

Abstract
We report here a relationship between intramolecular epitope spreading and the clinical onset of the endemic form of pemphigus foliaceus in a Brazilian community with a high prevalence and incidence of the disease. Also known as Fogo Selvagem (FS), this disease is characterized by severe skin blistering and pathogenic anti–desmoglein-1 (Dsg1) autoantibodies. These autoantibodies bind the Dsg1 ectodomain and trigger keratinocyte cell detachment, the hallmark of FS. We show that (a) sera from FS patients in the preclinical stage recognized epitopes on the COOH-terminal EC5 domain of Dsg1, (b) disease onset was associated with the emergence of antibodies specific for epitopes on the NH2-terminal EC1 and EC2 domains, (c) all sera from FS patients with active disease recognized the EC1 and/or EC2 domains, and (d) sera from FS patients in remission showed reactivity restricted to EC5. These results suggest that anti-Dsg1 autoantibodies in FS are initially raised against the COOH-terminal EC5 domain of Dsg1 in individuals without skin disease; in genetically predisposed subjects the autoimmune response may then undergo intramolecular epitope spreading toward epitopes on the NH2-terminal EC1 and EC2 domains of Dsg1 leading to disease onset. Moreover, intramolecular epitope spreading may also modulate remissions and relapses of FS.
Keywords: autoimmunity, autoantibodies, desmogleins, epitope spreading, pemphigus
Introduction
Pemphigus foliaceus (PF)* is a cutaneous autoimmune blistering disease mediated by autoantibodies to desmoglein-1 (Dsg1), an epidermal cell adhesion molecule. These autoantibodies are predominantly of the IgG4 subclass (1) and are pathogenic as demonstrated by passive transfer into neonatal mice (1). The pathogenic mechanism of skin blistering caused by these autoantibodies is not fully understood. Several mechanisms have been proposed, including antibody-induced transmembrane signal transduction activation (2) and direct blocking of the adhesive site of Dsg1 by bound antibody (3). Dsg1 is a 160-kD desmosomal core glycoprotein that belongs to the cadherin family of calcium-dependent cell adhesion molecules. Like other members of this family, its ectodomain is composed of five domains (EC1 to EC5). The first four domains (EC1-EC4) are cadherin homologous repeats that contain six putative calcium-binding sites; whereas, EC5 is a short membrane-proximal region with no significant homology to the other cadherin repeats. The epitopes recognized by PF sera are conformationally sensitive (4) and calcium-dependent (5, 6). Pathogenic anti-Dsg1 antibodies target the extracellular domain of Dsg1 (7, 8) and the majority of PF sera tested recognize the NH2-terminal region of the molecule (9, 10).
Endemic PF, also known as fogo selvagem (FS), occurs in rural populations of certain states of Brazil. The clinical, histological, and immunological features of FS are not distinguishable from those of nonendemic PF (11, 12); however, unlike nonendemic PF, FS exhibits geographic and familial clustering and the disease additionally affects young adults and children (13). Since 1994, we have been following a Brazilian Amerindian population (Terena tribe) exhibiting a high prevalence (3.4%) of FS (14). Representing a unique model to study etiology and pathogenesis of FS, this Terena tribe lives in the reservation of Limao Verde in the state of Mato Grosso do Sul, Brazil. More than 50% of Limao Verde FS patients have at least one genetically related family member affected with the disease. Using a highly sensitive ELISA technique, we detected low levels of anti-Dsg1 autoantibodies in 55% of normal subjects (n = 93) living in Limao Verde and in FS patients several years before the clinical onset of disease (15). Consistent with the hypothesis that FS is triggered by an unidentified environmental factor(s), the percentage of Dsg1 antibody-positive individuals decreases in populations with increasing distance from the endemic focus of Limao Verde. Subsequent IgG subclass analyses revealed that the transition from preclinical to clinical disease is associated with a marked increase of anti-Dsg1 autoantibodies, predominantly of the IgG4 subclass (16).
To understand the etiology and immunopathology of FS, we sought to compare the epitope profiles of anti-Dsg1 autoantibodies present in normal subjects living in Limao Verde and in patients at different clinical stages of FS including preclinical and clinical disease, remission, and relapse. Our data suggest that FS autoimmunity evolves from a nonpathogenic response against the EC5 domain of Dsg1 to a pathogenic response against the EC1 and EC2 domains of the molecule. In addition, disease remission and relapse are strongly correlated with anti-Dsg1 epitope specificities.
Materials and Methods
Patients and Sera.
We studied a total of 40 sera from 28 FS patients living in the Limao Verde Amerindian reservation (Mato Grosso do Sul, Brazil). The clinical, histological and serological features of most of these patients have been published previously (14). These patients showed skin lesions typical of FS and no mucosal lesions.
The 28 FS patients included 20 patients with active disease exhibiting typical skin lesions and 8 patients, with no skin lesions, considered to be in clinical remission. The 20 FS patients with active disease included six patients, early in the course of the disease, from whom serum samples were also available 1 to 7 yr before disease onset and another 14 patients. FS patients with active disease were treated initially with prednisone using doses that ranged from 30 to 60 mg per day. Once a patient entered clinical remission, the steroids were tapered over several months. Several of these patients relapsed during the course of their therapy. All serum samples were collected before the onset of therapy. The indirect immunofluorescence (IF) titers of anti-epidermal antibodies in patients with active disease ranged from negative to 1:640. Two patients in this group showed negative indirect IF titers, but showed anti-Dsg1 antibodies by a Dsg1-ELISA assay.
Of the eight patients in clinical remission, three patients received no therapy for over 2 yr and the remaining five patients were on maintenance therapy with prednisone (2.5 to 10 mg per day). Seven of these eight patient sera had negative indirect IF tests; in one patient (FS17) the serum showed titers below 1:40.
Sera from 12 normal donors from the Limao Verde reservation were also included in this study. Among the 12 donors, 5 were first-degree relatives to the patients and 7 were unrelated. All 12 donors had no evidence of skin disease, but contained anti-Dsg1 autoantibodies by ELISA (15). The ELISA index values of the 12 sera ranged from 1.1 to 24.1 (cut-off value 0.92) with a mean value of 7.7. The ELISA index and the cut-off value were as described previously (15). 11 normal donor sera from the US were also tested as controls.
The collected sera were stored at −20°C and then transported to our laboratories at the University of North Carolina, where the immunological assays were performed. The study had the approval of FUNAI, a body representing the interests of the Indians of Limao Verde, and the Institutional Review Boards of the University of North Carolina, Chapel Hill and the Universities of Sao Paulo and Mato Grosso do Sul, Brazil.
Construction and Expression of Recombinant Antigens.
A distinct desmosomal core glycoprotein is desmoglein-3 (Dsg3), an autoantigen of the related autoimmune skin blistering disease known as pemphigus vulgaris (PV) (17). Dsg1 and Dsg3 have similar molecular structures (Fig. 1 A), but exhibit distinct antigenic sites recognized by PF-IgG and PV-IgG (10). We have previously constructed and expressed in baculovirus the entire extracellular portion of the human Dsg1 and Dsg3 polypeptide containing a COOH-terminal histidine (His) tag (18, 19).
Figure 1.
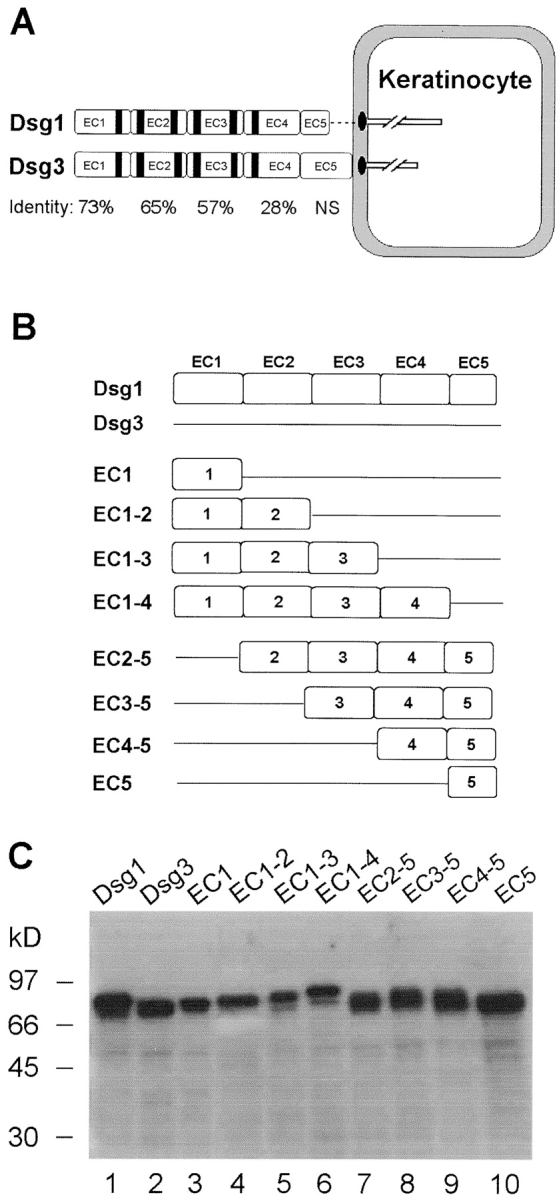
Schematic diagram and baculovirus production of Dsg1, Dsg3, and chimeric antigens. (A) Molecular structure of Dsg1 and Dsg3. Dsg1 and Dsg3 are structurally similar desmosomal transmembrane glycoproteins. The extracellular domain of the two molecules contains four cadherin-like domains (EC1 to EC4) and a membrane-proximal domain (EC5). Sequence identities between the cadherin repeats of Dsg1 and Dsg3 are indicated. NS indicates that sequence homology between the two molecules in the EC5 domain is not significant (reference 39). The black stripes represent six putative Ca2+ binding motifs found in the cadherin repeats. (B) Diagram of the recombinant Dsg1, Dsg3, and eight chimeric proteins. We constructed the full-length extracellular domains of Dsg1, Dsg3, and eight hybrid molecules that contained various combinations of Dsg1 extracellular domains (open box) with the corresponding Dsg3 (straight line) backbone. All recombinant proteins contain the endogenous signal sequence, a propeptide, and the extracellular domain of Dsg1/Dsg3 fused with a COOH-terminal His tag. (C) Production of the 10 recombinant proteins in the baculovirus expression system. The culture supernatants containing various secreted recombinant proteins (labeled on top of the figure) were collected and subjected to immunoblotting using HRP-labeled monoclonal antibody against the common His tag. Molecular weight standards are shown at left.
In the current study, we engineered various combinations of the extracellular domains (EC1 to EC5) of Dsg1 on the Dsg3 backbone (Fig. 1 B). Such domain swapping between homologous, but immune noncrossreactive proteins, has been used successfully for mapping many conformational epitopes (20, 21), including a recent study of Dsg1 and Dsg3 epitope mappings (10). Eight chimeric molecules of Dsg1 and Dsg3 were constructed from sixteen cDNA fragments encoding various domains of Dsg1 or Dsg3. These fragments were PCR amplified from human Dsg1 (GenBank accession no. X56654) and Dsg3 (GenBank accession no. M76482) cDNAs in vector PVL1393 (18, 19). The sequences of the PCR primers used are listed in Table I. BamHI and KpnI endonuclease sites were included in the forward and reverse primers, respectively, for amplifying the upstream cDNA fragments. Similarly, KpnI and NotI sites were introduced in the forward and reverse primers, respectively, for amplifying the downstream fragments. After digestion with appropriate restriction enzymes, each set of upstream and downstream cDNA fragments were ligated at the KpnI site and subcloned into the BamHI and NotI sites of the baculovirus expression vector pFastBac1 plasmid (Life Technologies). As a result of addition of the KpnI site, two extra residues of Glu and Ser were introduced at the junctions of Dsg1 and Dsg3 in these chimeric molecules. The coding region of the constructs begins with the endogenous initial Met followed by a signal sequence and a propeptide of Dsg1 or Dsg3, and a hybrid extracellular domain of Dsg1 and Dsg3. All constructs were fused with an in-frame COOH-terminal His tag, which enables us to detect or affinity purify the chimeric proteins.
Table I.
PCR Primers Used in Construction of Recombinant Chimeric Proteins
| Upstream fragments
|
Downstream fragments
|
|||||
|---|---|---|---|---|---|---|
| Primers
|
Primers
|
|||||
| PCR products |
Forward | Reverse | PCR products | Forward | Reverse | Dsg1 fragments on the Dsg3 backbone |
| Dsg1.EC1 | 1 | 2 | Dsg3.EC2-5 | 6 | 10 | EC1/Dsg3.EC2-5 |
| Dsg1.EC1-2 | 1 | 3 | Dsg3.EC3-5 | 7 | 10 | EC1-2/Dsg3.EC3-5 |
| Dsg1.EC1-3 | 1 | 4 | Dsg3.EC4-5 | 8 | 10 | EC1-3/Dsg3.EC4-5 |
| Dsg1.EC1-4 | 1 | 5 | Dsg3.EC5 | 9 | 10 | EC1-4/Dsg5 |
| Dsg3.EC1 | 11 | 12 | Dsg1.EC2-5 | 16 | 10 | EC2-5/Dsg3.EC1 |
| Dsg3.EC1-2 | 11 | 13 | Dsg1.EC3-5 | 17 | 10 | EC3-5/Dsg1.EC3-5 |
| Dsg3.EC1-3 | 11 | 14 | Dsg1.EC4-5 | 18 | 10 | EC4-5/Dsg3.EC1-3 |
| Dsg3.EC1-4 | 11 | 15 | Dsg1.EC5 | 19 | 10 | EC5/Dsg3.EC1-4 |
| Primer 1 | 5′-ATTCGGATCCCGCCATGGACTGGAGTTTCTTCAGAG | |||||
| Primer 2 | 5′-GCCGGTACCAAACACTGGAGGGTTGTCATT | |||||
| Primer 3 | 5′-GCCGGTACCCATGTAAGGGATATTATCATTGA | |||||
| Primer 4 | 5′-GCCGGTACCAAACACTGGGCCTTCAATTAC | |||||
| Primer 5 | 5′-GCCGGTACCAGTAATTTTAGTGTTCGGCTCT | |||||
| Primer 6 | 5′-GCCGGTACCTCACAACAAATTTTCATGGGTG | |||||
| Primer 7 | 5′-GCCGGTACCAGAGACTCTCAGTATTCAGCA | |||||
| Primer 8 | 5′-GCCGGTACCCCTGCTTCCAAGACATTTACT | |||||
| Primer 9 | 5′-GCCGGTACCACAGCTGTCCTCGAAAAAGAT | |||||
| Primer 10 | 5′-GCCGCGGCCGCCTAGCGCGCGTGATGGTG | |||||
| Primer 11 | 5′-CGGGGATCCCGCCATGGGGCTCTTCCCCAGAA | |||||
| Primer 12 | 5′-GCCGGTACCAAATACTGGAGGATTATCATTAAT | |||||
| Primer 13 | 5′-GCCGGTACCAAACATTGGGAAGTTATCGTTG | |||||
| Primer 14 | 5′-GCCGGTACCGAATGCAATTCCTTCTCTTACA | |||||
| Primer 15 | 5′-GCCGGTACCTGGACAATTGTCATTGAAATCG | |||||
| Primer 16 | 5′-GCCGGTACCTCAATGGCTACATTTGCAGGA | |||||
| Primer 17 | 5′-GCCGGTACCGAACAGTCTTCATATACCATAG | |||||
| Primer 18 | 5′-GCCGGTACCCGTCCAGGTTCAAAGACATAT | |||||
| Primer 19 | 5′-GCCGGTACCACCAATACTGGCAGACAAGAA | |||||
All chimeric cDNAs were sequenced to verify the expected hybrid sequences. The chimeric constructs were expressed as secreted glycoproteins in a baculovirus expression system (Bac-To-Bac; Life Technologies) according to the procedure provided by the manufacturer. High-five insect cells cultured in Express-Five serum-free medium (Life Technology) were used to produce the recombinant proteins. The yield of the recombinant proteins ranged from 2 to 10 μg/ml supernatant. The amounts of recombinant protein in these supernatants was estimated by comparing the intensities of the specific protein bands (shown by Western blots) produced by known amounts of purified recombinant Dsg1 and Dsg3. The concentration of purified recombinant Dsg1 and Dsg3 was determined by the Bradford assay (Bio-Rad Laboratories).
Immunoprecipitation Coupled with Immunoblotting.
The culture supernatants of recombinant proteins (∼0.5 μg recombinant protein) were incubated with each test serum (2 μl) in the presence of 5 mM Ca2+ overnight at 4°C. IgG-antigen immune complexes were precipitated by incubation with 20 μl of a 50% suspension of recombinant protein-G Sepharose 4B (Zymed Laboratories) for 1.5 h at 4°C. After 3 washes with Tris-buffered saline (pH 7.2) containing 5 mM Ca2+ and 0.1% Triton X-100, the pellet was extracted with 2× SDS sample buffer and boiled for 5 min. The eluted supernatant was then fractionated by 10% SDS-PAGE and analyzed by immunoblotting with an HRP-labeled anti-His monoclonal antibody (1:3000 dilution of Penta-His HRP Conjugate; QIAGEN). Antigen bands were detected by enhanced chemiluminesence (Amersham Biosciences).
To determine the spectrum of anti-Dsg1 autoantibody specificities, we analyzed the reactivity of each serum against a panel of 10 recombinant proteins (see Fig. 1 B). The entire extracellular domain of Dsg1 was included as a positive control. We also included the extracellular domain of Dsg3 to determine the background reactivity of each serum to the Dsg3 backbone.
Affinity-Purification and Pathogenicity of the Domain-specific Anti-Dsg1 Autoantibodies.
Total IgG fractions from a patient with FS exhibiting anti-Dsg1 antibodies specific for the EC1 and EC2 domains only were purified by protein G affinity chromatography. The anti-EC1–2 autoantibodies from the FS patient were affinity purified using the recombinant chimeric protein Dsg1-EC1–2 bound to the Ni-NTA resin (QIAGEN) as described by Ding et al. (19). Additionally, the IgG fraction was prepared from the serum of a normal individual from Limao Verde by Protein G affinity chromatography. This serum possessed high levels of anti-Dsg1 antibodies specific for the EC5 domain of the molecule. The specificity of the purified antibody fractions was confirmed by immunoprecipitation assays.
The pathogenicity of the anti-Dsg1 antibodies present in purified total IgG and purified anti-EC1-EC2 antibody fraction was tested by passive transfer experiments as we have described previously (1, 19). Briefly, BALB/c mice (24–36 h old with body weights 1.4 to 1.6 g) were injected intradermally with 100 μl aliquots of purified antibodies in PBS buffer. The doses of IgG are shown in Table II. The skin of neonatal mice was examined 18 h after the IgG injection. Skin sections were obtained for routine histopathological and direct immunofluorescence analysis.
Table II.
Pathogenicity of Anti-Dsg1 Autoantibodies Specific for the EC1-2 or EC5 Domains
| IgG | Dose injected (mg/g body weight) |
No. of mice tested |
Skin lesions |
Acantholysis (H&E) |
|---|---|---|---|---|
| Anti-EC1/EC2 | ||||
| Protein G purified | 1.5 | 3 | + | + |
| EC1-2 purified | 0.3 | 3 | + | + |
| Anti-EC5 | ||||
| Protein G purified | 1.5 | 3 | − | − |
| 15 | 3 | − | − |
Statistical Analysis.
Fisher's exact test was used to compare proportions in different groups of patients. The preonset versus post-onset comparison in the same patients was done with McNemar's test (exact P values reported).
Results
Construction and Baculovirus Expression of Recombinant Dsg1, Dsg3, and Chimeric Dsg1/Dsg3 Autoantigens.
We constructed eight chimeric molecules consisting of various ectodomain segments of Dsg1 (EC1, EC1–2, EC1–3, EC1–4, EC2–5, EC3–5, EC4–5, and EC5) grafted on the Dsg3 backbone and fused with a COOH-terminal His-tag (depicted in Fig. 1 B). 10 recombinant proteins were produced in the baculovirus system, including the full-length extracellular domain of Dsg1, the full-length extracellular domain of Dsg3, and eight chimeras containing various combinations of the extracellular domains of Dsg1 and Dsg3. Fig. 1 C shows anti-His tag immunoblot analysis of the 10 recombinant proteins secreted in the insect culture medium. It should be noted that the recombinant proteins were often expressed as a doublet, a phenomenon also noticed previously for the recombinant Dsg1 (8) and Dsg3 (22) produced by baculovirus. It has been suggested that the upper band represents the protein bearing the uncleaved propeptide due to the inefficiencies of proteolytic processing in the baculovirus expression system.
Sera from Normal Subjects Living in an Area Endemic for FS Recognize the EC5 Domain of Dsg1.
In the present epitope mapping study we selected 12 sera from normal subjects living in Limao Verde and showing relatively high Dsg1-ELISA index values. These sera were tested against a panel of 10 recombinant proteins (see Fig. 1, B and C) by immunoprecipitation (IP) assay. Representative IP results are shown in Fig. 2 . All sera tested reacted with the full-length extracellular domain of Dsg1 (Fig. 2, lane 1) and chimeras that harbored the EC5 domain of Dsg1, including EC2–5 (Fig. 2, lane 7), EC3–5 (Fig. 2, lane 8), EC4–5 (Fig. 2, lane 9), and EC5 (Fig. 2, lane 10). It should be noted that the reaction of the sera with the EC2–5 (Fig. 2, lane 7) and EC4–5 chimeras (Fig. 2, lane 9) was often weaker (Fig. 2, also seen in Fig. 3 , and Fig. 6, A and B below) than the reaction of the other two EC5-containing chimeras (Fig. 2, lanes 8 and 10). The lower reactivity of these Dsg1 chimeras may be due to a slightly altered folding of these hybrid molecules, thus making the EC5 domain less accessible.
Figure 2.

Analysis of anti-Dsg1 antibodies from normal subjects living in an area endemic for FS. Each serum sample was immunoprecipitated with 10 recombinant proteins (labeled on top of each panel), respectively, and subjected to immunoblotting using a monoclonal antibody against the His tag. IP results from three normal individuals living in the Limao Verde of Brazil are shown.
Figure 3.
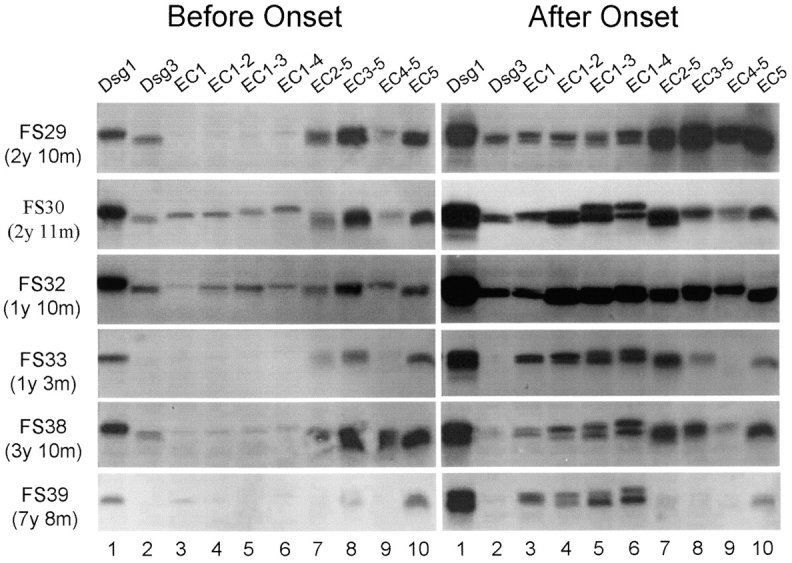
Antibody specificities from FS patients before and after the clinical onset of disease. Sera from six patients before (left panel) or after (right panel) clinical disease onset were analyzed by IP. Left panel: the time intervals between the preclinical sera and disease onset are indicated at left. y: year(s); m: months. Right panel: sera from patients FS29, FS32, and FS33 were collected after 3, 2, or 9 mo, respectively after the first symptoms of FS observed; sera from patients FS30, FS38, and FS39 were collected within the same month of disease onset.
Figure 6.
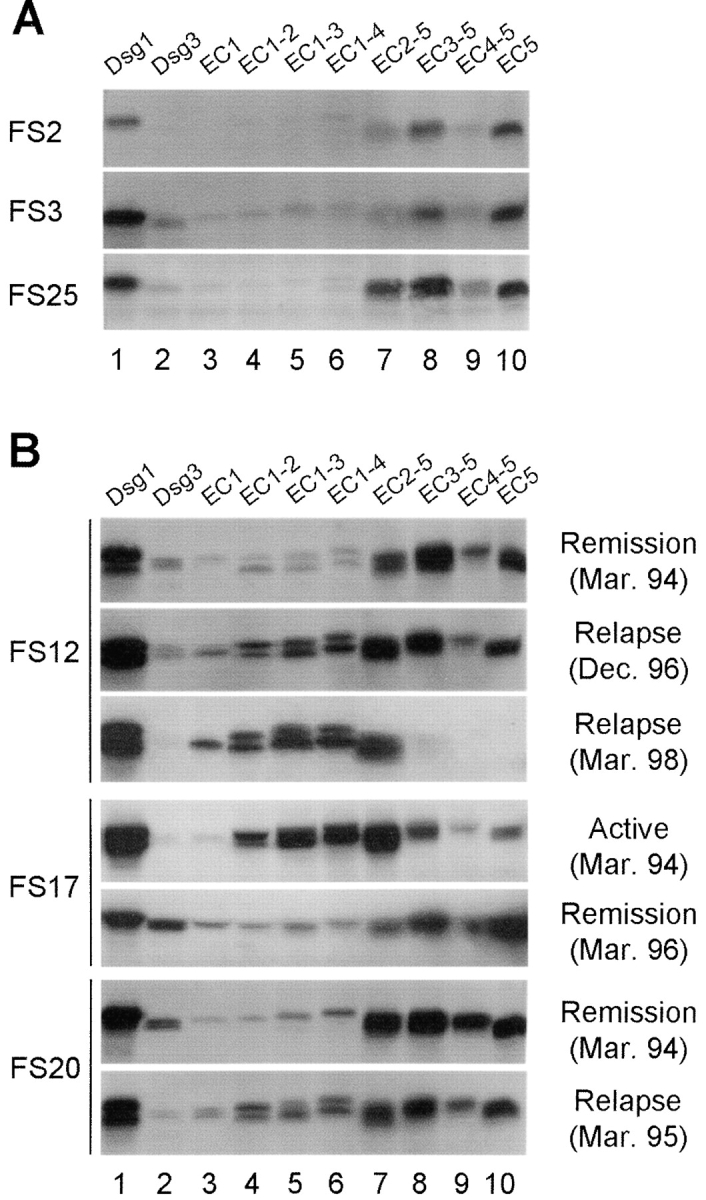
Antibody specificities from FS patients during the course of disease. (A) IP results from three FS patients in long-term clinical remission. (B) IP results from sequential sera samples obtained from three representative patients (FS12, FS17, and FS20) during the course of disease. Clinical outcomes and bleeding dates are labeled at right.
By contrast, the 11 normal sera from US donors showed only negative or background reactivities when tested against either the full-length extracellular domains of Dsg1 or the chimeric protein containing the EC5 domain of Dsg1 (data not depicted).
The Transition from the Preclinical to the Clinical Stage of FS Is Associated with Apparent Epitope Spreading from EC5 to EC1 and EC2 of Dsg1.
The observation that anti-Dsg1 autoantibodies from healthy individuals targeted the EC5 domain of Dsg1 suggests that anti-Dsg1 autoantibodies in FS might be initially raised against the EC5 domain of Dsg1. To test this hypothesis, we examined epitope specificities of anti-Dsg1 autoantibodies from six FS patients from whom sequential blood samples were available before and after the onset of disease. Fig. 3 shows the antibody reactivity of the sera obtained before (Fig. 3, left panel) and after (Fig. 3, right panel) disease onset. Low levels of anti-Dsg1 autoantibodies were present 1 to 7 yr before the clinical onset of FS (Fig. 3, left panel, lane 1). These preclinical sera bound the full-length extracellular domain of Dsg1 (Fig. 3, left panel, lane 1) and EC5 bearing chimeras (Fig. 3, lanes 7–10). Four sera (FS29, FS30, FS32, and FS38) from the preclinical stage bound the extracellular domain of Dsg3 (Fig. 3, lane 2), although the reaction was much weaker than the reactivity produced with Dsg1 (Fig. 3, lane 1).
A considerable increase of anti-Dsg1 autoantibodies was observed in sera obtained after the onset of clinical disease (right panel, lane 1). Interestingly, all serum samples at this stage reacted with chimeras containing the EC1 (right panel, lanes 3–6) and/or EC2 (lanes 4–7) domains of Dsg1. Three of the 4 sera, which reacted weakly with Dsg3 during the preclinical stage, continued recognizing Dsg3 during the active stage of FS (FS29, FS30, and FS32, right panel, lane 2).
Comparing the proportion of positive tests for anti-EC1 antibodies in the preclinical and clinical stage sera in six patients with pre and post-onset data, we found a statistically significant difference (P = 0.016).
Anti-EC5 Autoantibodies from Normal or Preclinical Individuals Do Not React with the Human Epidermis.
We further tested the binding of the anti-EC5 sera to human epidermis by indirect IF. As shown in Fig. 4 , FS sera possessing anti-EC1 and anti-EC2 reactivity stained human skin sections (Fig. 4, left panel). In contrast, anti-EC5 sera from all the 12 normal subjects tested and 6 preclinical FS patients sera showed negative staining. A representative result is shown on the right panel. This result suggests that anti-Dsg1 autoantibodies in normal or preclinical individuals do not bind to human epidermis, as assayed by indirect IF, although they are detectable by IP or ELISA.
Figure 4.

Indirect IF analysis of anti-EC1/EC2 and anti-EC5 sera on the human epidermis. Left panel: a serum from a FS patient with anti-Dsg1 antibodies specific for the EC1 and EC2 domains. Right panel: a normal serum with anti-Dsg1 antibodies specific for the EC5 domain of Dsg1.
All Sera from Patients with Active Disease Recognize the EC1 and/or EC2 Domains of Dsg1.
We then examined sera from 14 patients with active disease. Representative results are shown in Fig. 5 . These sera produced four epitope profiles or reaction patterns when tested with Dsg1 chimeras by IP. Pattern 1 was produced by nine sera (64%) and showed strong reactivity with chimeras containing the EC1 and EC2 domains (Fig. 5, lanes 3–7), but very little reactivity with chimeras that did not contain these domains (Fig. 5, lanes 8–10). A patient (FS12) during a severe clinical relapse of the disease showed the same reaction pattern (see Fig. 6 B below). Pattern 2 was produced by one serum only (7.1%). This serum recognized chimeras bearing only the EC1 domain in common (Fig. 5, lanes 3–6); it was barely reactive with chimeras lacking EC1 (Fig. 5, lanes 7–10). This pattern was also produced by the serum of patient FS39 at disease onset (see Fig. 3). Pattern 3 was seen with one serum (7.1%) that reacted with EC2 domain only (Fig. 5, lanes 4–7), but showed little or no reactivity with EC1 (Fig. 5, lane 3). The serum of patient FS17 also showed similar pattern during the active phase of the disease (see Fig. 6 B). Pattern 4 was produced by three sera (21.5%), which reacted with all Dsg1 chimeras tested (Fig. 5, lanes 3–10). This epitope profile was also observed when testing the sera of four patients shortly after disease onset (FS29, FS30, FS32, and FS38, Fig. 3) and the sera from two patients during clinical relapses (FS12-Dec. 96 and FS20-Mar. 95, in Fig. 6 B).
Figure 5.

Epitope profiles of anti-Dsg1 antibodies from patients with active disease. IP results from four representative FS patients with active disease are shown. The number of sera within this group (n = 14) showing similar reaction patterns is indicated at right.
Since all sera showing reaction pattern 4 recognized the EC1 (Fig. 5, lane 3) and the EC5 domains (Fig. 5, lane 10), it is likely that the reactivity observed with other chimeras (Fig. 5, lanes 4–9) are due to the presence of anti-EC1 (EC1 domain is common to chimeras of lanes 3–6) and anti-EC5 antibodies (EC5 domain is common to chimeras of lanes 4–10) in these sera. However, we cannot rule out the possibility that these sera may also recognize the EC2, EC3, or EC4 domains.
There was a significant difference between patients with active disease (n = 20) and normal subjects (n = 12) in the percentage of sera recognizing the EC1 (P < 0.0001), EC2 (P < 0.001), and EC5 reactivity (P < 0.001) domains.
Sera from Patients in Clinical Remission Reacted with the EC5 Domain Only.
Similar to the epitope profiles observed in sera from normal subjects (see Fig. 2) or from patients in the preclinical stage (see Fig. 3, left panel), the sera from patients in clinical remissions (n = 8) all predominantly recognized the EC5 domain. Fig. 6 A shows the epitope profile of the three FS patients in complete remission for more than 2 yr without therapy (FS2, FS3 and FS25). Fig. 6 B shows the reaction patterns produced by another three patients in clinical remission on low maintenance steroid therapy (FS12, FS17, and FS20). The remaining 2 sera showed similar results. There was a significant difference between patients with active disease (n = 20) and patients in remission (n = 8) in the percentage of sera recognizing the EC1 (P < 0.001), EC2 (P = 0.01), and EC5 reactivity (P = 0.003) domains.
Dynamic Changes of Epitope Specificities during Disease Remission and Relapses in Individual Patients.
To observe dynamic changing of epitope profiles, we determined the antibody specificities of sequential sera from FS patients (n = 5) during different clinical stages of the disease (active, remission, and relapse). Three representative patients (FS12, FS17, and FS20) are shown in Fig. 6 B.
Serum from patient FS12 during clinical remission (March 1994) predominantly reacted with EC5 containing chimeras (Fig. 6 B, lanes 7–10). A mild clinical relapse in December 1996 was heralded by the appearance of anti-EC1 and anti-EC2 antibodies (Fig. 6 B, lanes 3–7) and continuous anti-EC5 reactivity (Fig. 6 B, lanes 8–10). A severe clinical relapse in March 1998 was associated with an increase in the intensity of anti-EC1 and anti-EC2 reactivities and the disappearance of anti-EC5 antibodies.
Serum from patient FS17 during active disease (March 1994) reacted with EC2 containing chimeras (Fig. 6 B, lanes 4–7) and, to a lesser extent, with EC5 containing chimeras (Fig. 6 B, lanes 8–10). As the disease entered clinical remission (March 1996), anti-EC2 reactivity (Fig. 6 B, lanes 4–6) declined significantly. The weak reaction of FS17 (in remission) with EC1 containing chimeras (Fig. 6 B, lanes 3–6) is likely due to reactivity with the Dsg3 backbone since this serum also recognized Dsg3 (Fig. 6 B, lane 2).
Serum from patient FS20 in clinical remission (March 1994) predominantly reacted with EC5 containing chimeras (Fig. 6 B, lanes 7–10). In addition to reacting with Dsg1 (Fig. 6 B, lane 1), this serum also reacted weakly with Dsg3 (Fig. 6 B, lane 2). A mild clinical relapse in 1995 was associated with an increase in reactivity with EC1/EC2 containing chimeras (Fig. 6 B, lanes 3–6), and a decrease of reactivity with Dsg3, which declined to background (Fig. 6 B, lane 2).
Antibody Reactivity with Dsg3 in Different Groups of Sera Tested.
We found several sera from normal controls and FS patients that recognized the full-length ectodomain of Dsg3. The intensity of the Dsg3 IP reaction varied from weak, in the majority of sera, to fairly strong in a small group of sera.
We detected anti-Dsg3 antibodies by IP in the following groups of sera: (a) in 2 out of 12 sera from normal donors from Limao Verde (same group possessing anti-Dsg1 antibodies), (b) in the sera of 3 patients before and after the onset of FS (FS29, FS30, and FS32; shown in Fig. 3, left and right panel), (c) in one FS patient (FS38) 3 yr and 10 mo before the onset of the disease (shown in Fig. 3, the left panel), (d) in 2 of 20 patients with active FS, and (e) in 3 out of 8 patients in clinical remission (shown in Fig. 6 B, FS12, FS17, and FS20). The intensity of the IP reaction with Dsg3 was much weaker than that observed with Dsg1 in all groups of sera tested.
In summary, of the 28 FS patients tested, 5 sera (FS29, FS30 and FS32, shown in Fig. 3, right panel, and FS17 and FS20, shown in Fig. 6 B) demonstrated good IP reactivity with the full-length ectodomain of Dsg3.
It appears that the majority of sera showing anti-Dsg3 antibodies recognized chimeras expressing the EC5 domain of Dsg3 in common. This finding is shown in Fig. 3 (Fig. 6 B, lanes 3–6, left panel with FS30 and FS32) and also in sera from FS12, FS17 and FS20 in remission (see Fig. 6 B, lanes 3–6). Only one serum (from FS29 before disease onset) appears to bind the EC1 domain of Dsg3, as chimeras bearing the EC2–5, EC3–5 EC4–5 and EC5 domain of Dsg3 (Fig. 3, lanes 3–6, left panel) did not produce any reaction.
Autoantibodies to the EC1/EC2 Domains of Dsg1 Are Pathogenic, whereas the Anti-EC5 Autoantibodies Are Nonpathogenic.
We further tested the pathogenicity of the anti-EC1/EC2 and anti-EC5 autoantibodies by passive transfer experiments and the results are summarized in Table II. Mice injected with total IgG (affinity-purified by protein G) and EC1–2 domain-specific IgG (purified using EC1-EC2 chimeras) obtained from a FS patient with active disease developed skin lesions, which on histological examination revealed subcorneal acantholysis. In contrast, the IgG fraction (affinity-purified by protein G) from a normal unaffected donor from Limao Verde, possessing high titer anti-Dsg1 antibodies specific for the EC5 domain, failed to induce disease in neonatal mice. The lack of pathogenicity of this IgG fraction was observed with a dose 10 times higher than those injected with total pathogenic IgG from a FS patient (see Table II).
Discussion
Since 1994, our clinical and serological surveillance of the ∼1,200 individuals living in Limao Verde, Brazil has revealed a 3.4% prevalence of FS (14). Moreover, a significant pool of normal individuals (55%) living on the reservation possesses anti-Dsg1 antibodies (15). Follow up studies of this settlement have shown an average of 2–3 new cases of FS per year. In the present study, we have investigated the evolution of the anti-Dsg1 autoimmune response in these inhabitants. Our data showed that anti-Dsg1 antibodies from those normal individuals and patients with FS before the onset of clinical disease all recognized the EC5 domain (residues 453–496) of the molecule. Significantly, transition from the preclinical to clinical stage of FS was accompanied by emergence of autoantibodies specific for the EC1 (residues 1–108) and EC2 (residues 109–221) domains. Moreover, by passive transfer experiments we demonstrated that anti-EC1/EC2 autoantibodies were pathogenic using neonatal mice, whereas the IgG fraction containing anti-EC5 autoantibodies was incapable of inducing blisters in mice. These data suggest that the anti-Dsg1 autoimmune response in FS is initially raised against nonpathogenic epitopes located in the EC5 domain and later against pathogenic epitopes mapped to the EC1 and EC2 domains of the molecule. It is likely that anti-EC1/EC2 autoantibodies are generated through the mechanism of intramolecular epitope spreading (23, 24) from the initial anti-EC5 autoantibodies rather than through the mechanism of cross-reactivity, as there is no significant sequence homology between the EC5 domain and the cadherin-like domains (EC1 to EC4).
Epitope spreading is a phenomenon in which new epitopes, within the same or a different molecule, are recognized over time by T cell or B cell from an original noncross-reactive antigenic site (23, 24). This phenomenon has been well demonstrated in various experimentally induced or spontaneous animal models of autoimmunity at both the T cell and B cell levels (25). However, the evidence of epitope spreading in human autoimmune diseases is limited (25). Possible intramolecular epitope spreading has been observed recently in six patients with systemic lupus erythematosus in the anti-Sm B/B' antibody system (26) and patients with type 1 diabetes in the anti-GAD65 (27, 28) and anti–IA-2 (29) antibody systems. However, the epitope spreading seen in these patients was not specifically associated with onset of clinical disease.
The restricted anti-EC5 reactivity observed in normal individuals living in an endemic focus of FS and in FS patients before the clinical onset of disease strongly suggests a common immunological mechanism initiating Dsg1 autoimmune response. A unique feature of the anti-EC5 antibodies, as observed in this study, is that they do not bind Dsg1 in cryosections of human epidermis as demonstrated by indirect IF. The negative staining of anti-EC5 sera on human epidermis is unlikely due to the low titer of the antibodies as many of these sera showed very high titers by ELISA (15). We hypothesize that the EC5 domain, a very short region close to the cell membrane, is sequestered in vivo and consequently, unable to be bound by circulating anti-EC5 antibodies. The cryptic nature of the EC5 domain is consistent with the concept that molecular mimicry to cryptic self-peptides or exposure of cryptic self-peptides elicits autoimmunity because normally there is a lack of self-tolerance to cryptic epitopes.
Based on the current findings, we propose a two-phase model to encompass the immunopathogenic mechanisms of FS (Fig. 7) . In the first phase, an environmental antigen(s), bearing sequence homology with the EC5 domain of Dsg1, triggers an initial nonpathogenic antibody response to the EC5 domain of Dsg1. At this stage, individuals remain free of skin disease. In the second phase, in certain genetically susceptible individuals, intramolecular epitope spreading occurs, which leads to the production of pathogenic anti-Dsg1 antibodies against the EC1 and EC2 domains. In turn, these antibodies would bind to the EC1 and EC2 domain of Dsg1 and induce skin blistering. The IgG subclass of the anti-Dsg1 autoantibodies generated in the two phases is likely to be influenced by cytokines released during each stage of the immune response in these patients. The initial phase might be driven by Th0 or both Th1 and Th2 cytokines and therefore the anti-Dsg1 autoantibodies generated in this phase are IgG1 and IgG4 subclass (16, unpublished data). In the second phase, primed T cells are mainly Th2 type (30) and consequently, pathogenic autoantibodies are predominantly of IgG4 subclass (1). However, it appears that the EC1 or EC2 epitopes are the drivers of the pathogenicity of the anti-Dsg1 autoantibodies rather than the IgG subclass. Indeed, we have studied a PF patient with IgG1 anti-Dsg1 autoantibodies specific for the EC1 and EC2 domains, which are pathogenic when tested on the passive transfer mouse model (31).
Figure 7.

A model of the immunopathogenic pathway of FS. Environmental factor(s) trigger the production of nonpathogenic IgG1 and IgG4 antibodies against the EC5 domain of Dsg1. In certain genetically predisposed individuals, intramolecular epitope spreading results in the generation of predominantly IgG4 antibodies, against the EC1 and EC2 domains of Dsg1, that induce disease.
The two-phase model is supported by a recent study in our laboratory (unpublished data) that shows that a significant number of patients with diseases in which hematophagous vectors are involved in the transmission of the illness, i.e., onchocerciasis (black flies), leishmaniasis (sand flies), and Chagas disease (kissing bugs) possess antibodies against the EC5 domain of Dsg1. Onchocerciasis and Chagas disease are unknown in the Limao Verde reservation, where FS is prevalent, but it is well documented that inhabitants of this settlement are constantly bitten by black flies, kissing bugs, and sand flies (unpublished data). The authors hypothesize that a salivary antigen(s) from these blood-feeding insects may contain a cross-reactive molecule(s) that triggers the anti-Dsg1 EC5 antibody response.
The incubation time between the first and the second phase of FS could last as long as 7 yr as observed in one patient. Numerous factors may influence epitope spreading, including the HLA alleles (25, 32, 33). It is known that FS is strongly associated with certain HLA-DRB1 alleles, such as DRB1*0404, *1402, *1406, or *0102 (34, 35) alleles, which encode a shared peptide sequence at positions 67–74 in the P4 pocket of hypervariable DRβ1 chain. These FS-susceptibily alleles are not frequently found in normal individuals possessing anti-Dsg1 antibodies specific for the EC5 domain (unpublished data). It is possible that antigen-presenting cells expressing these susceptibility HLA-II alleles are capable of presenting Dsg1 peptides to T cells bearing specific receptors for EC1 or EC2 epitopes. These T cells in turn would stimulate EC1- and EC2-specific B cells to produce anti-EC1 and anti-EC2 autoantibodies.
One interesting finding in this study is the dynamic change in epitope specificities during the time course of the disease. Anti-EC1 and anti-EC2 autoantibodies disappeared with disease remission and reappeared with disease relapses. EC5-specific antibodies were present mainly in patients during disease onset or clinical relapse, and persisted during remission. These findings clearly demonstrate that the anti-Dsg1 immune response in FS is dynamic and that anti-EC1/EC2 autoantibodies correlate well with the clinical activity of the disease. The immunological mechanism responsible for the elimination of anti-EC1/EC2 antibodies in FS patients during drug-induced remission or spontaneous remission, reported to be around 10% (36), remains unknown. Although the mechanism regulating the recurrence of the anti-EC1 and anti-EC2 antibodies during disease relapses is not clear, it is possible that the reappearance these antibodies during disease relapse may result from reactivation of memory B and T cells by reexposure to self-Dsg1.
Although the majority of patients with active disease possess a mixture of anti-EC1 and anti-EC2 antibody populations, a few sera possessed antibodies to EC1 without EC2, or to EC2 without EC1. This observation indicates that autoantibodies against EC1 and EC2 recognized at least two distinct epitopes, and either one alone maybe capable of causing disease (31). This finding suggests that both the EC1 and EC2 domains of Dsg1 are relevant in the mechanism of acantholysis (cell–cell detachment) induced by anti-Dsg1 autoantibodies.
Another interesting finding of this study was the identification of several sera from FS patients and normal subjects living in the Limao Verde reservation that contained antibodies against the ectodomain of Dsg3. Patients and normal individuals exhibiting anti-Dsg3 antibodies in their sera did not have mucosal lesions, typical of pemphigus vulgaris, a disease characterized by anti-Dgs3 autoantibodies. It should be noted that the anti-Dsg3 reactivity of the majority of sera was weak. However, in 12% of the sera from FS patients with active disease (n = 25) we observed a fairly good IP reactivity with the ectodomain of Dsg3. These findings are in agreement with a recent study reported by Arteaga et al. (37) that found anti-Dsg3 antibodies in 18 out of 241 FS sera tested by an ELISA assay using the recombinant ectodomain of Dsg3 (7.5%). It is feasible that anti-Dsg3 and anti-Dsg1 autoantibodies in these sera represent distinct populations of antibodies or perhaps Dsg1/Dsg3 cross-reactive antibodies. This finding may also leave open the possibility that in some rare individuals the anti-Dsg3 response may lead to an endemic form of pemphigus vulgaris as suggested by a preliminary report from Brazilian investigators (38).
In conclusion, our data provide new insights into the immunopathogenesis of FS. These findings may be extended to the nonendemic form of PF and to other forms of pemphigus, and may also have implications to other antibody-mediated autoimmune diseases. Rather than using the entire Dsg1 ectodomain, the specific detection of anti-EC1/EC2 and anti-EC5 autoantibodies might be more reliable for the diagnosis of FS and for the monitoring of disease remission and relapse. This study may facilitate the identification of a subset of normal individuals living in Limao Verde who are at risk to develop FS, namely those carrying susceptible HLA class II alleles and possessing anti-Dsg1 EC5 autoantibodies. Finally, the results of these studies may enhance the chances of identifying the environmental trigger for FS.
Acknowledgments
We appreciate the support of Dr. Bahjat Qaqish, Department of Biostatistics, University of North Carolina, on the analysis of data.
This work was supported in part by U.S. Public Health Service Grants R01-AR30281, RO1-AR32599, and T32 AR07369 awarded to Dr. L.A. Diaz.
Footnotes
Abbreviations used in this paper: Dsg1, desmoglein-1; Dsg3, desmoglein-3; FS, fogo selvagem; His, histidine; IF, immunofluorescence; IP, immunoprecipitation; PF, pemphigus foliaceus; PV, pemphigus vulgaris.
References
- 1.Rock, B., C.R. Martins, A.N. Theofilopoulos, R.S. Balderas, G.J. Anhalt, R.S. Labib, S. Futamura, E.A. Rivitti, and L.A. Diaz. 1989. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). N. Engl. J. Med. 320:1463–1469. [DOI] [PubMed] [Google Scholar]
- 2.Seishima, M., Y. Iwasaki-Bessho, Y. Itoh, Y. Nozawa, M. Amagai, and Y. Kitajima. 1999. Phosphatidylcholine-specific phospholipase C, but not phospholipase D, is involved in pemphigus IgG-induced signal transduction. Arch. Dermatol. Res. 291:606–613. [DOI] [PubMed] [Google Scholar]
- 3.Mahoney, MyG., Z. Wang, K. Rothenberger, P.J. Koch, M. Amagai, and J.R. Stanley. 1999. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J. Clin. Invest. 103:461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kowalczyk, A.P., J.E. Anderson, J.E. Borgwardt, T. Hashimoto, J.R. Stanley, and K.J. Green. 1995. Pemphigus sera recognize conformationally sensitive epitopes in the amino-terminal region of desmoglein-1. J. Invest. Dermatol. 105:147–152. [DOI] [PubMed] [Google Scholar]
- 5.Eyre, R.W., and J.R. Stanley. 1987. Human autoantibodies against a desmosomal protein complex with a calcium-sensitive epitope are characteristic of pemphigus foliaceus patients. J. Exp. Med. 165:1719–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labib, R.S., B. Rock, M.A. Robledo, and G.J. Anhalt. 1991. The calcium-sensitive epitope of pemphigus foliaceus antigen is present on a murine tryptic fragment and constitutes a major antigenic region for human autoantibodies. J. Invest. Dermatol. 96:144–147. [DOI] [PubMed] [Google Scholar]
- 7.Emery, D.J., L.A. Diaz, J.A. Fairley, A. Lopez, A.F. Taylor, and G.J. Giudice. 1995. Pemphigus foliaceus and pemphigus vulgaris autoantibodies react with the extracellular domain of desmoglein-1. J. Invest. Dermatol. 104:323–328. [DOI] [PubMed] [Google Scholar]
- 8.Amagai, M., T. Hashimoto, K.J. Green, N. Shimizu, and T. Nishikawa. 1995. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J. Invest. Dermatol. 104:895–901. [DOI] [PubMed] [Google Scholar]
- 9.Olague-Alcala, M., G.J. Giudice, and L.A. Diaz. 1994. Pemphigus foliaceus sera recognize an N-terminal fragment of bovine desmoglein 1. J. Invest. Dermatol. 102:882–885. [DOI] [PubMed] [Google Scholar]
- 10.Sekiguchi, M., Y. Futei, Y. Fujii, T. Wasaki, T. Nishikawa, and M. Amagai. 2001. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J. Immunol. 167:5439–5448. [DOI] [PubMed] [Google Scholar]
- 11.Diaz, L.A., S.A. Sampaio, E.A. Rivitti, C.R. Martins, P.R. Cunha, C. Lombardi, F.A. Almeida, R.M. Castro, M.L. Macca, C. Lavrado, et al. 1989. Endemic pemphigus foliaceus (fogo selvagem). 1. Clinical features and immunopathology. J. Am. Acad. Dermatol. 20:657–659. [DOI] [PubMed] [Google Scholar]
- 12.Stanley, J.R., V. Klaus-Kovtun, and S.A. Sampaio. 1986. Antigenic specificity of fogo selvagem autoantibodies is similar to North American pemphigus foliaceus and distinct from pemphigus vulgaris autoantibodies. J. Invest. Dermatol. 87:197–201. [DOI] [PubMed] [Google Scholar]
- 13.Hans-Filho, G., V. Aoki, E. Rivitti, D.P. Eaton, M.S. Lin, and L.A. Diaz. 1999. Endemic Pemphigus Foliaceus (Fogo Selvagem)-1998. Clin. Dermatol. 17:225–235. [DOI] [PubMed] [Google Scholar]
- 14.Hans-Filho, G., V. dos Santos, J.H. Katayama, V. Aoki, E.A. Rivitti, S.A. Sampaio, H. Friedman, J.R. Moraes, M.E. Moraes, D.P. Eaton, et al. 1996. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. J. Invest. Dermatol. 107:68–75. [DOI] [PubMed] [Google Scholar]
- 15.Warren, S.J., M.S. Lin, G.J. Giudice, R.G. Hoffmann, G. Hans-Filho, V. Aoki, E.A. Rivitti, V. Santos, and L.A. Diaz. 2000. The prevalence of antibodies against desmoglein 1 in endemic pemphigus foliaceus in Brazil. N. Engl. J. Med. 343:23–30. [DOI] [PubMed] [Google Scholar]
- 16.Warren, S.J.P., and L.A. Arteaga. 2003. The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J. Invest. Dermatol. 120:104–108. [DOI] [PubMed] [Google Scholar]
- 17.Anhalt, G.J., and L.A. Diaz. 2001. Prospects for autoimmune disease: research advance in pemphigus. JAMA. 285:652–654. [DOI] [PubMed] [Google Scholar]
- 18.Ding, X., V. Aoki, J.M. Mascaro, A. Lopez-Swiderski, L.A. Diaz, and J.A. Fairley. 1997. Mucosal and mucocutaneous (generalized) pemphigus vulgaris show distinct autoantibody profiles. J. Invest. Dermatol. 109:592–596. [DOI] [PubMed] [Google Scholar]
- 19.Ding, X., L.A. Diaz, J.A. Fairley, G.J. Giudice, and Z. Liu. 1999. The antidesmoglein-1 autoantibodies in pemphigus vulgaris sera are pathogenic. J. Invest. Dermatol. 112:739–743. [DOI] [PubMed] [Google Scholar]
- 20.Henriksson, E.W., and I. Pettersson. 1997. Autoepitope-mapping of the U1-70K protein with human-Drosophila chimeric proteins. J. Autoimmun. 10:559–568. [DOI] [PubMed] [Google Scholar]
- 21.Netzer, K.-A., A. Leinonen, A. Boutaud, D.-B. Borza, P. Todd, S. Gunwar, J.P.M. Langeveld, and B.G. Hudson. 1999. The Goodpasture autoantigen. Mapping the major conformational epitope(s) of α(3IV) collagen to residues 17-31 and 127-141 of the NC1 domain. J. Biol. Chem. 274:11267–11274. [DOI] [PubMed] [Google Scholar]
- 22.Amagai, M., T. Hashimoto, N. Shimizu, and T. Nishikawa. 1994. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J. Clin. Invest. 94:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderlugt, C.J., and S.D. Miller. 1996. Epitope spreading. Curr. Opin. Immunol. 8:831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamula, M.J. 1998. Epitope spreading: the role of self peptide and autoantigen processing by B lymphocytes. Immunol. Rev. 164:231–239. [DOI] [PubMed] [Google Scholar]
- 25.McCluskey, J., A.D. Farris, C.L. Keech, A.W. Purcell, M. Rischmueller, G. Kinoshita, P. Reynolds, and T.P. Gordon. 1998. Determinant spreading: lesions from animal models and human disease. Immunol. Rev. 164:209–229. [DOI] [PubMed] [Google Scholar]
- 26.Arbuckle, M.R., M. Reichlin, J.B. Harley, and J.A. James. 1999. Shared early autoantibody recognition events in the development of anti-Sm B/B' in human lupus. Scand. J. Immunol. 50:447–455. [DOI] [PubMed] [Google Scholar]
- 27.Söhnlein, P., M. Müller, K. Syren, U. Hatmann, B.O. Böhm, H.M. Meinck, M. Knip, and H.K. Akerbom. 2000. Epitope spreading and a varying but not disease-specific GAD65 antibody response in Type I diabetes. Diabetologia. 43:210–217. [DOI] [PubMed] [Google Scholar]
- 28.Bonifacio, E., V. Lampasona, L. Bernasconi, and A.-G. Ziegler. 2000. Maturation of the humoral autoimmune response to epitopes of GAD in preclinical childhood Type 1 diabetes. Diabetes. 49:202–208. [DOI] [PubMed] [Google Scholar]
- 29.Naserke, H.E., A.-G. Ziegler, V. Lampasona, and E. Bonifacio. 1998. Early development and spreading of autoantibodies to epitopes of IA-2 and their association with progression to Type 1 diabetes. J. Immunol. 161:6963–6969. [PubMed] [Google Scholar]
- 30.Lin, M.S., C.L. Fu, V. Aoki, G. Hans-Fiho, E.A. Rivitti, J.R. Moraes, M.E. Moraes, A.M. Lazaro, G.J. Giudice, P. Stastny, and L.A. Diaz. 2000. Desmoglein-1-specific T lymphocytes from patients with endemic pemphigus foliaceus (fogo selvagem). J. Clin. Invest. 105:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, N., Z. Liu, and L.A. Diaz. 2002. Pemphigus foliaceus autoantibodies recognize two dominant pathogenic epitopes located in EC1 and EC2 domains of Desmoglein-1. J. Invest. Dermatol. 119:305. (Abst. 587). [Google Scholar]
- 32.Paisansinsup, T., U.S. Deshmukh, V.R. Chowdhary, H.S. Luthra, S.M. Fu, and C.S. David. 2002. HLA class II influences the immune response and antibody diversification to Ro60/Sjögren's syndrome-A: heightened antibody responses and epitope spreading in mice expressing HLA-DR molecules. J. Immunol. 168:5876–5884. [DOI] [PubMed] [Google Scholar]
- 33.James, J.A., and J.B. Harley. 1998. A model of peptide-induced lupus autoimmune B cell epitope spreading is strain specific and is not H-2 restricted in mice. J. Immunol. 160:502–508. [PubMed] [Google Scholar]
- 34.Moraes, M.E., M. Fernandez-Vina, A. Lazaro, L.A. Diaz, G.H. Filho, H. Friedman, E. Rivitti, V. Aoki, P. Stastny, and J.R. Moraes. 1997. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. 49:35–40. [DOI] [PubMed] [Google Scholar]
- 35.Moraes, J.R., M.E. Moraes, M. Fernandez-Vina, L.A. Diaz, H. Friedman, I.T. Campbell, R.R. Alvarez, S.A. Sampaio, E.A. Rivitti, and P. Stastny. 1991. HLA antigens and risk for development of pemphigus foliaceus (fogo selvagem) in endemic areas of Brazil. Immunogenetics. 33:388–391. [DOI] [PubMed] [Google Scholar]
- 36.Vieira, J.P. 1940. Pemphigus Foliaceus (Fogo Selvagem). An endemic disease of the state of Sao Paulo (Brazil). Arch. Dermatol. 41:858–863. [Google Scholar]
- 37.Arteaga, L.A., P.S. Prisayanh, S.J.P. Warren, Z. Liu, L.A. Diaz, and M.-S. Lin. 2002. A subset of pemphigus foliaceus patients exhibits pathogenic autoantibodies against desmoglein-1 and desmoglein-3. J. Invest. Dermatol. 118:806–811. [DOI] [PubMed] [Google Scholar]
- 38.Rocha-Alvarez, R., I.P. Campbel, H. Friedman, V. Aoki, and L.A. Diaz. 1995. Aspectos nâo usuais de Penfigo Vulgar em areas endemicas de Penfigo Foliaceo Endemico. 50° Congresso da Sociedade Brasileira de Dermatologia. Belem, Para, Brazil (Abstr).
- 39.Amagai, M., V. Klaus-Kovtun, and J.R. Stanley. 1991. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 67:869–877. [DOI] [PubMed] [Google Scholar]