

Abstract
Professional antigen-presenting cells (APCs) are capable of transporting self-antigens from peripheral tissues to secondary lymphoid organs where they are presented to potentially autoreactive CD8+ T cells. In the absence of an inflammatory response, this results in immune tolerance. The presence of activated, antigen-specific CD4+ T cells converts this tolerogenic encounter into an immunogenic one by promoting extensive proliferation of CD8+ T cells and their development into effectors. Surprisingly, activation of APCs with an agonistic antibody specific for CD40 could not substitute for CD4+ help in this task. Anti-CD40 induced recruitment of dendritic cells expressing high levels of B7 costimulatory molecules into the lymph nodes, which in turn, greatly enhanced activation and expansion of CD8+ T cells. However, these activated CD8+ cells did not demonstrate effector function. We conclude that proliferative potential and gain of effector function are separable events in the differentiation program of CD8+ T cells.
Keywords: cytotoxic T lymphocytes, peripheral tolerance, T cell activation, B7, CD40
Introduction
Not all potentially autoreactive T cells are deleted during thymic development and therefore, peripheral mechanisms of tolerance also exist to help prevent autoimmunity. Deletion and anergy of potentially autoreactive CD8+ T cells in the periphery has been demonstrated to occur for both self- and tumor antigens (1, 2). Dendritic cells (DCs)* play an important role in this process, as they are able to acquire, process, and present antigens expressed by parenchymal tissue and carry them to naive T cells transiting through draining LNs (3–5). Such presentation of exogenously acquired antigens by DCs is referred to as cross-presentation (6, 7). DCs also cross-present foreign antigens and pathogens and support the proliferation and differentiation of T cells into effectors (8, 9). The basis underlying how DCs can be responsible for both tolerance and immunity is still unclear. A variety of hypotheses have been proposed to explain this dual function of DCs. One such theory proposes that different subclasses of DCs are involved in presentation of toleragen versus antigen (10, 11). Another proposes it is the activation status of the DCs, which is determinative of the outcome (12). DCs that have been activated, directly or indirectly, through pathogens or adjuvants, convert from a tolerogenic to an immunogenic source of antigen (12). Yet another possibility relates to the different stages in the development of DCs. Immature DCs have been proposed to be tolerogenic, whereas activated, mature DCs could induce efficient CTL responses (13, 14). Evaluation of these possibilities is further complicated by evidence that DCs have the capacity to transfer antigen, such that the DCs responsible for carrying self-antigen to LNs may not be the same as those which present the same antigen to T cells (4, 14).
The importance of CD4+ T cell help in promoting an effective CD8+ T cell response is well established. Recent studies suggest that the mechanism by which CD4+ cells accomplish such ‘help’ is through activation of DCs by engagement of CD40 (15–17). Such activation in vitro results in enhanced expression of B7 costimulatory molecules and production of inflammatory cytokines, such as IL-12, which greatly augments CD8 responses (18, 19). In vivo, CD40 activated DCs also have an increased migratory potential toward secondary lymphoid organs, which would result in the enhanced transport of antigen to the draining LNs where they activate naive T cells (20). Engagement of CD40 by the in vivo administration of agonistic mAbs has been shown to prevent peptide-induced tolerance of CD8+ T cells (21, 22). Another way CD4+ cells assist CD8+ T cells is by providing IL-2, and systemic injection of IL-2 has been shown to reverse tumor-induced anergy of CD8+ T cell (23).
Our laboratory has been studying peripheral tolerance of CD8+ T cells in InsHA mice that express the hemagglutinin (HA) protein from the influenza virus under the control of the rat insulin promoter (24). InsHA mice are tolerant of HA that is expressed on the β cells of the pancreatic islets, such that the TCR repertoire is purged of T cells with high avidity for HA (24–26). We have demonstrated previously that HA specific CD8+ T cells can leave the thymus and become tolerized in the periphery (24, 27). To study the mechanism of tolerance induction in the periphery, we have used clone 4 TCR transgenic mice that express a TCR receptor specific for the major Kd restricted HA epitope (27). Upon transfer of naive clone 4 CD8+ T cells into InsHA recipients, the T cells proliferate in the draining LNs of the pancreas (28, 29). The proliferating clone 4 CD8+ T cells display a distinctive phenotype that distinguish them from either naive or effector cells (30). They do not exhibit cytolytic activity or production of IFN-γ, both of which are key effector activities of CD8+ T cells (30). The activated clone 4 T cells do not cause diabetes or even insulitis and appear to die within the lymphoid tissue (28–30). Since InsHA mice are also tolerant of HA in the CD4+ compartment (24), we have investigated the possibility of overcoming CD8+ T cell tolerance with an exogenous source of antigen specific help in the form of HA-specific CD4 cells from the HNT TCR transgenic line or with an agonistic anti-CD40 mAb.
Materials and Methods
Mice.
BALB/c mice were purchased from the breeding colony of The Scripps Research Institute. InsHA (24), clone 4 TCR (27), HNT TCR (31), and DO11 (provided by Susan Webb, The Scripps Research Institute; reference 35) transgenic mice lines had been backcrossed with BALB/c mice for at least eight generations. Clone 4 mice were then crossed with BALB/c Thy1.1+/+ for two generations to achieve homozygosity for Thy1.1. Mice were propagated and maintained under specific pathogen-free conditions in The Scripps Research Institute's animal facility. All mice used in these studies were between 8 and 12 wk of age. Experimental procedures were performed according to the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Preparation of Naive TCR Transgenic T Cells.
CD8+ Thy1.1+ T cells from clone 4 TCR were prepared as described previously (30). Briefly, single cell suspensions from spleen and LN of clone 4 TCR transgenic mice were sequentially subject to hypotonic red blood cell lysis, B cell depletion in a nylon wool column and mAb (clone RL172)-directed complement depletion of CD4+ T cells. HNT and DO11 CD4+ T cells were prepared in a similar way using anti-CD8 mAb (clone 3.155) for the complement depletion of CD8+ T cells instead of CD4+ T cells. T cell purity was >80%.
CFSE Labeling.
Purified T cells, at a maximum concentration of 5 × 107 cells/ml, were incubated in 5 μM CFSE (Molecular Probes) in HBSS for 10 min at 37°C. Cells were then washed with ice-cold HBSS.
In Vitro Activation of TCR Transgenic CD4+ T Cells.
Purified HNT or DO11 CD4+ T cells were seeded in 24-well plates, 2 × 106 per well, in RPMI 1640 media containing 10% FBS, 2 mM glutamine, 5 × 10−5 M β-mercaptoethanol, and 50 μg/ml gentamicin sulfate (complete media). For the priming of HNT cells, BALB/c spleen cells infected with influenza virus A/PR/8/34 H1N1 that was grown in the allantoic cavity of 10-d-old hen's eggs were used as APC. Cells from one spleen were irradiated (3,000 rads), mixed with 500 HA units of virus in 1 ml of serum free media and incubated at 37°C for 1 h. After washing, 6 × 106 cells per well were added to the cultures containing HNT cells. For the priming of DO11 cells, irradiated BALB/c spleen cells that had been pulsed for 2 h at 37°C with 1 μg/ml of OVA 323–339 peptide in complete media were used as APCs. In some experiments several wells were seeded with CFSE-labeled CD4+ T cells. Cultures were maintained at 37°C for 3 d and then viable cells were isolated by Ficoll-Paque separation.
Adoptive Transfer.
Recipient InsHA mice were injected intravenously with either 3 × 106 CFSE-labeled clone 4 TCR Thy 1.1+/+ CD8+ T cells or 3 × 106 CFSE-labeled HNT TCR CD4+ T cells in 200 μl of HBSS on day 0. In certain experiments, immediately after transfer, groups of mice were immunized intraperitoneally with 500 HA units of influenza virus. In other experiments, mice were coinjected with 3 × 106 CFSE-labeled clone 4 TCR Thy 1.1+/+ CD8+ T cells and 3 × 106 nonlabeled HNT TCR CD4+ T cells, 1.5 × 106 nonlabeled, in vitro–activated HNT CD4+ T cells or 1.5 × 106 nonlabeled, in vitro–activated DO11 CD4+ T cells.
Ab and Cytokine Treatment.
InsHA mice that received CFSE-labeled clone 4 T cells were injected intravenously on day 0 with 150 μg of the agonistic anti-CD40 mAb FGK45 (a gift from Stephen Schoenberger, La Jolla Institute for Allergy and Immunology, La Jolla, CA) in 200 μl of HBSS. Control mice received 150 μg purified rat IgG (Jackson ImmunoResearch Laboratory). Mice from these two groups where split and half of them received 5,000 U of recombinant human IL-2 (obtained from the National Cancer Institutes) intraperitoneally on days 1–3 in 200 μl of HBSS. Groups of mice that were killed on day 20 after cell transfer received Abs on days 0, 5, and 10 and IL-2 on days 1, 2, 3, 7, 8, 12, and 13. Alternatively, a single dose of 1 μg of IL-12 (a gift from Stanley Wolf, The Genetics Institute, Andover, MA) was administered intraperitoneal on day 1. Attempts to provide additional doses of IL-12 were hampered by increased morbidity.
In some experiments mice were treated with a combination of purified anti-B7.1 (clone 16–10A1; American Type Culture Collection) plus anti-B7.2 (clone GL1; ATCC) mAbs; 100 μg of each Ab per mouse per dose. Groups of mice treated with purified hamster IgG plus rat IgG (Jackson ImmunoResearch Laboratories) served as isotype controls. Abs were administered intraperitoneal in HBSS on days 0–3, and on day 5 in groups of mice killed on day 8.
Diabetes Monitoring.
Mice were monitored for diabetes by measuring blood glucose on days 4, 6, 8, and every 4 d thereafter for mice that were killed on day 20. They were considered diabetic when glucose levels were above 300 mg/dl.
Flow Cytometry.
4 or 8 d after adoptive transfer, pancreatic LNs, and a mixture of other LNs including inguinal, axillary, cervical, and mandibular were excised and processed separately to obtain single cell suspensions. After counting, all the cells from the pancreatic LNs and an equivalent number of cells from the other LNs were stained with the indicated Abs. LNs from three mice receiving the same treatment were pooled together for staining.
All mAbs and secondary reagents were purchased from BD PharMingen. Donor clone 4 T cells were detected and enumerated by virtue of their Thy1.1 expression. LN cells were incubated with anti-CD8α-PerCP and anti-Thy1.1-PE mAbs in HBSS, 0.1% BSA, 0.02% sodium azide for 30 min at 4°C. After washing, cells were analyzed with a FACSCalibur™ apparatus using CELLQuest™ software (BD Biosciences). In analyzing each sample, 1.5 × 106 events were collected.
For clone 4 CD8+ T cells, the intensity of CFSE fluorescence was analyzed in the CD8+ Thy1.1+ subpopulation of lymphocytes on day 4 after transfer. For HNT cells this was performed gating on CD4+ T cells after staining the LN cells with anti–CD4-PE mAb. CFSE fluorescence analyses on in vitro cultured CD4+ T cells were performed by propidium iodide exclusion of dead cells and gating on CD4+ T cells.
To assess production of IFN-γ in response to antigen, LN cells were incubated in complete media with 1 μg/ml of the Kd HA peptide (IYSTVASSL) and 1 μl/ml of brefeldin A containing Golgi-Plug solution (BD PharMingen) for 6 h at 37°C. An irrelevant peptide was used instead of the Kd HA peptide as a negative control. After washing, cells were stained to detect cell surface CD8 and Thy1.1 as described above. Cells were then permeabilized and stained to detect intracellular IFN-γ with anti–IFN-γ-APC mAb using the Cytofix/Cytoperm Plus kit (BD PharMingen) according to manufacturer's instructions.
For the detection and analysis of the activation status of DCs, single cell suspensions of pancreatic LNs on day 4 after treatment were stained with an anti–CD11c-FITC mAb plus anti–B7.1-PE or B7.2-PE for 20 min at 4°C. 105 events were collected in these experiments.
Immunohistochemisty.
Pancreas were excised on day 8 after transfer, fixed in a 10% formalin solution (Sigma-Aldrich), and processed for paraffin embedding. Serial sections were immunostained for the detection of insulin or stained with eosin (Sigma-Aldrich) as described previously (28). Finally slides, were counterstained with Mayer's Hematoxylin (Sigma-Aldrich).
Results
CD4 Help Prevents CD8+ T Cell Tolerance Induction.
HNT TCR transgenic mice express a TCR specific for an I-Ad–restricted epitope of the HA protein on CD4+ T cells (31). When labeled with CFSE and injected into InsHA mice, naive HNT CD4+ T cells behave much the same as HA-specific naive Clone 4 CD8+ T cells in this same situation (29). Small numbers of these cells become activated and proliferate in the pancreatic LNs but not elsewhere in the host (Fig. 1 A). Furthermore, these adoptively transferred HNT CD4+ T cells do not spontaneously induce diabetes (Table I).
Figure 1.


In vivo and in vitro proliferation of HNT CD4+ T cells. (A) 3 × 106 CFSE-labeled, purified, Thy1.1+ clone 4 CD8+ T cells (top) or HNT CD4+ T cells (bottom) were injected into InsHA hosts. Mice were killed on day 4 after transfer and cells from LNs were analyzed by FACS®. Histograms represent the amount of CFSE label gating on CD8+ Thy1.1+ lymphocytes (top) or CD4+ lymphocytes (bottom) of one representative experiment of five independent ones for clone 4 and three for HNT. The unstained cells in the bottom panels represent the endogenous CD4+ T cell repertoire. (B) Purified, CFSE-labeled HNT or DO11 CD4+ T cells were activated in vitro for 3 d and analyzed by FACS®. Histograms represent the amount of CFSE label gating on live CD4+ T cells.
Table I.
Diabetes Incidence in InsHA Mice
| Islet infiltrationa | |||||||
|---|---|---|---|---|---|---|---|
| Adoptive transfer and treatment | Clear | Periinsulitis | Insulitis | Islet destruction | Diabetes incidenceb | ||
| (%) | (%) | (%) | (%) | (%) | |||
| Clone 4 | n = 33 | 100 | 0 | 0 | 0 | n = 21 | 0 |
| Clone 4 plus HNTc | n = 27 | 93 | 7 | 0 | 0 | n = 11 | 0 |
| Clone 4 CD8+ plus activated DO11 | n = 18 | 89 | 11 | 0 | 0 | n = 6 | 0 |
| Clone 4 CD8+ plus activated HNT | n = 30 | 0 | 10 | 90 | 100 | n = 9 | 88 |
| Clone 4 plus activated HNT plus anti-B7.1 plus B7.2 |
n = 23 | 44 | 44 | 8 | 13 | n = 6 | 0 |
| Clone 4 plus IL-2c | n = 22 | 96 | 4 | 0 | 0 | n = 8 | 0 |
| Clone 4 plus anti-CD40c | n = 31 | 97 | 3 | 0 | 0 | n = 11 | 0 |
| Clone 4 plus anti-CD40 plus IL-2c | n = 24 | 96 | 4 | 0 | 0 | n = 9 | 0 |
| Clone 4 plus anti-CD40 plus IL-12 | n = 36 | 45 | 55 | 0 | 0 | ND | |
| Activated HNTc | n = 26 | 8 | 77 | 15 | 14 | n = 9 | 0 |
| HNTc | ND | ND | ND | ND | n = 6 | 0 | |
Groups of InsHA mice were treated as indicated and monitored for diabetes for 8 or 20 d. Mice were considered diabetic when the blood glucose levels were >300 mg/dl. Pancreas from mice sacrificed on day 8 were subject to histological analysis to determine the presence of islet infiltrates. Islet destruction was determined by the absence of insulin staining. ND, nondetermined.
Total numbers of islets examined in each group is indicated. Pancreatic sections from two to four mice per group were analyzed, except in the group that received clone 4 plus anti-CD40 plus IL-12 in which sections from seven mice were studied.
Total number of mice monitored for diabetes per group is indicated.
Conditions for which a group of three mice was monitored for 20 d.
We next coinjected clone 4 CD8+ T cells and HNT CD4+ T cells into InsHA recipients and monitored proliferation of the CFSE-labeled clone 4 CD8+ cells in the pancreatic LNs. The proliferation profile did not differ from that observed when Clone 4 CD8+ T cells were transferred alone (Fig. 2 A). Initially, the total numbers of clone 4 CD8+ T cells recovered from the pancreatic LNs was not substantially different from the control which did not receive HNT cells (Fig. 2 B). However, at a later time point, day 8, a slight increase was noticeable in these numbers of clone 4 CD8+ T cells recovered, relative to the mice injected with clone 4 CD8+ T cells alone (Fig. 2 B). No infiltrates were observed in the pancreatic islets on day 8 (Table I). Also, blood glucose levels remained normal, even in mice that were monitored for 3 wk after cotransfer (Table I). This demonstrates that in the conditions used in this experiment, HA-specific CD4+ T cells activated by cross-presented self-antigen are unable to cause diabetes, nor can they promote diabetes by HA-specific CD8+ cells.
Figure 2.
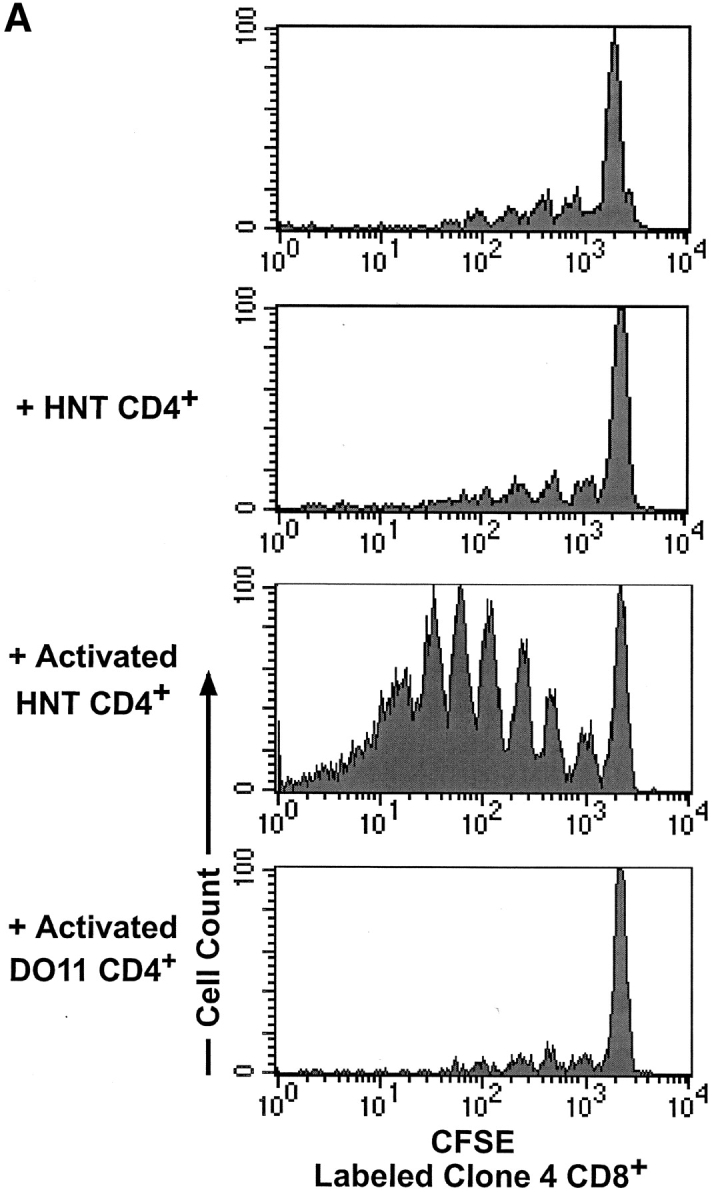


Activated HNT CD4+ T cells enhance proliferation and effector function of clone 4 CD8+ T cells in the pancreatic LNs of InsHA mice. 3 × 106 CFSE-labeled, purified, Thy1.1+ clone 4 CD8+ T cells were injected into InsHA hosts either alone or along with nonlabeled, 3 × 106 purified HNT CD4+ T cells, 1.5 × 106 in vitro activated HNT CD4+ T cells or 1.5 × 106 in vitro activated DO11 CD4+ T cells, as indicated. These data is representative of two to five independent experiments including a total of at least six mice per group per time point. (A) Mice were killed on day 4 after transfer and cells from pancreatic LNs were analyzed by FACS®. Histograms represent the amount of CFSE label gating on CD8+ Thy1.1+ lymphocytes. (B) Total numbers of CD8+ Thy1.1+ cells in the pancreatic LNs of host mice killed on days 4 and 8 after transfer. Data represent the mean of all experiments performed. Only negative standard deviation is depicted to achieve greater sensitivity in the graph. (C) On day 4 after transfer cells from pancreatic LNs were incubated with Kd HA peptide for 6 h and then analyzed by FACS® to detect accumulation of intracellular IFN-γ. Plots represent the amount of CFSE label versus the intensity of IFN-γ produced gating on lymphocytes CD8+ Thy1.1+. The percentage of clone 4 IFN-γ+ cells is indicated. The percentage of IFN-γ+ cells in controls that were stimulated with an irrelevant peptide was >1% in all the cases.
This finding was somewhat surprising, as there have been a number of reports demonstrating that CD4+ cells prevent tolerance and promote a vigorous immune response by CD8+ cells (32–34). However, in the HA model, very few CD4+ and CD8+ cells become activated at any given time, due to the limiting amount of cross-presented antigen. To determine if larger numbers of activated CD4+ cells could prevent tolerance of the clone 4 CD8+ T cells specific for HA, we coinjected naive clone 4 CD8+ T cells along with HNT CD4+ T cells that were previously activated in vitro with influenza virus (Fig. 1 B). A dramatic expansion of CSFE-labeled clone 4 CD8 + T cells was detected in the pancreatic LNs of the InsHA recipients receiving both the activated HNT CD4+ cells and the naive clone 4 CD8+ cells (Fig. 2 A). This was evident in both the proliferation profiles and in the total number of cells recovered from pancreatic LNs (Fig. 2 B). The majority of recipients (88%) became diabetic by day 8 after transfer (Table I). Immunohistological analysis of the pancreas showed massive infiltration of the islets and destruction of the β cells (Table I). Mice that received the same number of in vitro activated HNT cells alone, without receiving clone 4 CD8+ cells did not become diabetic (Table I). However, these cells did migrate into the pancreas, locating mainly in the periphery of the islets (Table I).
To determine if the CD4+ cells had to be specific for the islet antigen in order to promote activation of clone 4 CD8+ cells, CD4+ T cells from DO11 TCR transgenic mice, which express a TCR specific for an I-Ad–restricted OVA peptide (35), were activated in vitro (Fig. 1 B) and coinjected with clone 4 CD8+ T cells. No enhancement of proliferation or accumulation of clone 4 CD8+ T cells was observed (Fig. 2, A and B), suggesting a requirement for interaction between the activated CD4+ cells and the HA-presenting DCs in order to enhance the response by clone 4 CD8+ T cells.
We previously demonstrated that clone 4 CD8+ T cells proliferating in the pancreatic LNs of InsHA mice do not develop effector functions, such as production of IFN-γ (Fig. 2 C) and cytolytic activity (30). The experiments described above imply that the presence of activated HNT CD4+ cells not only enhanced activation of the naive clone 4 T cells, but also promoted effector function. To directly assess this issue, we monitored the ability of in vivo activated clone 4 T cells to produce IFN-γ. Clone 4 CD8+ T cells proliferating after coinjection with naive HNT produced little IFN-γ (Fig. 2 C). In contrast, a high proportion of the clone 4 CD8+ T cells that proliferated in the presence of activated HNT were able to efficiently produce IFN-γ. 42% of the clone 4 CD8+ T cells in the pancreatic LNs were IFN-γ+ after a brief period of activation in vitro with HA peptide (Fig. 2 C). This is almost comparable to the percentage of clone 4 CD8+ T cells producing IFN-γ when they are activated by priming in vivo with influenza virus (Fig. 2 C). These results indicate that activated HA-specific HNT CD4+ cells can promote vigorous proliferation of clone 4 CD8+ T cells and their differentiation into effector cells.
Comparison of the Effects of Anti-CD40 and CD4 Help on Clone 4 CD8+ T Cells.
CD4+ T cells promote activation and proliferation of CD8+ cells indirectly, through the activation of DCs by CD40-CD40L interaction, and also directly, by providing IL-2. To better understand the mechanisms by which activated HNT CD4+ cells prevent deletion of clone 4 CD8+ T cells in the pancreatic LNs, we examined separately the contribution of IL-2 and activation of DCs through CD40.
Systemic administration of IL-2 in InsHA mice that had received clone 4 CD8+ T cells induced a slight increase in the total numbers of these cells in the pancreatic LNs compared with mice that received Clone 4 CD8+ T cells alone (Fig. 3, A and B) . However, mice did not become diabetic and no infiltrates were found after histological examination of the pancreatic islets (Table I).
Figure 3.

Anti-CD40 treatment enhances proliferation but not effector function of clone 4 CD8+ T cells in the pancreatic LNs of InsHA mice. 3 × 106 CFSE-labeled, purified, Thy1.1+ clone 4 CD8+ T cells were injected into InsHA or BALB/c hosts. Mice were then treated with anti-CD40 mAb or purified rat IgG as isotype control. Anti-CD40 treated mice and isotype controls were then either left without further treatment or treated with IL-2 or IL-12 as described in Materials and Methods and as indicated in the figure. These data is representative of two to five independent experiments including a total of at least six mice per group per time point. (A) Proliferation profiles of clone 4 CD8+ T cells on day 4 after transfer were determined as in Fig. 2 A. (B) Total numbers of CD8+ Thy1.1+ cells in the pancreatic LNs of host mice killed on days 4 or 8 after transfer determined as in Fig. 2 B. (C) Production of IFN-γ on day 4 after transfer was determined as in Fig. 2 C.
Agonistic FGK45 anti-CD40 mAb has been shown to replace CD4+ T cell help in the development of CD8+ T cell responses (15–17). Consistent with these reports, we found that injection of this mAb into InsHA mice receiving clone 4 CD8+ T cells resulted in a dramatic enhancement of proliferation of these cells in the pancreatic LNs as compared with mice that received clone 4 CD8+ T cells and an isotype matched control Ab (Fig. 3). We observed a sixfold increase in the total numbers of clone 4 CD8+ T cells in the pancreatic LNs as compared with controls (Fig. 3 B). Surprisingly, none of the mice analyzed developed diabetes at day 8 (Table I). Mice that received serial injections of the agonistic mAb every five days were monitored for 3 wk and were found to display normal blood glucose levels. At this late time point almost all the clone 4 CD8+ T cells have disappeared from the lymphoid organs (unpublished data). A combined treatment with anti-CD40 mAb and IL-2 produced results similar to those obtained with anti-CD40 alone (Fig. 3, A and B and Table I). Again, only a slight increase in the total numbers of clone 4 CD8+ T cells was seen, as compared with mice that received anti-CD40 alone.
To rule out the possibility that the enhanced proliferation of HA-specific CD8+ T cells observed in InsHA hosts given anti-CD40 mAb is simply due to enhanced homeostatic division promoted by a generalized activation of DCs, we analyzed the proliferative potential of clone 4 cells in an antigen free environment. We had previously shown that clone 4 cells transferred into nonirradiated BALB/c recipients, where antigen is not present, do not proliferate (30). Clone 4 cells transferred into BALB/c mice that received anti-CD40 mAb did not undergo proliferation (Fig. 3, A bottom) indicating that the enhanced proliferation induced by anti-CD40 treatment is antigen dependent.
The surprising result that anti-CD40 resulted in massive proliferation yet did not promote development of diabetes, lead us to question whether the cells had acquired effector activity and whether they migrated to the pancreas. To determine if clone 4 CD8+ T cells responding to cross-presented self-antigen in the presence of anti-CD40 mAb developed into effectors, we tested their ability to secrete IFN-γ. Surprisingly, very few of the cells retrieved from the pancreatic LNs of anti-CD40–treated mice were able to efficiently produce IFN-γ (Fig. 3 C). When stimulated briefly in vitro with HA peptide, only 11% were IFN-γ+. This represents less than a threefold increase over the clone 4 CD8+ T cells injected alone in contrast to the 10-fold increase observed in the clone 4 CD8+ T cells coinjected with activated HNT cells (Fig. 2 C). To confirm the compromised functionality of these cells their cytolytic activity was assessed and very little if any was found (unpublished data) Furthermore, histological examination revealed that very few of the anti-CD40 activated cells could be found in the islets (Table I), despite the fact that by day 20 most of these cells had left the pancreatic LNs. These results indicate that enhancement of proliferative potential did not necessarily result in the development of effector functions by the clone 4 T cells and that anti-CD40 treatment could not replace activated HNT helpers in promoting the differentiation of clone 4 CD8+ T cells into effectors.
B7 Costimulation Is Required for Accumulation of Large Numbers of Activated Clone 4 CD8+ T Cells and for Induction of Diabetes.
We previously reported that clone 4 CD8+ T cells undergoing proliferation and tolerance in response to HA cross-presented in the pancreatic LNs did not require costimulation through CD28 (30). We wished to determine whether B7 was involved in the proliferation observed in response to anti-CD40 or activated HNT CD4+ cells and in the generation of clone 4 effector cells in the presence of activated HNT cells. InsHA mice that received a mixture of either naive clone 4 CD8+ cells and activated HNT CD4+ cells, or clone 4 CD8+ cells and anti-CD40 were also given anti-B7.1 plus anti-B7.2 mAbs to block CD28 costimulation. In both conditions, the antibodies were found to severely inhibit accumulation of clone 4 CD8+ T cells in the pancreatic LNs of InsHA mice (Fig. 4 A). Also, the number of IFN-γ–producing cells induced by the presence of activated HNT CD4+ cells was diminished threefold (compare Figs. 2 D and 4 B). Most importantly, the anti-B7 mAbs protected mice that received the mixture of activated HNT cells and naive clone 4 CD8+ cells from developing diabetes (Table I). Therefore, engagement of CD28 on clone 4 T cells is necessary for their accumulation in large numbers and also their differentiation into effectors.
Figure 4.


Blockade of B7 interaction inhibits accumulation and effector function of clone 4 CD8 T cells. (A) 3 × 106 CFSE-labeled, purified, Thy1.1+ clone 4 CD8+ T cells were injected into InsHA recipients along with anti-CD40 mAb or with 1.5 × 106 in vitro activated HNT CD4+ T cells. Half of the mice from the previous groups were treated with anti-B7.1 plus anti-B7.2 mAbs and the rest were treated with a mixture of rat and hamster IgG as isotype controls. On day 4 after transfer total numbers of CD8+ Thy1.1+ cells in the pancreatic LNs of host mice were determined as in Fig. 2 B. Results represent data from six mice per group analyzed in two independent experiments. The line represent the number of clone 4 CD8+ T cells in mice that received clone 4 CD8+ T cells without anti-CD40 or HNT cells. (B) Production of IFN-γ on day 4 after transfer was determined as in Fig. 2 C.
CD40 cross-linking has been shown to upregulate B7 expression on DCs and to promote their migration from the parenchyma to lymphoid tissue (20, 36, 37). A number of reports have implicated these cells as the ones responsible for cross-presentation of antigen (3, 5, 38). In fact, InsHA mice treated with anti-CD40 mAb showed a sixfold increase in the numbers of CD11c+ cells in the pancreatic LN (Fig. 5 A). Also, the expression of B7.1 and B7.2 was upregulated on these cells (Fig. 5 B). These results suggest that enhanced proliferation induced by anti-CD40 treatment is attributable to both the availability of more antigen and the upregulation of B7 costimulatory molecules. However, as the clone 4 CD8+ T cells did not develop effector function when stimulated in the presence of anti-CD40, the results indicate that B7 costimulation is necessary for optimal accumulation of activated Clone 4 CD8+ T cells, but it is not sufficient for their development into effectors.
Figure 5.


Anti-CD40 treatment promotes enhanced migration of activated CD11c+ cells to the pancreatic LNs of InsHA mice. Groups of InsHA mice were treated with anti-CD40 or rat IgG isotype control polyclonal Ab. On day 4, cells from the pancreatic LN were analyzed by FACS® for the expression of CD11c and B7.1 or B7.2 molecules. (A) Total numbers of CD11c+ cells in the pancreatic LNs. Data represent the mean and standard deviation of seven mice analyzed in two independent experiments. (B) Histograms represent the expression of B7 molecules gating in CD11c+ cells from the pancreatic LNs of representative treated InsHA mice. The geometrical mean fluorescence intensity is indicated in each panel.
IL-12 Can Trigger Effector Function in Activated CD8+ T Cells.
The results described above indicate that CD40 engagement is able to result in enhanced proliferation and accumulation of CD8+ T cells, but this does not promote development of effector function. One of the most potent signals for development of effector function by CD8+ cells is IL-12 (39). Therefore, we wished to determine if the clone 4 CD8+ T cells could develop effector activity when provided both anti-CD40 and IL-12. Recipients that received IL-12 and anti-CD40 along with the naive clone 4 CD8+ T cells showed a threefold increase in the proportion of activated CD8+ T cells that could produce IFN-γ respect to anti-CD40 alone (Fig. 3 C). Examination of the pancreas from these mice showed also a slight increase in the number of islets infiltrated (Table I). IL-12 alone was also able to enhance proliferation of clone 4 cells (Fig. 3 A). However, in the absence of CD40 engagement, IL-12 alone was not able to promote IFN-γ production by clone 4 CD8+ T cells (Fig. 3 C).
Discussion
One important function of DCs is the cross-presentation of antigens expressed in the parenchyma to naive T cells in secondary lymphoid tissue. This provides a mechanism by which potentially autoreactive CD8+ T cells that escape thymic deletion may be purged from the repertoire (2). This tolerance mechanism appears to be most useful in situations in which peripheral antigens are either not expressed in the thymus or expressed there at very low levels. Furthermore, there appears to be a requirement for a threshold level of expression of the self-antigen in the periphery in order for this mechanism to have an impact. If the level of antigen expression is too low it may simply be ignored (40–42).
We showed previously that clone 4 CD8+ cells undergoing activation in the pancreatic LNs of InsHA mice do not receive the benefit of B7 costimulatory signals and acquire only a limited proliferation potential. Furthermore, they do not develop effector function before deletion (30). This current study was undertaken to determine if provision of CD4+ cells specific for HA could prevent tolerance induction of clone 4 CD8+ cells in InsHA mice. Addition of naive HA-specific HNT CD4+ cells did increase somewhat the accumulation of activated clone 4 CD8+ T cells, suggesting some protection from deletion may occur, but they could not induce effector function by the clone 4 CD8+ cells. Only high numbers of in vitro activated HNT cells were capable of transforming a tolerizing signal into a signal resulting in the differentiation of naive clone 4 CD8+ T cells into effector CTL.
One possible mechanism by which activated CD4+ cells may lead to development of effector CTL is by initiating β cell damage and release of antigen. Although we do find evidence of insulitis with activated HNT CD4+ cells, we do not think that it is an increase in antigen that results in differentiation of CD8+ cells into effectors. In a previous study we showed that delivery of high levels of peptide antigen to clone 4 CD8+ cells in vivo resulted in strong proliferation, yet did not lead to effector function (30). For this reason, it is likely the development of effector function by the CD8+ cells is not simply due to greater antigen availability, but also reflects other proinflammatory signals provided to the APCs and CD8+ cells as a result of the presence of the activated HNT CD4+ cells.
In several previous studies CD4+ T cell help has been shown to prevent induction of CD8+ T cell tolerance (32, 33). However, the nature of the APC was unknown. In a recent study, the addition of naive OVA-specific CD4+ T cells was shown to hamper deletion of OVA-specific CD8+ T cells that occurs through cross-presentation of self-antigen by DCs (34). In our results, it was necessary to provide activated HA-specific CD4+ cells in order to prevent tolerance of the clone 4 CD8+ cells. This difference may simply reflect the fact that fewer T cells are activated in the InsHA than the RIP-OVA model. OVA is expressed at high levels in both the pancreas and kidneys in RIP-OVA mice, which results in vigorous T cell activation (43).
There are at least two mechanisms by which CD4+ cells are able to prevent tolerance of CD8+ cells. CD4+ cells provide IL-2, a cytokine that has multiple effects on CD8+ cells, including enhanced proliferation and the prevention of anergy (23). In our experiments, IL-2 enhanced only slightly the accumulation of clone 4 CD8 cells, but it did not enhance proliferation or effector function. This may be because the deletional mechanism of tolerance that occurs in InsHA mice differs from the classical anergic phenotype (30). The second pathway by which CD4+ T cells may prevent tolerance of CD8+ T cells is by activating the APC through interaction of the CD40L on CD4+ T cells with CD40 on the APC. It has been shown that activation of antigen-presenting DCs through CD40 using an agonistic anti-CD40 mAb can replace the requirement for CD4 help in the development of certain CTL responses, including those triggered by cross-presentation of antigen (15–17). To probe the mechanism by which activated HNT CD4+ cells lead to development of clone 4 CD8+ effectors, we attempted to replace the HNT cells with anti-CD40. This resulted in greatly enhanced proliferation and accumulation of the clone 4 CD8+ cells which corresponded with the recruitment of greater numbers of antigen presenting DCs expressing high levels of costimulatory molecules to the pancreatic LNs. Clone 4 CD8 + T cells proliferated extensively, dividing >7 times and accumulating in the LNs. However, unlike the clone 4 CD8+ cells that became stimulated in the presence of activated HNT CD4+ cells, they did not acquire effector function, and did not migrate into the islets. This result was surprising in light of previous studies that have reported the ability of anti-CD40 to augment induction of CTL that occurs through cross-presentation of antigen by DCs (16, 17). However, in these previous studies effector cell function was not assessed in vivo but rather after several days of in vitro stimulation of the in vivo–primed CD8+ cells. It is possible the in vitro activation conditions promoted effector cell function.
Numerous studies have shown that anti-CD40 activated DCs can produce IL-12, a potent inducer of IFN-γ production. This was one reason we were so surprised to find that anti-CD40 resulted in clonal expansion, but did not result in expression of IFN-γ by clone 4 CD8+ cells. However, in a recent study it was shown that although DCs activated in vitro with anti-CD40 are able to secrete IL-12, anti-CD40 was not sufficient to induce IL-12 production in vivo (44). It was suggested this difference may be due to additional activation signals received by DCs during the process of their purification and culture. In a recent report it has also been shown that anti-CD40 treatment accelerates the deletion of tumor-specific CD8+ T cells in the absence of antigen priming without development of effector function (45). Our results are consistent with these findings.
We have found that the combination of anti-CD40 and IL-12 promotes the generation of effector cells in vivo. Although this method of effector cell generation appears less efficient than with viral infection, we have observed that the proportion of IFN-γ–producing cells varies with time and dose of injection of these reagents (unpublished data). For this reason, we believe the method of delivery may be a major factor responsible for these differences. However, it is likely that viral infection would result in production of other proinflammatory cytokines and molecules that may potentiate production of IFN-γ by the activated CD8+ cells. Also, the fact that the combination of anti-CD40 and IL-12 did not promote insulitis, which occurs with activated HNT CD4+ cells, suggests that other proinflammatory signals are required to sustain effector function.
We do not know if the generation of effector cells was due to an effect by IL-12 on the CD8+ cells or the APCs. Not only does IL-12 activate DCs (46), but it also can act directly on CD8+ T cells, enhancing their clonal expansion and differentiation into effector CTL in vitro (47). IL-12 is also a powerful adjuvant in the development of CTL responses in vivo (48). Furthermore, it has been shown that IL-12 induces secretion of IFN-γ by T cells and enhances cytolytic activity by CD8+ T cells (48, 49). Normally, the major source of IL-12 in vivo is activated DCs. We do not yet know if production of IL-12 is responsible for clone 4 effector function as stimulated in the presence of activated HNT cells. Another cytokine that may be expressed under these conditions is IFN-γ. We have found that administration of anti-CD40 and IFN-γ also enhanced expression of IFN-γ by clone 4 CD8+ T cells (unpublished data). There may be a number of pathways that can promote the development of CD8+ effector function. Our results demonstrate a clear distinction between the signals involved in sustained proliferation and clonal expansion of CD8+ T cells and the signals required for the acquisition of effector function. As indicated by the effect of anti-CD40, the combination of signal 1 (antigen) and signal 2 (B7 costimulation) was sufficient to result in enhanced activation and accumulation of CD8 cells, however a third signal was required in order for these CD8+ cells to acquire effector function. It is generally accepted that cells that receive signal 1 plus 2 are committed to a differentiation program in which they will expand, undergoing multiple rounds of division, and become effectors. Indeed, there have been several reports that have argued that effector cell function occurred as a consequence of successful division (50–53). However, our observation that three signals, rather than two are required for production of CTL supports the findings of Mescher and colleagues, who proposed a similar three-signal model of CTL differentiation based on studies performed using a cell-free system of antigen presentation in vitro (47). Using antigen-coated beads as stimulators of CD8+ cells in vitro, they determined that in addition to signals 1 and 2, a third signal was required to induce CTL activity and that it may be provided, in part, by IL-12. Here we have validated this model in vivo and have extended this notion by assigning a clear role for each of these signals in the differentiation program of CD8+ cells. Furthermore, we have correlated these signals with situations in which there are differences in the activation of the DCs. We have found that in response to endogenous antigen cross-presented on APCs, signal 1 alone promotes a limited proliferation potential that leads to deletion (30). Signal 1 plus 2 (B7 costimulation) leads to enhanced clonal expansion and accumulation of CD8+ cells, as observed in the presence of anti-CD40. Signal 1 plus 2 plus 3 results in both clonal expansion, survival, and differentiation of the CD8+ cells into effectors. It is likely that activation of the innate immune system would normally provide signal 3 in vivo.
Of interest, it has been reported that DCs that mature in an IL-4 rich environment downregulate their expression of B7.1 and this prevents IFN-γ production by CD8+ T cells (54). However, the observation that anti-CD40 is not sufficient to promote effector function despite successful upregulation of both B7.1 and B7.2, suggests that other signals are involved in development of effector function. It will be of interest to examine how the conditions used for activation of the HNT CD4+ cells may alter the developmental program of the clone 4 CD8+ cells in the InsHA model.
Acknowledgments
We thank Dr. Stephen Schoenberger, Dr. Susan Webb, and Dr. Stanley Wolf for providing us with valuable reagents and mice.
This work was supported by grants DK50824 and CA57855 to L.A. Sherman.
J. Hernández's present address is the Dept. of Medicine and Cancer Center, University of California at San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0837.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; HA, hemagglutinin.
References
- 1.Stockinger, B. 1999. T lymphocyte tolerance: from thymic deletion to peripheral control mechanisms. Adv. Immunol. 71:229–265. [DOI] [PubMed] [Google Scholar]
- 2.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 3.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 4.Huang, F.P., N. Platt, M. Wykes, J.R. Major, T.J. Powell, C.D. Jenkins, and G.G. MacPherson. 2000. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurts, C., M. Cannarile, I. Klebba, and T. Brocker. 2001. Dendritic cells are sufficient to cross-present self-antigens to CD8 T cells in vivo. J. Immunol. 166:1439–1442. [DOI] [PubMed] [Google Scholar]
- 6.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heath, W.R., C. Kurts, J.F.A.P. Miller, and F.R. Carbone. 1998. Cross-tolerance: a pathway for inducing tolerance to peripheral tissue antigens. J. Exp. Med. 187:1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svensson, M., B. Stockinger, and M.J. Wick. 1997. Bone marrow-derived dendritic cells can process bacteria for MHC-I and MHC-II presentation to T cells. J. Immunol. 158:4229–4236. [PubMed] [Google Scholar]
- 9.Sigal, L.J., S. Crotty, R. Andino, and K.L. Rock. 1999. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 398:77–80. [DOI] [PubMed] [Google Scholar]
- 10.Suss, G., and K. Shortman. 1996. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kronin, V., K. Winkel, G. Suss, A. Kelso, W. Heath, J. Kirberg, H. von Boehmer, and K. Shortman. 1996. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 157:3819–3827. [PubMed] [Google Scholar]
- 12.Sallusto, F., and A. Lanzavecchia. 1999. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J. Exp. Med. 189:611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauter, B., M.L. Albert, L. Francisco, M. Larsson, S. Somersan, and N. Bhardwaj. 2000. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 191:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinman, R.M., S. Turley, I. Mellman, and K. Inaba. 2000. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 191:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridge, J.P., R.F. Di, and P. Matzinger. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ helper cell and T-killer cell. Nature. 393:474–478. [DOI] [PubMed] [Google Scholar]
- 16.Bennett, S.R.M., F.R. Carbone, F. Karamalis, R.A. Flavell, J.F.A.P. Miller, and W.R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 393:478–480. [DOI] [PubMed] [Google Scholar]
- 17.Schoenberger, S.P., R.E.M. Toes, E.I.H. van der Voort, R. Offringa, and J.M. Melief. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 393:480–483. [DOI] [PubMed] [Google Scholar]
- 18.Koch, F. 1996. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J. Exp. Med. 184:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cella, M., D. Scheidegger, K. Palmer-Lehmann, P. Lane, A. Lanzavecchia, and G. Alber. 1996. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 184:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moodycliffe, A.M., V. Shreedhar, S.E. Ullrich, J. Walterscheid, C. Bucana, M.L. Kripke, and L. Flores-Romo. 2000. CD40-CD40 ligand interactions in vivo regulate migration of antigen-bearing dendritic cells from the skin to draining lymph nodes. J. Exp. Med. 191:2011–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diehl, L., A.T. den Boer, S.P. Schoenberger, E.I. van der Voort, T.N. Schumacher, C.J. Melief, R. Offringa, and R.E. Toes. 1999. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 5:774–779. [DOI] [PubMed] [Google Scholar]
- 22.Garza, K.M., S.M. Chan, R. Suri, L.T. Nguyen, B. Odermatt, S.P. Schoenberger, and P.S. Ohashi. 2000. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 191:2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shrikant, P., A. Khoruts, and M.F. Mescher. 1999. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. 11:483–493. [DOI] [PubMed] [Google Scholar]
- 24.Lo, D., J. Freedman, J. Hesse, R.D. Palmiter, R.L. Brinster, and L.A. Sherman. 1992. Peripheral tolerance to an islet cell specific hemagglutinin transgene affects both CD4+ and CD8+ T cells. Eur. J. Immunol. 22:1013–1022. [DOI] [PubMed] [Google Scholar]
- 25.Morgan, D.J., T.C. Kreuwel, S. Fleck, H.I. Levitsky, D.M. Pardoll, and L.A. Sherman. 1998. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 160:643–651. [PubMed] [Google Scholar]
- 26.Nugent, C.T., D.J. Morgan, J.A. Biggs, A. Ko, I.M. Pilip, E.G. Pamer, and L.A. Sherman. 2000. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J. Immunol. 164:191–200. [DOI] [PubMed] [Google Scholar]
- 27.Morgan, D.J., R. Liblau, B. Scott, S. Fleck, H.O. McDevitt, N. Sarvetnick, D. Lo, and L.A. Sherman. 1996. CD8+ cell-mediated spontaneous diabetes in neonatal mice. J. Immunol. 157:978–983. [PubMed] [Google Scholar]
- 28.Morgan, D.J., C. Kurts, H.T.C. Kreuwel, H.K.L. Holst, W.R. Heath, and L.A. Sherman. 1999. Ontogeny of T cell tolerance to peripherally-expressed antigens. Proc. Natl. Acad. Sci. USA. 95:11377–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan, D.J., H.T.C. Kreuwel, and L.A. Sherman. 1999. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 163:723–727. [PubMed] [Google Scholar]
- 30.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo, D., C. Reilly, B. Scott, R. Liblau, H.O. McDevitt, and L. Burkly. 1993. Antigen-presenting cell in adoptively transferred and spontaneous autoimmune diabetes. Eur. J. Immunol. 23:1693–1698. [DOI] [PubMed] [Google Scholar]
- 32.Guerder, S., and P. Matzinger. 1992. A fail-safe mechanism for maintaining self-tolerance. J. Exp. Med. 176:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirberg, J., L. Bruno, and H. von Boehmer. 1993. CD4+8− helps prevent rapid deletion of CD8+ cells after a transient response to antigen. Eur. J. Immunol. 23:1963–1967. [DOI] [PubMed] [Google Scholar]
- 34.Kurts, C., F.R. Carbone, M. Barnden, E. Blanas, J. Allison, W.R. Heath, and J.F. Miller. 1997. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 186:2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+ CD8+ TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 36.Grewal, I.S., H.G. Foellmer, K.D. Grewal, J. Xu, F. Hardardottir, J.L. Baron, C.A. Janeway, Jr., and R.A. Flavell. 1996. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 273:1864–1867. [DOI] [PubMed] [Google Scholar]
- 37.Yang, Y., and J.M. Wilson. 1996. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 273:1862–1864. [DOI] [PubMed] [Google Scholar]
- 38.den Haan, J.M.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trinchieri, G. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 13:251–276. [DOI] [PubMed] [Google Scholar]
- 40.Ohashi, P.S., S. Oehen, K. Buerki, H. Pircher, C.T. Ohashi, B. Odermatt, B. Malissen, R.M. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 65:305–317. [DOI] [PubMed] [Google Scholar]
- 41.Oldstone, M.B.A., M. Nerenberg, P. Southern, J. Price, and H. Lewicki. 1991. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 65:319–331. [DOI] [PubMed] [Google Scholar]
- 42.Kurts, C., R.M. Sutherland, G. Davey, M. Li, A.M. Lew, E. Blanas, F.R. Carbone, J.F. Miller, and W.R. Heath. 1999. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc. Natl. Acad. Sci. USA. 96:12703–12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurts, C., H. Kosaka, F.R. Carbone, J.F.A.P. Miller, and W.R. Heath. 1997. Class I-restricted cross-presentation of exogenous self antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz, O., D.A. Edwards, M. Schito, J. Aliberti, S. Manickasingham, A. Sher, and C. Reis e Sousa. 2000. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 13:453–462. [DOI] [PubMed] [Google Scholar]
- 45.Kedl, R.M., M. Jordan, T. Potter, J. Kappler, P. Marrack, and S. Dow. 2001. CD40 stimulation accelerates deletion of tumor-specific CD8+ T cells in the absence of tumor-antigen vaccination. Proc. Natl. Acad. Sci. USA. 98:10811–10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grohmann, U., M.L. Belladonna, R. Bianchi, C. Orabona, E. Ayroldi, M.C. Fioretti, and P. Puccetti. 1998. IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity. 9:315–323. [DOI] [PubMed] [Google Scholar]
- 47.Curtsinger, J.M., C.S. Schmidt, A. Mondino, D.C. Lins, R.M. Kedl, M.K. Jenkins, and M.F. Mescher. 1999. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 162:3256–3262. [PubMed] [Google Scholar]
- 48.Schmidt, C.S., and M.F. Mescher. 1999. Adjuvant effect of IL-12: conversion of peptide antigen administration from tolerizing to immunizing for CD8+ T cells in vivo. J. Immunol. 163:2561–2567. [PubMed] [Google Scholar]
- 49.Nastala, C.L., H.D. Edington, T.G. McKinney, H. Tahara, M.A. Nalesnik, M.J. Brunda, M.K. Gately, S.F. Wolf, R.D. Schreiber, W.J. Storkus, et al. 1994. Recombinant IL-12 administration induces tumor regression in association with IFN-γ production. J. Immunol. 153:1697–1706. [PubMed] [Google Scholar]
- 50.Oehen, S., and K. Brduscha-Riem. 1998. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J. Immunol. 161:5338–5346. [PubMed] [Google Scholar]
- 51.Gudmundsdottir, H., A.D. Wells, and L.A. Turka. 1999. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J. Immunol. 162:5212–5223. [PubMed] [Google Scholar]
- 52.van Stipdonk, M.J., E.E. Lemmens, and S.P. Schoenberger. 2001. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2:423–429. [DOI] [PubMed] [Google Scholar]
- 53.Kaech, S.M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.King, C., R. Mueller Hoenger, M. Malo Cleary, K. Murali-Krishna, R. Ahmed, E. King, and N. Sarvetnick. 2001. Interleukin-4 acts at the locus of the antigen-presenting dendritic cell to counter-regulate cytotoxic CD8+ T-cell responses. Nat. Med. 7:206–214. [DOI] [PubMed] [Google Scholar]