

Abstract
Lymphoproliferative (lpr) mice, which lack functional Fas receptor expression and develop autoimmune lymphoproliferative disease, have an accumulation of T cell receptor-αβ+CD4−CD8− (double negative T cells [DNTC]) in the periphery. The function of the accumulating DNTC is not clear. In this study we demonstrate that B6/lpr DNTC can dose dependently kill syngeneic CD8+ and CD4+ T cells from wild-type B6 mice through Fas/Fas ligand interactions in vitro. We also demonstrate that B6/lpr DNTC that are activated and expand in vivo are able to specifically down-regulate allogeneic immune responses mediated by syngeneic Fas+CD4+ and CD8+ T cells in vivo. B6/lpr DNTC that have been preactivated in vivo by infusion of either class I– (bm1) or class II– (bm12) mismatched allogeneic lymphocytes are able to specifically enhance the survival of bm1 or bm12, but not third-party skin allografts when adoptively transferred into naive B6+/+ mice. These findings clearly demonstrate that B6/lpr DNTC have a potent immune regulatory function in vitro and in vivo. They also provide new insights into the mechanisms involved in the development of autoimmune disease in lpr mice.
Keywords: autoimmune, transplantation, immune tolerance, suppressor T lymphocytes, Fas ligand
Introduction
Compelling evidence indicate that regulatory T cells play an important role in the maintenance of immune tolerance to self- and foreign antigens (1–3). Various subsets of T lymphocytes have been isolated from peripheral lymphoid organs in mice and humans that have the ability to down-regulate the proliferation of autoimmune effector cells (4–6). CD4+CD25+ T cells are the most extensively studied regulatory T cells. Eliminating CD4+CD25+ T cells from the periphery of mice leads to the development of systemic autoimmune diseases and adding these T cells back ameliorates experimentally induced autoimmune symptoms (7). Other regulatory T cells, including CD4+CD45Rblow (1), CD4+DX5+ T cells (8), CD8+ T cells (9), γδ-TCR+ cells (10), and αβ-TCR+ CD3+CD4−CD8− double negative T cells (DNTC; references 11 and 12) have also been demonstrated in various models to have a potent role in down-regulating immune responses.
Lymphoproliferative (lpr) mice, which have a mutation in the gene responsible for the production of functional Fas molecules (13), develop systemic autoimmune disease concurrent with an accumulation of mature peripheral DNTC (14, 15). The development of autoimmune disease has been suggested to be caused by autoreactive T cells in the periphery from either incomplete negative selection in the thymus (16) or as more recent studies indicate, impaired peripheral deletion (17). In particular, T cells that are reactive to mouse endogenous superantigens are deleted in the thymus of Fas-null mice, but peripheral deletion in response to bacterial superantigen is impaired in the same mice (17). These data suggest that Fas mutation leads to the breakdown of peripheral tolerance. However, the mechanism leading to the impaired peripheral deletion remains unknown. Furthermore, the function of the accumulating DNTC in the periphery of lpr mice is still unclear.
We and others have demonstrated that DNTC and clones from transgenic mice can down-regulate immune responses by killing activated alloreactive CD8+ T cells that express functional Fas receptors but not CD8+ T cells from lpr mice that express nonfunctional Fas (11, 12). Recently, it was found that DNTC from normal mice can also down-regulate allogeneic immune responses in vitro and in vivo (11, 18). These data indicate that peripheral DNTC have a regulatory function in a variety of models. Because DNTC from lpr mice express similar cell surface markers to the regulatory DNTC clones (11, 19) and can kill syngeneic Fas+ thymocytes in vitro when stimulated by PMA and ionomycin (20), the accumulating DNTC in lpr mice may have a regulatory function. The goal of this study was to characterize the function of lpr DNTC. The results shown here demonstrate that lpr DNTC can down-regulate allogeneic immune responses mediated by syngeneic wild-type but not lpr CD4+ and CD8+ T cells both in vitro and in vivo. The inability of DNTC to kill activated lpr CD4+ and CD8+ T cells may contribute to the development of autoimmune disease in lpr mice.
Materials and Methods
Mice.
C57BL/6 (B6+/+), B6.MRL-Faslpr (B6/lpr), B6Smn. C3H-Faslgld (B6/gld), B6.C-H2bm1/By (bm1), B6.C-H2bm12/By (bm12), BALB/c-H-2-dm2 (dm2, a BALB/c Ld loss mutant), and BALB/c mice were purchased from Jackson ImmunoResearch Laboratories. All mice were maintained in the University Health Network (UNH; University of Toronto, Toronto, Canada) animal colonies. (B6Xdm2)F1 mice and the B6-P14 (P14) mice possessing a transgenic TCR specific for the lymphocytic choriomeningitis virus gp33 peptide (21) were bred in UHN animal facilities.
Isolation of DNTC.
Spleen and lymph node cells of B6/lpr mice were depleted of red blood cells and then passed through a nylon wool column to enrich the T cell population. The cells were then treated with depletion antibodies specific for murine CD4 (RL172, rat IgM; reference 22) and CD8 (3.168.8, rat IgM; reference 22) and rabbit complement (Cedarlane) to deplete CD4+ and CD8+ T cells.
Generation of Effector and Target T Cells for Killing Assays.
Alloreactive H-2d–specific DNTC CD8+ and CD4+ T cells were generated by incubating each purified cell type with irradiated (20 Gy) BALB/c splenocytes. gp33-specific CD8+ T cells were generated by incubating spleen cells from P14 TCR transgenic mice with gp33 (23). The cells were cultured in α-MEM media supplemented with 10% FCS and 50 U rIL-2. DNTC cultures also received 30 U rIL-4.
Killing Assays.
Primary activated B6/lpr or B6/gld DNTC were used as effector cells and plated in serial dilutions in a round-bottom 96-well plate. Target cells labeled with 5 μCi/ml [3H]TdR at 37°C for 18 h were washed three times and added in equal portions to each well. Each cell culture was also given 50 U rIL-2, 30 U rIL-4, and irradiated (20 Gy) allogeneic stimulator cells or peptide depending on the experiment. After coculture with effector cells at 37°C for 18 h, the cells were harvested and counted using a TOP COUNT™ cell harvester and plate reader (Packard Instrument Co.). Specific cell killing was calculated using this equation: % Specific killing = (SE)/S × 100, where E (experimental) is cpm of retained DNA in the presence of effector cells and S (spontaneous) is cpm of retained DNA in the absence of effector cells.
Cell Surface Marker Staining and Blocking.
B6/lpr spleen and lymph node cells were harvested 7 d after donor lymphocyte infusion (DLI) treatment and stained with fluorescence-conjugated monoclonal antibodies specifically recognizing CD3, CD4, and CD8 (BD Biosciences). Data were acquired and analyzed on an Epics XL-MCL flow cytometer (Coulter Corp.). Fas ligand (FasL) was blocked using recombinant mouse Fas-Fc chimera/TNFRSF6 (R&D Systems).
DLI, Adoptive Transfer of Lymphocytes, and Skin Grafting.
B6/lpr mice were injected with a mixture of 4.0 × 107 spleen and lymph node cells from either bm1 or bm12 mice. Spleen and lymph node cells from the B6/lpr mice were harvested 7 d after the infusion of allogeneic cells. Naive B6+/+ mice intravenously received either 4.0 × 107 full spleen and lymph node cells or DNTC isolated from DLI-treated lpr mice. 1 d later, all B6+/+ recipient mice were given skin grafts from either bm1 or bm12 mice and a third-party skin graft (BALB/c or [B6Xdm2]F1) as previously reported (24). Survival of the skin grafts was monitored and scored until rejection.
Statistics.
All skin grafting survival data were analyzed using the Student's t test.
Results
B6/lpr DNTC Kill Activated Syngeneic CD8+ T Cells through the Fas/FasL Pathway.
We and others have demonstrated that DNTC and clones derived from normal and transgenic mice can kill activated syngeneic CD8+ target cells through the Fas/FasL pathway (11, 12). Furthermore, mitogen-activated lpr DNTC can kill syngeneic Fas+ thymocytes (20). These findings suggest that lpr DNTC also have a regulatory role but are unable to execute their function due to the lack of functional Fas expression on their target cells. To test this hypothesis, B6/lpr DNTC were isolated from spleen and lymph nodes, allogeneically stimulated, and used as putative regulatory cells. CD8+ T cells from B6+/+ (Fas+/+) and B6/lpr (Fas-mutant) mice were similarly activated and used as targets in a JAM cytotoxicity assay (25). As shown in Fig. 1 A, B6/lpr DNTC killed B6+/+ (Fas+/+) CD8+ T cells much more effectively than the B6/lpr (Fas-mutant) CD8+ T cells. This result is consistent with the data obtained from non-lpr DNTC and indicates that allogeneically activated lpr DNTC can kill activated syngeneic CD8+ T cells from wild-type but not Fas-mutant lpr mice, which suggests that Fas–FasL interaction is an important mechanism in B6/lpr DNTC-mediated killing.
Figure 1.
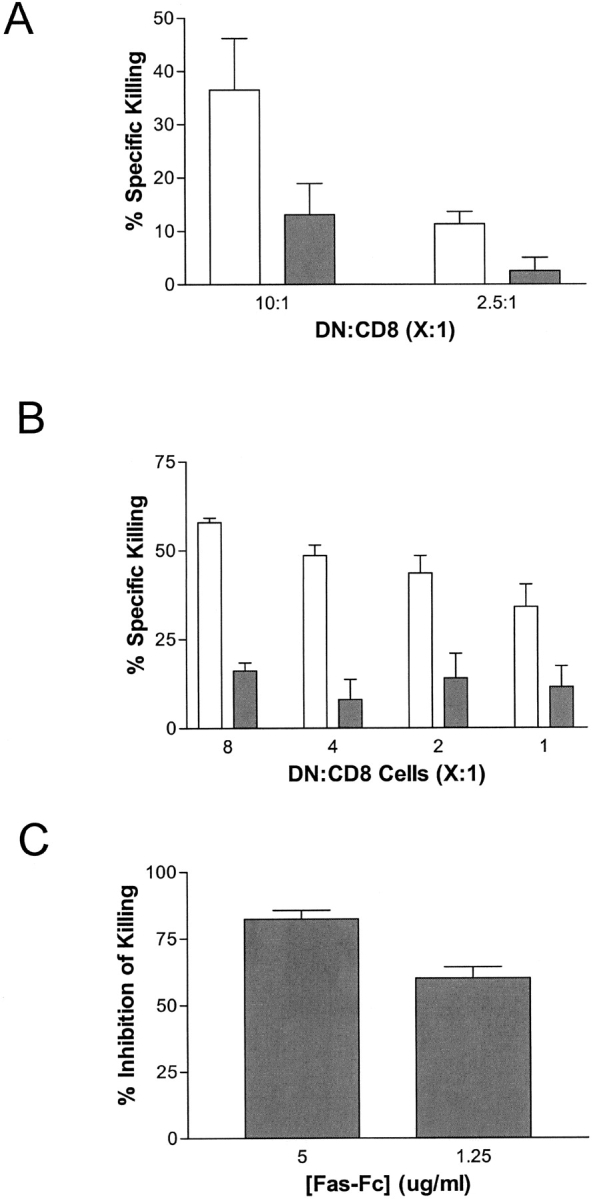
Lpr DNTC kill target cells through the Fas/FasL pathway. (A) H-2d–specific B6/lpr DNTC effectors were prepared as described in Materials and Methods and cocultured with primary activated CD8+ T cells from either B6+/+ (light bars), or B6/lpr (dark bars) mice. The percent-specific lysis of target cells was calculated. The data are expressed as mean percent killing of three replicate cultures. The experiment was repeated twice and similar results were obtained. (B) Activated B6 Fas+CD8+ T cells cocultured with DNTC isolated from either B6/lpr (light bars) or B6/gld (dark bars) mice. The percent-specific lysis of target cells was calculated. The data are expressed as mean percent killing of three replicate cultures. (C) 5 × 104/well of activated B6/lpr DNTC were incubated with Fas–Fc fusion protein in the indicated concentrations for 20 min and then cocultured with 104 primary activated B6+/+ CD8+ T cell targets. The mean percent inhibition of specific lysis of B6+/+ CD8 target cells compared with killing detected in the absence of Fas–Fc is shown. The data represent three replicate cultures. The experiment was repeated twice and representative data are shown.
Autoimmune-prone gld mice express FasL in a nonfunctional conformation that is not able to ligate Fas on target cells (13, 14). As seen in lpr mice, DNTC also accumulate in gld mice concomitant with the development of autoimmune disease symptoms (13, 14). To determine if the expression of FasL on DNTC is important for the killing of CD8+ T cells, DNTC from B6/gld (FasL-mutant) mice were purified and their ability to kill activated syngeneic Fas+CD8+ T cells was compared with DNTC from B6/lpr (FasL+/+) mice. As shown in Fig. 1 B, the cytotoxicity to activated syngeneic Fas+CD8+ T cells mediated by B6/gld DNTC was significantly reduced compared with that of B6/lpr DNTC. This result indicates that the expression of functional FasL on DNTC enhances the ability of DNTC to kill CD8+ T cell targets.
To further determine the specific contribution of FasL expression on lpr DNTC-mediated killing of syngeneic T cells, FasL on lpr DNTC was blocked with various concentrations of Fas–Fc fusion protein before coincubation with a fixed number of syngeneic Fas+CD8+ T cells. The ability of lpr DNTC to kill syngeneic Fas+CD8+ T cells was significantly inhibited when FasL was blocked on lpr DNTC by increasing concentrations of Fas–Fc fusion protein (Fig. 1 C). Taken together, these data demonstrate that lpr DNTC are able to down-regulate allogeneic immune responses mediated by syngeneic CD8+ T cells and the expression of functional FasL on DNTC and Fas on target CD8+ T cells is important for lpr DNTC-mediated killing.
In Vitro–activated B6/lpr DNTC Are Cytotoxic to B6+/+ CD4+ and CD8+ T Cells That Are Activated by a Different Antigen.
To further characterize the ability of lpr DNTC to regulate wild-type T cells, we first assessed the ability of H-2d–specific B6/lpr DNTC to kill activated syngeneic H-2d–specific B6+/+ CD4+, in addition to CD8+, T cells. Fig. 2 A shows that B6/lpr DNTC could dose dependently kill both activated syngeneic H-2d–specific CD8+ and CD4+ T cells from normal B6+/+ mice. Next, we addressed the question of whether the killing of syngeneic T cells mediated by lpr DNTC was antigen specific. Activated H-2d–specific B6/lpr DNTC were tested for their ability to kill syngeneic T cells that are activated by either the same or a different antigen. Specifically, CD8+ T cells from B6+/+ mice were activated against H-2d as described in Materials and Methods, or B6 transgenic P14 CD8+ T cells were stimulated with p33 peptide to create CD8+ T cell targets that carried TCR of different specificity from the B6/lpr DNTC. The ability of H-2d–specific B6/lpr DNTC to kill each cell type was compared. Fig. 2 B shows that H-2d–specific B6/lpr DNTC effectively killed both p33- and H-2d–specific CD8+ T cells. Together, these data indicate that lpr DNTC that were activated in vitro with allogeneic splenocytes can kill both activated syngeneic CD4+ and CD8+ T cells, and the cytotoxicity to syngeneic CD8+ T cells is not antigen specific in vitro.
Figure 2.
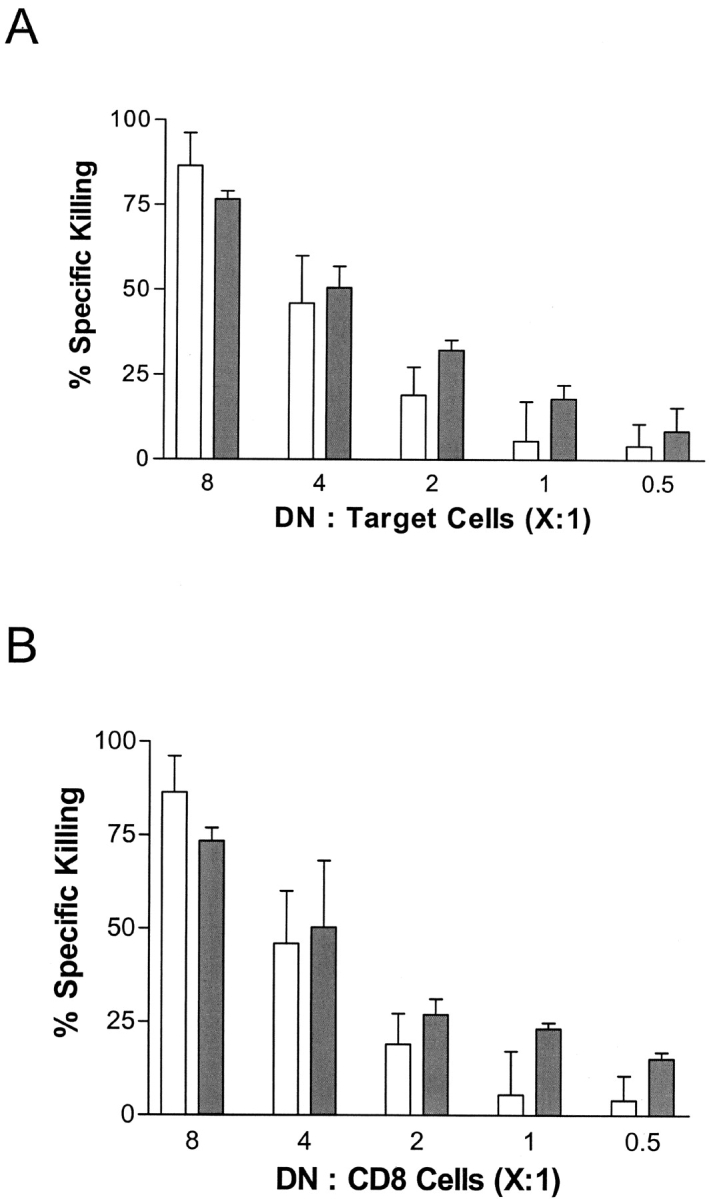
In vitro–activated B6/lpr DNTC are cytotoxic to B6+/+ CD4+ and CD8+ T cells that are activated by a different antigen. (A) DNTC were purified from the spleens of B6/lpr mice and cultured for 3.5 d in the presence of irradiated BALB/c splenocytes, rIL-2 and rIL-4, and used as effector cells. In vitro–activated H-2d–specific B6+/+ CD4+ (dark bars) and CD8+ (light bars) T cells were used as targets. After 18 h of co-culture, cells were harvested and counted, and the percent-specific killing was calculated. The data are expressed as mean percent killing of three replicate cultures. The experiment was repeated three times and representative data is shown. (B) Splenic CD8+ T cells from B6 mice or B6 anti–lymphocytic choriomeningitis virus-gp33 were activated in vitro with either irradiated BALB/c spleen cells or gp33 peptide as described previously (21, 23). H-2d–specific B6/lpr DNTC were stimulated independently by BALB/c spleen cells and cocultured with either H-2d–specific B6+/+ CD8+ T cells (light bars), or p33-specific B6 CD8+ T cells (dark bars). The percent-specific killing of each target cell culture was determined. The data are expressed as mean percent killing of three replicate cultures. The experiment was repeated three times and representative data is shown.
Infusion of Allogeneic Lymphocytes Leads to Expansion of B6/lpr DNTC.
DLI has been shown to enhance organ allograft survival in mice (24) and humans (26). Recently, we have demonstrated that pretransplant DLI induces the activation and expansion of recipient DNTC and the prolongation of allograft (18) and xenograft survival (unpublished data), and that adoptive transfer of activated DNTC clones can enhance donor-specific but not third-party skin graft survival (11).
To investigate the function of B6/lpr DNTC in vivo, we first studied whether B6/lpr DNTC can be activated and expand in vivo after the infusion of allogeneic lymphocytes. B6/lpr mice were given a DLI of bm1 (single class I–mismatched) spleen and lymph node cells. The recipient mice were killed 7 d later and the percentages of CD8+, CD4+, and DNTC in the lymph nodes and spleen were analyzed by flow cytometry. 1 wk after DLI there was no significant change in the percentage of CD8+ T cells and a significantly reduced proportion of CD4+ T cells in the spleen (P < 0.05) but not in the lymph nodes compared with age-matched B6/lpr non-DLI–treated controls. Interestingly, the percentage of DNTC in both the lymph nodes (Fig. 3 A) and spleen (Fig. 3 B) of DLI-treated B6/lpr mice was significantly increased compared with age-matched untreated controls (P < 0.04 and P < 0.02, respectively). This data indicates that single MHC class I locus–mismatched DLI can promote the activation and proliferation of lpr DNTC in vivo.
Figure 3.

Increase of DNTC in the lymph nodes and spleen of B6/lpr mice after infusion of bm1 cells. B6/lpr mice were injected intravenously with 4 × 107 bm1 lymphocytes. 7 d later, (A) lymph node and (B) spleen lymphocytes were harvested from bm1 cell-injected (dark bars) and age-matched control (light bars) mice. The percentage of CD3+CD4+CD8−, CD3+CD4−CD8+, and CD3+CD4−CD8− cell populations were analyzed by flow cytometry. The DNTC population was significantly increased in both the spleen (n = 5) and lymph nodes (n = 4) of DLI-treated mice when compared with age-matched nontreated control mice (n = 5; spleen, P < 0.02 and lymph node, P < 0.04).
Adoptive Transfer of In Vivo–activated B6/lpr DNTC Enhances Specific Skin Allograft Survival.
Our finding that in vitro–activated lpr DNTC can kill both CD4+ and CD8+ T cells suggests that DNTC might be able to inhibit immune responses in vivo. To study whether the in vivo–activated lpr DNTC could enhance donor-specific skin graft survival, the spleen and lymph node cells were collected from B6/lpr mice 1 wk after the infusion of bm1 lymphocytes. Naive B6+/+ mice were used as skin graft recipients and divided into two groups. One group was infused with a mixture of DLI-treated B6/lpr spleen and lymph node cells, and the other was left untreated as a control. 1 d later, both groups of mice were given tail skin grafts from bm1 (antigen-specific) and BALB/c (third-party) donors. Skin graft survival was monitored as previously described (11). Fig. 4 A shows that the mean survival time (MST) of bm1 skin grafts was significantly enhanced in mice that received B6/lpr lymphocytes that were preactivated by bm1 antigens (MST = 28.8 d, n = 8) when compared with that of the control group (MST = 15.7 d, n = 6, P = 0.046). All third-party BALB/c skin grafts were rejected within the normal time course by both groups, indicating that the enhancement of skin graft survival was donor specific.
Figure 4.
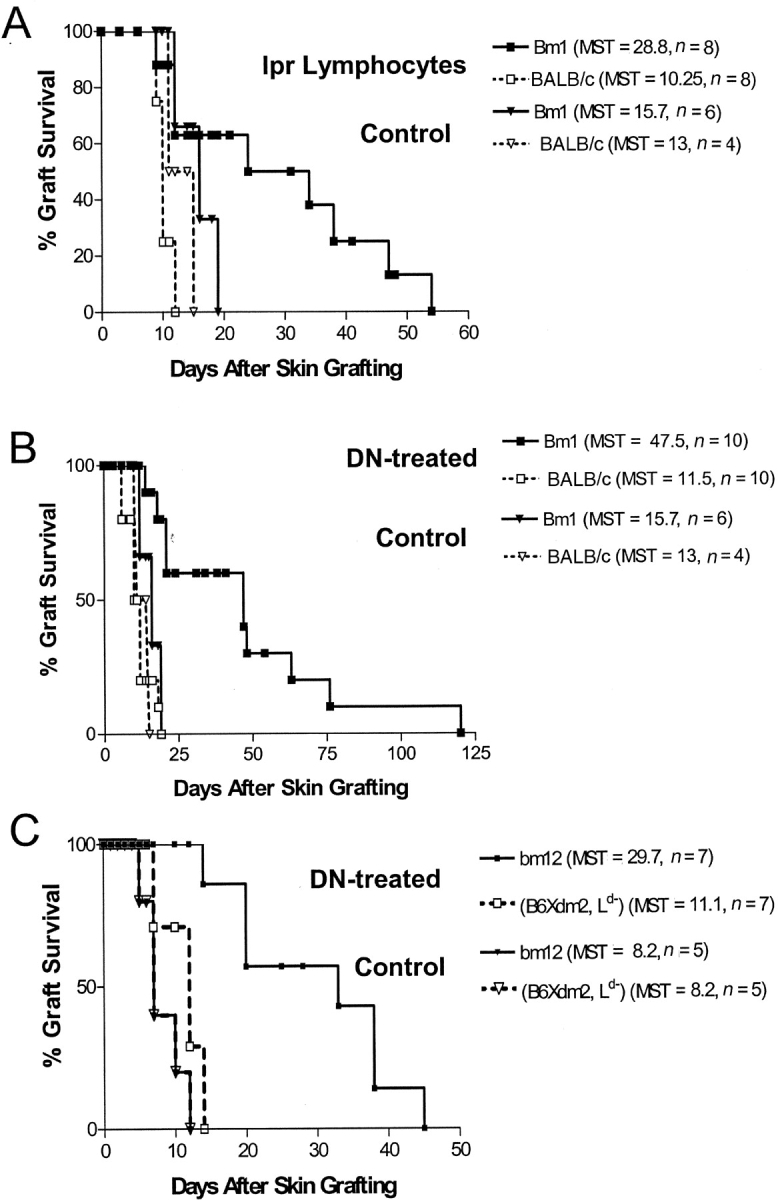
Adoptive transfer of bm1 or bm12 DLI-treated B6/lpr lymphocytes enhances bm1 or bm12 skin graft survival. (A) The spleen and lymph node cells were collected from B6/lpr mice that were injected intravenously with bm1 lymphocytes. B6+/+ mice were either left untreated as a control (▿ and ▾, n = 10) or injected with 4 × 107 bm1 DLI-treated B6/lpr lymphocytes per mouse (□ and ▪, n = 8) 1 d before transplantation. All recipient mice were then given tail skin grafts from both bm1 (solid lines) and BALB/c (dashed lines) donors. bm1 skin graft survival was significantly prolonged in mice that received lpr lymphocytes when compared with untreated controls (P < 0.05). (B) DNTC were purified from B6/lpr mice that had been infused with bm1 cells. Naive B6+/+ mice were either left untreated as a control (▿ and ▾, n = 10) or injected intravenously with 1.5 × 107 bm1 DLI-treated B6/lpr DNTC per mouse (□ and ▪, n = 10) 1 d before transplantation. Each mouse was transplanted with bm1 (solid lines) and BALB/c (dashed lines) skin grafts. bm1 skin graft survival was significantly prolonged in mice that received lpr DNTC when compared with untreated controls (P = 0.017). (C) DNTC were purified from B6/lpr mice that had been infused with bm12 cells. Naive B6+/+ mice were injected intravenously with 8.5 × 106 DNTC (□ and ▪, n = 7) 1 d before transplantation, or were left untreated as controls (▿ and ▾, n = 5). Each mouse was transplanted with bm12 (solid lines) and (B6Xdm2)F1 (dashed lines) skin grafts. bm12 skin graft survival was significantly prolonged in mice that received lpr DNTC when compared with untreated controls (P = 0.0012).
To further determine the specific contribution of B6/lpr DNTC in enhancing skin allograft survival, DNTC were isolated from the peripheral lymphocytes of bm1 DLI-treated B6/lpr mice. The in vivo–activated bm1-specific B6/lpr DNTC were adoptively transferred into naive B6+/+ mice 1 d before transplantation. Each recipient B6+/+ mouse was then given skin grafts from bm1 (donor-specific) and BALB/c (third-party) mice. Graft survival was compared with the B6+/+ mice that were given the same skin grafts without pretransplant infusion of B6/lpr DNTC. As shown in Fig. 4 B, the MST of bm1 skin grafts was significantly prolonged in the mice that received in vivo–activated bm1-specific DNTC when compared with the controls (MST = 47.5 d, n = 10 vs. MST 15.7 d, n = 6, P = 0.017). In contrast, both treatment groups rejected all third-party BALB/c skin grafts within 2 wk. These data demonstrate that the infusion of in vivo–activated B6/lpr DNTC can significantly enhance donor-specific bm1 skin allograft survival.
To test whether B6/lpr DNTC could also enhance the survival of MHC class II–mismatched skin grafts, B6/lpr mice were infused with bm12 (class II–mismatched) spleen and lymph node cells. The in vivo–activated bm12-specific B6/lpr DNTC were isolated and transferred into naive B6+/+ mice 1 d before transplantation or left untreated as a control. All B6+/+ recipient mice then received skin grafts from both bm12 mice (antigen-specific) and (B6xdm2)F1 (third-party) donors. Fig. 4 C shows that the mice that received bm12-specific B6/lpr DNTC had significant enhancement of bm12 skin graft survival (MST = 29.7, n = 7) when compared with control mice that did not receive any B6/lpr DNTC (MST = 8.2, n = 5, P = 0.0012). Third-party allogeneic (B6xdm2)F1 skin grafts were rejected at the same pace by both groups of recipients. These results demonstrate that in vivo–activated B6/lpr DNTC can also specifically enhance the survival of MHC class II–mismatched skin allograft survival. Together, these findings confirm and extend the results obtained from non-lpr mice (11, 24) and demonstrate that in vivo–activated B6/lpr DNTC can inhibit the immune response of syngeneic T cells toward MHC class I and II alloantigens in vivo, and prolong skin graft survival in an antigen-specific manner.
Discussion
The accumulation of TCR-αβ+ DNTC cells in the periphery of autoimmune lpr mice and patients has been observed for many years. However, the function and mechanism of these DNTC has remained elusive. Previous studies have shown that mature peripheral DNTC from transgenic (11, 12) and normal mice (11, 18) are able to down-regulate alloimmune responses mediated by syngeneic CD4+ and CD8+ T cells in vitro through the Fas–FasL pathway. Furthermore, several studies indicate that lpr DNTC may have a regulatory function. Watenabe et al. (15) demonstrated that the DNTC population in lpr mice could kill allogeneic tumor target cells through a FasL-dependent mechanism. Benihoud et al. (20) showed that lpr DNTC can kill syngeneic Fas-expressing T cells in vitro after stimulation with PMA and ionomycin. The results presented in this paper demonstrate for the first time that the DNTC that accumulate in autoimmune-prone lpr mice possess a potent ability to down-regulate allogeneic immune responses mediated by syngeneic CD4+ and CD8+ T cells from wild-type mice both in vitro and in vivo.
It is well known that regulatory T cells play an important role in preventing autoimmune diseases (4–6). Various studies have shown that autoimmune diseases develop because of a lack or a malfunction of regulatory cells (1, 2, 7, 8). In lpr mice, the number of DNTC is much higher than what is seen in normal mice, yet the animals develop autoimmune disease. Here we demonstrate that although DNTC from autoimmune-prone gld mice that lack functional FasL molecules cannot kill activated syngeneic Fas+ CD8+ T cells, lpr DNTC are able to kill both activated syngeneic CD4+ and CD8+ T cells that express functional Fas receptors. These results indicate that the regulatory function of DNTC in lpr mice is retained. Thus, we speculate that DNTC might accumulate partly as a failed attempt to suppress peripheral autoimmune T cells, and partly as a result of their own inability to undergo Fas-mediated activation-induced apoptosis. Because lpr DNTC-mediated suppression depends on interactions between FasL on the DNTC and Fas expressed on target cells, autoimmune disease may develop in lpr mice because the mutation of Fas on autoimmune CD4+ and CD8+ T cells renders them unsusceptible to DNTC-mediated regulation. Our data help explain the impaired peripheral clonal deletion observed in Fas null mice (17) and suggest that in addition to regulatory cells, defects in the target cells that are being regulated may lead to the development of autoimmune diseases.
Lpr DNTC-mediated regulation is similar to that previously observed in Ld-specific DNTC T regulatory clones (11). Both cell types are able to kill syngeneic cells in vitro using a Fas-dependent mechanism. However, the DNTC clones generated from 2C transgenic mice only killed syngeneic CD8+ T cell targets that were activated by the same antigen (11), whereas primary activated lpr DNTC were able to kill syngeneic T cells that were activated by either the same or a different antigen in vitro. This finding is similar to that found using some subsets of primary activated CD4+CD25+ T regulatory cells (20, 27), suggesting that cytotoxicity mediated by primary activated T regulatory cells is less specific than that mediated by transgenic DNTC clones. There are many possibilities that might explain the discrepancy between primary activated lpr DNTC and regulatory DNTC clone-mediated killing in vitro. First, lpr DNTC used in these experiments were generated by stimulation with fully allogeneic splenocytes, whereas DNTC clones were generated from T cells that carry TCR reactive to only one specific epitope of the Ld antigen. Thus, there is a higher possibility for TCR-MHC cross-reaction with the H-2d–specific lpr DNTC compared with single class I locus (Ld)-specific DNTC clones. Second, nonspecific cytotoxicity may result from high expression of FasL on lpr DNTC. Up-regulation of FasL on effector cells has been shown to induce nonspecific target cell death, termed bystander killing (28). It is known that lpr DNTC constitutively express a higher level of FasL mRNA compared with non-lpr mice (15). Primary activation in vitro with allogeneic cells and cytokines may induce overexpression of FasL on lpr DNTC resulting in bystander killing. In addition, in a recent study we have shown that the TCR on both DNTC and target T cells can influence non-lpr DNTC-mediated killing (unpublished data) and that in vitro antigen-specific killing can take place in many more conditions than was initially described using transgenic regulatory DNTC clones (11). Regardless of the in vitro discrepancy in antigen specificity, both regulatory DNTC clones (11) and lpr DNTC are able to suppress immune responses in an antigen-specific fashion in vivo. As seen in the DNTC clones, adoptive transfer of in vivo–activated lpr DNTC into wild-type syngeneic mice enhances donor-specific allogeneic skin graft survival. Additional studies are needed in order to understand what mechanism is responsible for the discrepancy between the functions of in vitro– and in vivo–activated lpr DNTC.
Although the ability of DNTC to inhibit allogeneic immune responses mediated by wild-type syngeneic CD4+ and CD8+ T cells was clearly demonstrated in this paper, it is not yet clear whether DNTC can also down-regulate syngeneic autoreactive T cells. The inability to identify the self-antigens that cause autoimmune disease in lpr mice makes it difficult to directly test the ability of lpr DNTC to suppress autoreactive T cells and prevent autoimmune disease in these mice. More investigations into the mechanism and specificity of both regulatory cells and their targets will lead to a better understanding of the development of autoimmune disease.
Acknowledgments
This work was supported by the Canadian Institute of Health Research (CIHR MOP-14431 to L. Zhang).
References
- 1.Shevach, E.M. 2000. Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 18:423–449. [DOI] [PubMed] [Google Scholar]
- 2.Chatenoud, L., B. Salomon, and J.A. Bluestone. 2001. Suppressor T cells-they're back and critical for regulation of autoimmunity! Immunol. Rev. 182:149–163. [DOI] [PubMed] [Google Scholar]
- 3.Zelenika, D., E. Adams, S. Humm, C.Y. Lin, H. Waldmann, and S.P. Cobbold. 2001. The role of CD4+ T-cell subsets in determining transplantation rejection or tolerance. Immunol. Rev. 182:164–179. [DOI] [PubMed] [Google Scholar]
- 4.Zhang, Z.X., K. Young, and L. Zhang. 2001. CD3+CD4− CD8− αβ-TCR+ T cell as immune regulatory cell. J. Mol. Med. 79:419–427. [DOI] [PubMed] [Google Scholar]
- 5.Roncarolo, M.G., and M.K. Levings. 2000. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr. Opin. Immunol. 12:676–683. [DOI] [PubMed] [Google Scholar]
- 6.Levings, M.K., R. Sangregorio, and M.G. Roncarolo. 2001. Human CD25+CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 193:1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor a-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 8.Gonzalez, A., I. Andre-Schmutz, C. Carnaud, D. Mathis, and C. Benoist. 2001. Damage control, rather than unresponsiveness, effected by protective DX5+ T cells in autoimmune diabetes. Nat. Immunol. 2:1117–1125. [DOI] [PubMed] [Google Scholar]
- 9.Jiang, H., and L. Chess. 2000. The specific regulation of immune responses by CD8+ T cells restricted by the MHC class Ib molecule, Qa-1. Annu. Rev. Immunol. 18:185–216. [DOI] [PubMed] [Google Scholar]
- 10.Hayday, A.C. 2000. γδ cells: a right time and a right place for a conserved third way of protection. Annu. Rev. Immunol. 18:975–1026. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, Z.X., L. Yang, K.J. Young, B. DuTemple, and L. Zhang. 2000. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat. Med. 6:782–789. [DOI] [PubMed] [Google Scholar]
- 12.Priatel, J.J., O. Utting, and H.S. Teh. 2001. TCR/self-antigen interactions drive double-negative T cell peripheral expansion and differentiation into suppressor cells. J. Immunol. 167:6188–6194. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe-Fukunaga, R., C.I. Brannan, N.G. Copeland, N.A. Jenkins, and S. Nagata. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 356:314–317. [DOI] [PubMed] [Google Scholar]
- 14.Cohen, P.L., and R.A. Eisenberg. 1992. The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunol. Today. 13:427–428 (erratum published 14:97). [DOI] [PubMed] [Google Scholar]
- 15.Watanabe, D., T. Suda, H. Hashimoto, and S. Nagata. 1995. Constitutive activation of the Fas ligand gene in mouse lymphoproliferative disorders. EMBO J. 14:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto, K., Y. Yoshikai, T. Asano, K. Himeno, A. Iwasaki, and K. Nomoto. 1991. Defect in negative selection in lpr donor-derived T cells differentiating in non-lpr host thymus. J. Exp. Med. 173:127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adachi, M., S. Suematsu, T. Suda, D. Watanabe, H. Fukuyama, J. Ogasawara, T. Tanaka, N. Yoshida, and S. Nagata. 1996. Enhanced and accelerated lymphoproliferation in Fas-null mice. Proc. Natl. Acad. Sci. USA. 93:2131–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang, Z.-X., W.L. Stanford, and L. Zhang. 2002. Ly6-A is critical for the function of DN T cells. Eur. J. Immunol. 32:1584–1592. [DOI] [PubMed] [Google Scholar]
- 19.Landolfi, M.M., N. Van Houten, J.Q. Russell, R. Scollay, J.R. Parnes, and R.C. Budd. 1993. CD2−CD4−CD8− lymph node T lymphocytes in MRL lpr/lpr mice are derived from a CD2+CD4+CD8+ thymic precursor. J. Immunol. 151:1086–1096. [PubMed] [Google Scholar]
- 20.Benihoud, K., D. Bonardelle, P. Bobe, and N. Kiger. 1997. MRL/lpr CD4− CD8− and CD8+ T cells, respectively, mediate Fas-dependent and perforin cytotoxic pathways. Eur. J. Immunol. 27:415–420. [DOI] [PubMed] [Google Scholar]
- 21.Pircher, H., K. Burki, R. Lang, H. Hengartner, and R.M. Zinkernagel. 1989. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 342:559–561. [DOI] [PubMed] [Google Scholar]
- 22.Kishimoto, H., and J. Sprent. 1999. Several different cell surface molecules control negative selection of medullary thymocytes. J. Exp. Med. 190:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garza, K.M., S.M. Chan, R. Suri, L.T. Nguyen, B. Odermatt, S.P. Schoenberger, and P.S. Ohashi. 2000. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 191:2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, L., T.B. Du, Q. Khan, and L. Zhang. 1998. Mechanisms of long-term donor-specific allograft survival induced by pretransplant infusion of lymphocytes. Blood. 91:324–330. [PubMed] [Google Scholar]
- 25.Matzinger, P. 1991. The JAM test. A simple assay for DNA fragmentation and cell death. J. Immunol. Methods. 145:185–192. [DOI] [PubMed] [Google Scholar]
- 26.van Twuyver, E., R.J. Mooijaart, I.J. ten Berge, A.R. van der Horst, J.M. Wilmink, W.M. Kast, C.J. Melief, and L.P. de Waal. 1991. Pretransplantation blood transfusion revisited. N. Engl. J. Med. 325:1210–1213. [DOI] [PubMed] [Google Scholar]
- 27.Thornton, A.M., and E.M. Shevach. 2000. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164:183–190. [DOI] [PubMed] [Google Scholar]
- 28.Badley, A.D., J.A. McElhinny, P.J. Leibson, D.H. Lynch, M.R. Alderson, and C.V. Paya. 1996. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J. Virol. 70:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]