

Abstract
Activation-induced cell death in macrophages has been observed, but the mechanism remains largely unknown. Activation-induced cell death in macrophages can be independent from caspases, and the death of activated macrophages can even be triggered by the pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD). Here, we show that this type of macrophage death can occur in the septic mouse model and that toll-like receptor (TLR)-2 or TLR4 signaling is required in this process. We conclude that Nur77 is involved in the macrophage death because Nur77 expression correlates with cell death, and cell death is reduced significantly in Nur77-deficient macrophages. The extracellular signal–regulated kinase pathway, which is downstream of TLR2 or TLR4, and myocyte-specific enhancer binding factor 2 (MEF2) transcription factor activity, which is up-regulated by zVAD, are required for Nur77 induction and macrophage death. Reporter gene analysis suggests that Nap, Ets, Rce, and Sp1 sites in the Nur77 promoter are regulated by TLR4 signaling and that MEF2 sites in the Nur77 promoter are regulated by zVAD treatment. MEF2 transcription factors are constitutively expressed and degraded in macrophages, and zVAD increases MEF2 transcription factor activity by preventing the proteolytic cleavage and degradation of MEF2 proteins. This paper delineates the dual signaling pathways that are required for Nur77 induction in macrophages and demonstrates a role of Nur77 in caspase-independent cell death.
Keywords: AICD, LPS, zVAD, MEF2
Introduction
Programmed cell death, often referred to as apoptosis, is a genetically regulated, self-destructive cellular process found in metazoans. It eliminates individual cells when they are no longer needed in development, tissue remodeling, or immune regulation, as well as in various diseases (1). In immune cells, apoptosis is a key phenomenon in what is referred to as activation-induced cell death (AICD).* During T cell maturation, AICD is a mechanism for T cells to negatively select thymocyte clones through TCR-mediated stimuli (2, 3). AICD in B cell lymphocytes is involved in antigen tolerance, which is initiated by antigen interaction with the B cell receptor (4). A similar term, activation-induced apoptosis, has been used to describe IFN-γ and zymosan- or phorbol ester–induced death of macrophages (5). AICD in T and B cells has been a focus of intense study and the underlying molecular mechanisms have been well-described (3, 4, 6, 7). However, little is known about the mechanism of AICD in macrophages.
Macrophage death occurs in vitro and in vivo, and has many different causes. Phagocytosis of infectious microorganisms by macrophages is a part of the host response to infection (8). Infectious microbial pathogens can be killed by macrophages, and their components are strong stimuli of macrophage activation. Activation of macrophages can lead to their own death if the macrophages are exposed to viruses, such as bovine herpes virus-1, or cytokines, such as IFN-γ and -α (2, 9–11). Priming macrophages with IFN-γ is known to be required for macrophages to eliminate highly resistant microorganisms (12). Although IFN-γ–primed macrophages are crucial for host defense, they are also dangerous because they can cause extensive local damage if uncontrolled (13, 14). Teleological reasoning holds that the evolution of susceptible death in these cells may be a mechanism to control activated macrophages. The cell death of activated macrophages could be a mechanism to control the level of inflammation.
Activation of macrophages with various stimuli, including the gram-negative bacterial cell wall product LPS at physiologically relevant concentrations, does not lead to cell death in vitro. Our recent work shows that treatment of macrophages with a pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD), which has no effect on the viability of resting macrophages, induces the death of activated macrophages (15). We have demonstrated that this cell death is a caspase-independent process. Macrophage death induced by LPS + zVAD and LPS + IFN-γ may share some common mechanisms because LPS + IFN-γ–induced macrophage death is also independent of the known proapoptotic caspases (unpublished data). LPS + IFN-γ–induced macrophage death appears to be a complicated process because both direct and indirect mechanisms are likely involved (9, 10, 16). Nitric oxide, induced by LPS + IFN-γ, is an indirect trigger of macrophage death, but a nitric oxide–independent mechanism also exists (9, 10, 16). LPS + zVAD–induced macrophage death was quicker and more effective than LPS + IFN-γ–induced cell death, and was triggered directly by targeting intracellular molecules. Because LPS + zVAD–induced cell death of macrophages could be simpler than other in vitro systems, such as LPS + IFN-γ–induced macrophage death, we have used this system to study the mechanism of macrophage death. In addition, this system may be used to address the mechanism of caspase-independent cell death. Studying zVAD-promoted cell death of activated macrophages will also help in understanding caspases as drug targets for treating inflammatory diseases (17). Recent papers have shown that a pan-caspase inhibitor improves survival in a murine sepsis model (18). Caspases, as targets of gene therapy in rheumatoid arthritis, have been reported to have a therapeutic advantage (19). The effect of zVAD on the viability of macrophages could influence the outcome of these therapeutic interventions.
There is no doubt that caspases play a key role in apoptosis of many different cells. However, caspases are not indispensable in promoting cell death, as evidenced by the failure of pan-caspase inhibitors (such as zVAD and boc-D) to prevent cell death in some cell death programs (20–23). In many cases, zVAD completely blocked the nuclear events associated with apoptosis, but failed to prevent cell death (20–22). In our work, zVAD even promoted cell death (15). Thus, there seems to be parallel caspase-dependent and -independent cell death pathways (24). The mechanism of caspase-independent cell death is poorly defined. Death of activated macrophages promoted by pan-caspase inhibitors could be an extreme case of caspase-independent cell death.
Apoptosis can be mediated through multiple pathways. Mitochondrial release of apoptogenic factors, such as cytochrome c, Smac/DIABLO, and apoptosis-inducing factor, occurs in many apoptotic processes (25). The release of these factors from mitochondria can be regulated by caspases, Bcl-2 family proteins, translocation of tumor suppressor p53, or the orphan nuclear receptor Nur77 (also known as TR3 or NGFI-B; reference 26). Nur77 was identified as an immediate early gene that is responsive to TCR signaling under conditions leading to thymocyte apoptosis, or serum or phorbol ester stimulation of quiescent fibroblasts (27, 28). Structurally, Nur77 belongs to the steroid receptor family (29). It is referred to as an “orphan” receptor because its ligand is unknown. It has been implied that Nur77 induction plays a crucial role in AICD of T cells. Both transcription activity and mitochondrial targeting of Nur77 has been suggested to be required for cell death (30, 31).
We reported previously that macrophage cell death could be triggered by costimulation with LPS and zVAD (15). In this paper, we explore the role of Nur77 and delineate its regulatory pathways in macrophage death induced by the costimulation LPS and zVAD.
Materials and Methods
Purified LPS from Escherichia coli O111.
B4 was purchased from List Biological Laboratories. Synthetic phosphodiester CpG (5′-AACGTTAACGTTAACGTT-3′) was obtained from Sigma-Genosys. Peptidoglycan (PG) was purchased from Sigma-Aldrich. Flagellin was a gift from Dr. A. Aderem (University of Washington, Seattle, WA). Polyclonal antibodies for myocyte-specific enhancer binding factor 2 (MEF2)-C and MEF2D were raised against peptides YGNPRNSPGLLVSPGNLNKNMQAKSP and LLEDKYRRASEELDGLFRRYGSTVPA, respectively.
Sepsis Model.
Polymicrobial sepsis was induced using cecal ligation and puncture (CLP) as described previously (32) with slight modifications. In brief, female ND4 mice weighing 20–25 g were anesthetized with an intramuscular injection of a mixture of 80 mg/kg/body weight ketamine and 16 mg/kg/body weight xylazine. The cecum was ligated and punctured twice with a 25-gauge needle. Sham-operated mice received cecal manipulation only, without ligation. The presence of sepsis was verified by checking the formation of bacterial colonies in agar plate from 30 μl of blood. Approximately 10–500 colonies were obtained from CLP mice, whereas none were obtained from sham-operated mice. 10 mg/kg of caspase inhibitor, zVAD (Enzyme System Products), or its diluent DMSO, was injected 60 min after CLP by intraperitoneal injection and repeated every 12 h. Peritoneal macrophages were obtained 26–30 h after CLP.
Nur77 and Toll-like Receptor (TLR)2/4 Knockout Mice.
Heterozygote Nur77−/+ knockout mice were provided by J. Milbrandt (Washington University, St. Louis, MO). Homozygote Nur77 wild-type(+/+) and knockout(−/−) mice were obtained from the same littermates by heterozygote mating. TLR4 knockout mice were provided by S. Akira (Osaka University, Osaka, Japan).
Preparations of Peritoneal Macrophages and FACS® Analysis.
Resident peritoneal macrophages were obtained from mice by normal saline lavage as described previously (33). Mice other than those used for the sepsis model received intraperitoneal injection of 3% thioglycollate 4 d before the preparation of peritoneal macrophages. Peritoneal macrophages were harvested by lavage of the peritoneal cavity with 5 ml of normal saline. The cells were washed twice in FACS® buffer (phosphate-buffered saline with 1% fetal bovine serum [vol/vol] and 0.01% sodium azide [wt/vol]), and ∼106 cells were labeled for 20 min with 1 μg each of APC-conjugated monoclonal rat anti–mouse CD11b (BD Biosciences), RPE-cy5–conjugated rat anti–mouse F4/80 (Serotec), and R-PE–conjugated hamster anti–mouse CD11c monoclonal antibodies (BD Biosciences). After washing with annexin labeling solution, the cells were incubated with FITC-conjugated annexin V (Roche Molecular Biochemicals) for 30 min. Macrophages were identified as CD11b-positive, F4/80-positive, and CD11c-negative cells, and the degree of cell death was quantified by annexin V–positive cells as described previously (15).
Cell Culture.
Peritoneal macrophages were plated on plastic tissue culture plates and incubated at 37°C for 2 h. The nonadherent cells were removed by repeated washing three times with fresh DMEM, and the adherent macrophage cells were cultured overnight in DMEM medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine (supplemented DMEM). The murine monocyte/macrophage cell line, RAW 264.7, was maintained in the supplemented DMEM. All experiments were performed in supplemented DMEM.
Cell Viability Assays.
The extent of cell death was measured using either crystal violet or propidium iodide (PI; Sigma-Aldrich) permeability analysis. The crystal violet uptake assays were performed in a 96-well plate as described previously (34). PI staining was assessed after incubation of the cells with 2 μg/ml PI, followed by FACS® analysis. The extent of cell death was analyzed using either a dot plot or a histogram against the PI fluorescence.
Total Cell Lysate Preparation, Western Blot Analysis, and Electrophoretic Mobility Shift Assay (EMSA).
Extraction of total cell lysate and Western blot analysis for MEF2s were performed as described previously (15). Antibody for MEF2A was purchased from Santa Cruz Biotechnology, Inc. and antibodies for MEF2C and MEF2D were raised in rabbit against their specific peptides. For EMSA, nuclear extracts of RAW 264.7 cells were incubated with a double-stranded, 32P-labeled oligonucleotide containing an MEF2 binding site as a probe (35).
Plasmids.
The wild-type and mutant pGL2-TR3/Nur77 (−93 to +17) reporter genes were provided by X. Liu (Danish Cancer Society, Copenhagen, Denmark). Reporter genes for wild-type and mutant MEF2-response Nur77 promoter (−307 to −242; provided by J. Liu [Center for Cancer Research, Massachusetts Institute of Technology, Cambridge, MA]) reporters were constructed by cutting wild-type TR3/Nur77 (−93 to +17) reporter gene with KpnI and MluI, and inserting the promoter sequences into KpnI site. The dominant active form of TR3/Nur77 mutant (lacking the DNA binding domain) plasmid was provided by X-K. Zhang (Burnham Institute, La Jolla, CA).
Transfection of Plasmids and Reporter Gene Analysis.
Raw 264.7 cells were transiently transfected by electrophoration (300 V, 950 μF). After electrophoration, cells were washed and plated in 6-well plates and stabilized for 12–16 h in supplemented DMEM before starting treatments. Luciferase was extracted according to the manufacturer's instructions (Promega) and activity was determined by a bioluminescent assay. Activities in individual samples were normalized on the basis of protein content.
RT-PCR and Real-time PCR.
Total RNA from RAW 246.7 cells was prepared as per the manufacturer's instructions (RNeasy Mini kit; QIAGEN). RT-PCR was performed using 0.5 μg of total RNA as a template to detect the expression of Nur77, Nurr1, and Nor1, using One-step RT-PCR kit (QIAGEN). The primers used were as follows: for Nur77, 5′-CTCTCCGAACCGTGACACTT-3′ (5′-primer) and 5′-GGAAGGAGAGCGGAAGAGAT-3′ (3′-primer); for Nurr1, 5′-CAGCACAGGCTACGACGTCAA-3′ (5′-primer) and 5′-AGCCCTCACAAGTGCGAACAC-3′ (3′-primer); and for Nor1, 5′-GAGAGGTCGTCTGCCTTCCAA-3′ (5′-primer) and 5′-GTCCCTTCCTCTGGTGGTCCT-3′ (3′-primer). For the quantitative PCR analysis, 0.5 μg of total RNA from RAW 246.7 cells was used to prepare cDNA using Omniscript RT kit (QIAGEN) with oligo(dT12) as a primer, which was quantified by real-time PCR using SYBR green PCR master mix kit (Applied Biosystems). The primers used for Nur77, Nurr1, Nor1, and MEF2s were as follows: for Nur77, 5′-CTCGCCATCTACACCCAACT-3′ (5′ primer) and 5′-AGCCTTAGGCAACTGCTCTG-3′ (3′ primer); for Nurr1, 5′-CATGGACCTCACCAACACTG-3′ (5′ primer) and 5′-ACAGGGGC-ATTTGGTACAAG-3′ (3′ primer); for Nor1, 5′-TCAGCCTTTTTGGAGCTGTT-3′ (5′-primer) and 5′-TGAAGTCGATGCAGGACAAG-3′ (3′ primer); for MEF2A primers, 5′-TTGAGCACTACAGACCTCACG-3′ (5′ primer) and 5′-TGCACCAGTATTTCCAATCAA-3′ (3′ primer); for MEF2C primer, 5′-GGATGAGCGTAACAGACAGGT-3′ (5′ primer) and 5′-ATCAGTGCAATCTCACAGTCG-3′ (3′ primer); and for MEF2D primer, 5′-GAGAAGATGGGGAGGAAAAAG-3′ (5′ primer) and 5′-GCCTTCTTCATCAGTCCAAAC-3′ (3′ primer).
Results
Pan-caspase Inhibitor, zVAD, Promotes Cell Death of Activated Macrophages.
We demonstrated previously that costimulation of macrophages with LPS and pan-caspase inhibitor zVAD results in cell death (15). Because this cell death occurs quickly and is a direct result of LPS + zVAD treatment, we believe this system can be used to study the mechanism of macrophage death. To further characterize this system, we first examined whether zVAD promoted activated macrophage death in vivo. Macrophages activated in septic models were used in our experiments. Sepsis was induced in mice with CLP, as described in Materials and Methods. 10 mg/kg body weight zVAD or control vehicle (DMSO) was injected peritoneally in CLP and sham mice. Sepsis induced by CLP was verified by the presence of bacteria in the blood by counting the bacterial colony formation in blood cultures. Peritoneal macrophages were harvested 26–30 h after CLP or sham surgery. FACS® analysis was used to identify macrophages (CD11b+, F4/80+, and CD11c−) in the peritoneal isolates and to analyze the level of cell death in these macrophages. Because apoptotic cells are cleared quickly in vivo, we needed to measure early markers of cell death to detect the apoptotic cells. Translocation of phosphatidylserine from the inner to the outer layer of the plasma membrane is an early event in apoptosis; therefore, we used annexin V staining of the cell surface phosphatidylserine to identify the macrophages that underwent apoptotic cell death. In sham-operated mice, ∼1% of the peritoneal macrophages were annexin V–positive (Fig. 1 A). Administration of zVAD had no effect on the viability of the macrophages in these mice. Similarly, there were ∼1% apoptotic macrophages isolated from CLP mice. However, zVAD significantly increased annexin V–positive cells to ∼15% in CLP mice. Therefore, zVAD-enhanced macrophage cell death in sepsis could contribute to the improved survival observed in zVAD-treated septic mice (18).
Figure 1.

Induction of cell death by the pan-caspase inhibitor zVAD in septic and LPS-activated macrophages. (A) CLP or sham-operated ND4 mice were treated with zVAD or its vehicle (DMSO) for 26–30 h. Macrophages in peritoneal isolates were gated by CD11b+, F4/80+, and CD11c− using flow cytometry, and the cell death of macrophages was determined by annexin V staining. (B) Peritoneal macrophages of wild-type, TLR2−/−, or TLR4−/− mice and (C) RAW264.7 cells were treated for 12 h with 10 ng/ml LPS, 10 μg/ml PG, 10 μg/ml flagellin, or 10 μg/ml CpG oligonucleotide together with or without 50 μM zVAD as indicated. The extent of cell death was measured by crystal violet uptake of live cells as described in Materials and Methods. Results represent the means ± SE (n = 3–4).
We showed previously that zVAD promotes cell death in LPS or lipoprotein-activated macrophages, but did not promote cell death in flagellin- or CpG oligonucleotide–treated macrophages (15). Because LPS and PG activate macrophages via TLRs 4 and 2, respectively, we examined whether TLR2 and TLR4 were required for macrophage death in our system. Peritoneal macrophages were isolated from wild-type, TLR2−/−, and TLR4−/− mice, and challenged with LPS or PG in the presence or absence of zVAD. As expected, LPS and PG stimulation led to macrophage death in the presence of zVAD (Fig. 1 B). TLR2−/− macrophages were resistant to PG + zVAD, but not LPS + zVAD–induced cell death, and TLR4−/− macrophages were resistant to LPS + zVAD, but not PG + zVAD–induced cell death. Thus, TLR2- or TLR4-mediated signaling is required for zVAD-induced cell death in the presence of their respective ligands. zVAD did not promote death of flagellin (a TLR5 ligand)- or CpG oligonucleotide (a TLR9 ligand)–treated macrophages from the wild-type, TLR2−/−, or TLR4−/− mice (unpublished data). In summary, zVAD, together with signaling initiated from TLR2 or TLR4, can lead to macrophage cell death.
The murine macrophage cell line, RAW 264.7, has many of the features of peritoneal macrophages, including LPS responsiveness and cytokine production. LPS, PG, flagellin, and CpG exerted the same profile of killing effects on RAW 264.7 cells as on peritoneal macrophages in the presence of zVAD (Fig. 1 C). Thus, RAW 264.7 cells were used to study the mechanisms of zVAD-promoted cell death in activated macrophages.
Induction of Nur77 Expression Correlated to the LPS + zVAD-induced Macrophage Cell Death.
Cell death can be mediated by different mechanisms, including induction of death genes, whose products, in turn, lead to cell death. Because zVAD-promoted macrophage death is not mediated by the classic caspase pathways, we asked whether there is death gene induction during the stimulation of RAW 264.7 cells with LPS + zVAD, but not with LPS or zVAD alone. We analyzed mRNA and protein levels of several genes (e.g., Nur77, p53, Rb, Bad, and Bax), whose inductions are known to promote cell death. Of these, only Nur77 was strongly induced in LPS + zVAD–treated RAW 264.7 cells (Fig. 2, A and B) . Fig. 2 A shows an RT-PCR product of Nur77 that was detected in cells treated with LPS + zVAD, but not in cells treated only with LPS or zVAD. By using real-time PCR, we found the level of Nur77 mRNA in LPS + zVAD–treated cells is ∼30–40-fold higher than in the control, or LPS- or zVAD-treated cells (Fig. 2 B). We compared the time courses of Nur77 induction (Fig. 2 B) and cell death (Fig. 2 C) after LPS + zVAD treatment, and found that Nur77 induction is a prelude to cell death, suggesting that Nur77 could be a trigger of cell death.
Figure 2.

Induction of Nur77 mRNA and its involvement in cell death in LPS + zVAD–treated RAW 264.7 cells. RAW 264.7 cells were treated with 10 ng/ml LPS, 50 μM zVAD, or both for 9 h or indicated time periods. (A) Total mRNA extraction and RT-PCR of Nur77 mRNA were performed as described in Materials and Methods. An expected 1,494-bp band was detected only in LPS + zVAD–treated cells. (B) Real-time RT-PCRs of Nur77 mRNA were performed as described in Materials and Methods. The results show a means ± SE (n = 4). (C) PI-permeable cells were separated by FACS® and the percentage of PI-positive (dead) cells was plotted for different times. The results shown are the means ± SE (n = 3). (D) RAW 264.7 cells were electrophoretically transfected with vector control or expression vector of a putative Nur77-dominant active (Nur77-DA) mutant together with a GFP expression vector. Cells were harvested 48 h after transfection. GFP expression was used to identify the transfected cells using FACS®, and cell death in transfected cells was analyzed by PI staining using FACS®. Results show the means ± SE (n = 3). (E) Peritoneal macrophages from Nur77+/+ and Nur77−/− mice were treated with 50 μM zVAD, 10 ng/ml LPS, or both for 12 h. The cells were examined under microscope and photomicrographs of two experiments are shown. (F) The extent of cell death of Nur77+/+ or Nur77−/− macrophages induced by zVAD + LPS was measured using crystal violet uptake by living cells. The results show the means ± SE (n = 3).
Nur77 as a Causative Factor in the LPS + zVAD-induced Macrophage Cell Death.
To investigate if Nur77 contributes to cell death in LPS + zVAD–treated RAW 264.7 cells, we first examined whether Nur77 can cause death of RAW 264.7 cells. It was reported that a Nur77 mutant, lacking DNA binding domain (Nur77-DA), does not localize in the nucleus as the wild-type Nur77 does and can directly target mitochondria to trigger cell death (31). Expression of the GFP–Nur77-DA fusion protein in LNCaP cells was reported to cause death of ∼35% cells (31). Expression of this putative dominant active mutant in RAW 264.7 cells led to ∼25% cell death, and cell death was not inhibited by zVAD (Fig. 2 D). This finding indicates that Nur77 promotes caspase-independent cell death in RAW 264.7 cells. Because zVAD has no effect on Nur77-mediated cell death, Nur77 could be located downstream of zVAD.
A role for Nur77 in negative selection of T cells has been demonstrated using dominant mutant transgenic mice (27). The Nur77 knockout mouse has been generated, but T cell apoptosis remains normal (36). This is probably due to Nur77 family members compensating for the loss of Nur77 (37). To determine whether Nur77 is required for LPS + zVAD–induced macrophage death, we isolated peritoneal macrophages from wild-type and Nur77−/− mice, and tested their sensitivity to LPS + zVAD–induced cell death. Unlike the situation with T cells, macrophage death was influenced by Nur77 deletion. As shown in Fig. 2 E, LPS + zVAD–induced cell death of Nur77−/− macrophages was significantly less than that of wild-type macrophages. About sixfold increase in survival rate was found in Nur77−/− macrophages in comparison with wild-type macrophages (Fig. 2 F). We suspect that the functional redundancy of Nur77 family members may also affect macrophage death because Nur77 deletion did not completely prevent LPS + zVAD–induced cell death. These data demonstrate that Nur77 is involved in LPS + zVAD–induced cell death of macrophages.
Expression of Other Nur77 Family Members in LPS + zVAD–treated Macrophages.
Because a functional redundancy of Nur77 family members has been suggested (37), we investigated whether other Nur77 family members are also induced by LPS + zVAD in peritoneal macrophages. The proteins that exhibit a close structural relationship to the orphan nuclear receptor, Nur77, are Nurr1 and Nor1 (38). Nur77, Nurr1, and Nor1 are not expressed, or are expressed at very low levels, in resting macrophages because RT-PCR did not detect their mRNA (Fig. 3) . As in the case of Nur77, Nurr1 mRNA was induced in the macrophages treated with LPS + zVAD, but not in the macrophages treated with LPS or zVAD alone (Fig. 3, A and B). Nor1 is different from Nur77 and Nurr1 in that LPS alone is sufficient for its mRNA induction (Fig. 3 C). Real-time PCR analysis reveals that Nur77 and Nurr1 mRNA were increased ∼75-fold and ∼60-fold, respectively, after treatment with LPS + zVAD (Fig. 3, A and B, right). Nor1 mRNA was induced ∼50-fold by either LPS alone or LPS + zVAD (Fig. 3 C, right). These results indicate that Nurr1 expression also correlates with macrophage death induced by LPS + zVAD.
Figure 3.

mRNA level of Nur77 family members in peritoneal macrophages before and after stimulation with LPS, zVAD, or LPS + zVAD. (A–C) Peritoneal macrophages were treated with 10 ng/ml LPS, 50 μM zVAD, or both for 2 h. Total RNA extraction and RT-PCR were performed as described in Materials and Methods. The expected 1,494-, 647-, and 611-bp cDNA bands amplified from Nur77, Nurr1, and Nor1 mRNA, respectively, were detected. Real-time RT-PCRs were performed. The results are shown in right panels with the means ± SE (n = 3).
The Role of zVAD in Nur77 Induction Is to Prevent Degradation of MEF2 Transcription Factors Whose Activity Is Required for Nur77 Expression.
To further investigate the mechanisms underlying LPS + zVAD–induced Nur77 expression, we first tried to identify the zVAD-mediated changes that contributed to Nur77 induction. Because zVAD is a pan-caspase inhibitor, one possible role of zVAD in Nur77 induction is to prevent cleavage of the protein(s) required for Nur77 induction. Caspases can cleave a number of proteins including MEF2 transcription factors (39, 40). MEF2 activity has been well-documented as being required for the expression of Nur77 proteins in T cells (41, 42). Therefore, we tested whether zVAD has any effect on MEF2 activity in RAW 264.7 cells. As shown in Fig. 4 A, treatment with zVAD or zVAD + LPS, but not LPS, quickly increased MEF2 activity, which was measured by EMSA using a 32P-labeled double-stranded probe containing the consensus sequence of the MEF2 site. Thus, zVAD has an effect on MEF2 transcription factors. There are four members of the MEF2 family. We used real-time PCR to measure the mRNA of MEF2A, MEF2B, MEF2C, and MEF2D before and after zVAD treatment, showing that there is no detectable increase in mRNA levels of these MEF2 transcription factors (Fig. 4 B). MEF2B mRNA was barely detectable and is not depicted. We also examined samples treated with LPS + zVAD and obtained the same result (unpublished data). These data indicate that zVAD does not affect transcription or the mRNA stability of MEF2 transcription factors. We examined protein levels of the MEF2 transcription factors and found that MEF2A, MEF2C, and MEF2D proteins were dramatically increased in zVAD or LPS + zVAD, but not in samples treated with LPS alone (Fig. 4 C). This indicates that zVAD itself can increase MEF2A, MEF2C, and MEF2D proteins. The induction of the MEF2 proteins occurred quickly and reached their maximum within 45 min (Fig. 4 C). MEF2B was not detected in RAW 264.7 cells (unpublished data). zVAD was able to significantly increase the MEF2 isoforms at concentrations as low as 25 μM, but maximally between 50 and 100 μM (Fig. 4 D).
Figure 4.

zVAD increases MEF2 proteins to induce MEF2 transcription activity. (A) RAW 264.7 cells were incubated with 10 ng/ml LPS, 50 μM zVAD, or both, for the indicated time periods. MEF2 activity was measured by EMSA. (B) RAW 264.7 cells were incubated with 50 μM zVAD for the indicated time periods. Real-time RT-PCRs for MEF2 isoforms were performed as described in Materials and Methods. The results shown are the means ± SE (n = 3). (C) RAW 264.7 cells were incubated with LPS, zVAD, or both, for the indicated time periods. Total cell extracts (20 μg of protein/lane) were separated on SDS-PAGE and immunoblotted with antibodies specific to MEF2 family members. (D) RAW 264.7 cells were incubated with different concentrations of zVAD for 45 min, and the cell extracts (20 μg of protein/lane) were separated on SDS-PAGE and immunoblotted with antibodies specific to MEF2 family members.
We next explored the mechanism by which zVAD induces MEF2 protein. Because there is no change at the mRNA level, the increased protein level should be mediated by increased protein translation or increased protein stability. We treated RAW 264.7 cells with a protein synthesis inhibitor, 10 μM cyclohexamide, together with or without zVAD. As shown in Fig. 5 A, MEF2A, MEF2C, and MEF2D proteins were reduced after protein synthesis was blocked. However, protein levels were maintained when zVAD was included, suggesting that zVAD stabilizes the MEF2 proteins. Caspases have been shown to cleave many proteins (1). It seems that zVAD inhibits caspase activity to prevent the degradation of MEF2 proteins. This possibility is also suggested in recent reports that MEF2C and MEF2D can be cleaved by caspases, but their subsequent degradation was not reported (40). We used a large amount of cell lysate from resting RAW 264.7 cells and found trace amounts of a short fragment of MEF2A, referred to as MEF2A(f) (Fig. 5 B). The cleavage is most likely mediated by a caspaselike protease since zVAD treatment inhibited the appearance of this fragment. To confirm that cleavage of MEF2A occurs in macrophages, we used peritoneal macrophages to repeat this experiment. As shown in Fig. 5 C, MEF2A is cleaved in the absence of zVAD, but this cleavage is prevented when zVAD is present. Thus, the MEF2 proteins are constitutively expressed and quickly degraded in resting macrophages, the degradation is initiated by a caspaselike protease, and zVAD inhibits caspase activity and prevents degradation of MEF2 proteins.
Figure 5.
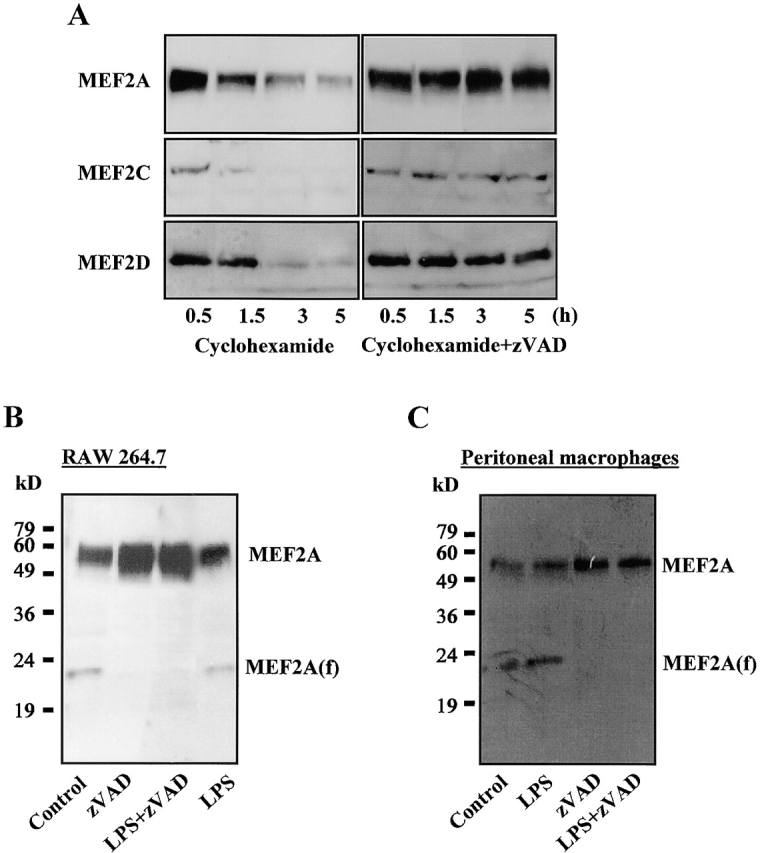
zVAD induces MEF2 protein level by blocking the degradation. (A) RAW 264.7 cells were treated with 10 μg/ml cyclohexamide for 10 min, and then were treated with nothing or with 50 μM zVAD for the indicated time periods. Total cell extracts (40 μg of protein/lane) were separated on SDS-PAGE and immunoblotted with antibodies specific to MEF2 family members. (B and C) RAW 264.7 cells (B) or peritoneal macrophages (C) were incubated with LPS, zVAD, or both for 45 min. Total cell extracts (100 μg of protein/lane) were separated on SDS-PAGE and immunoblotted with antibodies specific to MEF2A. An ∼23-kD fragment of MEF2A was indicated as MEF2A(f). Films were exposed for a prolonged time period to visualize the MEF2 proteins and MEF2A(f).
Two MEF2 binding sites were identified in sequences between −307 and −242 in the Nur77 promoter (Fig. 6 A). To demonstrate that MEF2 activity is indeed required for Nur77 expression in RAW 264.7 cells, we used luciferase reporters. One luciferase reporter is driven by a portion of the Nur77 promoter, which contains MEF2 sites, and another is an identical reporter except that the MEF2 sites have been mutated. The structure of the reporter constructs is shown in Fig. 6 A. RAW 264.7 cells were transfected with the wild-type or mutated reporter plasmid, and then stimulated with zVAD, LPS, or LPS + zVAD. Neither reporter was affected by zVAD treatment (Fig. 6 B). LPS stimulation induced approximately fivefold increase of the expression of both reporters. LPS alone did not induce endogenous Nur77 expression, but induced the Nur77 promoter driven reporter. This phenomenon is often seen. One possible interpretation is that the chromatin structure influences endogenous gene expression that cannot be mimicked in transiently transfected reporter genes. Nevertheless, LPS stimulation alone did not discriminate mutated MEF2 sites from the wild type. Similar to the endogenous gene, expression of the Nur77 reporter gene was significantly induced in cells costimulated with LPS and zVAD. An important finding is that mutation of MEF2 sites in the promoter eliminated the effect of zVAD, indicating that zVAD's effect is through MEF2 transcription factors. Thus, MEF2 transcription factors are the targets of zVAD, and MEF2 activity is required, but not sufficient, for Nur77 induction in macrophages.
Figure 6.

Mutation of MEF2 sites in the promoter of Nur77 abolished the effect of zVAD on gene expression. (A) Illustration of the sequence of the Nur77 reporter constructs. The putative MEF2 binding sequences in the Nur77 promoter and their mutated sequences were shown. The portion of Nur77 promoter (−307 to −242 nucleotide) was conjugated to 5′-end of the minimal Nur77 promoter sequences. Nucleotides are numbered relative to the transcriptional start site of genes. (B) Wild-type and me2f mut reporter plasmids were transfected into RAW 264.7 cells. After 24 h, cells were treated with 50 μM zVAD, 10 ng/ml LPS, or LPS + zVAD for another 12 h. Transient expression of the reporter gene was determined as described in Materials and Methods.
Extracellular Signal-regulated Kinase (ERK) Pathway Activated by LPS Is Required for Nur77 Induction.
LPS stimulation in macrophages leads to activation of multiple signaling pathways. To find out which LPS-activated signaling pathways participate in LPS + zVAD–induced Nur77 expression, our initial approach was to use various inhibitors to search for the candidate(s) involved. Bisindolylmaleimide I, a protein kinase C inhibitor, and Ly294002, a phosphatidylinositol-3 (PI3) kinase inhibitor, had no effect on LPS + zVAD–induced cell death (Fig. 7 A). SB203580 (25 μM), a p38 inhibitor, enhanced LPS + zVAD–induced cell death. U0126 (25 μM), a Mek inhibitor, significantly blocked ERK activation by LPS + zVAD (unpublished data) and blocked the cell death induced by LPS + zVAD in RAW 264.7 cells (Fig. 7 A). To examine whether these inhibitors have the same effect on LPS + zVAD–induced Nur77 expression, we measured Nur77 mRNA level in RAW 264.7 cells treated with LPS + zVAD in the presence of different inhibitors. The effects of these inhibitors on Nur77 mRNA levels correlate well with their effect on cell death (Fig. 7 B). These results suggest that the ERK pathway is required for LPS + zVAD–induced Nur77 induction and cell death.
Figure 7.

Effects of various inhibitors in LPS + zVAD–induced Nur77 mRNA expression and cell death in RAW 264.7 cells. (A) RAW 264.7 cells were treated with 10 ng/ml LPS or 50 μM LPS + zVAD in the absence or presence of 25 μM SB203580, 25 μM U0126, 10 μM bisindolylmaleimide I, or 10 μM Ly294002 for 12 h. PI-positive dead cells were counted by FACS®. Results show the means ± SE (n = 4). (B) RAW 264.7 cells were treated with LPS + zVAD for 9 h in the absence or presence of various inhibitors as above. The levels of mRNA for Nur77 were analyzed by real-time PCR. Results show the means SE (n = 2–3). The effectiveness of U0126 in inhibition of LPS-induced ERK activation was confirmed by Western blotting with anti–phospho-ERK1/2 antibody (not depicted).
The ERK pathway can regulate a number of transcription factors. There are several cis elements located in a conserved region of the Nur77 promoter that might possibly respond to ERK activation (Fig. 8 A; references 43, 44). To study the role of LPS in LPS + zVAD–induced Nur77 expression, we transiently transfected a reporter driven by this portion of Nur77 promoter in RAW 264.7 cells. The cells were stimulated with LPS, zVAD, or LPS + zVAD in the presence or absence of the U0126, and reporter gene expression was analyzed. As shown in Fig. 8 B, an approximately eightfold increase of the reporter gene activity was detected in LPS- or LPS + zVAD–stimulated cells. The induction of this reporter expression was abolished by the U0126, indicating that the induction is dependent on the ERK pathway. To further investigate which promoter motif is involved in the expression of the reporter gene, each cis element was mutated and its effect on reporter gene expression was analyzed. Mild reduction was observed in mutants of Ets (ets m), Rce (rce m), and Sp1 (sp1 m; Fig. 8 C). The most significant reduction was found in the Nap mutant (nap m). The level of reduction in reporter gene expression by the Nap sequence mutation is similar to that by the U0126 (Fig. 8, B and C). In addition, U0126 only had a slight inhibitory effect on nap m reporter expression in LPS-treated RAW 264.7 cells. This data suggests that Nap is a major target of the ERK pathway. Activation of Ets- and Sp1-dependent transcription by ERK has been reported in various cell types, including RAW 264.7 cells (45), and is most likely also involved in Nur77 induction.
Figure 8.

The effect of the mutation of different cis elements in the Nur77 promoter on LPS-induced Nur77 reporter gene expression. (A) Illustration of the sequence of the Nur77 promoter and its mutants. (B) Wild-type Nur77 promoter reporter plasmids were transfected into RAW 264.7 cells. After 24 h, cells were treated with or without 10 ng/ml LPS, 50 μM zVAD, or 25 μM LPS + zVAD in the presence or absence of U0126 for another 12 h. The expression of the reporter gene was determined as described in Materials and Methods. (C) Wild-type and mutated Nur77 promoter plasmids were transfected into RAW 264.7 cells. After 24 h, cells were treated with or without 10 ng/ml LPS for another 12 h. The expression of the reporter gene was determined.
Discussion
A role of Nur77 in T cell death has been well-documented. Here, we show that Nur77 also has a role in the caspase-independent cell death of macrophages. Similar to T cells, Nur77 induction in macrophages requires costimulatory signals. Due to the difference between macrophages and T cells, the death stimuli of these cells are also different. Macrophages are activated when exposed to bacterial wall components, such as LPS. A costimulus that leads to cell death of LPS-activated macrophages is the pan-caspase inhibitor zVAD. Nur77 is clearly involved in the cell death of macrophages for several reasons: its induction closely correlates with LPS + zVAD–induced cell death (Fig. 2 A–C); the putative dominant active Nur77 mutant causes macrophage death (Fig. 2 D); and LPS + zVAD–induced cell death is decreased significantly in macrophages from Nur77 knockout mice (Fig. 2, E and F). We found that activation of the ERK pathway by LPS is required for Nur77 induction and cell death (Figs. 7 and 8). Elevation in the level of MEF2 transcription factors is a cellular event resulting from zVAD treatment (Fig. 4), and is required for Nur77 induction and cell death. The integration of MEF2 activity with other transcription factors downstream of the ERK pathway on the Nur77 promoter is essential for Nur77 induction and cell death. ERK activation in activated macrophages is well-documented, involving Ras, raf, and MEK (15, 46, 47). These results show for the first time that MEF2 transcription factors are short-lived proteins in macrophages and can be regulated at the level of protein stability (Figs. 4 and 5). zVAD prevents cleavage of MEF2 proteins, and, therefore, increases MEF2 activity. The mechanism of LPS + zVAD–induced macrophage death is summarized in Fig. 9 .
Figure 9.

The proposed mechanism of LPS + zVAD–induced macrophage cell death. The transcription factor downstream of the LPS-activated ERK pathway cooperates with zVAD-stabilized MEF2 transcription factor to induce Nur77 expression. The Nur77 induction leads to the cell death of activated macrophages.
Because macrophages and T cells are different types of cells, both in their origin and function, induction of the same protein to elicit AICD in these two types of immune cells was unexpected. Given that the same molecules are involved in the cell death of macrophages and T cells, there may be some common execution steps in each cell death process. In support of this idea, both macrophage death and negative selection of T cells have been shown to be caspase-independent (15, 48, 49). Nur77 induction could be a broadly common mechanism in caspase-independent cell death. Although the activation signals in T cells and macrophages are initiated from different cell surface receptors, there is still some similarity of the intracellular signaling events between T cells and macrophages that lead to Nur77 induction. Activation of the ERK pathway and increased activity of MEF2 transcription factors are required for Nur77 induction in both T cells and macrophages, although the activation mechanisms are different (41, 42). The activation of ERK in T cells and macrophages is certainly initiated through different ligand–receptor interactions. MEF2 is activated in T cells through the release of the MEF2 protein from repressor Cabin1 by a calcium-dependent mechanism (42), whereas the activation of MEF2 transcription factors in macrophages is due to the increase of the MEF2 by protein stabilization. An increase in macrophage intracellular calcium concentration has no effect on MEF2 activity, Nur77 expression, or cell death (unpublished data). The zVAD effect on the MEF2 protein in T cells did not appear to be the same as in macrophages. Therefore, the effect of zVAD on protein stabilization may be cell-type specific.
Both the transcription activity of Nur77 and its translocation to the mitochondria are implicated in cell death (30, 31). Phosphorylation of Nur77 on Ser-350 by Akt was shown to inhibit T cell death by suppressing the DNA binding activity of Nur77 and stimulating Nur77 association with 14-3-3 (50). Activation of Akt in macrophages by LPS also has been reported (51). It is possible that Akt prevents death signals in LPS-activated macrophage death. We treated LPS-activated cells with inhibitors of PI3 kinase, the upstream kinase of Akt, and did not find any influence on cell viability. We compared phosphorylation of Akt in macrophages stimulated with LPS and LPS + zVAD, and found no difference (unpublished data), suggesting that zVAD did not influence the Akt pathway to promote cell death. Direct phosphorylation of Nur77 by ERK was reported (52), but its physiological relevance is unclear. The ERK pathway may not only be required for Nur77 induction, but could also influence Nur77 function by direct phosphorylation. Nuclear factor (NF)-κB is another survival pathway activated by LPS in macrophages (53). It has been shown that in short time periods (within 2 h), IκB-α degradation and nuclear translocation of NF-κB, induced by LPS in RAW 264.7 cells, are not influenced by zVAD at 50 μM concentration (15). Yet in another report, it is documented that the NF-κB reporter gene activity is inhibited at a later time (8 h) by zVAD at 100-μM concentration (54). These contrasting observations may be due to the different doses or exposure time of zVAD used. The “survival pathways” do not appear to play a role in macrophage death in our system.
Nurr1 and Nor1 are orphan nuclear receptors that belong to the same family as Nur77. These proteins exhibit a close structural relationship and may function redundantly (38). Because Nur77 knockout cannot completely prevent LPS + zVAD–induced macrophage death (Fig. 2 F), the other Nur77 family members may be able to compensate for the function of Nur77 in LPS + zVAD–induced macrophage death. Nor1 may not be involved in LPS + zVAD–induced cell death because its expression does not need dual stimuli (Fig. 3 C). Nurr1 is a candidate that compensates the function of Nur77 in Nur77 knockout cells because its induction also requires dual stimuli (Fig. 3 B). Sequence analysis reveals that there are cis elements such as CREB, CarG box, and Sp-1 in the Nurr1 promoter that can be regulated by LPS signaling (55). However, we were unable to find a typical MEF2 site, suggesting a different mechanism was used by zVAD to costimulate Nurr1 expression. Determining the target of zVAD in Nurr1 induction is a subject for further investigation.
Macrophage death has been observed in a number of inflammatory diseases. Although the lifespan of macrophages is modulated during inflammation, there are not many studies addressing the effect of macrophage death on the outcome of inflammatory diseases. Administration of zVAD has been shown to significantly increase the survival rate of septic mice (18), but zVAD's effect on macrophages was not evaluated. Our data show that zVAD-enhanced macrophage death in septic mice provides important information for understanding the in vivo role of caspase inhibition. Inducible activation of caspases is a current focus in studies of caspase-mediated cellular responses. Our data indicate that caspaselike proteins also function in resting cells by maintaining MEF2 proteins at low levels. Inhibition of constitutive turnover of MEF2 transcription factors in macrophages by zVAD suggests that caspaselike proteases have roles in controlling the stability of some short-lived proteins. The basal activity of caspase(s) probably plays a key role in controlling the susceptibility of macrophages to AICD. It would be very interesting to find the identity of this caspaselike protease and study its role in the fate of macrophages.
Acknowledgments
We thank Drs. X. Liu and J. Liu for providing Nur77 reporter plasmids; Drs. X-K. Zhang and A. Winoto (University of California, Berkeley) for providing Nur77-DBD and Nur77-ZnCT plasmid; and Drs. J. Milbrandt and S. Akira for providing Nur77−/− and TLR4−/− mice, respectively. We also thank Dr. S. Stanton for critical reading of the manuscript and T. Thompson for excellent secretarial assistance.
This work was supported in part by National Institutes of Health grant AI-41637, GM67101, and a grant from the California Cancer Research Program (to J. Han). S.O. Kim was supported by a Canadian Institutes of Health Research Fellowship. This is publication no. 15057-IMM from the Department of Immunology, The Scripps Research Institute.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; CLP, cecal ligation and puncture; EMSA, electrophoretic mobility shift assay; ERK, extracellular signal–regulated kinase; MEF2, myocyte-specific enhancer binding factor 2; NF, nuclear factor; PG, peptidoglycan; PI, propidium iodide; TLR, toll-like receptor; zVAD, benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone.
References
- 1.Rathmell, J.C., and C.B. Thompson. 1999. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 17:781–828. [DOI] [PubMed] [Google Scholar]
- 2.Conroy, L.A., and D.R. Alexander. 1996. The role of intracellular signalling pathways regulating thymocyte and leukemic T cell apoptosis. Leukemia. 10:1422–1435. [PubMed] [Google Scholar]
- 3.Winoto, A. 1997. Genes involved in T-cell receptor-mediated apoptosis of thymocytes and T-cell hybridomas. Semin. Immunol. 9:51–58. [DOI] [PubMed] [Google Scholar]
- 4.Donjerkovic, D., and D.W. Scott. 2000. Activation-induced cell death in B lymphocytes. Cell Res. 10:179–192. [DOI] [PubMed] [Google Scholar]
- 5.Munn, D.H., A.C. Beall, D. Song, R.W. Wrenn, and D.C. Throckmorton. 1995. Activation-induced apoptosis in human macrophages: developmental regulation of a novel cell death pathway by macrophage colony–stimulating factor and interferon-γ. J. Exp. Med. 181:127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumann, S., A. Krueger, S. Kirchhoff, and P.H. Krammer. 2002. Regulation of T cell apoptosis during the immune response. Curr. Mol. Med. 2:257–272. [DOI] [PubMed] [Google Scholar]
- 7.Kroesen, B.J., B. Pettus, C. Luberto, M. Busman, H. Sietsma, L. de Leij, and Y.A. Hannun. 2001. Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. J. Biol. Chem. 276:13606–13614. [DOI] [PubMed] [Google Scholar]
- 8.van Rooijen, N., and A. Sanders. 1997. Elimination, blocking, and activation of macrophages: three of a kind? J. Leukoc. Biol. 62:702–709. [DOI] [PubMed] [Google Scholar]
- 9.Gotoh, T., and M. Mori. 1999. Arginase II downregulates nitric oxide (NO) production and prevents NO-mediated apoptosis in murine macrophage-derived RAW 264.7 cells. J. Cell Biol. 144:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brune, B., C. Gotz, U.K. Messmer, K. Sandau, M.R. Hirvonen, and E.G. Lapetina. 1997. Superoxide formation and macrophage resistance to nitric oxide-mediated apoptosis. J. Biol. Chem. 272:7253–7258. [DOI] [PubMed] [Google Scholar]
- 11.Adler, B., H. Adler, T.W. Jungi, and E. Peterhans. 1995. Interferon-alpha primes macrophages for lipopolysaccharide-induced apoptosis. Biochem. Biophys. Res. Commun. 215:921–927. [DOI] [PubMed] [Google Scholar]
- 12.Adams, D.O., and T.A. Hamilton. 1984. The cell biology of macrophage activation. Annu. Rev. Immunol. 2:283–318. [DOI] [PubMed] [Google Scholar]
- 13.Ricevuti, G. 1997. Host tissue damage by phagocytes. Ann. NY Acad. Sci. 832:426–448. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan, G.W., I.J. Sarembock, and J. Linden. 2000. The role of inflammation in vascular diseases. J. Leukoc. Biol. 67:591–602. [DOI] [PubMed] [Google Scholar]
- 15.Kim, S.O., K. Ono, and J. Han. 2001. Apoptosis by pan-caspase inhibitors in lipopolysaccharide-activated macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L1095–L1105. [DOI] [PubMed] [Google Scholar]
- 16.Xaus, J., M. Cardo, A.F. Valledor, C. Soler, J. Lloberas, and A. Celada. 1999. Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity. 11:103–113. [DOI] [PubMed] [Google Scholar]
- 17.Talanian, R.V., K.D. Brady, and V.L. Cryns. 2000. Caspases as targets for anti-inflammatory and anti-apoptotic drug discovery. J. Med. Chem. 43:3351–3371. [DOI] [PubMed] [Google Scholar]
- 18.Hotchkiss, R.S., K.C. Chang, P.E. Swanson, K.W. Tinsley, J.J. Hui, P. Klender, S. Xanthoudakis, S. Roy, C. Black, E. Grimm, et al. 2000. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat. Immunol. 1:496–501. [DOI] [PubMed] [Google Scholar]
- 19.Rabinovich, G.A. 2000. Apoptosis as a target for gene therapy in rheumatoid arthritis. Mem. Inst. Oswaldo Cruz. 95 (Suppl.):225–233. [DOI] [PubMed] [Google Scholar]
- 20.Bouchon, A., P.H. Krammer, and H. Walczak. 2000. Critical role for mitochondria in B cell receptor-mediated apoptosis. Eur. J. Immunol. 30:69–77. [DOI] [PubMed] [Google Scholar]
- 21.Kawahara, A., Y. Ohsawa, H. Matsumura, Y. Uchiyama, and S. Nagata. 1998. Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mateo, V., L. Lagneaux, D. Bron, G. Biron, M. Armant, G. Delespesse, and M. Sarfati. 1999. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat. Med. 5:1277–1284. [DOI] [PubMed] [Google Scholar]
- 23.Drenou, B., V. Blancheteau, D.H. Burgess, R. Fauchet, D.J. Charron, and N.A. Mooney. 1999. A caspase-independent pathway of MHC class II antigen-mediated apoptosis of human B lymphocytes. J. Immunol. 163:4115–4124. [PubMed] [Google Scholar]
- 24.Holler, N., R. Zaru, O. Micheau, M. Thome, A. Attinger, S. Valitutti, J.L. Bodmer, P. Schneider, B. Seed, and J. Tschopp. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1:489–495. [DOI] [PubMed] [Google Scholar]
- 25.Du, C., M. Fang, Y. Li, L. Li, and X. Wang. 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 102:33–42. [DOI] [PubMed] [Google Scholar]
- 26.Hunot, S., and R.A. Flavell. 2001. Apoptosis. Death of a monopoly? Science. 292:865–866. [DOI] [PubMed] [Google Scholar]
- 27.Woronicz, J.D., B. Calnan, V. Ngo, and A. Winoto. 1994. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature. 367:277–281. [DOI] [PubMed] [Google Scholar]
- 28.Liu, Z.G., S.W. Smith, K.A. McLaughlin, L.M. Schwartz, and B.A. Osborne. 1994. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature. 367:281–284. [DOI] [PubMed] [Google Scholar]
- 29.Winoto, A. 1994. Molecular characterization of the Nur77 orphan steroid receptor in apoptosis. Int. Arch. Allergy Immunol. 105:344–346. [DOI] [PubMed] [Google Scholar]
- 30.Kuang, A.A., D. Cado, and A. Winoto. 1999. Nur77 transcription activity correlates with its apoptotic function in vivo. Eur. J. Immunol. 29:3722–3728. [DOI] [PubMed] [Google Scholar]
- 31.Li, H., S.K. Kolluri, J. Gu, M.I. Dawson, X. Cao, P.D. Hobbs, B. Lin, G. Chen, J. Lu, F. Lin, Z. Xie, J.A. Fontana, J.C. Reed, and X. Zhang. 2000. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 289:1159–1164. [DOI] [PubMed] [Google Scholar]
- 32.Baker, C.C., I.H. Chaudry, H.O. Gaines, and A.E. Baue. 1983. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 94:331–335. [PubMed] [Google Scholar]
- 33.Border, J.R. 1988. Hypothesis: sepsis, multiple systems organ failure, and the macrophage. Arch. Surg. 123:285–286. [DOI] [PubMed] [Google Scholar]
- 34.Mathison, J.C., E. Wolfson, and R.J. Ulevitch. 1988. Participation of tumor necrosis in the mediation of gram-negative bacterial lipopolysaccharide-induced injury in rabbits. J. Clin. Invest. 81:1925–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kravchenko, V.V., Z. Pan, J. Han, J.M. Herbert, R.J. Ulevitch, and R.D. Ye. 1995. Platelet-activating factor induces NF-kappa B activation through a GX protein-coupled pathway. J. Biol. Chem. 270:14928–14934. [DOI] [PubMed] [Google Scholar]
- 36.Lee, S.L., R.L. Wesselschmidt, G.P. Linette, O. Kanagawa, J.H. Russell, and J. Milbrandt. 1995. Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77). Science. 269:532–535. [DOI] [PubMed] [Google Scholar]
- 37.Cheng, L.E., F.K. Chan, D. Cado, and A. Winoto. 1997. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J. 16:1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winoto, A., and D.R. Littman. 2002. Nuclear hormone receptors in T lymphocytes. Cell. 109(Suppl.):S57–S66. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto, S., Z. Li, C. Ju, M.N. Scholzke, E. Mathews, J. Cui, G.S. Salvesen, E. Bossy-Wetzel, and S.A. Lipton. 1999. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA. 99:3974–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li, M., D.A. Linseman, M.P. Allen, M.K. Meintzer, X. Wang, T. Laessig, M.E. Wierman, and K.A. Heidenreich. 2001. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J. Neurosci. 21:6544–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Youn, H.D., L. Sun, R. Prywes, and J.O. Liu. 1999. Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science. 286:790–793. [DOI] [PubMed] [Google Scholar]
- 42.Youn, H.D., and J.O. Liu. 2000. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 13:85–94. [DOI] [PubMed] [Google Scholar]
- 43.Chen, X., V. Zachar, C. Chang, P. Ebbesen, and X. Liu. 1998. Differential expression of Nur77 family members in human T-lymphotropic virus type 1-infected cells: transactivation of the TR3/nur77 gene by Tax protein. J. Virol. 72:6902–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu, X., X. Chen, V. Zachar, C. Chang, and P. Ebbesen. 1999. Transcriptional activation of human TR3/nur77 gene expression by human T-lymphotropic virus type I Tax protein through two AP-1-like elements. J. Gen. Virol. 80:3073–3081. [DOI] [PubMed] [Google Scholar]
- 45.Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C.J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406–17409. [DOI] [PubMed] [Google Scholar]
- 46.Zhu, W., J.S. Downey, J. Gu, F. di Padova, H. Gram, and J. Han. 2000. Regulation of TNF Expression by multiple mitogen-activated protein kinase pathways. J. Immunol. 164:6349–6358. [DOI] [PubMed] [Google Scholar]
- 47.Sanghera, J.S., S.L. Weinstein, M. Aluwalia, J. Girn, and S.L. Pelech. 1996. Activation of multiple proline-directed kinases by bacterial lipopolysaccharide in murine macrophages. J. Immunol. 156:4457–4465. [PubMed] [Google Scholar]
- 48.Bidere, N., and A. Senik. 2001. Caspase-independent apoptotic pathways in T lymphocytes: a minireview. Apoptosis. 6:371–375. [DOI] [PubMed] [Google Scholar]
- 49.Hara, H., A. Takeda, M. Takeuchi, A.C. Wakeham, A. Itie, M. Sasaki, T.W. Mak, A. Yoshimura, K. Nomoto, and H. Yoshida. 2002. The apoptotic protease-activating factor 1-mediated pathway of apoptosis is dispensable for negative selection of thymocytes. J. Immunol. 168:2288–2295. [DOI] [PubMed] [Google Scholar]
- 50.Masuyama, N., K. Oishi, Y. Mori, T. Ueno, Y. Takahama, and Y. Gotoh. 2001. Akt inhibits the orphan nuclear receptor Nur77 and T-cell apoptosis. J. Biol. Chem. 276:32799–32805. [DOI] [PubMed] [Google Scholar]
- 51.Salh, B., R. Wagey, A. Marotta, J.S. Tao, and S. Pelech. 1998. Activation of phosphatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide production. J. Immunol. 161:6947–6954. [PubMed] [Google Scholar]
- 52.Slagsvold, H.H., A.C. Ostvold, A.B. Fallgren, and R.E. Paulsen. 2002. Nuclear receptor and apoptosis initiator NGFI-B is a substrate for kinase ERK2. Biochem. Biophys. Res. Commun. 291:1146–1150. [DOI] [PubMed] [Google Scholar]
- 53.Tebo, J.M., W. Chaoqun, Y. Ohmori, and T.A. Hamilton. 1994. Murine inhibitory protein-kappa B alpha negatively regulates kappa B-dependent transcription in lipopolysaccharide-stimulated RAW 264.7 macrophages. J. Immunol. 153:4713–4720. [PubMed] [Google Scholar]
- 54.Chakravortty, D., Y. Kato, T. Sugiyama, N. Koide, M.M. Mu, T. Yoshida, and T. Yokochi. 2001. Inhibition of caspase 3 abrogates lipopolysaccharide-induced nitric oxide production by preventing activation of NF-kappaB and c-Jun NH2-terminal kinase/stress-activated protein kinase in RAW 264.7 murine macrophage cells. Infect. Immun. 69:1315–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Castillo, S.O., Q. Xiao, M.S. Lyu, C.A. Kozak, and V.M. Nikodem. 1997. Organization, sequence, chromosomal localization, and promoter identification of the mouse orphan nuclear receptor Nurr1 gene. Genomics. 41:250–257. [DOI] [PubMed] [Google Scholar]