

Abstract
Nonobese diabetic (NOD) mice develop spontaneous autoimmune diabetes that results from the destruction of insulin secreting β cells by diabetogenic T cells. The time and location of the encounter of autoantigen(s) by naive autoreactive T cells in normal NOD mice are still elusive. To address these issues, we analyzed diabetes development in mice whose spleen or pancreatic lymph nodes (panLNs) had been removed. Excision of panLNs (panLNx) at 3 wk protected mice against insulin autoantibodies (IAAs), insulitis, and diabetes development almost completely, but had no effect when performed at 10 wk. The protection afforded by panLNx at weaning was not due to modifications of the immune system, the absence of autoreactive T cells, or the increase in the potency of regulatory T cells. That panLNs are dispensable during adult life was confirmed by the capacity of 10-wk-old panLNx irradiated recipients to develop diabetes upon transfer of diabetogenic T cells. In contrast, splenectomy had no effect at any age. Partial excision of mesenteric LN at 3 wk did not prevent accelerated diabetes by cyclophosphamide as panLNx did. Thus, in normal NOD mice, autoreactive T cell initial priming occurs in LNs draining the target organ of the disease from 3 wk of age.
Keywords: insulin-dependent diabetes mellitus, lymph node excision, splenectomy, T cell activation, autoimmunity
Introduction
Nonobese diabetic (NOD)* mice develop spontaneous autoimmune diabetes that shares many features with human type 1 diabetes. The disease results from the destruction of insulin secreting β cells mediated by autoreactive T cells. Several findings have suggested that the development of the disease comprises several stages. Until around 3–4 wk of age all the islets are free from lymphocytic infiltration, suggesting that the thymus-derived autoreactive T cells remain naive during this period. From 3–4 wk of age the first signs of a nondestructive insulitis appear, indicating that activation and proliferation of self-reactive T cells is initiated. This age is also critical for the establishment/maintenance of a transient dominant tolerance by protective T cells that prevent β cell attack. The breaking down of self-tolerance, a consequence of a disequilibrium between regulatory and effector T cells, occurs from around 12 wk of age and results in β cell destruction and diabetes onset (1, 2). A key event in the development of the disease is the activation of naive autoreactive T cells by APCs bearing diabetogenic Ag: when and where does this activation occur? Regarding the site of the initial priming of naive anti-β cell T cells, three possibilities can be envisaged. First, the presence of APCs loaded with diabetogenic peptides inside the islets may indicate that Ag presentation takes place inside the islets (3). It can be objected that naive T cells essentially migrate into secondary lymphoid organs, whereas activated T cells preferentially home to nonlymphoid tissues (4–6). In this line, we showed that upon transfer into NOD-scid recipients only activated/memory CD62L− T cells enter inside the islets. However, some naive CD62L+ T cells from young animals migrate to the periphery of the islets (7) where macrophages and dendritic cells are normally present and may exert antigen-presenting function (8). Second, the spleen provides an appropriate environment for the priming of T cells by APCs presenting blood-borne Ags. Such Ags may be islet Ags themselves, as shown after β cell injury (3) or molecules from diverse pathogens that cross-react with diabetogenic Ags. Deduced from what is known on T cell activation by locally introduced foreign Ag, priming of naive autoreactive T cells in the LN draining the pancreas is a third possibility. Transfer of unmanipulated dendritic cells from young NOD mice did prevent diabetes only if they were originating from pancreatic LNs (panLNs), possibly by altering the balance between regulatory and effector T cells (9). This indicates that APCs from panLN and not from other LN bear peptides related to the disease. Activation of autoaggressive cells was also shown to occur in the panLN of mice with β cells expressing the nuclear SV40T Ag (10) and in NOD mice transgenic for a diabetogenic TCR (BDC2.5/NOD; reference 11).
In BDC2.5/NOD, ∼90% of the T cells are CD4+ Tg cells and are easily detectable, whereas in normal NOD autoreactive T cells mediating the disease are rare cells belonging to the CD4 and CD8 lineages and cannot be phenotypically analyzed. Expression of autoimmunity requires activation of the self-reactive T cells, and thus the onset of diabetes in regular NOD mice, the rapid transfer of diabetes in immunosuppressed animals by spleen T cells (1), and the acceleration of the disease in young animals by cyclophosphamide (12) reveal the presence of primed anti-β T cells. Therefore, to gain information on the site and time of initial activation of β cell reactive T cells in normal NOD mice, diabetes development was analyzed after excision of the spleen and the panLN at different ages. Our results showed that initial activation of anti-β cell T cells occurs around 3 wk of age in panLN.
Materials and Methods
Mice.
NOD-Nk, NOD-NON-Thy1a, NOD.scid, BDC2.5/NOD, BDC2.5 Cα0/0, and C57BL/6 mice were bred in our own facilities under specific pathogen-free conditions. BDC2.5/NOD and BDC2.5 Cα0/0 mice were a gift from D. Mathis and C. Benoist (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Strasbourg, France).
The cumulative incidence of diabetes in our NOD-Nk and BDC2.5/NOD colonies reaches 80 and 30% in females and 40 and 20% in males, respectively, by 8 mo of age. In the BDC2.5 Cα0/0 colony, diabetes developed in 75% of the mice between 3 and 5 wk of age.
Cell Preparation.
Single cell suspensions of spleen, peripheral, pancreatic, periaortic, and mesenteric LNs (perLNs, panLNs, PALNs, and MLNs, respectively) were prepared in RPMI 1640 using a homogenizer. After one wash, cell viability was determined by trypan blue exclusion.
Antibodies.
The following mAbs were used purified or conjugated to biotin or FITC: anti-Thy 1.1 (HO.22), anti-Thy-1 (42–21), anti-Thy 1.2 (30-H.12), anti-CD44 (IM-7), and anti-CD62L (MEL-14). PE–anti-B220 (RA3–6B2), –anti-CD62L, –anti-CD8 and –streptavidin, and APC–anti-CD4 were purchased from BD Biosciences. Extravidin-FITC was from Sigma-Aldrich.
Immunofluorescence Staining.
Cells were pelleted in 96-well plates and stained with 20 μl of medium (PBS supplemented with 2% FCS and 5 mM sodium azide) or biotin-, PE-, or FITC-labeled antibodies at an appropriate concentration 30 min on ice, followed by Extravidin-FITC or PE-streptavidin. Cells were then washed twice and resuspended in PBS containing 1% formaldehyde. Annexin-V–FITC/propidium iodide (PI) staining to detect apoptotic cells was performed using an apoptosis detection kit from Beckman Coulter. Flow cytometric analysis was performed using a FACScan™ and CELLQuest™ software (Becton Dickinson). Data were collected from analysis of 5,000–10,000 cells.
Surgical Excision of panLN (panLNx), MLN (MLNx), and Splenectomy (Sx).
Mice were anesthetized (0.01 ml/g body weight of a 2.5% solution of Avertin [Aldrich Chimie], intraperitoneally) and surgery was performed under a dissection microscope. The animal was placed on its right flank and a dorsoventral incision in the skin and muscles was made over the spleen. Lateral retraction of the spleen, the pancreas, the stomach, and the intestines allowed clearing the cranial side of the kidney. The two panLNs were exposed by careful blunt dissection. The vessels emerging from the hilus of each LN were grasped with two pairs of forceps and crushed. The LN was excised by gentle tractions of the forceps nearest the node. The proximal forceps were maintained a few seconds before being released. Incisions were closed with wound clips.
To remove the posterior end of the MLN, a ventral incision was made. The intestine loops were gently reflected to the right to expose the mesentery. The small posterior node of the mesenteric chain was excised as described for panLNs.
To splenectomize mice, the spleen was exposed, gently lifted on the tips of curved forceps, and blood vessels were sectioned with an ophthalmic cautery (Moria).
Sham-operated animals were anesthetized and their abdomen incised and closed as described.
Migration of Lymphocytes in Lymphnodectomized (panLNx) Mice.
Splenocytes from congenic NOD-NON-Thy1a mice (9-wk-old) containing 30 × 106 T cells were injected intravenously to NOD mice (Thy-1b) panLNx or sham-operated at 3 wk of age, ≥2 mo after surgery. 16 h later, mice were killed and cell suspensions of their spleen, perLN, and MLN were individually prepared. Each cell population was labeled with biotin-anti-Thy-1.1 and FITC-anti-Thy-1.2 antibodies followed by PE-streptavidin. The frequency of donor type Thy-1.1+ cells was determined.
T Cell Proliferation Assays.
Responses to KLH and anti-CD3 were tested in mice panLNx and sham-operated at 3 wk of age, 3 mo after surgery. Complete culture medium consisted in RPMI 1640 containing 7% FCS, 1% penicillin/streptomycin, 4 mM-glutamine, 10 mM Hepes, 5 × 10−5 M 2-ME.
For KLH responses, mice were immunized with 200 μg KLH in CFA, in the tail base. PALNs and MLNs were removed 5 d later. T cells were enriched by panning on Petri dishes coated with goat anti–mouse Ig antibodies. 2 × 105 T cells of each population were cultured with 2 × 105 control or antigen-pulsed APCs prepared as follows. Normal spleen cells depleted of T cells with anti–Thy-1 (clone 42–21) + C (Low-Tox rabbit complement; Cedarlane), used as a source of APCs, were then cultured for 18 h in complete medium alone or with 100 μl/ml KLH, before irradiation with 30 Gy.
For anti-CD3-induced T cell response, 105 T cell–enriched suspensions of perLN and spleen cells were cultured with 105 irradiated control APCs, in the presence of 10 μg/ml purified anti-CD3 antibody.
All cultures were set in triplicate in 96-well culture plates and pulsed for the last 18 h of a 3 d culture with 1 μCi 3H TdR/well (5 Ci/mmol; Amersham Biotech) before being harvested for liquid scintillation counting.
Adoptive Transfer of Diabetes.
NOD/Nk male mice aged 8 wk were irradiated with 7.5 Gy the day before transfer of cells. NOD.scid recipients were used at 4–6 wk of age.
To detect the presence of diabetogenic T cells in sham-operated and panLNx mice, spleen cell suspensions were prepared individually, and split equally into two parts. Each spleen cell aliquot was injected into one recipient. Whether diabetes developed into only one or both recipients, the donor spleen was considered as containing anti-β cell T cells.
To determine whether panLNs are required for transfer of diabetes by primed anti-β cell T cells, 8-wk-old male NOD mice were subjected to panLNx 2 wk before irradiation. These recipients were transferred with 5 × 106 T cells (85% pure after passage on nylon wool column) from spleens of regular diabetic donors.
Mice were tested twice a week for glycosuria. After a positive urine test, blood glucose levels were determined with test strips and by a quantitative colorimetric assay (Reflolux; Boehringer). Animals showing glycemia higher than 300 mg/dl were classified as overtly diabetic.
Cyclophosphamide Treatment.
Cyclophosphamide (CY; Endoxan-Asta; Laboratoires Lucien) was injected intraperitoneally twice at a 2-wk interval at a dose of 200 mg/kg (1 mg/ml in saline). Mice were monitored every 2–3 d for 4 wk as described above.
Insulin Autoantibody Assay.
A 96-well filtration plate micro insulin autoantibody (IAA) assay was performed as described previously (13). Briefly, 125I-insulin (Amersham Biotech) of 24,000 cpm was incubated with 5 μl of serum with and without cold human insulin, respectively, at a 1:5 dilution of serum for 3 d at 4°C in an appropriate buffer. 50 μl of 50% protein A/8% protein G Sepharose (Amersham Biotech) were added to the incubation in a Multi-Screen 96-well filtration plate (Millipore). The plate was shaken for 60 min at 4°C. After washing, scintillation liquid was added to each well, and then counted. The results were calculated on the basis of the difference in cpm between the well without cold insulin and the well with cold insulin (Δ cpm) and expressed as an index: index = (sample Δ cpm − negative control)/(positive control Δ cpm − negative control) × 100. Serum samples were collected at 8 and 12 wk of age.
Histopathology.
Pancreases and salivary glands were excised, fixed in Bouin's solution, and processed for paraffin embedding. Four sections (5 μm) taken at 100 μm intervals were stained according to the hematein-eosin-safranin method. The proportion of infiltrated islets was based on the observation of at least 20 islets per specimen.
Statistical Analysis.
Pooled data were computed as mean ± SEM and were compared using Student's t test. When appropriate results were analyzed using Chi-square test, Mann Whitney test, or two-way analysis of variance (ANOVA). The Kaplan-Meier estimation was used to calculate diabetes incidence, and the log-rank test was used for the evaluation of significance.
Results
Excision of PanLN at Weaning Prevents Diabetes Development.
To determine the relative importance of spleen, panLN, and islets in the primary activation of diabetogenic T cells, the incidence of diabetes was analyzed in mice whose spleen or panLN were surgically removed at different ages.
Splenectomy, performed at 3 or 10 wk of age, had no effect on diabetes prevalence (P > 0.05; Fig. 1 A).
Figure 1.

Incidence of spontaneous diabetes in female NOD mice Sx (A) or panLNx (B) at different ages.
After panLNx at weaning (3 wk), diabetes development was greatly reduced (P < 0.0001) compared with sham-operated animals (20 and 100% were diabetic at 28 wk of age, respectively). panLNx performed at 4 wk of age slightly reduced diabetes incidence (P > 0.05), and had no effect in adult (10 wk) mice (P > 0.05; Fig. 1 B).
Histological analysis of the pancreas over a 38 wk period showed that, in mice panLNx at weaning, insulitis remained moderate, with 50–80% of the islets showing no signs of infiltration (Fig. 2 A). In sham-operated mice the frequency of intact islets significantly decreased with time, and was lower than in panLNx mice at 18 and 38 wk of age. This frequency was overestimated from 18 wk of age because insulitis was evaluated in nondiabetic mice only, while >50% and >85% of control mice were diabetic at 18 and 38 wk of age, respectively. Thus, the difference in insulitis severity between control and panLNx mice is greater than that illustrated on Fig. 2 A. No sign of pancreatitis was observed in panLNx mice.
Figure 2.

Histopathological analysis of islets in the pancreas of NOD mice after panLNx at 3 wk of age. (A) Severity of insulitis was determined at various times after surgery. Sham-operated animals served as controls. (B) Insulitis in mice panLNx at 3 wk, injected with CY at 8 and 10 wk and killed at 12 wk of age.
Interestingly, excision of panLNs did not prevent development of sialitis, indicating that inhibition of autoimmune response is specific for β cells.
These data indicate that diabetes development requires the presence of panLNs for at least the first 4 wk of life.
panLNx at Weaning Had No Consequence on the Localization and TCR-mediated Responses of the T Cell Pool.
We verified whether excision of panLNs at weaning resulted in modifications of the T cell pool behavior that could explain the reduced diabetes prevalence.
The cellularity of spleen and LN was not modified (data not shown). The frequency of B (not shown) and T cells (Table I) in secondary lymphoid organs remained unchanged. Lymphocytes from normal NOD mice congenic at the Thy-1 locus injected intravenously into panLNx and control mice homed to the spleen, perLN, and MLN with equal efficiency (not shown).
Table I.
Lymphoid Organ T Cell Contents after Excision of panLN at 3 Wk of Age
| Percentage of cells
|
||||
|---|---|---|---|---|
| Groups | Time after panLN |
Spleen | perLN | MLN |
| Sham | 1 mo | 41.4 ± 2.2 | 75.5 ± 1.5 | 70.8 ± 1 |
| panLNx | 35.2 ± 3.4 | 68.6 ± 1.7 | 74.9 ± 2 | |
| Sham | 3 mo | 46.9 ± 4.7 | 67.4 ± 1.9 | – |
| panLNx | 47.3 ± 4.7 | 68.9 ± 2.9 | – | |
| Control | 8 mo | 41.3 | 67.2 | 34.2 |
| panLNx | 38.2 ± 1.3 | 60.8 ± 3.9 | 26.3 ± 2.4 | |
The percentage of T cells was determined after labeling with anti-Thy 1.2 FITC. Control mice were sham-operated animals at 1 and 3 mo after surgery and age-matched female NOD mice at 8 mo. Mean ± SEM of three animals or mean of two mice (P > 0.05 control vs. LNxpanLNx).
In panLNx mice the responses of T cells to anti-CD3 in vitro and the generation of KLH-specific primary effector CD4 cells in vivo, were indistinguishable from those of control mice (P > 0.05) in two independent experiments (Fig. 3) . Thus, panLNx does not seem to perturb T cell production and functions.
Figure 3.

Proliferative responses of T cells to anti-CD3 and KLH after panLNx at 3 wk. T cells were tested 3 mo after surgery. Anti-CD3 response was performed on antibody coated dishes (top). T cells were primed in vivo 5 d before restimulation in vitro by KLH-pulsed APC (bottom).
Protection Does Not Result from the Absence of β Cell–specific T Cells in panLNx Mice.
Excision of panLNs at weaning did not prevent the production of anti–β cell T cells by the thymus and their settlement in lymphoid organs. This was demonstrated in adoptive transfer of diabetes experiments where 57 and 46% of the spleens from panLNx mice, 4–6 mo after surgery, contained T cells able to induce the disease in irradiated and NOD.scid recipients, respectively. After injection of sham-operated mouse spleen cells the transfer of diabetes occurred earlier in 100 and 75% of irradiated and NOD.scid recipients, respectively. Statistical analysis showed that the frequencies of positive spleens in both groups were not significantly different (P > 0.05; Table II). The late and slow diabetes onset in recipients of panLNx mouse cells likely corresponded to the time required for priming of naive autoreactive T cells in an animal with intact panLNs. In contrast, antigen-experienced anti–β cells from control mice could exert their function rapidly.
Table II.
Spleens from panLNx Mice Contain Cells Capable of Transferring Diabetes
| Incidence of diabetes (at weeks)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Donor | Recipients | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Control | Irrad. | 5/7 | 5/7 | 6/7 | 7/7 | ||||||
| (%) | 71 | 71 | 86 | 100 | |||||||
| panLNx | 0/7 | 0/7 | 2/7 | 3/7 | 3/7 | 3/7 | 3/7 | 3/7 | 4/7a | ||
| (%) | 0 | 0 | 29 | 43 | 43 | 43 | 43 | 43 | 57 | ||
| Control | scid | 0/8 | 0/8 | 1/8 | 2/8 | 3/8 | 4/8 | 6/8 | 6/8 | 6/8 | |
| (%) | 0 | 0 | 12 | 25 | 37 | 50 | 75 | 75 | 75 | ||
| panLNx | 0/13 | 0/13 | 0/13 | 0/13 | 0/13 | 4/13 | 4/13 | 5/13 | 6/13b | ||
| (%) | 0 | 0 | 0 | 0 | 0 | 31 | 31 | 38 | 46 | ||
Splenocytes from each donor were equally injected intravenous in two recipients. The results are from one experiment with irradiated mice and from two independent experiments with NOD.scid recipients, and are expressed as the number of spleens transferring diabetes/the total number of spleens tested. Control mice comprised sham-operated mice in two experiments, and age-matched mice in a third one.
χ2 analysis: P = 0.0507.
χ2 analysis: P > 0.05.
CY Fails to Induce Diabetes in panLNx Mice.
CY, an immunosuppressive drug, accelerates diabetes in NOD mice in which insulitis is installed, that is from around 5 wk of age. The disease results from regulatory T cell elimination that allows autoreactive T cells to exert their function (12). After two injections, 2 wk apart, of CY administered 4–5 wk after surgery (when mice were aged 7–8 wk), diabetes developed in only 20% of panLNx mice, whereas 94% of sham-operated animals became diabetic during the following 3–4 wk (P < 0.0001) (Fig. 4 A). Interestingly, these results are similar to the incidence of spontaneous diabetes in the two groups of mice at 28 wk of age. Analysis of insulitis in 12-wk-old mice resistant to CY treatment showed that 50% of the islets in panLNx mice were free of infiltrate versus only 6% in control mice (Fig. 2 B). To verify the specific effect of panLNx, mice MLNx at 3 wk of age were treated with CY when 8 wk old. Diabetes developed in MLNx mice as efficiently as in control mice (P > 0.05; Fig. 4 B).
Figure 4.

CY injection failed to induce spontaneous diabetes in mice panLNx at 3 wk, whereas it accelerated diabetes in mice MLNx at 3 wk. CY was administered 5 and 7 wk after excision of panLN or MLN. Incidence of the disease was followed for 28 d.
These results suggest that the protection afforded by panLNx does not result from an increased inhibitory control of the pathogenic T cells, nor the absence of autoreactive T cells, as shown above, but is rather consequential to the lack of priming of these cells that remain harmless.
Pancreatic LNs Are Not Required for the Migration of Primed Anti–β Cell T Cells into the Islets.
The above results indicate that panLNs are required for the initial priming of anti–β cell T cells. The fact that panLNx at 10 wk had no effect suggests that once primed diabetogenic T cells are present, panLNs are dispensable. To strengthen this notion we transferred diabetogenic T cells into irradiated recipients whose panLN had been excised. All mice became diabetic with a kinetic of disease induction similar to that observed in irradiated control recipients (Fig. 5) . Thus, once self-reactive T cells have been primed in the donors' panLNs the latter are not anymore required, and in their absence diabetogenic T cells probably expand in other recipients' lymphoid organs and/or the islets themselves.
Figure 5.

panLNs are dispensable for the adoptive transfer of diabetes. NOD mice were panLNx before irradiation and injection of splenocytes from diabetic mice (▪, normal recipients; ♦, Sham-operated recipients; ○, panLNx recipients).
panLNx at Weaning Prevents IAA Development.
IAA level analysis showed that 50% of NOD mice 12 wk old were positive. The same score was found with serum samples of mice panLNx at 10 wk. In contrast, only 10 and 0% of the sera collected from mice panLNx at 3 and 4 wk of age, respectively, were positive (the panLNx-4wk mice tested were still free of diabetes at 20 wk). The IAA levels in control and MLNx mice aged 8 wk were similar (Fig. 6) .
Figure 6.

Levels of insulin autoantibodies are greatly reduced in mice LNx at weaning. Serum samples were collected at 8 or 12 wk of age. +/n, number of positive samples/total samples.
Thus, panLNx at 3–4 wk of age greatly affected the production of one of the first autoantibodies detected in NOD, but had no effect when panLNx was performed in adults.
T Cell Activation in panLNs Is Detectable in BDC2.5 but Not in Normal NOD Mice.
In normal NOD mice, the low frequency of and lack of specific markers for autoreactive T cells in panLNs impedes the direct visualization of their activation. However, diabetogenic cells are detectable in BDC2.5/NOD mice in which activated transgenic CD4 cells were shown to reside specifically in the pancreatic islets and their draining LNs (11). To test whether any sign of activation could be detected in normal mice, we analyzed the frequency of memory/activated CD62LloCD44hi cells among CD4 cells from spleen, MLN, and panLN in NOD, BDC2.5/NOD, and BDC2.5 Cα0/0 at different ages. We confirmed that this population is increased in the panLNs of transgenic mice only, and show that this increase is detectable from 3 wk of age, but not earlier (Table III). In addition, we observed in the panLN of BDC2.5/NOD transgenics a 2–3-fold increase in the frequency of CD4 cells expressing an intermediary phenotype (CD44+CD62Lint), characteristic of the transition stage between naive and activated cells (Fig. 7) . These intermediary cells were absent from perLNs. This indicates that the stimulation of cells does occur in panLNs and the increase in activated cells does not result from a preferential migration of activated cells into panLNs compared with other LNs.
Table III.
Frequency of CD44hiCD62Llo Cells Among CD4 Cells in Different Organs at Various Ages
| Organs | Age (wk) | NOD | n | BDC | n | BDC Cα0/0 | n | 2-way ANOVAaeffect of | |
|---|---|---|---|---|---|---|---|---|---|
| Spleen | 2 | 17.9 ± 2.6 | 3 | 13.3 ± 0.7 | 5 | 13.3 ± 0.9 | 3 | Age: | P = 0.0008 |
| 3 | 23.8 ± 1.4 | 10 | 28.9 ± 2.3 | 8 | 20.4 ± 3.3 | 7 | Strain: | NS | |
| 5 | 20.9 ± 1.9 | 4 | 26.6 ± 1.6 | 8 | 24.2 ± 4.3 | 6 | Interaction: | NS | |
| MLN | 2 | 8.3 ± 0.3 | 5 | 9.4 ± 0.7 | 5 | 11.0 ± 1.3 | 6 | Age: | NS |
| 3 | 8.3 ± 1.0 | 7 | 9.2 ± 0.9 | 8 | 12.3 ± 1.1 | 11 | Strain: | NS | |
| 5 | 9.6 ± 0.9 | 4 | 12.6 ± 1.4 | 8 | 10.7 ± 2.5 | 4 | Interaction: | NS | |
| panLN | 2 | 8.9 ± 2.0 | 2 | 10.3 ± 1.6 | 5 | 10.0 ± 0.6 | 3 | Age: | P = 0.0002 |
| 3 | 13.4 ± 1.2 | 5 | 25.5 ± 0.7 | 8 | 28.4 ± 3.7 | 11 | Strain: | P = 0.008 | |
| 5 | 14.9 ± 1.6 | 3 | 34.5 ± 2.8 | 8 | 29.2 ± 3.1 | 4 | Interaction: | NS | |
| 7–8 | 10.7 ± 1.7 | 3 | 29.7 ± 1.7 | 6 | |||||
n = number of tested animals.
Two-way analysis of variance showed an effect, on the percentage of CD4+CD44+CD62Llo cells, of age in both spleen and panLNs, and an effect of strains in panLNs only. These effects were not interdependent.
Figure 7.
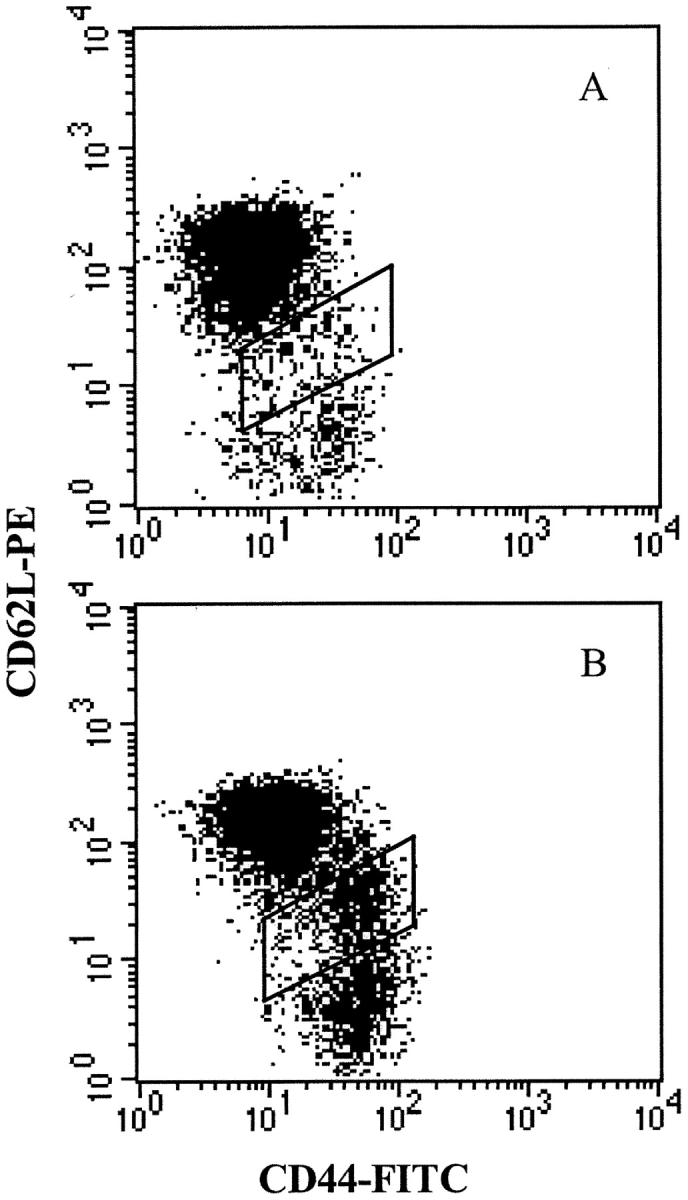
Surface expression of CD44 and CD62L on CD4 T cells from panLN of normal NOD (A) and BDC2.5/NOD mice (B). In BDC2.5/NOD mice a subset with an intermediate phenotype CD44+CD62Lint is highly increased compared with NOD mice, showing the transition step between naive CD44loCD62L+ and activated/memory CD44hiCD62L− cells. Cells were successively gated on side vs. forward scatter, to select alive cells, and on CD4 expression.
In the three strains of mice the percentage of CD4 CD62LloCD44hi cells was increased in the spleen of mice 3-wk-old, compared with 2-wk-old mice, but cells with the transitional phenotype were absent. This, together with the lack of effect of splenectomy, favor accumulation rather than activation of primed cells inside the spleen. Indeed, activated CD4 cells exit responding LNs as early as 2 d after antigen encounter, and primarily recirculate through the spleen (14).
The phenotypic changes observed in panLNs came with an increase in the fraction of early apoptotic CD4+Annexin-V+PI− cells in the panLNs of regular and transgenic NOD mice, compared with BALB/c mice. However, this increase was greater in the panLNs of BDC2.5 transgenics than in normal NOD mice, and was not observed in perLNs and MLNs of any mice (Table IV). Although different pathways induce apoptosis, it is tempting to relate this observation to activation-induced cell death (15).
Table IV.
Distribution of Early Apoptotic CD4 Cells in LNs
| Percentage of apoptotic CD4 cells
|
||||
|---|---|---|---|---|
| perLN | MLN | panLN | ||
| BALB/c | 3 wk | 5.5 (2) | 5.6 (2) | 3.9 ± 0.8 (3) |
| 5–6 wk | 2.1 (2) | 2.0 ± 0.2 (5) | 3.0 ± 0.6 (5) | |
| NOD | 3 wk | 2.5 ± 0.2 (4) | 3.8 ± 0.6 (5) | 6.5 ± 0.7 (7) |
| 5–6 wk | 2.2 ± 0.3 (4) | 3.8 ± 0.3 (7) | 8.1 ± 1.7 (9) | |
| BDC | 3 wk | – | 3.0 ± 0.2 (3) | 7.8 ± 0.3 (6) |
| 5–6 wk | 2.2 ± 0.4 (3) | 4.2 ± 0.5 (5) | 16.0 ± 6.0 (5) | |
The percentage of Annexin V+PI− cells among CD4 cells is shown. The number of animals tested is indicated in parentheses. Two-way analysis of variance showed an effect of strains (P = 0.015) without an effect of age (P > 0.05) on the percentage of apoptotic cells in the panLN.
Although phenotype analysis could not evidenced an increase in T cell activation in normal NOD mice, the increase in apoptotic cells likely points panLNs as distinct from other LNs in NOD mice.
Discussion
β cell–reactive T cells are generated in the thymus and recirculate through peripheral lymphoid organs until antigen is encountered. The time and location of this encounter in normal NOD mice are still elusive. To address these issues, we analyzed diabetes development in mice whose spleen, part of the MLN, or panLNs had been removed. Our data indicate that panLNs play a pivotal role in autoreactive T cell initial priming that takes place from 3 wk of age. This age, referred to as the first check point in the progression of the disease (16), corresponds to the first signs of insulitis (8), and to the emergence of the regulation of the autoimmune process (17,18).
Ablation of panLNs at 3 wk protected mice against diabetes almost completely. The protection afforded by panLNx at weaning was not due to modifications of the immune system. First, removal of panLNs at 3 wk of age did not modify the number, composition and reactivity of T cells, as shown by the proliferation of the latter in response to anti-CD3 in vitro and to KLH in vivo. Second, panLNx did not result in the absence of autoreactive T cells or the increase in the potency of regulatory T cells. Thus, it appears that panLNx at weaning induces a long-lasting prevention of β cell–specific T cell activation. When panLNs were removed at 4 wk the protection was incomplete, indicating that priming had already taken place in some mice. At 10 wk of age excision of panLNs did not influence diabetes progression, showing that at this time panLNs were dispensable. This was confirmed by the capacity of 10-wk-old panLNx irradiated recipients to develop diabetes upon transfer of T cells from diabetic mice. On the contrary, splenectomy had no effect at any age, and MLNx at 3 wk of age did not prevent acceleration of diabetes by CY. This indicates that initial priming of islet-reactive T cells does not occur in the spleen, although this organ is the largest reservoir of pathogenic T cells (19), nor in other abdominal LNs. That islets were the primary site of anti–β cell priming could not be directly tested, but if it were the case, ablation of panLNs would had no effect. These observations show that out of the three locations where antigen encounter can occur, that is the spleen, the panLN and the islets, panLNs are the site of initial priming of β cell–specific T cells. However, primed anti–β cells were not readily detectable in panLNs of young mice, as already reported for panLNs of diabetic mice (20). Indeed, panLN cells from mice aged 4–5 wk transferred diabetes in only 20% of recipients (data not shown). This is likely due to the fact that primed T cells leave rapidly responding LNs and do not return to LNs (14). In addition, the presence of insulitis in panLNx mice suggests either that some priming in panLNs may occur before 3 wk of age but does not include all the T cell reactivities required for diabetes development, or that priming of some autoreactive T cells may occur in other lymphoid organs but without the contribution of panLNs disease does not occur.
The present results fit with the notion that antigen outside organized lymphoid tissues is ignored (21, 22), notably because naive cells do not migrate into non-lymphoid tissues due to the absence of expression of appropriate homing receptors on endothelium (6). The establishment of insulitis requires the expression on endothelial cells of vascular addressins involved in homing of activated as well as naive cells. The events that induce the expression of these adhesion molecules on postcapillary venules are unknown but critical to understanding diabetes progression. After infiltration is initiated, lymphocytes, including both naive and activated T cells, get organized in the vicinity of these venules (23). There, intraislet APCs that bear antigen can eventually prime naive T cells and allow the diabetogenic process to proceed. Intraislet proliferation of lymphocytes was suggested by the findings that the infiltrate from each individual islet was generated by oligoclonal expansion of a few precursors (24). Thus, when panLNx was performed in 10-wk-old mice, effector T cells were already antigen experienced, insulitis was present and priming of naive autoreactive T cells could possibly occur inside the islets. However, such intraislet activation is not necessary since thymectomy at 6 wk of age that stops production of naive autoreactive T cells has no effect on diabetes incidence (18).
Excision of panLNs, which provide the required environment for β cell antigens encounter, impeded, not only the development of the Th1 arm of the autoimmune response, but also humoral immunity. In mice panLNx at 3–4 wk, IAA levels were drastically decreased. The notion that an immune response to insulin is in place early in life in the majority of NOD mice and children developing type 1 diabetes has emerged (13). Our results show that humoral immunity to insulin in NOD requires the LNs draining the pancreas.
Absence of activation of autoreactive T cells in mice less than 3 wk of age was already demonstrated in two experimental models of mice expressing antigens transgenically in the pancreatic islets (10, 25), and in BDC2.5/NOD mice expressing an unknown natural islet protein (11), and NOD mice injected with a diabetogenic CTL clone (26). This results from the unavailability of autoantigens in the panLNs of very young animals (11). Later on, pancreatic auto antigens were shown to be first presented to autoreactive T cells in the panLNs (10, 11). Upon injection, T cells from diabetic mice located in the panLNs before entering other LNs and rapidly expressed CD25 (27). Similarly, shortly after transfer of BDC2.5 T cells, proliferating cells localized specifically in panLNs (11). Antigens are presented to naive T cells by dendritic cells, which, when they originate from panLNs, prevent diabetes after injection into young NOD mice. Other LN dendritic cells could not achieve this goal, confirming the presence in panLNs of disease-related antigens (9).
It was important to confirm in standard NOD mice conclusions drawn from mice expressing self-antigens transgenically in the pancreatic islets. This work shows that panLNs are a requisite for the initiation of autoimmune diabetogenic activation which takes place from 3 wk of age, the first checkpoint in the diabetogenic process.
Acknowledgments
We thank Mireille Dardenne and André Herbelin for critical comments on the manuscript, Diane Mathis and Christophe Benoist for the gift of mice, Gérard Pivert and Olivier Babin for performing the tissue sections, Isabelle Cisse for managing the mouse colonies, and Catherine Slama for help with manuscript preparation.
This work was supported by the Centre National de la Recherche Scientifique and the Université Paris V.
J.J. Luan's present address is Genset, Route Nationale 7, 91000 Evry, France.
Footnotes
Abbreviations used in this paper: CY, cyclophosphamide; IAA, insulin autoantibody; MLN, mesenteric LN; MLNx, excision of MLN; NOD, nonobese diabetic; panLN, pancreatic LN; panLNx, excision of panLN; perLN, peripheral LN.
References
- 1.Bach, J.F. 1995. Insulin-dependent diabetes mellitus as a β-cell targeted disease of immunoregulation. J. Autoimmun. 8:439–463. [DOI] [PubMed] [Google Scholar]
- 2.Delovitch, T.L., and B. Singh. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: Immune dysregulation gets the NOD. Immunity. 7:727–738. [DOI] [PubMed] [Google Scholar]
- 3.Shimizu, J., E. Carrasco-Marin, O. Kanagawa, and E.R. Unanue. 1995. Relationship between β cell injury and antigen presentation in NOD mice. J. Immunol. 155:4095–4099. [PubMed] [Google Scholar]
- 4.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413–2417. [DOI] [PubMed] [Google Scholar]
- 5.Lee Reinhardt, R., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 6.Von Andrian, U.H., and C.R. MacKay. 2000. T-cell function and migration. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 7.Lepault, F., and M.C. Gagnerault. 2000. Characterization of the peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J. Immunol. 164:240–247. [DOI] [PubMed] [Google Scholar]
- 8.Jansen, A., F. Homo-Delarche, H. Hooijkaas, P.J. Leenen, M. Dardenne, and H.A. Drexhage. 1994. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes. 43:667–675. [DOI] [PubMed] [Google Scholar]
- 9.Clare-Salzler, M.J., J. Brooks, A. Chai, K. Van Herle, and C. Anderson. 1992. Prevention of diabetes in nonobese mice by dendritic cell transfer. J. Clin. Invest. 90:741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forster, I., and I. Lieberam. 1996. Peripheral tolerance of CD4 T cells following local activation in adolescent mice. Eur. J. Immunol. 26:3194–3202. [DOI] [PubMed] [Google Scholar]
- 11.Hoglund, P., J. Mintern, C. Waltzinger, W. Heath, C. Benoist, and D. Mathis. 1999. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 189:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasunami, R., and J.F. Bach. 1988. Anti-suppressor effect of cyclophosphamide on the development of spontaneous diabetes in NOD mice. Eur. J. Immunol. 18:481–484. [DOI] [PubMed] [Google Scholar]
- 13.Yu, L., D.T. Robles, N. Abiru, P. Kauer, M. Rewers, K. Kelemen, and G.S. Eisenbarth. 2000. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc. Natl. Acad. Sci. USA. 97:1701–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradley, L.M., J. Harbertson, and S.R. Watson. 1999. Memory CD4 cells do not migrate into peripheral lymph nodes in the absence of antigen. Eur. J. Immunol. 29:3273–3284. [DOI] [PubMed] [Google Scholar]
- 15.Newton, K., and A. Strasser. 2001. Cell death control in lymphocytes. Adv. Immunol. 76:179–226. [DOI] [PubMed] [Google Scholar]
- 16.Andre, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bendelac, A., C. Carnaud, C. Boitard, and J.F. Bach. 1987. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J. Exp. Med. 166:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dardenne, M., F. Lepault, A. Bendelac, and J.F. Bach. 1989. Acceleration of the onset of diabetes in NOD mice by thymectomy at weaning. Eur. J. Immunol. 19:889–895. [DOI] [PubMed] [Google Scholar]
- 19.Lepault, F., and M.C. Gagnerault. 1998. L-selectin-/lo and diabetogenic T cells are similarly distributed in prediabetic and diabetic NOD mice. Lab. Invest. 78:551–558. [PubMed] [Google Scholar]
- 20.Lepault, F., C. Faveeuw, J.J. Luan, and M.C. Gagnerault. 1993. Lymph node T cells do not optimally transfer diabetes in NOD mice. Diabetes. 42:1823–1825. [DOI] [PubMed] [Google Scholar]
- 21.Ochsenbein, A.F., P. Klenerman, U. Karrer, B. Ludewig, M. Pericin, H. Hengartner, and R.M. Zinkernagel. 1999. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl. Acad. Sci. USA. 96:2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohashi, P.S., S. Ochen, K. Buerki, H. Pircher, C.T. Ohashi, B. Odermatt, B. Malissen, R.M. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in vitro antigen transgenic mice. Cell. 95:305–317. [DOI] [PubMed] [Google Scholar]
- 23.Faveeuw, C., M.C. Gagnerault, and F. Lepault. 1994. Expression of homing and adhesion molecules in infiltrated islets of Langerhans and salivary glands of NOD (Nonobese diabetic) mice. J. Immunol. 152:5969–5978. [PubMed] [Google Scholar]
- 24.Sarukhan, A., P. Bedossa, H.J. Garchon, J.F. Bach, and C. Carnaud. 1995. Molecular analysis of TCR junctional variability in individual infiltrated islets of non-obese diabetic mice: Evidence for the constitution of largely autonomous T cell foci within the same pancreas. Int. Immunol. 7:139–146. [DOI] [PubMed] [Google Scholar]
- 25.Morgan, D.J., C. Kurts, H.T.C. Kreuwel, K.L. Holst, W.R. Heath, and L.A. Sherman. 1999. Ontogeny of T cell tolerance to peripherally expressed antigens. Proc. Natl. Acad. Sci. USA. 96:3854–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang, Y., B. O'Brien, J. Trudeau, R. Tan, P. Santamaria, and J.P. Dutz. 2002. In situ β cell death promotes priming of diabetogenic CD8 T lymphocytes. J. Immunol. 168:1466–1472. [DOI] [PubMed] [Google Scholar]
- 27.Fabien, N., I. Bergerot, V. Maguer-Satta, J. Orgiazzi, and C. Thivolet. 1995. Pancreatic lymph nodes are early targets of T cells during adoptive transfer of diabetes in NOD mice. J. Autoimmun. 8:323–334. [DOI] [PubMed] [Google Scholar]