

Abstract
Hematopoietic cell growth, differentiation, and chemotactic responses require coordinated action between cytokines and chemokines. Cytokines promote receptor oligomerization, followed by Janus kinase (JAK) kinase activation, signal transducers and transactivators of transcription (STAT) nuclear translocation, and transcription of cytokine-responsive genes. These include genes that encode a family of negative regulators of cytokine signaling, the suppressors of cytokine signaling (SOCS) proteins. After binding their specific receptors, chemokines trigger receptor dimerization and activate the JAK/STAT pathway. We show that SOCS3 overexpression or up-regulation, stimulated by a cytokine such as growth hormone, impairs the response to CXCL12, measured by Ca2+ flux and chemotaxis in vitro and in vivo. This effect is mediated by SOCS3 binding to the CXC chemokine receptor 4 receptor, blocking JAK/STAT and Gαi pathways, without interfering with cell surface chemokine receptor expression. The data provide clear evidence for signaling cross-talk between cytokine and chemokine responses in building a functional immune system.
Keywords: SOCS3, JAK-STAT activation, chemokine signaling, cytokine signaling
Introduction
The chemokines comprise a large family of low molecular weight (8–10 kD) cytokines, with chemotactic and pro-activatory effects on different leukocyte lineages (1–3). Studies have established the central role of chemokines in a number of physiological situations, including T helper responses, hematopoiesis, angiogenesis, and homeostasis, as well as in pathological conditions such as asthma, tumor rejection, HIV-1 infection, and arteriosclerosis (4–8).
Chemokines mediate their biological effects after binding to specific receptors, members of the seven-transmembrane domain G protein–coupled receptor family (1, 2). Chemokine receptors are promiscuous, as each can bind more than one chemokine; expression is heterogeneous among different cells of the leukocyte lineage and is transcriptionally regulated (3, 5). An exception is the CXC chemokine receptor 4 (CXCR4)* receptor, which binds only stromal cell–derived factor (SDF)-1α (CXCL12) (1, 2), a chemokine isolated from stromal cell culture supernatants (9). Its chemotactic properties have been described in PBLs (10), CD34+ progenitor cells (11), and pre- and pro-B cell lines (12). Knockout mice lacking the CXCL12 protein (10) and mice lacking the CXCR4 receptor (13, 14) display similar phenotypes: animals die before birth, displaying abnormalities in B cell lymphopoiesis and bone marrow myelopoiesis, lack of blood vessel formation in the gut, severe ventricular septal defects, and altered cerebellar neuron migration (13–15).
After binding to their receptors, chemokines trigger receptor association of guanine nucleotide-binding proteins (G proteins; reference 16); this activates signaling molecules that mediate changes in the cytoskeletal apparatus and transcription factors that regulate cell growth (17–19). We have shown that CXCL12 (16), as well as other chemokines such as monocyte chemotactic protein (MCP)-1 (CCL2) and regulated upon activation, normal T cell–expressed and secreted (RANTES; CCL5) (20, 21), exert their effects via dimerization of their receptors and activation of Janus kinase (JAK) kinases, a protein family originally implicated in cytokine signaling (22). This in turn phosphorylates the chemokine receptors in tyrosine residues and activates signal transducers and transactivators of transcription (STAT) transcription factors (23).
The suppressors of cytokine signaling (SOCS) family proteins have been identified as feedback regulators of JAK/STAT activation, through their binding to JAK kinases or cytokine receptors (24–28). The majority of cytokines analyzed to date, such as LIF, IL-2, IL-3, IL-6, growth hormone (GH), IFN-γ, and leptin, induce several SOCS family members in a tissue-specific manner (24, 28). The regulatory role of SOCS molecules is not limited to the cytokine receptor superfamily, however, as interaction has been described between SOCS and other cellular targets, such as the IGF-I receptor, suggesting that SOCS proteins may have an extensive role in receptor signaling (29).
Here we show that through JAK/STAT activation, CXCR4 occupancy by CXCL12 upregulates a member of the SOCS protein family, SOCS3. Cytokine-stimulated overexpression or upregulation of SOCS3 interferes with signaling by certain chemokines, including CXCL12. By binding to CXCR4, SOCS3 blocks JAK/STAT complex activation and association to CXCR4, inhibiting CXCL12-mediated responses both in vivo and in vitro. These data, discussed here in the context of cross-talk between cytokine and chemokine responses, will aid in understanding the functional role of this chemokine/chemokine receptor pair and add a new element to the complex function of the immune system.
Materials and Methods
Biological Materials.
IM-9 (ATCC CCL159) and HEK-293 cells (ATCC TIB202) were from the American Type Culture Collection. Antibodies used include anti-CXCR4 (CXCR4–01) and anti-hGH receptor (hGHR-05) mAb generated in our laboratory (16, 30), horseradish peroxidase (PO)-labeled anti-PTyr mAb (4G10; Upstate Biotechnology), anti-phosphothreonine and -phosphoserine mAb (Calbiochem), anti-β2 microglobulin mAb (BD Biosciences), anti-Flag M2 mAb (Sigma-Aldrich), anti-CD71 mAb (Zymed Laboratories), FITC-labeled anti-CD3 (Southern Biotechnology Associates, Inc.), FITC-anti-CD11b and PE-b220 (BD Biosciences); rabbit anti-PTyr (Promega); anti-JAK1, -JAK2, and -JAK3 (Upstate Biotechnology); anti-Gαι, -Gαs, anti-STAT1 to -STAT6, anti-pol II (Santa Cruz Biotechnology, Inc.), and anti–β-actin (Sigma-Aldrich). For immunostaining, we used anti-STAT3 and -phospho-STAT3 antibody (New England Biolabs, Inc.). AG490 and AG9 were from Calbiochem and pertussis toxin (Ptx) was from Sigma-Aldrich.
Flow Cytometry Analysis.
Cells were centrifuged (250 g, 10 min, room temperature), plated in V-bottom 96-well plates (2.5 × 105 cells/well), and incubated with biotin-labeled hGHR-05 mAb (1 μg/50 μl/well, 60 min, 4°C). Cells were washed twice in PBS with 2% BSA and 2% FCS and centrifuged (250 g, 5 min, 4°C). Fluorescein isothiocyanate-labeled streptavidin (Southern Biotechnology Associates, Inc.) was added, incubated (30 min, 4°C), plates washed twice, and cell-bound fluorescence determined in a Profile XL flow cytometer at 525 nm (Beckman Coulter).
Immunocytochemical Analysis.
IM-9 cells were cultured in RPMI with 1% BSA for 2 h, alone or CXCL12-stimulated (50 nM), washed with PBS at room temperature (RT), fixed with 100% methanol (10 min, −20°C), and permeabilized with 0.2% Triton X-100 in TBS (15 min, RT). After washing with TBS (3×, 5 min, RT), nonspecific binding sites were blocked with 5% goat serum in TBS (45 min, RT). Cells were washed with TBS, then incubated with anti-STAT3 or anti-phospho-STAT3 mAb diluted in TBS, 2% BSA, 2% normal goat serum, followed by Cy3-labeled anti–rabbit IgG antibody (Jackson ImmunoResearch Laboratories). Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) was added and cells analyzed in a Leica fluorescence microscope.
Cell Migration.
Cells were placed (0.25 × 106 cells in 0.1 ml) in the upper well of 24-well transmigration chambers (5 μm pore; Transwell; Costar Corp.) whose membrane was precoated with type VI collagen (20 μg/ml; Sigma-Aldrich); 50 nM CXCL12 or 10 nM CCL20 (in 0.6 ml RPMI with 0.25% BSA; PeproTech) was then added to the lower well. Plates were incubated (120 min, 37°C) and cells that migrated to the lower chamber were counted as described (23). When recombinant hGH (Genotropin; Pharmacia AB) treatment was used, cells (106/ml) were incubated in RPMI 1640 with 0.1% BSA and 10 μg/ml hGH for the time indicated.
To evaluate primary cells, mouse spleen, lymph node, and bone marrow cells (0.25 × 106 cells in 0.1 ml) were placed in the upper well of 24-well transmigration chambers (3 μm pore; Transwell) as described above. In spleen and bone marrow cell preparations, erythrocytes were lysed with NH4Cl (5 min, 37°C).
In assays in which depletion of fresh isolated primary cells was necessary, cells were plated (106 cells in 1 ml) in RPMI 1640 supplemented with 0.1% BSA and cultured (2 h, 37°C). After washing, migration in response to CXCL12 was evaluated as above.
HEK-293 cell migration was studied in a 96-well microchamber (NeuroProbe Inc.; reference 31). Chemokines at several concentrations were loaded into lower wells (30 μl/well), and cells (200 μl/well, 3 × 106 cells/ml) in upper wells. Polyvinylpyrrolidone-free filters (10 μm pore; NeuroProbe Inc.) were precoated (2 h, 37°C) with 20 μg/ml type VI collagen (Sigma-Aldrich). The chamber was incubated (5 h, 37°C), after which filters were removed and the cells on the upper part wiped off. Cells on the filters were fixed and stained (0.5% crystal violet, 20% methanol). Blue spots developed at positions at which cell migration had occurred, allowing densitometric quantitation of migration (National Institutes of Health Image software). The migration index was calculated by mean spot intensity.
In Vivo Biological Activity of CXCL12.
3-mo-old bGH-transgenic (Tg) and non-Tg littermate mice received intraperitoneal injection of CXCL12 (1 μg in 400 μl sterile PBS) or PBS alone. Mice were killed 6 h after injection and migrated cells extracted from the peritoneal cavity by injecting 6 ml of sterile PBS. Cells were counted, centrifuged (250 g, 10 min), and the distinct cell populations enumerated by flow cytometry analysis using specific cell surface markers. Migration is calculated for each cell population from CXCL12-treated mice as the x-fold increase over negative control (PBS-treated mice).
Immunoprecipitation, SDS-PAGE, and Western Blot Analysis.
CXCL12-stimulated cells (10 × 106) were lysed in detergent buffer (20 mM triethanolamine, pH 8.0, 300 mM NaCl, 2 mM EDTA, 20% glycerol, 1% digitonin, with 10 μM sodium orthovanadate, 10 μg/ml leupeptin, and 10 μg/ml aprotinin; 30 min, 4°C, continuous rocking), then centrifuged (15,000 g, 15 min). Immunoprecipitation was as described (16); protein G-Sepharose was used to analyze SOCS3 immunoprecipitates. Precipitates or protein extracts were separated in sodium dodecyl sulfate PAGE (SDS-PAGE) and transferred to nitrocellulose membranes. Western blot analysis was as described (16), using 2% BSA in TBS as blocking agent for anti-phosphotyrosine analyses. To strip, membranes were incubated (60 min, 60°C) with 62.5 mM Tris-HCl, pH 7.8, containing 2% SDS and 0.5% β-mercaptoethanol. Protein loading was controlled using a protein detection kit (Pierce Chemical Co.) and, when necessary, by reprobing the membrane with the immunoprecipitating antibody. For drug treatment, cells were incubated (16 h, 37°C) with Ptx (0.1 μg/ml) and AG490 or AG9 (50 μM). When necessary, quantitation was performed using the National Institutes of Health Image software.
Preparation of Nuclear Extracts.
Nuclear extracts were prepared from CXCL12-stimulated IM-9 cells (10 × 106; reference 16). Cells were washed with ice-cold PBS, resuspended in 1 ml of buffer A (50 mM NaCl, 0.5 M sucrose), incubated (2 min, 4°C), and pelleted (4,500 g, 3 min, 4°C). They were then resuspended in 1 ml of buffer B (50 mM NaCl, 25% glycerol), pelleted (4,500 g, 3 min, 4°C), and incubated in 60 μl of buffer C (350 mM NaCl, 25% glycerol) for 30 min at 4°C with continuous rocking. After centrifugation (20,000 g, 20 min, 4°C), supernatants containing nuclear extracts were aliquoted and stored at −80°C. All buffers contained 0.5 mM spermidine, 0.15 mM spermine, 0.1 mM EDTA, 10 mM HEPES, pH 8, 0.5 mM PMSF, 2 μg/ml leupeptin, 3 μg/ml pepstatin, 0.2 IU/ml aprotinin, 1.75 mM β-mercaptoethanol, 1 mM orthovanadate, and 10 mM NaF. Nuclear extract (20 μg) was separated in SDS-PAGE and transferred to nitrocellulose membranes for Western blot analysis, performed as above, and developed with anti-STAT3 and -STAT5b antibodies.
Electrophoretic Mobility Shift Assay.
Nuclear extracts (10 μg), prepared as above from untreated or chemokine-treated cells, were analyzed in binding reactions. Extracts were incubated with 0.5 ng 32P-end-labeled double-stranded oligodeoxynucleotides containing the sis-inducible element (SIE) of the c-fos promoter sequence (GGGGTGCATTTCCCGTAAATCTTGTCT) (wild type; wt-SIE); where indicated, a mutant version was used that is unable to bind STAT proteins (GGGGTGCATCCACCGTAAATCTTGTCT) (mut-SIE). The binding reaction was performed in electrophoretic mobility shift assay (EMSA) buffer (30 min, RT; final volume 10 μl) with 1.5 μg poly(dI-dC) and, where indicated, a 20-fold molar excess of unlabeled SIE or nonspecific oligonucleotide competitor. Protein–DNA complexes were resolved on a 4.5% polyacrylamide gel using 0.5× Tris-borate-EDTA running buffer. EMSA buffer contained 10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM dithiothreitol, 1 nM EDTA, and 5% glycerol. Protein amount in nuclear extracts was confirmed before Western blot or EMSA using a protein detection kit (Pierce Chemical Co.).
Cell Transfection.
Human embryonic kidney cells (HEK-293) were transiently transfected with pEF-Flag-I/mSOCS1, mSOCS2, or mSOCS3 constructs (donated by Dr. T. Willson, Walter and Eliza Hall Institute of Medical Research, Victoria, Australia) using lipofectamine (GIBCO BRL) according to the manufacturer's protocol. When required, IM9 cells or CCR6-stably transfected BaF/6 cells (32) were transiently transfected by electroporation (12 × 10−6 cells/500 μl Optimem, 280 V, 975 mF).
RNA Interference.
We synthesized a silent RNA (siRNA) sequence targeting SOCS3 position 80–101 relative to the start codon, and a scrambled siRNA duplex as control (Dharmacon Research). For transfection, we used 1.2 μM of the siRNA or 20 μg of the pEF-Flag-I/mSOCS3 construct. Cell migration was evaluated as described 48 h after electroporation. Transfection efficiency was controlled by evaluating SOCS3 mRNA in Northern blot.
Northern Hybridization.
Total RNA from 3-mo-old bGH Tg and non-Tg littermate mouse tissues and IM-9 cells, untreated or treated (60 min, 37°C) with 60 nM CXCL12 or 10 μg/ml GH, was extracted using Tri-reagent (Sigma-Aldrich). RNA samples were resolved on denaturing formaldehyde-agarose gels and transferred to nylon membrane (Hybond N+; Amersham Biotech). Membranes were hybridized with 32P-labeled cDNA from the pEF-Flag-I/mSOCS3 construct.
Results
CXCL12-induced SOCS3 Up-regulation Inhibits Functional CXCR4 Activity.
The human IM-9 B cell line responds to CXCL12 through CXCR4-mediated Ca2+ mobilization and cell migration (see below). As for other cell lineages (16), pretreatment with AG490, a specific JAK inhibitor, completely abrogates CXCL12-mediated responses (Fig. 1 A), suggesting JAK involvement. CXCL12 promotes JAK1 and JAK3 association with CXCR4 (Fig. 1 B), followed by their rapid tyrosine phosphorylation (Fig. 1 C). As a consequence, STAT1, -2, -3, and -5, but not STAT4 or -6, are also activated, indicated by their association to CXCR4 (not shown) and their phosphorylation pattern (Fig. 2, A and B) . As in the case of the cytokines (33), CXCL12 also promotes Ser/Thr phosphorylation of STAT. Using specific antibodies, we found that STAT3 is Ser/Thr phosphorylated after stimulation with CXCL12, and that the kinetics correlate with its tyrosine phosphorylation (Fig. 2 C); in both cases, maximum phosphorylation occurs 15 min after stimulation.
Figure 1.
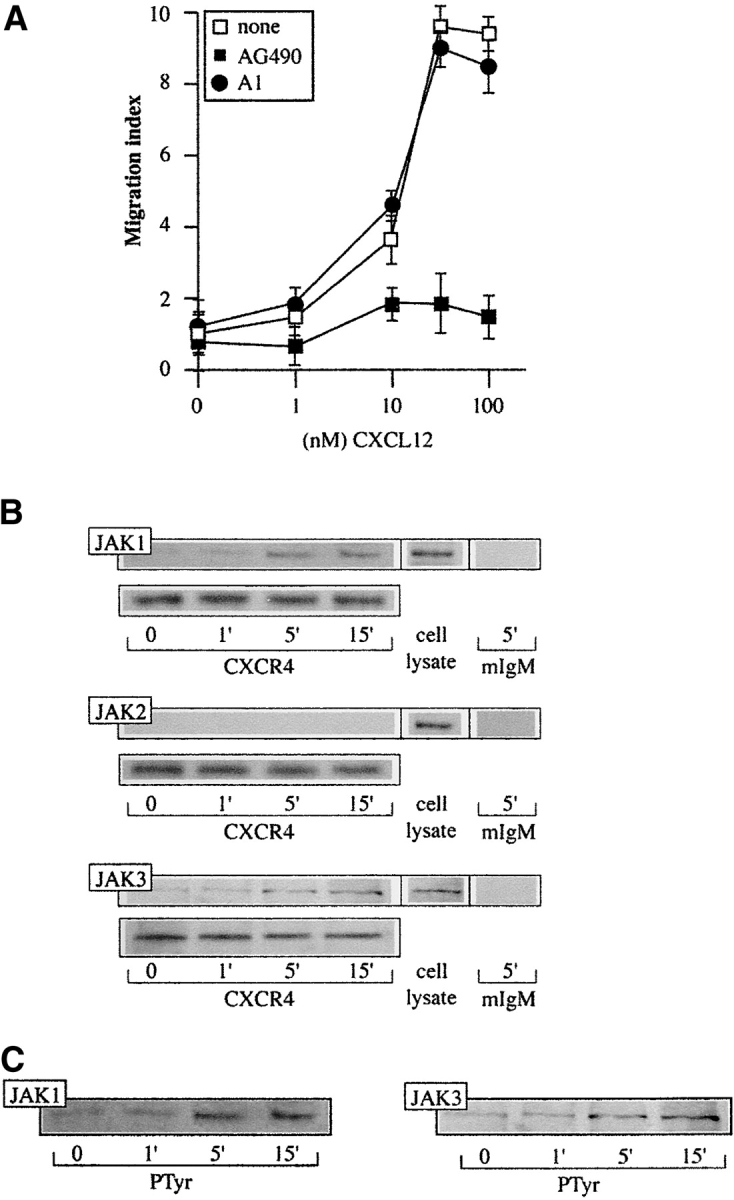
CXCL12 induces JAK activation in IM-9 cells. (A) CXCL12-induced chemotaxis of IM9 cells, untreated or pretreated with AG490 or tyrphostin A1, was assayed in transwell chemotaxis chambers. Results are expressed as a migration index, calculated as the x-fold increase over the negative control. Mean ± SD of five experiments is shown. (B) CXCL12 (50 nM)-induced IM-9 cell lysates were immunoprecipitated with anti-CXCR4 mAb or mouse IgM as a control, and tested in Western blot with anti-JAK1, -JAK2, or -JAK3 antibodies, as indicated. Equivalent receptor loading was controlled by reprobing each membrane with CXCR4–01 mAb. As a positive control, unprecipitated IM-9 cell lysates were tested in Western blot with anti-JAK antibodies. (C) CXCL12-activated IM-9 cell lysates as in B were immunoprecipitated with anti-PTyr antibodies and analyzed in Western blot with anti-JAK1 and -JAK3 antibodies. Equivalent protein loading in each lane was controlled.
Figure 2.

CXCL12 induces activation of STAT transcription factors in IM-9 cells. (A) STAT phosphorylation was determined in stimulated cell lysates immunoprecipitated with anti-Ptyr as in Fig. 1 C, and assayed in Western blot with anti-STAT3 or STAT5b antibodies. Equal protein loading was controlled. (B) Immunohistochemical analysis of IM-9 cells, untreated or treated with CXCL12, using anti-STAT3 and anti-PTyr-STAT3 antibodies. Nuclear staining is shown (insets). (C) STAT2 and STAT3 Ser/Thr phosphorylation was measured in lysates of CXCL12-activated IM-9 cells immunoprecipitated with anti-PSer/PThr and assayed in Western blot with anti-STAT2 and -3 antibodies. (D) IM-9 cells were stimulated with CXCL12 at the times indicated and nuclear extracts analyzed in Western blot using anti-STAT2 and –5b antibodies. Membranes were developed with anti-pol II antibodies to control equivalent protein loading. (E) Nuclear extracts of untreated or CXCL12-treated IM-9 cells were incubated with 32P-end-labeled wt-SIE in EMSA buffer. Where indicated, a 20-fold molar excess of unlabeled wt-SIE or mut-SIE oligonucleotide was added.
Nuclear extracts of CXCL12-stimulated IM-9 cells were analyzed in Western blot using anti-STAT antibodies, and maximum nuclear translocation was observed 30 min after activation (Fig. 2 D, top). Nuclear extracts analyzed in Western blot using anti-pol II antibody as a nuclear marker (Fig. 2 D, bottom) were free of cytoplasmic contamination, confirmed by developing with an anti–β-actin cytoplasmic marker antibody (not shown). As predicted, SIE binding activity was detected after chemokine treatment, measured by EMSA (Fig. 2 E). Protein levels in nuclear extracts were controlled using a protein detection kit. We thus concluded that CXCL12 triggers JAK/STAT pathway activation and the nuclear translocation of STAT.
Cytokine activation of the JAK/STAT pathway leads to SOCS up-regulation, implicated in turn in the negative feedback of cytokine signaling. We tested whether SOCS are up-regulated as a consequence of CXCL12-induced STAT translocation in IM-9 cells. Only SOCS3 was detected in lysates of CXCL12-stimulated IM-9 cell analyzed in Western blot using anti-SOCS antibodies (Fig. 3 A, left; maximum at 60 min). Pretreatment of IM-9 cells with the JAK kinase inhibitor AG490 (10 nM, overnight, 37°C), but not with control AG9 (not shown), abrogated CXCL12-mediated SOCS3 up-regulation (Fig. 3 A, center); nonetheless, PTx treatment (0.1 μg/ml, overnight, 37°C) had no effect (Fig. 3 A, right). These data indicate that chemokine-induced SOCS3 up-regulation is independent of Gi activation, and confirm previous results showing independence between JAK/STAT activation and the G protein pathway in chemokine signaling (23).
Figure 3.
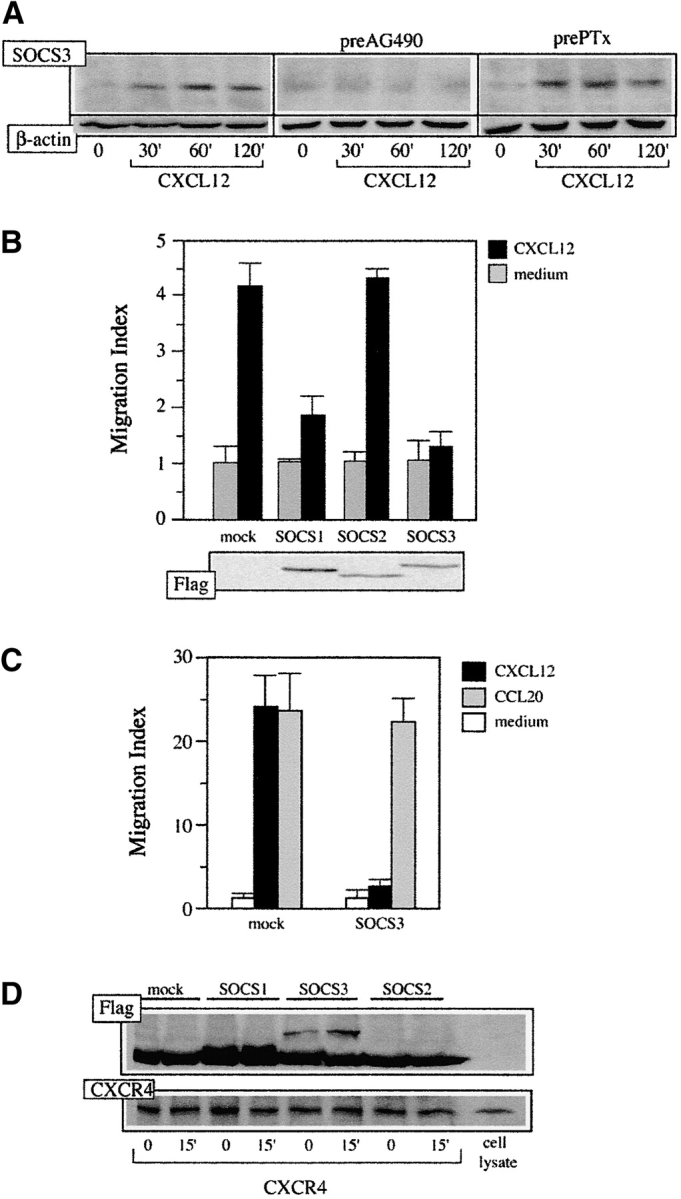
CXCL12-induced SOCS expression blocks CXCL12-induced cell responses. (A) Lysates from IM-9 cells, untreated or pretreated with AG490 or PTx and stimulated with 50 nM CXCL12, were immunoblotted with anti-SOCS3 antibody. Protein loading was controlled by reprobing the membrane with anti-β actin mAb. (B) pEF-Flag-I/mSOCS1-, mSOCS2-, and mSOCS3-transfected HEK-293 cells or mock-transfected controls were allowed to migrate in response to CXCL12. A representative experiment is shown (mean ± SD, n = 3 replicates) of five performed (top). SOCS expression in migrating cells was confirmed by Western blot of transfected cell lysates with anti-Flag antibody (bottom). (C) CCR6-BaF/3 cells were transfected with pEF-Flag-I/mSOCS3 and allowed to migrate in response to CXCL12 or CCL20. Mock-transfected cells were used as a control. A representative experiment is shown (mean ± SD, n = 3 replicates) of four performed. (D) Lysates of pEF-Flag-I/mSOCS1-, mSOCS2-, and mSOCS3-transfected HEK-293 cells or mock-transfected controls, untreated or CXCL12-stimulated, were immunoprecipitated using CXCR4–01 mAb and analyzed in Western blot with the anti-Flag mAb. As control, the membrane was reprobed using the CXCR4–01 mAb.
To evaluate the role of SOCS3 up-regulation in the control of CXCL12 responses, HEK-293 cells transiently transfected with pEF-Flag-I/mSOCS1, /mSOCS2, or /mSOCS3 constructs were allowed to migrate in response to a CXCL12 gradient. Whereas there was no influence on migration of SOCS2-expressing cells, we observed a clear reduction in the migration index in SOCS1- and SOCS3-expressing cells (Fig. 3 B, top). SOCS expression was controlled in each experiment by Western blot of cell lysates with anti-Flag antibody (Fig. 3 B, bottom). Potential toxic effects of SOCS overexpression were discarded by analyzing cell incorporation of propidium iodide in flow cytofluorometry (not shown). To test the specificity of SOCS effects on chemokine receptors, SOCS3 was overexpressed in CCR6-stably transfected BaF/3 cells (32). Whereas CXCL-12-mediated migration of these cells was completely abrogated by SOCS overexpression, migration through a CCL20 gradient was unaffected (Fig. 3 C). As a control, SOCS3 expression was determined as before in each experiment by Western blot (not shown). These data indicate that SOCS3 and SOCS1 are negative regulators of CXCL12 signaling, opening a route for cross-talk between chemokine and cytokine signals. Although SOCS3 expression does not alter CCR6-mediated migration, we cannot exclude potential control of CCR6 or other chemokine receptors by other SOCS proteins. To evaluate the mechanism involved in these SOCS effects, HEK-293 cells transiently transfected with pEF-Flag-I/mSOCS1, /mSOCS2, or /mSOCS3 constructs, untreated or CXCL12-stimulated, were lysed and cell extracts immunoprecipitated using CXCR4–01 mAb. Western blot analysis of the immunoprecipitates with an anti-Flag antibody showed that SOCS1 and SOCS3, but not SOCS2, associate to CXCR4; this association increases when cells are activated by CXCL12 (Fig. 3 D). As a protein loading control, membranes were reprobed with the CXCR4–01 mAb. The data indicate that SOCS interfere with chemokine-mediated responses by binding to their receptors, blocking JAK/STAT pathway activation. These results, and the fact that CXCL12 up-regulation of SOCS3 requires long stimulation periods, exclude SOCS involvement in CXCR4 desensitization, a rapid process involving GRK and arrestin (34).
GH Blocks CXCL12-mediated Signaling.
We examined whether cytokine-induced SOCS3 upregulation affects CXCL12 signaling. GH mediates SOCS3 up-regulation by activating the JAK2/STAT5b pathway, which modulates later effects of the hormone (35). As IM-9 cells have surface GH receptors (Fig. 4 A), we used GH as a model to evaluate time-dependent SOCS3 up-regulation. Up-regulation was observed in lysates of GH-treated IM-9 cells analyzed in Western blot using anti-SOCS antibodies, with maximum effect at 60 min of treatment (band intensity 4.9-fold greater than control values), which concurs with the period observed for other cytokines (36; Fig. 4 B). As a consequence of GH activation, SOCS3 up-regulation again has functional consequences, as Ca2+ and migratory responses to CXCL12 are seriously impaired in GH-pretreated IM-9 cells (Fig. 4, C and D), although GH does not promote Ca2+ or migratory responses. As in the case of HEK-293 cells, transient SOCS3 transfection in IM-9 cells also abolished CXCL12-mediated responses (Fig. 4 D, left). To confirm these data, we specifically silenced SOCS3 gene expression using RNA interference (37; Fig. 4 D, bottom right). Under these conditions, GH treatment does not induce SOCS3 up-regulation, and CXCL12-mediated responses are thus unaffected (Fig. 4 D, top right).
Figure 4.

GH treatment affects CXCL12-mediated responses. (A) hGHR levels were measured by flow cytometry in IM-9 cells incubated with biotin-labeled hGHR-05 mAb, followed by FITC-streptavidin. mAb binding is compared with that of a negative control (mIgM). (B) GH-activated IM-9 cell lysates were analyzed in Western blot with anti-SOCS3 antibody. Protein loading was controlled by reprobing the membrane with anti-β actin mAb. Data were quantitated using NIH software, normalized using the signal of an unrelated band, and represented as the x-fold increase compared with unstimulated cells. (C) CXCL12- or GH-induced Ca2+ mobilization in IM-9 cells, untreated or GH-pretreated, was measured in flow cytometry. (D) Untreated or GH-treated IM-9 cells were allowed to migrate in response to 50 nM CXCL12 (left). Cells that migrated to the lower chamber were counted and expressed as a migration index. Data represent the mean of triplicate determinations, with SD indicated. IM-9 cells transfected with pEF-Flag-I/mSOCS3, a siRNA sequence targeting SOCS3, or a scrambled siRNA, were left untreated or GH-treated (60 min, 37°C) and allowed to migrate in response to 50 nM CXCL12 as before (top right). To control SOCS3 silencing, mRNA was extracted from untreated or GH-treated cells previously transfected with a siRNA sequence targeting SOCS3, or with scrambled siRNA. SOCS3 mRNA was analyzed in Northern blot (bottom right).
GH treatment does not modify surface CXCR4 levels, shown by flow cytometry analysis using anti-CXCR4 mAb (Fig. 5 A). GH pretreatment also prevents CXCL12-mediated CXCR4 internalization (Fig. 5 A), indicating that GH interferes with CXCL12 signaling rather than with surface CXCR4 levels. To test the mechanism by which SOCS3 interferes with chemokine signaling in a system using nontransfected cells, anti-SOCS3 immunoprecipitates of IM-9 cells, untreated or GH-pretreated (60 min, 37°C), were analyzed in Western blot using anti-CXCR4 mAb. GH treatment up-regulates SOCS3, shown by Western blot analysis using anti-SOCS3 antibodies (Fig. 5 B, bottom), and SOCS3 binds the chemokine receptor (Fig. 5 B, top). This association increases following CXCL12 binding, indicating that the chemokine promotes a conformational change in the CXCR4 that exposes or stabilizes a SOCS3 binding site (Fig. 5 B, top). As a control, the membrane was reprobed with an anti-transferrin receptor mAb (CD71), showing specificity of the SOCS3/CXCR4 association (Fig. 5 B, center). As JAK and Gi protein activation are two of the earliest events in chemokine signaling, we analyzed CXCL12-induced association of these two proteins to CXCR4 under conditions in which SOCS3 is up-regulated. Serum-starved IM-9 cells, untreated or GH-treated, were CXCL12-stimulated at different times; cell lysates were immunoprecipitated using anti-CXCR4 antibody and immunoblotted with specific anti-Gαi (Fig. 5 C) and -JAK3 antibodies (Fig. 5 D). This experiment showed that GH-induced SOCS3 up-regulation blocks both JAK3 and Gαi association to the CXCR4. Protein loading equivalence was controlled by reprobing membranes with anti-CXCR4 antibody. The data indicate that SOCS3 affects CXCL12-mediated responses; by binding to CXCR4, SOCS3 blocks JAK/STAT pathway activation without affecting cell surface CXCR4 levels.
Figure 5.

GH treatment blocks CXCL12-mediated signaling without affecting surface CXCR4 levels. (A) Surface CXCR4 levels were analyzed in IM-9 cells treated with GH, CXCL12, or GH followed by CXCL12. CXCR4 was measured by flow cytometry with CXCR4–01 mAb and PE-anti–mouse IgM, and is expressed as a percentage of receptor in untreated cells (none) with SD indicated. (B) Lysates of CXCL12-activated IM-9 cells, untreated (medium) or pretreated for 60 min with GH (10 μg/ml) were immunoprecipitated with anti-SOCS3 antibody and the Western blot developed with anti-CXCR4 mAb, anti-transferrin receptor (CD71), or anti-SOCS3 antibody. CXCR4 and CD71 protein expression was controlled in unprecipitated IM-9 cell lysates with the corresponding antibody. (C) Lysates of CXCL12-activated IM-9 cells, untreated or GH-treated, were immunoprecipitated with CXCR4–01 or control mIgM mAb and the Western blot developed with anti-Gαi antibody. Unprecipitated IM-9 cell lysates were controlled with anti-Gαi antibody. To verify equal protein loading, the membrane was reprobed with CXCR4–01 mAb. (D) Cell lysates treated and precipitated as in C were immunoblotted with anti-JAK3 antibody. Unprecipitated IM-9 cell lysates were controlled with anti-JAK3 antibody; equal protein loading was verified as in C.
SOCS3 Blocks CXCL12-mediated Responses In Vivo.
We then explored whether CXCL12-mediated responses are affected in cells that coexpress GHR and CXCR4, obtained from transgenic mice expressing a fusion gene coding for bovine GH (bGH-Tg; reference 38). It was previously determined that B cell numbers in bGH-Tg mouse spleen increased two- to fourfold, with a lesser increase in T cells. Some differences were also found in peripheral lymph nodes, in which B cells increased fivefold in bGH-Tg mice. Analysis of bone marrow cells showed that bGH-Tg mice are deficient in pre–B cells, whereas B cell numbers are preserved (38). CXCL12 was injected into the peritoneal cavity of bGH-Tg mice and control littermates; after 6 h, migrating cells were recovered and analyzed by flow cytometry using specific cell markers. In CXCL12-injected bGH-Tg mice, B cell and granulocyte/macrophage recovery is impaired compared with wild-type mice, shown by double staining with B220/CD45 and CD11b/CD45, respectively (Fig. 6 A). In contrast, T lymphocyte recovery is unimpaired, shown by CD3/CD45 double staining (not shown).
Figure 6.


bGH-Tg mice have an impaired chemotactic response to CXCL12. (A) CXCL12 (1 μM) was injected intraperitoneally in bGH-Tg mice or control littermates. After 60 min, cells that had migrated into the peritoneal cavity were recovered and the populations characterized by flow cytometry using specific cell markers. The figure shows specific staining of b220/CD45 and CD11b/CD45 cells; the percentage of each population is also indicated. Results are shown of one representative experiment of five performed. (B) CXCL12-mediated cell migration of spleen, lymph node, and bone marrow cells from bGH-Tg mice or wild-type littermates were evaluated in 24-well transmigration chambers; migration was calculated as in Fig. 1 A. Data represent the mean ± SD of triplicate determinations. (C) Northern analysis of SOCS3 mRNA from spleen, lymph node, and bone marrow of bGH-Tg mice compared with control littermates. As a control, the membrane was rehybridized with eF1α. (D) mRNA was extracted from bone marrow cells of bGH-Tg mice and control littermates, undepleted or GH-depleted in vitro, and SOCS3 analyzed in Northern blot. Loading was controlled as in C. (E) Bone marrow cells from mice as in D, undepleted (open symbols) or GH-depleted (filled symbols), were allowed to migrate in response to CXCL12. Migration was calculated as in Fig. 1 D. Data represent the mean ± SD of triplicate determinations.
The percentage of B220lowCD45+ cells recovered from the peritoneal cavity of wild-type mice is markedly increased after CXCL12 treatment (5 ± 0.3-fold) compared with PBS-treated controls. In contrast, no increase was observed in this cell population in CXCL12-treated bGH-Tg mice (0.73 ± 0.06-fold). Analysis showed that CD11b+ CD45+ cells also accumulate in the peritoneal cavity of CXCL12-treated wild-type mice (3.8 ± 0.2-fold), whereas moderate migration was observed in treated bGH-Tg mice (2.07 ± 0.2-fold). No notable differences were found for B220highCD45+ cells from CXCL12-treated bGH-Tg mice (2.8 ± 0.2-fold) and wild-type mice (3.2 ± 0.3-fold; Fig. 6 A). A similar effect is observed when spleen, lymph node, and bone marrow cells from 3-mo-old bGH-Tg mice are allowed to migrate in vitro toward a CXCL12 gradient. Whereas control non-Tg mouse cells respond normally to CXCL12, bGH-Tg cells showed a marked decrease in migration (Fig. 6 B). Northern blot analysis showed that bGH-Tg mouse spleen, lymph node, and bone marrow cells express higher SOCS3 levels than those from non-Tg littermates (Fig. 6 C). When SOCS3 levels were reduced by in vitro depletion (120 min) of bone marrow cells from bGH-Tg mice in GH-free medium (Fig. 6 D), their migratory response to CXCL12 was restored compared with undepleted cells (Fig. 6 E), indicating that SOCS3 has a critical role in chemokine response regulation. All together, these results assign a direct role to the SOCS proteins in controlling chemotactic responses to CXCL12 and expose new aspects of the relationship between cytokines and chemokines in immune response control.
Discussion
Functional lymphoid microenvironments, organogenesis, and leukocyte patrolling are established by cell migration. This requires the integrated action of cell surface ligands such as integrins and selectins, with soluble mediators including cytokines, chemokines, and their receptors. Whereas the former group participates in rolling and cell adhesion, the latter has a key role in the lymphohematopoiesis and cell polarization required for cell motility (39). In addition to their selective role under physiological conditions, chemokines also have an important function in inflammation, wound healing, and angiogenesis (6).
In recent years it has been established that hematopoietic cell growth, differentiation, and function are controlled by the coordinated action of the cytokine–chemokine network (40). After cytokine binding to its receptor on the cell surface, receptor oligomerization takes place, inducing JAK kinase activation. The activated JAK kinases then phosphorylate the cytokine receptors, leading to recruitment and subsequent activation of other signaling molecules, such as the STAT family proteins. Activated STAT proteins translocate to the nucleus and mediate transcription of a range of cytokine-responsive genes (22), including those that code for the SOCS proteins. These molecules have recently attracted interest, as they exercise their effect directly on the JAK/STAT pathway. Indeed, the majority of cytokines analyzed to date, such as LIF, IL-2, IL-3, IL-6, GH, IFN-γ, and leptin, induce several SOCS family members in a tissue-specific manner (28).
Much like cytokines, the chemokines trigger oligomerization of their receptors and activation of the JAK/STAT pathway (31, 41, 42). The similarity among chemokine receptors, including conservation of the DRY motif, suggests that oligomerization and JAK/STAT pathway activation are not exclusive to CCR2, in which they were first described (20, 23); CCR5 and CXCR4 both activate several JAK/STAT family members in a cell lineage–dependent fashion. Agonist-induced dimerization has been described for other GPCR receptors, including the β2-adrenergic (43), opioid (44), and γ-amino butyric acid (GABA) receptors (45); furthermore, the angiotensin II (46), TSH (47), and α-melanocyte-stimulating hormone (α-MSH) receptors (48) not only dimerize but also activate the JAK/STAT pathway.
As described for cytokines (25, 26), CXCL12-mediated STAT activation and nuclear translocation promote SOCS3 protein up-regulation. By binding to the CXCR4, overexpressed SOCS1 or SOCS3 proteins prevent a chemotactic response to CXCL12 in HEK-293 cells. This is comparable to previous reports of SOCS3 association to other transmembrane receptors (49–51); here, the consequence is blockade of JAK and Gi association to CXCR4 and impaired chemotactic responses. CXCL12-mediated SOCS3 up-regulation depends directly on JAK/STAT pathway activation, as AG490 treatment completely eliminates SOCS3 in lysates of CXCL12-stimulated cells. PTx treatment does not affect SOCS3 up-regulation, however, indicating that JAK/STAT activation is G protein pathway independent. These data, together with the observation that both PTx and AG490 treatments block CXCL12-mediated responses, suggest that JAK activation is a very early event in chemokine signaling. The data confirm our previous finding that AG490 treatment of Mono Mac-1 cells inhibits JAK and Gi association to the CCR2b receptor after CCL2 binding, whereas PTx does not affect JAK association (23). The time course of CXCL12-mediated SOCS3 up-regulation excludes the role of these suppressor molecules in the rapid ligand-triggered desensitization of chemokine receptors, which requires GRK and arrestin recruitment (34).
SOCS blockade of chemokine responses may thus be environmentally influenced, as the presence of other chemokines and/or cytokines alters these responses. Although more exhaustive study is required, preliminary data show that, as for cytokines (28), SOCS3 blockage selectively affects the responses of certain chemokines without interfering with others. We nonetheless cannot exclude the involvement of other SOCS family members in the control of chemokine signals, in which case the specificity of the effect would depend on differential SOCS protein expression in diverse cell types. A distinct SOCS expression pattern has been described that correlates with differentiation into Th1 or Th2 phenotype (52).
GH belongs to the cytokine family, which also comprises placental lactogen and prolactin; its biological effects vary widely, and include skeletal growth during childhood and regulation of a variety of anabolic processes in adult life. Lymphocytes also have GH receptors, as defined by biochemical, molecular, and functional evidence, and GH has been implicated as a growth and differentiation factor in the hematopoietic system (53). After binding GH, the receptor dimerizes and signals through JAK2 kinase (35). This pathway includes tyrosine phosphorylation of several proteins, among them the latent cytoplasmic transcription factors, STATs. This activation leads to upregulation of a variety of genes in vivo and in vitro, including SOCS2, SOCS3, and CIS mRNA (54). SOCS inhibit receptor signaling to STAT5b via phosphotyrosine-dependent binding interactions with the tyrosine kinase JAK2 (SOCS1) and/or the cytoplasmic tail of GHR (CIS and SOCS3; reference 55).
Given the ability of both GH and CXCL12 to stimulate the JAK/STAT pathway and to upregulate SOCS3, we studied the relationship between chemokines and cytokines using CXCL12 and GH as a model. We show that the IM-9 cell line does not migrate in response to GH, but does so in response to CXCL12, a process that is blocked by pretreatment with the JAK2-specific inhibitor AG490. When cells were pretreated with GH under conditions that up-regulate SOCS3, cell migration to a CXCL12 gradient was impaired; when SOCS3 was downregulated or its expression silenced by RNA interference (37), however, the chemotactic response was recovered. These data concur with the absence of a chemotactic response to CXCL12 in SOCS3-transfected HEK-293 cells. GH does not affect membrane CXCR4 levels, shown by anti-CXCR4 staining of GH-treated cells. SOCS3 is nonetheless associated to CXCR4 under conditions in which it is up-regulated by GH treatment of IM9 cells. This association increases after CXCL12 stimulation, indicating that the ligand may promote conformational changes in the CXCR4, which then exposes the SOCS3 binding site or stabilizes the complex. As a consequence, neither JAK nor Gi associates to the CXCR4, blocking chemotactic responses; a model is outlined in Fig. 7 . The data thus indicate that CXCL12 is able to activate CXCR4 under conditions in which SOCS3 is up-regulated, not only in vitro but also in vivo, as observed in bGH-Tg mice. These mice, which have altered hematopoiesis, show a defective CXCR4 response in cells expressing the GH receptor, compatible with the high SOCS3 levels in these cells. As predicted, similar cell populations in wild-type littermates show no SOCS3 up-regulation and thus have normal responses to CXCL12. Furthermore, when bGH-Tg mouse cells are depleted of growth factor in vitro, SOCS3 levels diminish and CXCL12-mediated responses are restored.
Figure 7.

Model for cytokine intervention in chemokine responses. Through interaction with its receptor (GHR), GH induces JAK/STAT pathway activation, resulting in STAT nuclear translocation and SOCS3 up-regulation. SOCS3 binds to the chemokine receptor (CXCR4); this interaction is facilitated by receptor dimerization induced by ligand (CXCL12) binding. As a consequence, chemokine-mediated activation of JAK/STAT and Gi pathways are impaired and chemokine responses are blocked.
Identification of the chemokine-activated JAK/STAT/SOCS pathway has opened a new avenue in signal transduction research, by integrating this pathway with those of cytokine signaling. It is now of interest to identify the cytokine/chemokine combination and the SOCS expression pattern required by each lineage during development, as well as those of cells mobilized in normal immune responses and the inflammatory response. In vivo experiments indicate a possible role for such interactions, as neutrophils purified from acromegalic or hyperprolactinemic individuals show decreased in vitro chemotaxis to an N-formylmethionyl-phenylalanine gradient (56). Chemokine response defects may also collaborate in the reduced B cell lymphopoiesis, reduced myelopoiesis in fetal liver, and virtual absence of myelopoiesis in bone marrow described in bGH-Tg mice (38). These in vivo and in vitro studies could thus lead to the development of specific pharmacological inhibitors of cytokine and chemokine signaling that are able to interfere with inflammatory responses.
Acknowledgments
We thank Drs. T. Willson, R. Starr, and D. Hilton (Walter and Eliza Hall Institute of Medical Research) for the gift of SOCS expression vectors, Dr. J.P. García-Ruiz and E. Delgado for bovine GH transgenic mice, Dr. L. Kremer for the gift of CCR6-transfected BaF/3 cells, J. Palacín and L. Gómez for expert animal care, Ma C. Moreno-Ortíz and Dr. I. López-Vidiero for help with FACS® analysis, and C. Bastos and C. Mark for secretarial and editorial assistance, respectively.
CEC-P is supported by the Brazilian Coordenaçao de Aperfeiçoamento de Pessoal de Nivel Superior Foundation and by the Universidade Federal Fluminense, Niteroi-Rio de Janeiro, Brazil. This work was partially supported by grants from the Spanish CICyT and the Comunidad de Madrid. The Department of Immunology and Oncology was founded and is supported by the Spanish Council for Scientific Research (CSIC) and by the Pharmacia Corporation.
Footnotes
Abbreviations used in this paper: CXCR, CXC chemokine receptor; EMSA, electrophoretic mobility shift assay; GH, growth hormone; JAK, Janus kinase; RT, room temperature; SIE, sis-inducible element; siRNA, silent RNA; SOCS, suppressors of cytokine signaling; STAT, signal transducers and transactivators of transcription; Tg, transgenic.
References
- 1.Baggiolini, M., B. Dewald, and B Moser. 1997. Human chemokines: an update. Ann. Rev. Immunol. 15:675–705. [DOI] [PubMed] [Google Scholar]
- 2.Rollins, B.J. 1997. Chemokines. Blood. 90:909–928. [PubMed] [Google Scholar]
- 3.Mackay, C.R. 2001. Chemokine: immunology's high impact factors. Nat. Immunol. 2:95–101. [DOI] [PubMed] [Google Scholar]
- 4.Schall, T.J., and K.B Bacon. 1994. Chemokines, leukocyte trafficking and inflammation. Curr. Opin. Immunol. 6:865–873. [DOI] [PubMed] [Google Scholar]
- 5.Rossi, D., and A. Zlotnik. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242. [DOI] [PubMed] [Google Scholar]
- 6.Gerard, C., and B.J. Rollins. 2001. Chemokines and diseases. Nat. Immunol. 2:108–115. [DOI] [PubMed] [Google Scholar]
- 7.Moser, B., and P. Loetscher. 2001. Lymphocyte traffic control by chemokines. Nat. Immunol. 2:123–128. [DOI] [PubMed] [Google Scholar]
- 8.Luther, S.A., and J.G. Cyster. 2001. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2:102–107. [DOI] [PubMed] [Google Scholar]
- 9.Bleul, C.C., R.C. Fuhlbrigge, J.M. Casasnovas, A. Aiuti, and T.A. Springer. 1996. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 SDF-1. J. Exp. Med. 184:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagasawa, T., S. Hirota, K. Tachibana, N. Takakura, S. Nishikawa, Y. Kitamura, N. Yoshida, H. Kikutani, and T. Kishimoto. 1996. Defects of B cell lymphopoiesis and bone marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 382:635–638. [DOI] [PubMed] [Google Scholar]
- 11.Aiuti, A., I.J. Webb, C. Bleul, T.A. Springer, and J.C. Gutierrez-Ramos. 1997. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 185:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Apuzzo, M., A. Rolink, M. Loetscher, J.A. Hoxie, I. Clark-Lewis, F. Melchers, M. Baggiolini, and B. Moser. 1997. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 27:1788–1793. [DOI] [PubMed] [Google Scholar]
- 13.Zou, Y.-R., A.H. Kottmann, M. Kuroda, I. Taniuchi, and D.R. Littman. 1998. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 393:595–599. [DOI] [PubMed] [Google Scholar]
- 14.Tachibana, K., S. Hirota, H. Iizasa, H. Yoshida, K. Kawabata, Y. Kataoka, Y. Kitamura, K. Matsushima, N. Yoshida, S. Nishikawa, et al. 1998. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 393:591–594. [DOI] [PubMed] [Google Scholar]
- 15.Ma, Q., D. Jones, P.R. Borghesani, RA. Segal, T. Nagasawa, T. Kishimoto, R.T. Bronson, and T.A. Springer. 1998. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA. 95:9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vila-Coro, A.J., J.M. Rodríguez-Frade, A.M. Martín de Ana, M.C. Moreno-Ortíz, C. Martínez-A., and M. Mellado. 1999. The chemokine SDF-1α triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 13:1699–1710. [PubMed] [Google Scholar]
- 17.Wang, J.F., I.W. Park, and J.E. Groopman. 2000. Stromal cell-derived factor-1α stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide-3 kinase and protein kinase C. Blood. 95:2505–2513. [PubMed] [Google Scholar]
- 18.Ganju, R.K., S.A. Brubaker, J. Meyer, P. Dutt, Y. Yang, S. Qin, W. Newman, and J.E. Groopman. 1998. The α-chemokine, stromal cell-derived factor-1α, binds to the transmembrane G-protein-coupled CXCR4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 273:23169–23175. [DOI] [PubMed] [Google Scholar]
- 19.Vicente-Manzanares, M., M. Rey, D.R. Jones, D. Sancho, M. Mellado, J.M. Rodriguez-Frade, M.A. del Pozo, M. Yañez-Mó, A. Martín de Ana, C. Martínez-A., et al. 1999. Involvement of phosphatidylinositol 3-kinase in stromal cell-derived factor-1α-induced lymphocyte polarization and chemotaxis. J. Immunol. 163:4001–4012. [PubMed] [Google Scholar]
- 20.Rodríguez-Frade, J.M., A. Vila-Coro, A.M. Martín de Ana, J.P. Albar, C. Martínez-A., and M. Mellado. 1999. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc. Natl. Acad. Sci. USA. 96:3628–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodríguez-Frade, J.M., A.J. Vila-Coro, A.M. Martín de Ana, M. Nieto, F. Sánchez-Madrid, A.E.I. Proudfoot, T.N.C. Wells, C. Martínez-A., and M. Mellado. 1999. Similarities and differences in RANTES- and (AOP)-RANTES-triggered signals: implications for chemotaxis. J. Cell Biol. 144:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonard, W.J., and J.J. O'Shea. 1998. JAK and STATs: biological implications. Annu. Rev. Immunol. 16:293–322. [DOI] [PubMed] [Google Scholar]
- 23.Mellado, M., J.M. Rodríguez-Frade, A.M. Aragay, G. del Real, A. Vila-Coro, A. Martin de Ana, A. Serrano, F. Mayor, Jr., and C. Martínez-A. 1998. The chemokine MCP-1 triggers tyrosine phosphorylation of the CCR2B receptor and the JAK2/STAT3 pathway. J. Immunol. 161:805–813. [PubMed] [Google Scholar]
- 24.Yoshimura, A., T. Ohkubo, T. Kiguchi, N.A. Jenkins, D.J. Gilbert, N.G. Copeland, T. Hara, and A. Miyajima. 1995. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14:2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starr, R., T.A. Willson, E.M. Viney, L.J. Murray, J.R. Rayner, B.J. Jenkins, T.J. Gonda, W.S. Alexander, D. Metcalf, N.A. Nicola, and D.J. Hilton. 1997. A family of cytokine-inducible inhibitors of signalling. Nature. 387:917–921. [DOI] [PubMed] [Google Scholar]
- 26.Endo, T.A., M. Masuhara, M. Yokouchi, R. Suzuki, H. Sakamoto, K. Mitsui, A. Matsumoto, S. Tanimura, M. Ohtsubo, H. Misawa, et al. 1997. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 387:921–924. [DOI] [PubMed] [Google Scholar]
- 27.Naka, T., M. Narazaki, M. Hirata, T. Matsumoto, S. Minamoto, A. Aono, N. Nishimoto, T. Kajita, T. Taga, K. Yoshizaki, et al. 1997. Structure and function of a new STAT-induced STAT inhibitor. Nature. 387:924–929. [DOI] [PubMed] [Google Scholar]
- 28.Chen, X.P., J.A. Losman, and P. Rothman. 2000. SOCS proteins, regulators of intracellular signaling. Immunity. 13:287–290. [DOI] [PubMed] [Google Scholar]
- 29.Dey, B.R., R.W. Furlanetto, and P. Nissley. 2000. Suppressor of cytokine signaling (SOCS-3) protein interacts with the insulin-like growth factor-I receptor. Biochem. Biophys. Res. Commun. 278:38–43. [DOI] [PubMed] [Google Scholar]
- 30.Mellado, M., J.M. Rodríguez-Frade, L. Kremer, and C. Martínez-A. 1996. Characterization of monoclonal antibodies which specific for the human growth hormone 22K and 20K isoforms. J. Clin. Endocrinol. Metab. 81:1613–1618. [DOI] [PubMed] [Google Scholar]
- 31.Mellado, M., J.M. Rodríguez-Frade, S. Mañes, and C. Martínez-A. 2001. Chemokine signaling and functional responses: The role of receptor dimerization and TK pathway activation. Annu. Rev. Immunol. 19:397–421. [DOI] [PubMed] [Google Scholar]
- 32.Kremer, L., L. Carramolino, I. Goya, A. Zaballos, J. Gutierrez, M.C. Moreno-Ortiz, C. Martínez-A., and G. Márquez. 2001. The transient expression of C-C chemokine receptor 8 in thymus identifies a thymocyte subset committed to become CD4+ single-positive T cells. J. Immunol. 166:218–225. [DOI] [PubMed] [Google Scholar]
- 33.Decker, T., and P. Kovarik. 2000. Serine phosphorylation of STATs. Oncogene. 19:2628–2637. [DOI] [PubMed] [Google Scholar]
- 34.Orsini, M.J., J.L. Parent, S.J. Mundell, J.L. Benovic, and A. Marchese. 1999. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J. Biol. Chem. 274:31076–31086. [DOI] [PubMed] [Google Scholar]
- 35.Herrington, J., and C. Carter-Su. 2001. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol. Metab. 12:252–257. [DOI] [PubMed] [Google Scholar]
- 36.Zhang, J.G., A. Farley, S.E. Nicholson, T.A. Willson, L.M. Zugaro, R.J. Simpson, R.L. Moritz, D. Cary, R. Richardson, G. Hausmann, et al. 1999. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA. 96:2071–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp, P.A. 2001. RNA interference. Genes Dev. 15:485–490. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalo, J.A., R. Mazuchelli, M. Mellado, J.M.R. Frade, A.C. Carrera, C. von Kobbe, I. Mérida, and C. Martínez-A. 1996. Enterotoxin septic shock protection and deficient T helper 2 cytokine production in growth hormone transgenic mice. J. Immunol. 157:3298–3304. [PubMed] [Google Scholar]
- 39.von Andrian, U.H., and C.R. Mackay. 2000. T cell function and migration: two sides of the same coin. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 40.Youn, B.S., C. Mantel, and H.E. Broxmeyer. 2000. Chemokines, chemokine receptors and hematopoiesis. Immunol. Rev. 177:150–174. [DOI] [PubMed] [Google Scholar]
- 41.Mellado, M., J.M. Rodríguez-Frade, A.J. Vila-Coro, S. Fernández, A.M. Martín de Ana, D.R. Jones, J.L. Torán, and C. Martínez-A. 2001. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 20:2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodríguez-Frade, J.M., M. Mellado, and C. Martínez-A. 2001. Chemokine receptor dimerization in cell signaling: two are better than one. Trends Immunol. 22:612–617. [DOI] [PubMed] [Google Scholar]
- 43.Hebert, T.E., S. Moffett, J.-P. Morello, T.P. Loisel, D.G. Bichet, C. Barret, and M. Bouvier. 1996. A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 271:28612–28616. [DOI] [PubMed] [Google Scholar]
- 44.Cvejic, S., and L.A. Devi. 1997. Dimerization of the delta opioid receptor: implication for a role in receptor internalization. J. Biol. Chem. 272:26959–26964. [DOI] [PubMed] [Google Scholar]
- 45.Kuner, R., G. Köhr, S. Grünewald, G. Eisenhardt, A. Bah, and H.-C. Kornau. 1999. Role of heteromer formation in GABAB receptor function. Science. 283:74–77. [DOI] [PubMed] [Google Scholar]
- 46.Marrero, M.B., B. Schieffer, W.G. Paxton, L.H. Eerdt, B.C. Berk, P. Delafontaine, and K.E. Bernstein. 1995. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature. 375:247–250. [DOI] [PubMed] [Google Scholar]
- 47.Park, E.S., H. Kim, J.M. Suh, S.J. Park, S.H. You, H.K. Chung, K.W. Lee, O.Y. Kwon, B.Y. Cho, Y.K. Kim, et al. 2000. Involvement of JAK/STAT (Janus kinase/signal transducer and activator of transcription) in the thyrotropin signaling pathway. Mol. Endocrinol. 114:662–670. [DOI] [PubMed] [Google Scholar]
- 48.Buggy, J.J. 1998. Binding of a melanocyte-stimulating hormone to its G protein-coupled receptor on B lymphocytes activates the JAK/STAT pathway. Biochem. J. 331:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emanuelli, B., P. Peraldi, C. Filloux, D. Sawka-Verhelle, D. Hilton, and E. Van Obberghen. 2000. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 275:15985–15991. [DOI] [PubMed] [Google Scholar]
- 50.Pezet, A., H. Favre, P.A. Kelly, and M. Edery. 1999. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J. Biol. Chem. 274:24497–24502. [DOI] [PubMed] [Google Scholar]
- 51.Sporri, B., P.E. Kovanen, A. Sasaki, A. Yoshimura, and W.J. Leonard. 2001. JAB/SOCS1/SS-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. 97:221–226. [DOI] [PubMed] [Google Scholar]
- 52.Egwuagu, C.E., C.R. Yu, M. Zhang, R.M. Mahdi, S.J. Kim, and I. Gery. 2002. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and th2 cells: implications for th cell lineage commitment and maintenance. J. Immunol. 168:3181–3187. [DOI] [PubMed] [Google Scholar]
- 53.Dorshkind, K., and N.D. Horseman. 2000. The roles of prolactin, growth hormone, insulin-like growth factor-I and thyroid hormones in lymphocyte development and function: insights from genetic models of hormone and hormone receptor deficiency. Endocr. Rev. 21:292–312. [DOI] [PubMed] [Google Scholar]
- 54.Tollet-Egnell, P., A. Flores-Morales, A. Stavreus-Evers, L. Sahlin, and G. Norstedt. 1999. Growth hormone regulation of SOCS-2, SOCS-3, and CIS messenger ribonucleic acid expression in the rat. Endocrinology. 140:3693–3704. [DOI] [PubMed] [Google Scholar]
- 55.Ram, P.A., and D.J. Waxman. 2000. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J. Biol. Chem. 275:39487–39496. [DOI] [PubMed] [Google Scholar]
- 56.Fornari, C., F.M. Palacios, R.A. Diez, and A.D. Intebi. 1994. Decreased chemotaxis of neutrophils in acromegaly and hyperprolactinemia. Eur. J. Endocrinol. 130:463–468. [DOI] [PubMed] [Google Scholar]