

Abstract
B cell linker (BLNK) protein is a component of the B cell receptor (BCR) signaling pathway and BLNK−/− mice have a block in B lymphopoiesis at the pro-B/pre-B cell stage. To study the effect of BLNK mutation at later stages of B cell development, we introduce an innocuous transgenic BCR into BLNK−/− mice and show that two populations of immature B cells distinguishable by their IgMlow (lo) and IgMhigh (hi) phenotypes are found in the bone marrow of these mice in contrast to a single population of IgMhi cells found in control BCR-transgenic BLNK+/+ mice. The mutant IgMlo and IgMhi cells are at an earlier developmental stage compared with the control IgMhi cells as indicated by their differential expression of CD43, B220, and major histocompatibility complex class II antigens and their timing of generation in culture. Thus, in the absence of BLNK the differentiation of immature B cells is delayed. Furthermore, mutant IgMlo cells produce equivalent level of immunoglobulin (Ig) μ but less Ig κ proteins than control and mutant IgMhi cells and this defect is attributed to a decrease in the amount of κ transcripts being generated. Finally, splenic B cells in BCR-transgenic BLNK−/− mice are predominantly of the transitional B cell phenotype and are rapidly lost from the peripheral B cell pool. Taken together, the data suggest a role for BLNK and perhaps BCR signaling, in the regulation of κ light chain expression and continued immature B cell differentiation.
Keywords: B lymphocytes, B cell development, B cell receptor, signal transduction, adaptor protein
Introduction
B lymphopoiesis occurs through discrete stages, starting first from the pro-B and progressing to the pre-B, immature B and finally, mature B cells. Signal originating from the pre-B cell receptor (preBCR), and possibly the B cell receptor (BCR)* are thought to be required for this highly regulated process to occur (1). The preBCR is made up of the Ig heavy chain and the surrogate light chains while the BCR is composed of the surface Ig, and both forms of the receptors are complexed to the signal-transducing subunits Igα and Igβ (2). The importance of the preBCR in B cell development is evident in gene knockout studies in mice in which the inactivation of its various components results in the arrest of B cell differentiation at the very early pro-B cell stage (1). Although the role of preBCR signaling in early B cell development is well defined, it is less clear whether BCR signaling plays a role in the intermediate stages of B cell maturation other than the maintenance (3) and activation of mature B cells (2).
BCR signaling has been studied extensively. It is known that the engagement of the BCR activates various cytoplasmic protein tyrosine kinases such as Syk, Lyn, Blk, and Bruton's tyrosine kinase (Btk), and this leads ultimately to the activation of transcription factors such as nuclear factor of activated T (NF-AT) and NF-κB (4). However, it is not clear how the activation of the proximal protein tyrosine kinases is coupled to the activation of the terminal transcription factors, and this in turn is translated into specific cellular responses such as the activation, proliferation, differentiation, and death of B lymphocytes. Recently, adaptor proteins with modular domains but no intrinsic enzymatic activities have been shown to play a major role in BCR signaling by interfacing tyrosine kinase activation with selective downstream molecules (5). Thus, they may play the critical role of channeling signaling pathways and directing specific cellular responses. One such adaptor proteins in B lymphocytes is the B cell linker protein (BLNK) (6), which is also known as SLP-65 (7) or BASH (8). Upon BCR stimulation, BLNK couples activated Syk to PLC-γ, Vav, Grb2, and Nck (9). In addition, it binds Btk (10, 11) and is required for the activation of the transcription factor NF-κB (12).
We and others have generated mice lacking BLNK (13–16). BLNK−/− mice have a major block in B cell development at the pro-B to pre-B cell stage where the preBCR is first expressed. This mutant phenotype is consistent with a role for BLNK in the preBCR signaling of early B lymphopoiesis. As few peripheral B cells are generated in BLNK−/− mice (13–16), this makes the analysis of the role of BLNK in the later stages of B cell differentiation difficult. Although BLNK is known to be involved in the activation of mature B cells as evidenced by the inability of BLNK−/− mice to respond to a T cell–independent antigen (13), its role (if any) in regulating some aspects of B cell physiology at the immature B cell stage is less clear. Thus, to further examine the role of BLNK in the intermediate stage of B cell differentiation, we now introduced an innocuous transgenic BCR into BLNK−/− mice.
Materials and Methods
Mice.
BLNK−/− (13), B1–8H/+ (3) and 3–83κ/+ (17) mice had been reported previously. All mice were bred and used at 6–8 wk of age according to institutional guidelines.
Cell Preparation and Culture.
Bone marrow cells were obtained by injecting PBS containing 3% FCS and 0.1% NaN3 into the femurs and tibia of mice. Splenic cells were obtained by dissociating the tissue with a plastic mesh and rubber-stopper from a 5-ml syringe. All cells were treated with red blood cell lysing solution (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA) to eliminate erythrocytes. For cell culture experiments, IgM+ cells were removed from bone marrow samples using anti-IgM mAb-coupled MACS beads (Miltenyi Biotec) in a negative selection protocol. The postselected bone marrow cells were cultured in RPMI 1640 supplemented with 10% FCS. In some experiments, cells were cultured for 48 h in the presence of chemical inhibitors (Calbiochem) of signaling molecules. The inhibitors and the range of concentrations used are: BAPTA/AM (10–50 μM); EGTA (0.75–500 μM); SN50 (18–36 μM); PD98059 (20–100 μM); LY294002 (2–10 μM) and PP2 (50–200 μM).
Flow Cytometry.
For FACS® analyses, cells were stained with optimal amounts of FITC-, PE-, and biotin-conjugated mAbs for 10 min on ice and washed twice with PBS. Biotin-conjugated mAbs were revealed with Streptavidin-CyChrome. For intracellular staining, cells were fixed in 2% PBS-buffered paraformaldehyde for 20 min at 4°C, washed, and stained with either anti-μ or anti-κ mAb for 20 min in PBS containing 0.5% saponin (Sigma-Aldrich). FACS® analyses were performed on a FACScan™ (Becton Dickinson) using CELLQuest™ software. The following mAbs were obtained from BD PharMingen: anti-IgM (R6–60.2), anti-B220 (RA3–6B2), anti-CD21 (7G6), anti-CD24 (HSA), anti-CD43 (S7), anti-κ (R5–240), anti-λ (LS136), and anti-MHC class II (2G9). The Ac146 anti-idiotypic antibody was obtained from Dr. K. Rajewsky (Harvard Medical School, Boston, MA).
5-Bromo-2-Deoxyuridine Incorporation Studies.
To study the rate of turnover of peripheral B cells, mice were fed continuously with water containing 1 mg/ml of 5-bromo-2-deoxyuridine (BrdU) over a 1-wk period. Cells were recovered from the animals and stained with anti-B220 and anti-IgM antibodies to identify B cells and later fixed for 30 min in 1 ml ice-cold 70% ethanol and overnight in PBS containing 2% paraformaldehyde and 0.02% Tween-20. Thereafter, cells were washed once in PBS and incubated for 10 min at room temperature in a solution containing 150 mM MgCl2, 10 μM HCl, and 300 μg/ml Dnase 1. Finally, cells were stained intracellularly with anti-BrdU antibody (Becton Dickinson) for 20 min at room temperature, washed twice in PBS, and analyzed on a FACScan™.
Semiquantitative Reverse Transcription PCR Analysis of κ Expression.
Bone marrow IgM+ cells from BCR transgenic BLNK+/+ mice were purified via positive selection using anti-IgM mAb-coupled MACS beads (Miltenyi Biotec). IgMlo and IgMhi cells from TG1 BLNK−/− mice were FACS®-sorted using anti-IgM and anti-B220 mAbs. The purity of the cell population obtained as assessed by FACS® is >90%. Total RNA was extracted from 5 × 105 cells with TRIZOL reagent (GIBCO BRL) and reverse transcribed into cDNA in a reaction of 20 μl using the Superscript Preamplification System (GIBCO BRL). The following primers were used in the amplification: the constant region of immunoglobulin μ heavy chain (Cμ1: 5′-GTCTGATAAGAATCTGGTGGCC-3′, Cμ2: 5′-TTCTTCAAGAAGGTGAGACCCC-3′); constant region of κ light chain (Cκ1: 5′-ATGCTGCACCAACTGTATCC-3′, Cκ 2: 5′-GTTGAAGCTCTTGACAATGGG-3′); and the control G protein α subunit (GαS) (GαS1: 5′-ATTGAAACCATTGTGGCCGCCATGAGC-3′, GαS2: 5′-GAAGACACGGCGGATGTTCTCAGTGTC-3′). Reverse transcription (RT)-PCR was performed in a 50-μl reaction mixture that contained 1 μl of threefold serially diluted cDNA template. PCR was performed at 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min for a total of 40 cycles. RT-PCR products were loaded onto 1.2% agarose gel and visualized by Southern blotting using specific radioactive probes.
Results
Appearance of IgMlo Cells in the Bone Marrow of BLNK−/− Mice Expressing an Innocuous Transgenic BCR.
Mice lacking the adaptor protein BLNK have a major block in B cell development at the pro-B to pre-B cell transition stage where the preBCR is first expressed (13–16). This developmental stage arrest is likely due to defective signaling by the preBCR in the absence of BLNK. However, the impairment in B cell differentiation in BLNK−/− mice is not absolute as some peripheral B cells are generated (13–16) and the reason for this leakiness is unknown. To determine if the expression of a transgenic BCR can bypass the early developmental block in BLNK−/− mice, we crossed wild-type and BLNK−/− mice with mice bearing the VHB1–8 VDJ and Vκ3–83 VJ gene segments that were inserted by gene-targeting into the physiological Ig heavy (H) and light (L) chain loci, respectively (3, 17). The resulting BLNK−/− mice bearing the VHB1–8/Vκ3–83 BCR are designated as TG1 BLNK−/− mice. The VHB1–8 gene segment was derived from an antibody directed against the hapten 4-hydroxy-3-nitrophenyl acetyl (18) while the Vκ3–83 gene segment was derived from an anti-H-2b antibody (19). However, the VHB1–8/Vκ3–83 BCR is of unknown but innocuous specificity (20). Mice bearing single alleles of VHB1–8 and Vκ3–83 are used in all subsequent experiments to ensure that the level of expression of the transgenic H and L chains is physiological and mimics that of the μ and κ proteins found in normal mice.
As shown in Fig. 1 A, wild-type mice have a distinct population of B220+IgM− pre-B cells in their bone marrow in addition to a population of immature B cells that express a continuum level of IgM on their cell surfaces (Fig. 1 A, top panel). In contrast, BLNK−/− mice have few immature B cells as B cell development in these mice is arrested at the pro-B/pre-B cell stage (13–16). On the other hand, BLNK+/+ B cells that express the transgenic VHB1–8/Vκ3–83 BCR can bypass the pre-B cell stage of development and directly differentiate into IgMhigh (hi) immature B cells (Fig. 1 A, third panel). This is in agreement with previously published data (20) and is not surprising as the pre-B cell stage is where the rearrangement of L chain gene segments takes place but these transgenic B cells already possessed and expressed a functional L chain knock-in V region gene segment and thus exhibited expedited development. The IgMhi phenotype of immature B cells found in these mice probably reflects the development of a monoclonal population of B cells in contrast to the development of a polyclonal B cell population in wild-type mice. Interestingly, BLNK−/− mice bearing the VHB1–8/Vκ3–83 BCR generate a larger fraction of IgM+ cells (>2–3-fold) in their bone marrow compared with BLNK+/+ mice bearing the same transgenic BCR. Moreover, two distinct B cell populations are evident in the bone marrow of TG1 BLNK−/− mice (Fig. 1 A, fourth panel). In addition to the IgMhi population that is seen in the TG1 BLNK+/+ mice, TG1 BLNK−/− mice also have an IgMlow (lo) B cell population. The presence of this population of B cells that expresses lower level of BCR on the cell surface is also confirmed by anti-κ staining (Fig. 1 B, top panel).
Figure 1.

B cell development in BLNK−/− mice bearing an innocuous knock-in BCR. FACS® analysis of bone marrow B cells in wild-type and BLNK−/− as well as BCR transgenic BLNK+/+ and BLNK−/− mice bearing a knock-in VHB1–8/Vκ3–83 BCR (TG1). Mice used are heterozygous with respect to the knock-in Ig heavy and light chain alleles. The bone marrow samples were stained for (A) B220 and IgM, and (B) B220 and Igκ- or Igλ-expressing cells. (C) Splenic B cells from wild-type, TG1 BLNK+/+, and TG1 BLNK−/− mice were analyzed for the expression of the VHB1–8/Vκ3–83 idiotype using the anti-Ac146 Ab. Numbers indicate percentage of total cells and the figure shown is representative of five independent analyses.
As BLNK is directly involved in BCR signaling (6), it is conceivable that the presence of this IgMlo B cell population in TG1 BLNK−/− mice is indicative of the occurrence of some form of receptor editing. Our previous finding that BLNK-deficient B cells maintain intact allelic exclusion of the Ig loci (21) suggests that this is unlikely the case. However, to definitively rule out this possibility, we stained for Igλ-expressing cells in TG1 BLNK−/− mice. It has been shown previously that as a consequence of receptor editing, there is an increase in the population of Igλ-expressing B cells in autoreactive BCR transgenic mice (22). However, FACS® analyses of bone marrow (Fig. 1 B, bottom panel) or splenic (data not shown) B cells indicate that only a small fraction of the B cells express the λ L chain in TG1 BLNK−/− mice and this is comparable to the fraction found in TG1 BLNK+/+ mice. Furthermore, staining of B cells with the anti-idiotypic mAb Ac146, which recognizes the VHB1–8/Vκ3–83 combination, indicates that virtually all the B cells in the spleen (Fig. 1 C) and bone marrow (data not shown) of TG1 BLNK−/− mice express the knock-in H and L chains. Taken together, the data suggest that there is no evidence of receptor editing occurring in TG1 BLNK−/− mice.
Thus, the data suggest that the introduction of an innocuous transgenic BCR can bypass the early developmental block in BLNK−/− mice and leads to the generation of a large population of IgM+ cells in their bone marrow. However, a substantial fraction of these cells (∼50%) is of the IgMlo phenotype and appears to represent cells in an as yet undefined stage of differentiation.
Delayed Cellular Maturation of IgM+ Cells in TG1 BLNK−/− Mice.
To gain insight into the population of IgMlo cells in TG1 BLNK−/− mice, we analyze these cells with respect to a series of maturation markers. As shown in Fig. 2 , TG1 BLNK+/+ B cells express high levels of the pan-B cell marker B220 and high levels of the heat stable antigen CD24 but low levels of CD43 on their cell surfaces. Thus, they resemble immature B cells found in normal mice. Interestingly, both the IgMhi and IgMlo cells in TG1 BLNK−/− B cells express lower levels of B220 and CD24 but higher levels of CD43 on their cell surfaces suggesting that they resemble more of the pro-B/pre-B than the immature B cell phenotype (23). On closer examination, the level of CD43 expression on the mutant IgMlo cells is even higher than that found on the mutant IgMhi cells. Thus, the IgMlo and IgMhi cells in TG1 BLNK−/− mice appear to be less “mature” than the IgM+ cells found in TG1 BLNK+/+ mice.
Figure 2.

Phenotypic analysis of IgM+ cells in the bone marrow of TG1 BLNK−/− mice. Bone marrow cells from TG1 BLNK+/+ and TG1 BLNK−/− were stained with anti-B220 and anti-IgM and with either anti-CD43, anti-CD24, or anti-MHC class II mAbs. Figure shown is representative of four independent analyses.
To confirm this observation, we stained the cells with anti-MHC class II antibodies. The expression of the MHC class II antigens is regulated during B lymphopoiesis such that there is a progressive increase in their expression from the pre-B cell stage onwards with a higher level of expression on immature and mature B cells (24–26). Indeed, FACS® analyses indicate that both the IgMlo and IgMhi cells in TG1 BLNK−/− mice express significantly lower levels of MHC class II antigens on their cell surfaces compared with the IgM+ cells in TG1 BLNK+/+ mice (Fig. 2). In addition, the level of MHC class II antigens on mutant IgMlo cells is even lower than that found on mutant IgMhi cells. Thus, the FACS® analyses suggest that the immature B cells in TG1 BLNK−/− mice are at an earlier differentiation stage compared with the immature B cells in TG1 BLNK+/+ mice, and furthermore that the mutant IgMlo cells may be even less differentiated than the mutant IgMhi cells.
As the mutant IgMlo and IgMhi cells are relatively immature compared with the control BLNK+/+IgM+ cells, we decided to examine the cell cycle status of these cells. However, analysis of the intracellular DNA content of wild-type IgM+ and mutant IgMlo and IgMhi cells by 7-AAD staining revealed no significant differences among these cells and that they are all noncycling (data not shown).
To further distinguish between the differentiation of IgMlo and IgMhi cells in TG1 BLNK−/− mice, we purified and cultured bone marrow B220−IgM− cells for various times. As seen in Fig. 3 , B220−IgM− cells from control TG1 BLNK+/+ mice give rise mainly to IgMhi cells after 16–24 h of culture and the same population of cells is recovered even after the 72 h time point. In contrast, B220−IgM− cells from TG1 BLNK−/− mice generate mainly IgMlo cells after 16–24 h in culture. However, an increasing fraction of IgMhi cells is recovered at the 48–72 h time points. This, together with the in vivo staining data (Fig. 2), suggest that the IgMlo cells likely arise before the IgMhi cells in TG1 BLNK−/− mice. Taken together, the overall data indicate that the absence of BLNK appears to delay the maturation of developing B cells in the bone marrow.
Figure 3.

In vitro culture of Ig-negative bone marrow cells from TG1 BLNK−/− mice. Bone marrow cells were depleted of IgM+ cells using anti-IgM mAb-coupled magnetic beads before culture. Cells were stained with anti-B220 and anti-Igκ mAb at the 0 h time point to determine the purity of the cells and with anti-B220 and anti-IgM mAbs to examine the differentiation of the cells at various time points in culture. Figure shown is representative of five independent analyses. Numbers indicate percentage of total IgM+ cells.
Decreased Intracellular κ Light Chain Expression in IgMlo Cells of TG1 BLNK−/− Mice.
We next examined the reason for the lower level of BCR expression on IgMlo cells in the bone marrow of TG1 BLNK−/− mice. It is possible that these cells produce lesser amount of μ and/or κ proteins or alternatively, these IgMlo cells could produce equal amount of cytoplasmic μ and κ proteins as the IgMhi cells but they express lower levels of IgM on their cell surfaces. To distinguish between these possibilities, we stain for intracellular μ and κ proteins in the IgM+ cells of both TG1 BLNK+/+ and TG1 BLNK−/− mice. As shown in Fig. 4 , the IgMhi cells in TG1 BLNK−/− mice have equivalent amount of cytoplasmic μ and κ compared with the IgMhi cells in TG1 BLNK+/+ mice. Interestingly, the IgMlo cells in TG1 BLNK−/− mice produce equivalent amounts of μ but much lesser amounts of κ compared with the IgMhi cells in TG1 BLNK+/+ or BLNK−/− mice. This indicates that the lower levels of surface BCR expression on IgMlo cells is likely due to the decreased production of κ proteins.
Figure 4.

Expression levels of intracellular μ and κ proteins in IgM+ cells from TG1 BLNK+/+ and IgMhi and IgMlo cells from TG1 BLNK−/− mice. Bone marrow cells obtained from TG1 BLNK+/+ and TG1 BLNK−/− mice were stained for the extracellular expression of B220 and IgM and the intracellular expression of μ and κ via FACS®. Figure shown is representative of five independent analyses. Abs used are FITC-anti-IgM, PE-anti-B220 for the extracellular staining, and bio-anti-IgM or bio-anti-Igκ for the intracellular staining, the latter two of which are revealed with streptavidin-Cychrome.
The decrease in the production of κ light chain proteins in the IgMlo cells could be due to a decrease in the generation of the κ transcripts or in their translation into proteins. If the latter were the reason, then equivalent amount of transcripts should be found in the cells of TG1 BLNK+/+ and TG1 BLNK−/− mice. To examine these possibilities, we isolated RNA from equal number of purified IgM+ cells from TG1 BLNK+/+ and IgMlo and IgMhi cells from TG1 BLNK−/− mice and subject them to RT-PCR analyses. As shown in Fig. 5 , semiquantitative RT-PCR analyses of serially diluted samples indicate that compared with the IgM+ cells from TG1 BLNK+/+ mice, much less κ transcripts are found in both IgMhi and IgMlo cells of TG1 BLNK−/− mice and with the mutant IgMlo cells having even much lesser κ transcripts compared with the mutant IgMhi cells. As control and consistent with the intracellular μ staining data (Fig. 4), equivalent amounts of μ transcripts is found in all three samples. Hence, the data suggest that the decrease in κ protein in the mutant IgMlo cells is likely due to a decrease in the amount of transcripts being generated.
Figure 5.

Semiquantitative RT-PCR analysis of μ and κ RNA transcripts in bone marrow IgM+ cells of TG1 BLNK+/+ and IgMhi and IgMlo cells of TG1 BLNK−/− mice. Bone marrow IgM+ cells from TG1 BLNK+/+ mice were purified via positive selection using anti-IgM–coupled magnetic beads while IgMhi and IgMlo cells from TG1 BLNK−/− mice were FACS® sorted. The purity of cells obtained is >90%. RNA is prepared from equal numbers of purified cells and reverse transcribed into cDNA. The cDNA template is serially diluted threefold and subjected to PCR analyses to detect the expression level of κ and μ transcripts. GαS expression is used as a control for the amount of cDNA template used.
Surface IgM Expression Levels on Differentiating B Cells Can Be Modulated by Inhibitors of Signaling Proteins.
As BLNK is an adaptor protein in BCR signal transduction (6) and is known to activate numerous signaling pathways (6–12), we decided to investigate the signaling pathways that might be involved in signaling κ expression. The small number of B cells in the bone marrow makes it difficult for us to obtain enough cells from either TG1 BLNK+/+ or TG1 BLNK−/− mice for Western blot analysis of biochemical pathways. Besides, low level basal signaling in the absence of deliberate BCR stimulation may not be detectable using this conventional methodology. Last but not least, it is impossible for us to isolate mutant IgMhi and IgMlo cells from TG1 BLNK−/− mice without stimulating their BCR, as we will have to FACS-sort these cells using fluorochrome-coupled anti-IgM antibodies to identify their phenotype.
We thus employed the alternative method of blocking specific signaling pathways using chemical inhibitors. Bone marrow cells depleted of IgM+ cells from TG1 BLNK+/+ mice were cultured for 48 h in the presence of varying concentrations of various inhibitors and developing B cells were analyzed for their cell surface expression of IgM. As can be seen in Fig. 6 A, disruption of calcium signaling using calcium chelators such as EGTA at 0.75–500 μM or BAPTA/AM at 10–50 μM did not affect the level of surface IgM expression on B cells. Similarly, inhibiting the NF-κB pathway using SN50 (18–36 μM) or the extracellular signal–regulated kinase (ERK) pathway using PD98059 (20–100 μM) did not lead to a lower level of surface IgM expression. On the other hand, B cells that differentiated in the presence of LY294002 (2–10 μM) that inhibits the activation of PI3K or PP2 (50–200 μM) that inhibits src-family tyrosine kinases did have much lower expression of surface IgM. Moreover, the decrease in surface IgM expression on B cells in the presence of LY294002 or PP2 is dosage dependent (data not shown). Finally, as shown in Fig. 6 B, B cells differentiating in the presence of LY294002 or PP2 but not BAPTA/AM and PD98059 also have a lesser amount of intracellular κ proteins. Thus, it appears that a signaling pathway involving Src-family kinase, PI3K, and BLNK is involved in signaling κ light chain expression in B cells.
Figure 6.

Expression level of surface IgM and intracellular κ on differentiating bone marrow B cells from TG1 BLNK+/+ mice cultured in the presence of chemical inhibitors of signaling molecules. Bone marrow cells depleted of B220+IgM+ cells from TG1 BLNK+/+ mice were cultured in the presence of various inhibitors for 48 h. Differentiating B220+ B cells were analyzed for their expression level of surface IgM (A); and intracellular level of κ (B) via FACS®. B cells from TG1 BLNK+/+ and TG1 BLNK−/− B cells cultured in medium alone or in the presence of 0.1% DMSO that was used to dissolve some of the chemical inhibitors are used as controls. Figure shown is representative of three independent analyses.
Splenic B Lymphocytes in TG1 BLNK−/− Mice Are Predominantly Early Stage Transitional B Cells.
As seen in Figs. 1 and 7 , the introduction of a transgenic BCR leads to a greater than twofold increase in the number of IgM+ (IgMlo and IgMhi) cells in the bone marrow of TG1 BLNK−/− mice compared with control animals. This suggests that the expression of a transgenic BCR can bypass the developmental block in BLNK−/− mice. However, such an accumulation of B cells is not apparent in the peripheral lymphoid tissues of these mice. Instead, there is a two- to threefold reduction in the number of B cells in the spleen of TG1 BLNK−/− mice compared with the control animals (Fig. 7). This suggests that BLNK deficiency may also perturb the further differentiation of peripheral B cells.
Figure 7.

Number of B cells in the bone marrow and spleen of TG1 BLNK+/+ and BLNK−/− mice. Comparison of the number of IgM+ cells in the bone marrow (two hind limbs) and spleen of TG1 BLNK+/+ (white circle) and TG1 BLNK−/− (black circle) mice. Each circle represents a single animal. Calculation of the number of B cells is based on total cell count and the percent of B220+IgM+ cells as assayed by FACS® staining. Statistical significance is determined by a paired two-tailed Student's t test.
Given the finding that the B cells in the bone marrow of TG1 BLNK−/− mice are at an earlier differentiation stage compared with those in control mice, we next examined the differentiation of the B cells in the peripheral lymphoid tissues of TG1 BLNK−/− mice. Previously, we have shown that the peripheral B cells in BLNK−/− mice do not differentiate into the mature IgMloIgDhi fraction (13). Consistent with that observation, the splenic B cells in TG1 BLNK−/− mice are mainly of the IgMhi transitional B cell phenotype (Fig. 8) . In addition, we now show that a large fraction of these cells express low level of the MHC class II antigens on their cell surfaces in contrast to the high level expression of these antigens on splenic B cells found in the control TG1 BLNK+/+ mice. Similarly, a large fraction of the splenic B cells in TG1 BLNK−/− mice are also CD43+ compared with the cells found in control mice. These CD43+ splenic B cells are not B-1 cells, as the CD5+ B cell subset is not found in BLNK−/− mice (13–16). These data would again suggest that the splenic B cells in TG1 BLNK−/− mice are less mature compared with those found in the spleen of control mice.
Figure 8.
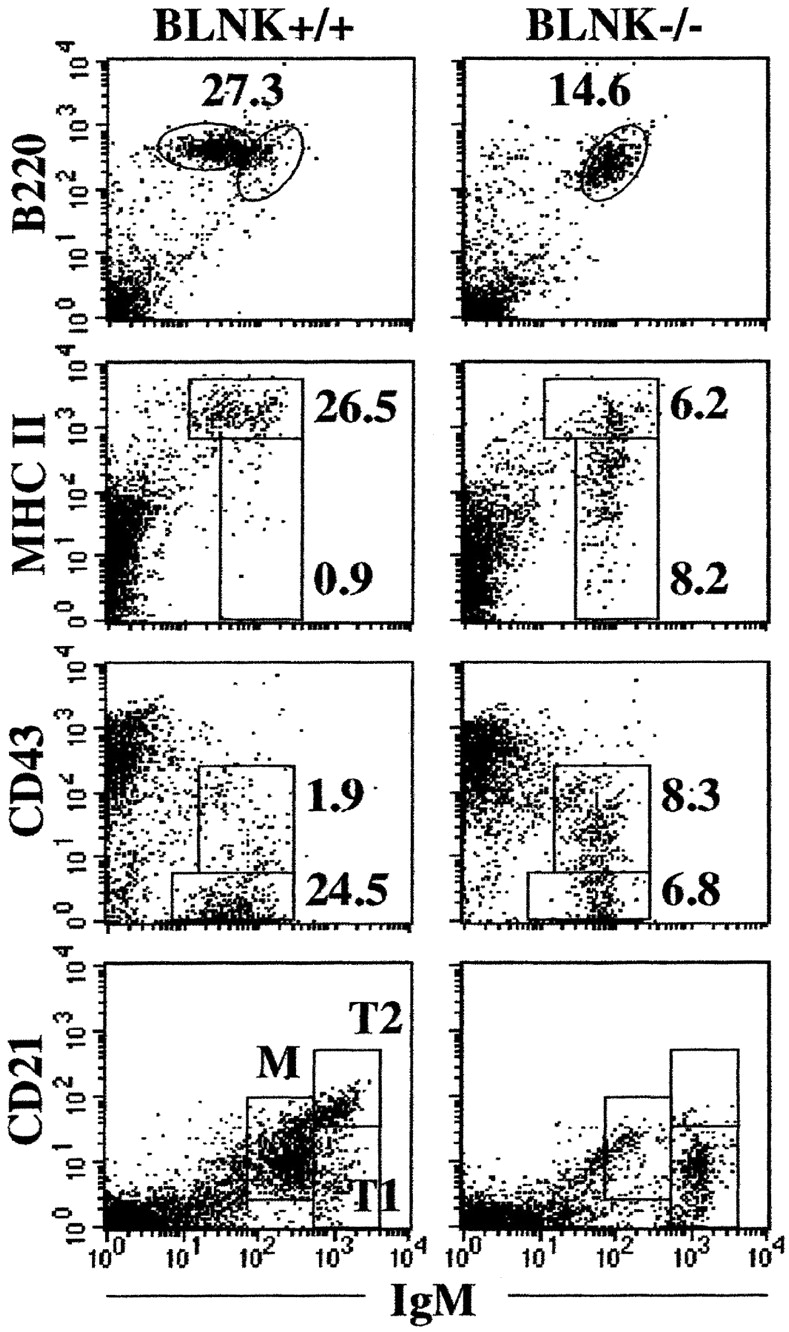
Phenotypic analysis of splenic B cells in TG1 BLNK−/− mice. Spleen cells from TG1 BLNK+/+ and TG1 BLNK−/− mice were stained with anti-B220 and anti-IgM and with either anti-MHC class II or anti-CD43 mAbs (top three panels). Transitional (T) stage 1 and 2 and mature (M) B cells are resolved using anti-IgM and anti-CD21 mAbs (bottom panel). Figure shown is representative of five independent analyses. Numbers indicate percent of total cells.
To determine the precise stage in which BLNK deficiency affects B cell differentiation in the periphery, we examined in detail the transitional B cell population in TG1 BLNK−/− mice. Transitional (T) B cells can be further resolved into the earlier T1 and the later T2 cell stages on the basis of CD21 expression (27, 28). T1 cells are IgMhiCD2llo while T2 cells are IgMhiCD21hi and mature B cells are IgMintermediate (int) CD21int. As shown in Fig. 8, the splenic B cells in TG1 BLNK−/− mice are predominantly T1 cells, suggesting that in the absence of BLNK, the cells failed to develop into the T2 cell stage.
As B cells in TG1 BLNK−/− mice are arrested at the transitional T1 cell stage, they are likely to be short-lived (27) and not selected into the long-lived mature B cell pool (29). Indeed, one would expect a higher rate of turnover of peripheral B cells in the mutant mice, and this might account for the loss of cells in these animals. To determine if this is the case, we examine the rate of turnover of the peripheral B cells in the mutant mice. TG1 BLNK+/+ and TG1 BLNK−/− mice were fed with BrdU in drinking water and the fraction of splenic B cells that had incorporated BrdU over a 1-wk period was determined. As shown in Fig. 9 , the fraction of IgM+ B cells that had incorporated BrdU in the mutant mice is approximately twice that of the wild-type mice, suggesting that there is a greater loss of peripheral B cells in the absence of BLNK.
Figure 9.

Turnover of splenic B cells in TG1 BLNK−/− mice. Groups of five TG1 BLNK+/+ and TG1 BLNK−/− mice were continuously fed with BrdU in drinking water for a period of 1 wk and splenic B220+IgM+ cells were stained for their intracellular BrdU content. Statistical significance is determined by a paired two-tailed Student's t test.
Discussion
BLNK−/− mice have a major block in B cell development at the pro-B/pre-B cell stage and few peripheral B cells are generated (13–16). This makes the analysis of the role of BLNK in the later stages of B cell development difficult. We show here that the introduction of an innocuous transgenic BCR into BLNK−/− mice leads to the generation of a large population of IgM+ cells in the bone marrow and thus could circumvent the problem. Our data further indicate that BLNK plays a role in the intermediate stages of B cell differentiation, in particular at the immature and transitional B cell stages. BLNK−/− mice bearing an innocuous transgenic BCR develop two populations of immature B cells in their bone marrow that is distinguishable by their surface IgM expression level (Fig. 1). The IgMlo cells in these mutant mice are apparently at an earlier developmental stage compared with the IgMhi cells and both these cell populations are less “mature” than the immature IgMhi cells found in control BLNK+/+ mice bearing the same transgenic BCR (Fig. 2). Furthermore, the IgMlo cells in BCR transgenic BLNK−/− mice express less Ig κ light chain proteins compared with the IgMhi cells in both BLNK+/+ and BLNK−/− mice (Figs. 4 and 5). Thus, in the absence of BLNK the production of κ light chain is decreased and the cellular maturation of developing B cells is delayed. In addition, BLNK also play a role in the further differentiation of B cells in the periphery (Fig. 8) as almost all the peripheral B cells in the mutant mice are transitional stage 1 (T1) cells.
B cell development occurs in discrete stages and the role of preBCR signaling in driving B lymphopoiesis is well documented as the inactivation of various signaling molecules such as Igβ (30), Btk (31, 32), and BLNK (13–16) all lead to an arrest of B cell development at the pro-B cell stage. In addition, mice with mutations in the cytoplasmic regions of Igα (33) or Igβ (34) have impairment in the generation of immature B cells. However, it is less clear whether BCR signaling is required for continued B cell differentiation at the immature B cell stage other than for the maintenance (3) and activation (2) of mature B cells. In particular, it is not known whether the assembly and expression of a functional BCR per se is sufficient for immature B cell development or that its signaling is also required. Our current data from the TG1 BLNK−/− mice would suggest the latter since BLNK is an adaptor protein in the BCR signal transduction pathway.
The data presented here suggest that the BCR, upon assembly and presumably initial low level expression on the cell surface, can signal its own increased expression by regulating the transcription of the Ig light chain. Apparently, in the absence of BLNK, basal BCR signaling is perturbed and this leads initially to a decrease in Ig κ production and subsequently results in the generation of a population of IgMlo cells in the mutant mice. Later, as the κ transcripts and protein accumulate, the cells then acquire the IgMhi phenotype. In addition, as a consequence of continued impairment in BCR signaling due to the absence of BLNK, the IgMlo mutant cells did not down-regulate CD43 and up-regulate B220 and MHC class II antigens. Gradual changes to these cell surface antigens and hence further differentiation occur only after the mutant cells acquire the IgMhi phenotype but the change is still delayed compared with the control IgMhi cells, presumably due to a weaken signal. This would suggest that the strength of the BCR signaling is the determining factor in the further differentiation of immature B cells. A weak signal leads to a delay in the maturation of developing B cells (this paper) while excessive BCR signaling leads to their deletion as in the case of autoreactive B cells (19). It remains to be determined whether BCR signaling directly regulates the expression level of some of the antigens associated with the differentiation of B cells, in particular the MHC class II antigens. This is particularly interesting as the expression level of MHC class II antigens correlates with the expression level of IgM on BCR transgenic BLNK−/− cells (Fig. 2) and furthermore, their level is decreased upon the induced deletion of BCR on mature B cells (3).
BLNK is a central molecule in the BCR signaling pathway (6) and is involved in the activation of Rac-JNK (9), Vav and Nck (6), and NF-κB (12) signaling. Hence, it remains to be determined if any of these molecules and pathways are also involved in the various aspects of immature B cell differentiation described here, in particular, in the regulation of Ig κ expression. Our cell culture studies using chemical inhibitors suggest that a Src-family tyrosine kinase and phosphoinositide 3-kinase (PI3K) are involved in the latter aspect (Fig. 6). It is reasonable to assume that a Src family tyrosine kinase is involved as Lyn is proximal to BCR signaling and together with Syk, initiates the whole cascade of BCR signal transduction (2, 4). The role of PI3K is far less clear although incidentally, PI3K−/− mice (35, 36) have similar defects compared with BLNK−/− mice (13–16). PI3K is activated upon BCR stimulation and generates lipid products that recruit Btk and phospholipase C-γ2 (PLCγ2) to the plasma membrane (37, 38). As BLNK is part of the B cell “signalosome” that also includes Btk and PLCγ2 (37, 38), it will be interesting to determine if Btk−/− (31, 32), PLCγ2−/− (39, 40), and PI3K−/− (35, 36) mice bearing transgenic BCR also have similar defects in Ig κ expression and immature B cell differentiation. These experiments are currently underway. Last but not least, as a note of precaution, our inhibition studies in no way rule out the involvement of other signaling pathways, in particular that of NF-κB signaling and Ca2+ fluxes, in the regulation of κ light chain expression and much work remains to be done in these aspects.
The finding that the majority of the peripheral B cells in TG1 BLNK−/− mice are blocked at the transitional B cell stage is also intriguing (Fig. 8). Presumably, BCR signaling involving BLNK may be required for T1 cells to differentiate into T2 cells and later, enter the long-lived mature B cell pool. Many other mutations that affect BCR signaling, for example those of Igα, Btk, Syk, and CD45, have also been shown to interfere with transitional B cell maturation (27). However, the exact mechanism that regulates transitional to mature B cell differentiation remains to be determined. Recently, it has been shown that T2 cells can be distinguished from T1 cells by their ability to up-regulate the cell survival Bcl-xL and cell cycle cyclin D2 proteins upon BCR stimulation (28). We have previously shown that BLNK-deficient B cells do not up-regulate these cell survival and cell cycle proteins and have a defect in NF-κB signaling (12). Thus, basal or induced NF-κB signaling may be required for the differentiation of transitional B cells. Consistent with this argument, mice lacking IκB kinase α and therefore unable to activate NF-κB signaling do not generate mature B cells (41).
In conclusion, the data presented in this paper indicate that the adaptor protein BLNK plays a critical role in immature and transitional B cell differentiation. Although BLNK may be involved in signaling by other cell surface receptors, the phenotype described here is more consistent with its role in BCR signal transduction. Our data would suggest that once the BCR is assemble in immature B cell, it is capable of signaling and the signals regulate both the increased production of Ig light chains at the transcriptional level as well as the continued cellular maturation of lymphocytes. Elucidation of the signaling pathways associated with BLNK may shed light on how these processes are regulated.
Acknowledgments
We thank Weng-Keong Chew and Koon-Guan Lee for technical assistance and the Institute of Molecular and Cell Biology In Vivo Model Unit for the care and maintenance of mice. The work is supported by grants from the Biomedical Research Council of the Singapore Agency for Science, Technology and Research (A*Star).
Footnotes
Abbreviations used in this paper: BCR, B cell receptor; BLNK, B cell linker; BrdU, 5-bromo-2-deoxyuridine; Btk, Bruton's tyrosine kinase; NF-AT, nuclear factor of activated T; PI3K, phosphoinositide 3-kinase; RT, reverse transcription.
References
- 1.Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. [DOI] [PubMed] [Google Scholar]
- 2.Reth, M., and J. Wienands. 1997. Initiation and processing of signals from the B cell antigen receptor. Annu. Rev. Immunol. 15:453–479. [DOI] [PubMed] [Google Scholar]
- 3.Lam, K.-P., R. Kuhn, and K. Rajewsky. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 90:1073–1083. [DOI] [PubMed] [Google Scholar]
- 4.Satterthwaite, A., and O. Witte. 1996. Genetic analysis of tyrosine kinase function in B cell development. Annu. Rev. Immunol. 14:131–154. [DOI] [PubMed] [Google Scholar]
- 5.Peterson, E.J., J.L. Clements, N. Fang, and G.A. Koretzky. 1998. Adaptor proteins in lymphocyte antigen-receptor signaling. Curr. Opin. Immunol. 10:337–344. [DOI] [PubMed] [Google Scholar]
- 6.Fu, C., C.W. Turck, T. Kurosaki, and A.C. Chan. 1998. BLNK: a central linker protein in B cell activation. Immunity. 9:93–103. [DOI] [PubMed] [Google Scholar]
- 7.Wienands, J., J. Schweikert, B. Wollscheid, H. Jumma, P.J. Nielsen, and M. Reth. 1998. SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J. Exp. Med. 188:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goitsuka, R., Y. Fujimura, H. Mamada, A. Umeda, T. Morimura, K. Uetsuka, K. Doi, S. Tsuji, and D. Kitamura. 1998. Cutting edge: BASH, a novel signaling molecule preferentially expressed in B cells of the bursa of Fabricius. J. Immunol. 161:5804–5808. [PubMed] [Google Scholar]
- 9.Ishiai, M., M. Kurosaki, R. Pappu, K. Okawa, I. Ronko, C. Fu, M. Shibata, A. Iwamatsu, A.C. Chan, and T. Kurosaki. 1999. BLNK required for coupling Syk to PLCγ2 and Rac1-JNK in B cells. Immunity. 10:117–125. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto, S., A. Iwamatsu, M. Ishiai, K. Okawa, T. Yamadori, M. Matsushita, Y. Baba, T. Kishimoto, T. Kurosaki, and S. Tsukada. 1999. Identification of the SH2 domain binding protein of Bruton's tyrosine kinase as BLNK—functional significance of Btk-SH2 domain in B-cell antigen receptor-coupled calcium signaling. Blood. 94:2357–2364. [PubMed] [Google Scholar]
- 11.Su, Y., Y. Zhang, J. Schweikert, G. Koretzky, M. Reth, and J. Wienands. 1999. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 29:3702–3711. [DOI] [PubMed] [Google Scholar]
- 12.Tan, J.E.-L., S.-C. Wong, S.K.-E. Gan, and K.-P. Lam. 2001. The adaptor protein BLNK is required for B cell antigen-receptor-induced activation of nuclear factor-κB and cell cycle entry and survival of B lymphocytes. J. Biol. Chem. 276:20055–20063. [DOI] [PubMed] [Google Scholar]
- 13.Xu, S., J.E.-L. Tan, E.P.-Y. Wong, A. Manickam, S. Ponniah, and K.-P. Lam. 2000. B cell development and activation defects resulting in xid-like immunodeficiency in BLNK/SLP-65-deficient mice. Int. Immunol. 12:397–404. [DOI] [PubMed] [Google Scholar]
- 14.Jumma, H., R. Wollescheid, M. Mitterer, J. Wienands, M. Reth, and P.J. Nielsen. 1999. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity. 11:547–554. [DOI] [PubMed] [Google Scholar]
- 15.Pappu, R., A.M. Cheng, B. Li, Q. Gong, C. Chiu, N. Griffin, M. White, B.P. Sleckman, and A.C. Chan. 1999. Requirement for B cell linker protein (BLNK) in B cell development. Science. 286:1949–1954. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi, K., R. Nittono, N. Okamoto, S. Tsuji, Y. Hara, R. Goitsuka, and D. Kitamura. 2000. The B cell-restricted adaptor BASH is required for normal development and antigen receptor-mediated activation of B cells. Proc. Natl. Acad. Sci. USA. 97:2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelanda, R., S. Schaal, R.M. Torres, and K. Rajewsky. 1996. A prematurely expressed Igκ transgene, but not a VκJκ gene segment targeted into the Igκ locus, can rescue B cell development in λ5-deficient mice. Immunity. 5:229–239. [DOI] [PubMed] [Google Scholar]
- 18.Reth, M., T. Imanishi-Kari, and K. Rajewsky. 1978. Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. I. Characterization of antibody families in the primary and hyperimmune response. Eur. J. Immunol. 8:393–400. [DOI] [PubMed] [Google Scholar]
- 19.Nemazee, D.A., and K. Burki. 1989. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 337:562–566. [DOI] [PubMed] [Google Scholar]
- 20.Pelanda, R., S. Schwers, E. Sonoda, R.M. Torres, D. Nemazee, and K. Rajewsky. 1997. Receptor editing in a transgenic mouse model: site, efficiency, and role in B cell tolerance and antibody diversification. Immunity. 7:765–775. [DOI] [PubMed] [Google Scholar]
- 21.Xu, S., S.-C. Wong, and K.-P. Lam. 2000. Cutting edge: B cell linker protein is dispensable for the allelic exclusion of immunoglobulin heavy chain locus but required for the persistence of CD5+ B cells. J. Immunol. 165:4153–4157. [DOI] [PubMed] [Google Scholar]
- 22.Tiegs, S.L., D.M. Russell, and D. Nemazee. 1993. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardy, R.R., C.E. Carmack, S.A. Shinton, J.D. Kemp, and K. Hayakawa. 1991. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 173:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarlinton, D. 1993. Direct demonstration of MHC class II surface expression on murine pre-B cells. Int. Immunol. 5:1629–1635. [DOI] [PubMed] [Google Scholar]
- 25.Hayakawa, K., D. Tarlinton, and R.R. Hardy. 1994. Absence of MHC class II expression distinguishes fetal from adult B lymphopoiesis in mice. J. Immunol. 152:4801–4807. [PubMed] [Google Scholar]
- 26.Lam, K.-P., and A.M. Stall. 1994. Major histocompatibility complex class II expression distinguishes two distinct B cell developmental pathways during ontogeny. J. Exp. Med. 180:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carsetti, R., G. Kohler, and M.C. Lamers. 1995. Transitional B cells are the target of negative selection in the B cell compartment. J. Exp. Med. 181:2129–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su, T.T., and D.J. Rawlings. 2002. Transitional B lymphocyte subsets operate as distinct checkpoints in murine splenic B cell development. J. Immunol. 168:2101–2110. [DOI] [PubMed] [Google Scholar]
- 29.Forster, I., W. Muller, B. Schittek, and K. Rajewsky. 1991. Generation of long-lived B cells in germ-free mice. Eur. J. Immunol. 21:1779–1782. [DOI] [PubMed] [Google Scholar]
- 30.Gong, S., and M.C. Nussenzweig. 1996. Regulation of an early developmental checkpoint in the B cell pathway by Ig beta. Science. 272:411–414. [DOI] [PubMed] [Google Scholar]
- 31.Khan, W.N., F.W. Alt, R.M. Gerstein, B.A. Malynn, I. Larsson, G. Rathbun, L. Davidson, S. Muller, A.B. Kantor, and L.A. Herzenberg. 1995. Defective B cell development and function in Btk-deficient mice. Immunity. 3:283–299. [DOI] [PubMed] [Google Scholar]
- 32.Kerner, J.D., N.W. Appleby, R.N. Mohr, S. Chien, D.J. Rawlings, C.R. Maliszewski, O.N. Witte, and R.M. Perlmutter. 1995. Impaired expansion of mouse progenitors lacking Btk. Immunity. 3:301–312. [DOI] [PubMed] [Google Scholar]
- 33.Torres, R.M., H. Flaswinkel, M. Reth, and K. Rajewsky. 1996. Aberrant B cell development and immune response in mice with a compromised BCR complex. Science. 272:1802–1804. [DOI] [PubMed] [Google Scholar]
- 34.Reichlin, A., Y. Hu, E. Meffre, H. Nagaoka, S. Gong, M. Kraus, K. Rajewsky, and M.C. Nussenzweig. 2001. B cell development is arrested at the immature B cell stage in mice carrying a mutation in the cytoplasmic domain of immunoglobulin beta. J. Exp. Med. 193:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki, H., Y. Terauchi, M. Fujiwara, S. Aizawa, Y. Yazaki, T. Kadowaki, and S. Koyasu. 1999. Xid-like immunodeficiency in mice with disruption of the p85α subunit of phosphoinositide 3-kinase. Science. 283:390–392. [DOI] [PubMed] [Google Scholar]
- 36.Fruman, D.A., S.B. Snapper, C.M. Yballe, L. Davidson, J.Y. Yu, F.W. Alt, and L.C. Cantley. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science. 283:393–397. [DOI] [PubMed] [Google Scholar]
- 37.Kurosaki, T. 2002. Regulation of B cell fates by BCR signaling components. Curr. Opin. Immunol. 14:341–347. [DOI] [PubMed] [Google Scholar]
- 38.Fruman, D., A. Satterthwaite, and O. Witte. 2000. Xid-like phenotypes: a B cell signalosome takes shape. Immunity. 13:1–3. [DOI] [PubMed] [Google Scholar]
- 39.Wang, D., J. Feng, R. Wen, J. Marine, M. Sangster, E. Parganas, A. Hoffmeyer, C. Jackson, J. Cleveland, P. Murray, and J. Ihle. 2000. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 13:25–35. [DOI] [PubMed] [Google Scholar]
- 40.Hashimoto, A., K. Takeda, M. Inaba, M. Sekimata, T. Kaisho, S. Ikehara, Y. Homma, S. Akira, and T. Kurosaki. 2000. Cutting edge: essential role of phospholipase C-gamma 2 in B cell development and function. J. Immunol. 165:1738–1742. [DOI] [PubMed] [Google Scholar]
- 41.Kaisho, T., K. Takeda, T. Tsujimura, T. Kawai, F. Nomura, N. Terada, and S. Akira. 2001. IkB kinase alpha is essential for mature B cell development and function. J. Exp. Med. 193:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]