcription of CXCR3-A and CXCR3-B and Their Related Functions.
The expression of CXCR3-A and CXCR3-B mRNA in different cell types, known to express CXCR3, was then investigated. Assessment of CXCR3-A and CXCR3-B mRNA by real-time quantitative RT-PCR revealed the presence of both CXCR3-A and CXCR3-B mRNAs in activated peripheral blood T lymphocytes, CXCR3-A expression being consistently prevalent (Fig. 5 a). By contrast, primary cultures of HMC expressed CXCR3-A, but not CXCR3-B, mRNA, whereas CXCR3-B was the only type of CXCR3 present in primary cultures of HMVEC (Fig. 5 a). In agreement with previous reports (22), CXCL10 strikingly up-regulated HMC proliferation with a peak response at 36 h to a concentration of 1 nM (Fig. 5 b), an effect which was completely inhibited by the anti-CXCR3 mAb 49801.111 (Fig. 5 b). By contrast, CXCL10 induced growth inhibition of both HMVEC (Fig. 5 c) with a peak response occurring at 72 h at a concentration of 100 nM–1 μM. This inhibitory effect was reversed by the addition of the anti-CXCR3 mAb 49801.111 (Fig. 5, c and d). CXCL4 induced comparable inhibitory effects on the growth of HMVEC (Fig. 5, c and d), which were also inhibited by the anti-CXCR3 mAb 49801.111 (Fig. 5 c), whereas CXCL9 and CXCL11 usually had lower inhibitory effects (unpublished data).
Figure 5.

Cellular expression of CXCR3-A (solid bars) and CXCR3-B (hatched bars) and their opposite effects on the proliferation of different cell types. (a) CXCR3-A and CXCR3-B expression by peripheral blood–activated T cells, HMC, and HMVEC as assessed by real-time quantitative RT-PCR. Columns represent mean values (±SD) of eight separate experiments. (b) Up-regulation of HMC growth by CXCL10 and its inhibition by an anti-CXCR3 mAb. (c) Inhibitory effect of CXCL4 and of CXCL10 on the proliferation of HMVEC. The inhibitory effects of both CXCL4 and CXCL10 are reverted by an anti-CXCR3 mAb. (d) Selective expression of CXCR3-B mRNA by the ACHN cell line as detected by quantitative RT-PCR. (e) Surface expression of CXCR3 on the membrane of ACHN cells as detected by FACS® analysis performed with the 49801.111 mAb. (f) Demonstration of a single type of surface receptor in ACHN cells with an IC50 for CXCL10 and CXCL4 comparable to that observed for CXCR3-B. (g) Inhibitory effect of CXCL4 and of CXCL10 on the proliferation of ACHN cells. The inhibitory effects of both CXCL4 and CXCL10 are reverted by an anti-CXCR3 mAb. (h) Effect of CXCL10 (2 μM) on basal cAMP production and of CXCL10 (2 μM) on forskolin (1 μM)-stimulated cAMP production in CXCR3-B transfectants. Columns represent mean values (±SD) of six separate experiments. *, P < 0.05; **, P < 0.001. (i) Up-regulation in CXCR3-B transfectants of p21Cip1/Waf1, but not of p53, mRNA levels by increasing concentrations of CXCL4 as assessed by real-time quantitative RT-PCR. Symbols represent mean values (±SD) of four separate experiments. *, P < 0.01. ▪, CXCL4 plus 20 μg/ml isotype mAb; □, CXCL4 plus 20 μg/ml anti-CXCR3 mAb; •, CXCL10 plus 15 μg/ml isotype mAb; ○, CXCL10 plus 15 μg/ml anti-CXCR3 mAb (clone 49801.111).
To provide evidence that CXCR3-B was expressed and functional on other cell types, mRNA levels for CXCR3-A and CXCR3-B were assessed by quantitative RT-PCR on different human cell lines. Among the cell types analyzed, the highest levels of CXCR3-B were observed in the renal carcinoma ACHN cell line, where CXCR3-A mRNA was not detectable (Fig. 5 d). Interestingly, the renal carcinoma ACHN cell line is well known for its property to undergo growth inhibition in response to IFNs (33). FACS® analysis using the 49801.111 mAb demonstrated surface expression of CXCR3 on ∼30% of the cells (Fig. 5 e). The binding assay performed on the ACHN cell line using CXCL9, CXCL10, CXCL11, and CXCL4 demonstrated a single class of high affinity binding sites for CXCL10 (IC50 = 2.30 ± 1.44 nM; Fig. 5 f), CXCL4 (IC50 = 6.83 ± 4.37 nM; Fig. 5 f), CXCL9 (IC50 = 88.45 ± 69.3 nM; unpublished data), and CXCL11 (IC50 = 49.58 ± 11.27 nM; unpublished data), characterized by an IC50 comparable to the one showed by the same ligands for CXCR3-B. CXCL10 and CXCL4 induced growth inhibition of the ACHN cell line with a peak response occurring at 72 h at micromolar concentrations (Fig. 5 g). The inhibitory effect of both CXCL10 and CXCL4 was reversed by the addition of anti-CXCR3 mAb 49801.111 (Fig. 5 g). Additional evidence that ACHN cells selectively expressed functional CXCR3-B was provided by the demonstration that they responded to 2 μM CXCL10, CXCL9, CXCL11, or CXCL4 with a significant increase of intracellular cAMP levels as well as potentiation of forskolin-induced cAMP levels (Fig. 5 h). Finally, treatment of the ACHN cell line with CXCL10 and CXCL4 induced a significant p53-independent up-regulation of p21 mRNA transcription (Fig. 5 i).
CXCR3-B Protein Expression by Endothelial Cells of Human Neoplastic Tissues.
To investigate the expression of CXCR3-B in vivo, mAbs able to selectively recognize CXCR3-B apart from CXCR3-A were raised against a peptide corresponding to the NH2-terminal region of the CXCR3-B receptor. When tested by immunohistochemistry, two mAbs (PL1 and PL2) stained CXCR3-B, but not mock nor CXCR3-A, transfectants (Fig. 6 , a–c). Furthermore, primary cultures of HMC did not react with the same anti–CXCR3-B mAbs (Fig. 6 d), whereas primary cultures of HMVEC (Fig. 6 e), as well as the ACHN cell line (Fig. 6 f), were positively stained. The anti–CXCR3-B mAbs also stained vessels from neoplastic tissues (Fig. 6 h, k, m–o, and r), but poorly or not from their normal counterparts (Fig. 6, g and p). Preadsorption with the peptide used to generate mAbs completely abolished the staining (Fig. 6 l). Moreover, comparison of immunostaining on the same tumor specimens between the anti–CXCR3-B mAb and the anti-CXCR3 mAb 49801.111, which reacts with a portion of the receptor shared by both CXCR3-B and CXCR3-A (Fig. 6, h vs. i and q vs. r), as well as double label immunohistochemistry with anti–CXCR3-B plus vWf (Fig. 6 o), demonstrated that staining of neoplastic tissues was due to CXCR3-B expression by their endothelial cells.
Figure 6.
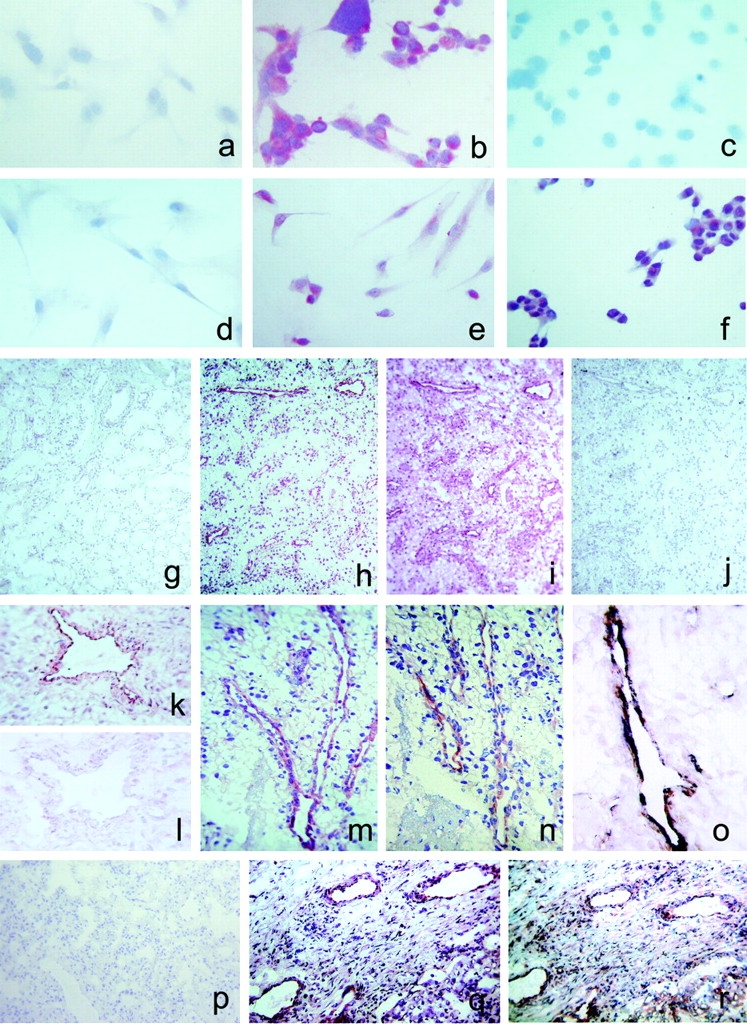
Detection of CXCR3-B protein expression by different types of cell cultures and by endothelial cells of human neoplastic tissues by CXCR3-B–specific mAbs. (a) Absence of reactivity in mock transfectants stained with an anti-CXCR3-B mAb. ×100. (b) Intense staining of CXCR3-B transfectants with an anti–CXCR3-B mAb. ×100. (c) Absence of reactivity in CXCR3-A transfectants stained with an anti–CXCR3-B mAb. ×100. (d) Absence of reactivity in primary cultures of HMC stained with an anti–CXCR3-B mAb. ×100. (e) Positive staining of primary cultures of HMVEC as well as the ACHN cell line (f) with an anti–CXCR3-B mAb. ×100. (g) Absence of reactivity in normal human renal tissue stained with PL1 anti–CXCR3-B mAb. ×10. (h) Staining with the same mAb of endothelial cells in a specimen of renal cell carcinoma. (i) Staining of both endothelial and tumor cells with the anti-CXCR3 mAb 49801.111 tested in an adjacent section. (j) Absence of reactivity in an adjacent section stained with an isotype-matched control mAb. (k and l) Reactivity of endothelial cells from the same renal carcinoma specimen before and after adsorption of PL1 mAb with the peptide used for mouse immunization. (m) Reactivity with PL1 mAb of endothelial cells from a group of vessels in a renal cell carcinoma specimen as detected at a higher power magnification. ×250. (n) Staining of an adjacent section with PL2 anti–CXCR3-B mAb. (o) Double label immunohistochemistry for CXCR3-B (red) and vWf (blue-gray), showing costaining (brown). (p) Absence of CXCR3-B reactivity in normal human lung tissue stained with PL1 anti–CXCR3-B mAb. (q) Staining with the same mAb of endothelial cells in a specimen of NSCLC. (r) Staining of both endothelial cells and other cell types with the anti-CXCR3 mAb 49801.111 in an adjacent section.
Discussion
The chemokines CXCL9, CXCL10, and CXCL11 exhibit several activities, including the induction of proliferation of HMC (22, 23) and the inhibition of endothelial cell growth (4–10). The results of this study provide evidence that these opposite functions can be explained by the interaction of these chemokines with two distinct receptors. In addition to the classic form of CXCR3 receptor, here renamed CXCR3-A, a novel CXCR3 receptor variant, named CXCR3-B, derived from an alternative splicing between the same donor site used for CXCR3-A and a novel acceptor site localized within the intron of the CXCR3 gene, was identified. Transfection of the two receptors in an HMEC-1 resulted in differences in cell morphology, viability, proliferative activity, apoptosis, and angiogenic capability. CXCR3-A overexpression enhanced cell viability and proliferation, induced rescue from apoptotic cell death, and increased ability of vessel formation in vitro. By contrast, overexpression of CXCR3-B resulted in altered growth properties and massive apoptotic cell death. These opposite effects were related to constitutive expression of CXCL11 on the surface of all transfectants, as demonstrated by the fact that the addition of an anti-CXCL11 mAb reduced the spontaneous proliferation of CXCR3-A–transfected cells and enhanced the proliferation of CXCR3-B–transfected cells. However, despite the high constitutive expression of CXCL11 on their surface, CXCR3-A– and CXCR3-B–transfected cells still responded to high concentrations of CXCL10 with further increase or decrease of their proliferation, respectively. These findings provide a convincing explanation for both the proliferation induced by CXCL9, CXCL10, and CXCL11 on HMC, which selectively express CXCR3-A (22, 23), and the inhibitory activity exerted by the same chemokines on the proliferation of HMVEC (11, 12, 14, 16), which selectively express CXCR3-B. CXCR3-A showed higher affinity than CXCR3-B for all three chemokines, thus explaining why, in the case of coexpression of the two receptors on the same cell type, CXCR3-A–mediated effects become prevalent. These data, together with the demonstration that presently available anti-CXCR3 Abs react with both receptors (unpublished data), account for why the existence of the second receptor had not previously been recognized.
An additional and unexpected finding emerging from this study is the demonstration that CXCR3-B acts as a functional receptor for CXCL4. CXCL4 is also a member of CXC chemokines released in high concentrations from activated platelets (35), which is involved in the control of proliferation of different cell types, such as endothelial cells or hematopoietic progenitors (10, 14–16). Because of their heparin-binding motifs, CXCL4 and CXCL10 have been shown to interfere with the binding of FGF-2 or VEGF165 to their receptors by impairing the growth factor's ability to form homodimers, competing with heparan sulfate proteoglycans on the cell surface, or by directly binding to these growth factors (36–37). Yet, a large body of evidence indicates that another, still unidentified, receptor-mediated mechanism exists. CXCL4 can antagonize EGF-mediated endothelial cell proliferation by inducing an impairment of p21CIP1/WAF1 down-regulation and this effect occurred in a model system in which the stimulation is glycosaminoglycan independent (38). Furthermore, the interference by CXCL10 and CXCL4 with EGF-mediated calpain activation led to down-modulation of EGF-mediated migration of fibroblasts without affecting early postreceptor events, but up-regulating cAMP levels (39). More importantly, the endothelial cell stimulatory activity of VEGF121, an endothelial cell mitogen that lacks both heparin affinity and the property to bind CXCL4, was susceptible to CXCL4 inhibition (29). CXCL4 can act even on cells lacking heparan sulphates on their surface membranes (37), and CXCL4 peptides made unable to bind heparan sulphates still inhibited endothelial (40) and hematopoietic cell growth (41), whereas CXCL4 peptides showing heparin-binding properties did not consistently exhibit inhibitory activity (42). Finally, at concentrations inhibiting cell proliferation, CXCL4 significantly reduced FGF2-induced ERK activation, whereas the PI3K pathway was not affected, thus excluding a simply competitive mechanism (37). Taken together, these findings suggest that CXCL4 delivers its specific inhibitory signals through a not yet identified receptor. It has also been reported that like CXCR3 (11), the putative CXCL4 receptor is expressed by microvascular endothelial cells only when they proliferate (43). The results of this study provide evidence that CXCR3-B may represent the not yet identified functional receptor for CXCL4. By contrast, CXCR3-B did not react with any of the other 17 chemokines tested, including CCL21, which reacts with both CCR7 and CXCR3 in the mouse (44), as also shown by its activity on microglial cells in CCR7 knockouts (45). However, differently from its murine equivalent, human CCL21 was found to be unable to bind CXCR3 and exert any angiostatic activity (13, 27). Our results are consistent with these findings and support the concept that CCL21 binding to CXCR3 is a mouse-restricted phenomenon. Thus, at least in humans, CXCR3-B seems to act as the functional receptor for the four angiostatic chemokines, CXCL9, CXCL10, CXCL11, and CXCL4.
The opposite biological activities mediated by CXCR3-A and CXCR3-B suggest that the two receptors may trigger different signal transduction pathways. Chemokine receptors are G protein–coupled receptors (GPCRs) that mediate chemotactic activity and Ca++ flux induction through the activation of PTX-sensitive Gi proteins (1, 2). Accordingly, increased proliferative activity in CXCR3-A transfectants could be reduced by PTX treatment. However, PTX had no effect on the proliferation and survival of CXCR3-B transfectants. Furthermore, only CXCR3-A transfectants exhibited calcium influx in response to CXCR3 ligands. This finding is consistent with the results of previous reports showing that both activated lymphocytes and HMC respond with calcium influx to CXCL9, CXCL10, and CXCL11 (1, 2, 19, 22), whereas endothelial cells do not (12). Multifunctional coupling is common to many GPCRs and has been previously described for the putative CXCL10 receptor (46). Together, the lack of calcium influx in response to ligands and PTX insensitivity suggests the coupling of CXCR3-B to other types of G proteins. This hypothesis was further supported by the selective activation of adenylyl cyclase and consequent increase of intracellular cAMP levels in response to CXCL10, CXCL9, CXCL11, and CXCL4 in CXCR3-B, but not in CXCR3-A, transfectants, a property usually related to linkage of GPCR to Gs proteins (47). Of note, the increase of cAMP levels in response to CXCL10 and CXCL4 interaction with an unidentified receptor had previously been found to be associated with the inhibition of cellular proliferation and chemotaxis in hematopoietic progenitors and fibroblasts (16, 39). More recent data configure the adenylate cyclase pathway as an important pharmacological target for the inhibition of angiogenesis, because cAMP up-regulation in endothelial cells suppresses their migration and proliferation through the enhancement of apoptosis (48). Accordingly, CXCL10 increases intracellular cAMP levels and inhibits the angiogenic activity of the GPCR-Kaposi's sarcoma-associated herpes virus, a property which is related to a specific sequence of the NH2-terminal region of the viral receptor (49, 50). CXCR3-A and CXCR3-B differ for 52 amino acids at the NH2 end, leading to a change of extracellular conformation that can account for the coupling of CXCR3-B to different type(s) of G proteins (47). Indeed, the active conformation of GPCR likely consists of multiple subspecies that may favor different signaling pathways or regulatory events (47). Finally, CXCR3-B, but not CXCR3-A, transfectants responded to CXCL10 and CXCL4 with increased transcription of the cell cycle inhibitor p21CIP1/WAF-1. This is of importance because a p53-independent up-regulation of p21 levels is responsible for the antiangiogenic property of CXCL4 (38) as well as the growth inhibitory effects of IFNs on different cell types (51). Interestingly, the induction of p21 transcription is one of the mechanisms involved in the antiproliferative effect induced by Gs-mediated intracellular cAMP increase (52).
The results of this study also provide evidence for the in vivo expression of CXCR3-B and thus for its possible physiological relevance in angiogenesis. Indeed, two mAbs selectively recognizing CXCR3-B were able to react with endothelial cells from neoplastic tissues, but not with those from their normal tissue counterparts. In conclusion, our findings not only describe a novel chemokine receptor, but also demonstrate that it is the previously unidentified functional receptor of CXCL4 and a common mediator of the effects of the four angiostatic CXC chemokines. Further characterization of the CXCR3 gene regulation may refine the ability to inhibit or promote blood vessel growth and allow the development of new effective therapeutic strategies.
Acknowledgments
We thank Dr. Francisco J. Candal, Dr. Edwin Ades (Centers for Disease Control), and Dr. Thomas Lawley (Emory University) for providing the HMEC-1 cell line and Dr. Chung-Her Jenh (Department of Immunology, Schering-Plough Research Institute) for providing us with the pCEP4 expression vector containing the CXCR3-A sequence. We thank Dr. Mario Maggi and Dr. Gabriella Torcia (Florence, Italy) for critical review of the manuscript.
The experiments reported in this paper were supported by funds from Associazione Italiana per la Ricerca sul Cancro, from Ministero della Sanità, and from Ministero dell'Università e della Ricerca Scientifica.
Note added in proof: The clone ID19600412004045 obtained from the cDNA library NFLHAL1 is now available at http://www.celeradiscoverysystem.com
Footnotes
Abbreviations used in this paper: CXCR3, CXC chemokine receptor 3; GPCR, G protein–coupled receptor; HMC, human mesangial cells; HMEC-1, human microvascular endothelial cell line-1; HMVEC, human microvascular endothelial cells; PTX, pertussin toxin; RACE, rapid amplification of cDNA ends; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
References
- 1.Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity. 12:121–127. [DOI] [PubMed] [Google Scholar]
- 2.Rossi, D., and A. Zlotnik. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242. [DOI] [PubMed] [Google Scholar]
- 3.Grone, H.J., C.D. Cohen, E. Grone, C. Schmidt, M. Kretzler, D. Schlondorff, and P.J. Nelson. 2002. Spatial and temporally restricted expression of chemokines and chemokine receptors in the developing human kidney. J. Am. Soc. Nephrol. 13:957–967. [DOI] [PubMed] [Google Scholar]
- 4.Belperio, J.A., M.P. Keane, D.A. Arenberg, C.L. Addison, J.E. Ehlert, M.D. Burdick, and R.M. Strieter. 2000. CXC chemokines in angiogenesis. J. Leukoc. Biol. 68:1–8. [PubMed] [Google Scholar]
- 5.Homey, B., A. Müller, and A. Zlotnik. 2002. Chemokines: agents for the immunotherapy of cancer? Nat. Rev. Immunol. 2:175–184. [DOI] [PubMed] [Google Scholar]
- 6.Strieter, R.M., J.A. Belperio, and M.P. Keane. 2002. CXC chemokines in angiogenesis related to pulmonary fibrosis. Chest. 122:298S–301S. [DOI] [PubMed] [Google Scholar]
- 7.Arenberg, D.A., S.L. Kunkel, P.J. Polverini, S.B. Morris, M.D. Burdick, M.C. Glass, D.T. Taub, M.D. Iannettoni, R.I. Whyte, and R.M. Strieter. 1996. Interferon-γ–inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J. Exp. Med. 184:981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sgadari, C., J.M. Farber, A.L. Angiolillo, F. Liao, J. Teruya-Feldstein, P.R. Burd, L. Yao, G. Gupta, C. Kanegane, and G. Tosato. 1997. Mig, the monokine induced by interferon-gamma, promotes tumor necrosis in vivo. Blood. 89:2635–2643. [PubMed] [Google Scholar]
- 9.Tanaka, T., Y. Manome, P. Wen, D.W. Kufe, and H.A. Fine. 1997. Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat. Med. 3:437–442. [DOI] [PubMed] [Google Scholar]
- 10.Maione, T.E., G.S. Gray, J. Petro, A.J. Hunt, A.L. Donner, S.I. Bauer, H.F. Carson, and R.J. Sharpe. 1990. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science. 247:77–79. [DOI] [PubMed] [Google Scholar]
- 11.Romagnani, P., F. Annunziato, L. Lasagni, E. Lazzeri, C. Beltrame, M. Francalanci, M. Uguccioni, G. Galli, L. Cosmi, L. Maurenzig, et al. 2001. Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J. Clin. Invest. 107:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salcedo, R., J.H. Resau, D. Halverson, E.A. Hudson, M. Dambach, D. Powell, K. Wasserman, and J.J. Oppenheim. 2000. Differential expression and responsiveness of chemokine receptors (CXCR1-3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J. 14:2055–2064. [DOI] [PubMed] [Google Scholar]
- 13.Arenberg, D.A., A. Zlotnick, S.R. Strom, M.D. Burdick, and R.M. Strieter. 1999. The murine CC chemokine, 6C-kine, inhibits tumor growth and angiogenesis in a human lung cancer SCID mouse model. Cancer Immunol. Immunother. 49:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luster, A.D., S.M. Greenberg, and P. Leder. 1995. The IP-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J. Exp. Med. 182:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strieter, R.M., P.J. Polverini, S.L. Kunkel, D.A. Arenberg, M.D. Burdick, J. Kasper, J. Dzuiba, J. Van Damme, A. Walz, D. Marriott, et al. 1995. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 270:27348–27357. [DOI] [PubMed] [Google Scholar]
- 16.Aronica, S.M., C. Mantel, R. Gonin, M.S. Marshall, A. Sarris, S. Cooper, N. Hague, X.F. Zhang, and H.E. Broxmeyer. 1995. Interferon-inducible protein 10 and macrophage inflammatory protein-1 alpha inhibit growth factor stimulation of Raf-1 kinase activity and protein synthesis in a human growth factor-dependent hematopoietic cell line. J. Biol. Chem. 270:21998–22007. [DOI] [PubMed] [Google Scholar]
- 17.Bonecchi, R., G. Bianchi, P.P. Bordignon, D. D'Ambrosio, R. Lang, A. Borsetti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, et al. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sallusto, F., D. Lenig, C.R. Mackay, and A. Lanzavecchia. 1998. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 187:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loetscher, M., B. Gerber, P. Loetscher, S.A. Jones, L. Piali, I. Clark-Lewis, M. Baggiolini, and B. Moser. 1996. Chemokine receptor specific for IP-10 and Mig: structure, function, and expression in activated T lymphocytes. J. Exp. Med. 184:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janatpour, M.J., S. Hudak, M. Sathe, J.D. Sedgwick, and L.M. McEvoy. 2001. Tumor necrosis factor–dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J. Exp. Med. 194:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penna, G., S. Sozzani, and L. Adorini. 2001. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J. Immunol. 167:1862–1866. [DOI] [PubMed] [Google Scholar]
- 22.Romagnani, P., C. Beltrame, F. Annunziato, L. Lasagni, M. Luconi, G. Galli, L. Cosmi, E. Maggi, M. Salvadori, C. Pupilli, et al. 1999. Role for interactions between IP-10/Mig and their receptor (CXCR3) in proliferative glomerulonephritis. J. Am. Soc. Nephrol. 10:2518–2526. [DOI] [PubMed] [Google Scholar]
- 23.Romagnani, P., E. Lazzeri, L. Lasagni, C. Mavilia, C. Beltrame, M. Francalanci, M. Rotondi, F. Annunziato, L. Maurenzig, L. Cosmi, et al. 2002. IP-10 and Mig production by glomerular cells in human proliferative glomerulonephritis and regulation by nitric oxide. J. Am. Soc. Nephrol. 13:53–64. [DOI] [PubMed] [Google Scholar]
- 24.Bonacchi, A., P. Romagnani, R.G. Romanelli, E. Efsen, F. Annunziato, L. Lasagni, M. Francalanci, M. Serio, G. Laffi, M. Pinzani, et al. 2001. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J. Biol. Chem. 276:9945–9954. [DOI] [PubMed] [Google Scholar]
- 25.Pupilli, C., L. Lasagni, P. Romagnani, F. Bellini, M. Mannelli, N. Misciglia, C. Mavilia, U. Vellei, D. Villari, and M. Serio. 1999. Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. J. Am. Soc. Nephrol. 10:245–255. [DOI] [PubMed] [Google Scholar]
- 26.Romagnani, P., M. Rotondi, E. Lazzeri, L. Lasagni, M. Francalanci, A. Buonamano, S. Milani, P. Vitti, L. Chiovato, M. Tonacchera, et al. 2002. Expression of IP-10/CXCL10 and MIG/CXCL9 in the thyroid and increased levels of IP-10/CXCL10 in the serum of patients with recent-onset Graves' disease. Am. J. Pathol. 161:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenh, C.H., M.A. Cox, H. Kaminski, M. Zhang, H. Byrnes, J. Fine, D. Lundell, C.C. Chou, S.K. Narula, and P.J. Zavodny. 1999. Cutting edge: species specificity of the CC chemokine 6Ckine signaling through the CXC chemokine receptor CXCR3: human 6Ckine is not a ligand for the human or mouse CXCR3 receptors. J. Immunol. 162:3765–3769. [PubMed] [Google Scholar]
- 28.Ades, E.W., F.J. Candal, R.A. Swerlick, V.G. George, S. Summers, D.C. Bosse, and T.J. Lawley. 1992. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 99:683–690. [DOI] [PubMed] [Google Scholar]
- 29.Gengrinovitch, S., S.M. Greenberg, T. Cohen, H. Gitay-Goren, P. Rockwell, T.E. Maione, B.Z. Levi, and G. Neufeld. 1995. Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J. Biol. Chem. 270:15059–15065. [DOI] [PubMed] [Google Scholar]
- 30.Pugliese, G., F. Pricci, G. Romeo, F. Pugliese, P. Mene, S. Giannini, B. Cresci, G. Galli, C.M. Rotella, H. Vlassara, et al. 1997. Upregulation of mesangial growth factor and extracellular matrix synthesis by advanced glycation end products via a receptor-mediated mechanism. Diabetes. 46:1881–1887. [DOI] [PubMed] [Google Scholar]
- 31.De Lean, A., P.J. Munson, and D. Rodbard. 1978. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 235:E97–E102. [DOI] [PubMed] [Google Scholar]
- 32.Van Riper, G., S. Siciliano, P.A. Fischer, R. Meurer, M.S. Springer, and H. Rosen. 1993. Characterization and species distribution of high affinity GTP-coupled receptors for human RANTES and monocyte chemoattractant protein 1. J. Exp. Med. 177:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan, A.I., and P.C. Keng. 1987. Potentiation of radiation cytotoxicity by recombinant interferons, a phenomenon associated with increased blockage at the G2-M phase of the cell cycle. Cancer Res. 47:4338–4341. [PubMed] [Google Scholar]
- 34.Romagnani, P., F. Annunziato, R. Manetti, C. Mavilia, L. Lasagni, C. Manuelli, G.B. Vannelli, V. Vanini, E. Maggi, C. Pupilli, et al. 1998. High CD30 ligand expression by epithelial cells and Hassal's corpuscles in the medulla of human thymus. Blood. 91:3323–3332. [PubMed] [Google Scholar]
- 35.Files, J.C., T.W. Malpass, E.K. Yee, J.L. Ritchie, and L.A. Harker. 1981. Studies of human platelet α-release in vivo. Blood. 58:607–618. [PubMed] [Google Scholar]
- 36.Perollet, C., Z.C. Han, C. Savona, J.B. Caen, and A. Bikfalvi. 1998. Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2 dimerization. Blood. 91:3289–3299. [PubMed] [Google Scholar]
- 37.Sulpice, E., M. Bryckaert, J. Lacour, J.O. Contreres, and G. Tobelem. 2002. Platelet factor 4 inhibits FGF2-induced endothelial cell proliferation via the extracellular signal-regulated kinase pathway but not by the phosphatidylinositol 3-kinase pathway. Blood. 100:3087–3094. [DOI] [PubMed] [Google Scholar]
- 38.Gentilini, G., N.C. Kirschbaum, J.A. Augustine, R.H. Aster, and G.P. Visenti. 1999. Inhibition of human umbilical vein endothelial cell proliferation by the CXC chemokine, platelet factor 4 (PF-4), is associated with impaired downregulation of p21Cip1/WAF1. Blood. 93:25–33. [PubMed] [Google Scholar]
- 39.Shiraha, H., A. Glading, K. Gupta, and A. Wells. 1999. IP-10 inhibits epidermal growth factor–induced motility by decreasing epidermal growth factor receptor–mediated calpain activity. J. Cell Biol. 146:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maione, T.E., G.S. Gray, A.J. Hunt, and R.J. Sharpe. 1991. Inhibition of tumor growth in mice by an analogue of platelet factor 4 that lacks affinity for heparin and retains potent angiostatic activity. Cancer Res. 51:2077–2083. [PubMed] [Google Scholar]
- 41.Lecomte-Raclet, L., M. Alemany, A. Sequeira-Le Grand, J. Amiral, G. Quentin, A.M. Vissac, J.P. Caen, and Z.C. Han. 1998. New insights into the negative regulation of hematopoiesis by chemokine platelet factor 4 and related peptides. Blood. 91:2772–2780. [PubMed] [Google Scholar]
- 42.Jouan, V., X. Canron, M. Alemany, J.P. Caen, G. Quentin, J. Plouet, and A. Bikfalvi. 1999. Inhibition of in vitro angiogenesis by platelet factor-4-derived peptides and mechanism of action. Blood. 94:984–993. [PubMed] [Google Scholar]
- 43.Hansell, P., T.E. Maione, and P. Borgstrom. 1995. Selective binding of platelet factor 4 to regions of active angiogenesis in vivo. Am. J. Physiol. 269:H829–H836. [DOI] [PubMed] [Google Scholar]
- 44.Soto, H., W. Wang, R.M. Strieter, N.G. Copeland, D.J. Gilbert, N.A. Jenkins, J. Hedrick, and A. Zlotnik. 1998. The CC chemokine 6Ckine binds the CXC chemokine receptor CXCR3. Proc. Natl. Acad. Sci. USA. 95:8205–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rappert, A., K. Biber, C. Nolte, M. Lipp, A. Schubel, B. Lu, N.P. Gerard, C. Gerard, H.W. Boddeke, and H. Kettenmann. 2002. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl-current and chemotaxis in murine microglia. J. Immunol. 168:3221–3226. [DOI] [PubMed] [Google Scholar]
- 46.Maghazachi, A.A., B.S. Skalhegg, B. Rolstad, and A. Al-Aoukaty. 1997. Interferon-inducible protein-10 and lymphotactin induce the chemotaxis and mobilization of intracellular calcium in natural killer cells through pertussis toxin-sensitive and -insensitive heterotrimeric G-proteins. FASEB J. 11:765–774. [DOI] [PubMed] [Google Scholar]
- 47.Marinissen, M.J., and J.S. Gutkind. 2001. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22:368–376. [DOI] [PubMed] [Google Scholar]
- 48.Kim, S., M. Bakre, H. Yin, and J.A. Varner. 2002. Inhibition of endothelial cell survival and angiogenesis by protein kinase A. J. Clin. Invest. 110:933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho, H.H., D. Du, and M.C. Gershengorn. 1999. The N terminus of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor is necessary for high affinity chemokine binding but not for constitutive activity. J. Biol. Chem. 274:31327–31332. [DOI] [PubMed] [Google Scholar]
- 50.Couty, J.P., E. Geras-Raaka, B.B. Weksler, and M.C. Gershengorn. 2001. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor signals through multiple pathways in endothelial cells. J. Biol. Chem. 276:33805–33811. [DOI] [PubMed] [Google Scholar]
- 51.Zhou, Y., S. Wang, B.G. Yue, A. Gobl, and K. Oberg. 2002. Effects of interferon alpha on the expression of p21cip1/waf1 and cell cycle distribution in carcinoid tumors. Cancer Invest. 20:348–356. [DOI] [PubMed] [Google Scholar]
- 52.Lee, T.H., L.Y. Chuang, and W.C. Hung. 2000. Induction of p21WAF1 expression via Sp1-binding sites by tamoxifen in estrogen receptor-negative lung cancer cells. Oncogene. 19:3766–3773. [DOI] [PubMed] [Google Scholar]