

Abstract
Transforming growth factor (TGF)-β inhibits T cell proliferation and differentiation. TGF-β has been shown to inhibit the expression of transcription factors such as GATA-3 and T-bet that play important roles in T cell differentiation. Here we show that TGF-β inhibits T cell differentiation at a more proximal step. An early event during T cell activation is increased intracellular calcium levels. Calcium influx in activated T cells and the subsequent activation of transcription factors such as NFATc, events essential for T cell differentiation, are modulated by the Tec kinases that are downstream of the T cell receptor and CD28. We show that in stimulated CD4+ T cells, TGF-β inhibits phosphorylation and activation of the Tec kinase Itk, increase in intracellular Ca2+ levels, NFATc translocation, and activation of the mitogen-activated protein kinase ERK that together regulate T cell differentiation. Our studies suggest that by inhibiting Itk, and consequently Ca2+ influx, TGF-β limits T cell differentiation along both the Th1 and Th2 lineages.
Keywords: TGF-β, T cell, NFAT, Itk, calcium
Introduction
TGF-β inhibits the function of macrophages, T cells, B cells, and NK cells (1). Mice deficient in TGF-β have been shown to develop multi-organ inflammatory immune infiltrate at the age of 2–3 wk (2). Also, it has been shown that TGF-β signaling is critical for controlling T cell homeostasis as well as T cell differentiation by using transgenic mice which express a dominant-negative TGF-β receptor type 2 under a T cell–specific promoter (3). The major effect of TGF-β on mature T cells was though to be at the level of inhibition of T cell proliferation and IL-2 production (4). But it has been reported that TGF- β can interfere with T cell polarization even under conditions in which T cell proliferation is normal (5).
Because of its potent immunoinhibitory effects, some studies have focused on the role of TGF-β on allergic airway inflammation (6, 7). Transfer of antigen-specific CD4+ T cells expressing latent TGF-β into mice resulted in profound inhibition of Th2-induced airway inflammation and airway hyperreactivity (7). Conversely, in another study, overexpression of Smad7, an intracellular antagonist of TGF-β signaling, resulted in enhanced airway reactivity and inflammation (8). These studies strongly suggest a potential regulatory role of TGF-β in asthma. Asthma has been characterized as a Th2-mediated airway inflammation (9). Several molecules are now known to be essential for Th2 differentiation including STAT6, c-Maf, NFATc, and GATA-3 (9–11). Recently, we demonstrated that by regulating GATA-3 expression, nuclear factor (NF)*-κB also plays a critical role in Th2 differentiation (12). The activation of these factors appears to be regulated by signaling through TCR, costimulatory molecules, and the IL-4 receptor (10, 13, 14), indicating the requirement for multiple signaling events for efficient Th2 differentiation. T cell differentiation along the Th1 lineage is similarly regulated by specific transcription factors, among which T-bet, expressed in a lineage-specific fashion, plays an essential role in IFN-γ production (15, 16). Although it is well recognized that TGF-β suppresses T cell–mediated immune responses, whether TGF-β targets upstream pathways that prime for T cell differentiation along either lineage, has not been investigated. Here, we show that TGF-β can interfere with critical upstream signaling events such as Tec kinase phosphorylation and Ca2+ mobilization that are critical for T cell differentiation.
Materials and Methods
Antibodies and Reagents.
Anti-CD28 (37.51), anti-CD3ɛ (145–2C11) were purchased from BD Biosciences. Anti-STAT6, anti-GATA-3, anti-NFATc1, anti Txk (Rlk), anti-Oct-1, and anti-CREB-1 were obtained from Santa Cruz Biotechnology, Inc. Anti-Itk (2F12, monoclonal antibody), anti-Itk (rabbit polyclonal), and anti-phosphotyrosine (4G10) were purchased from Upstate Biotechnology. Anti-phospho-ZAP-70, anti-ZAP-70, anti-phospho-Lck, anti-Lck, anti-phospho–extracellular signal–regulated kinase (ERK), anti-ERK, anti-phospho-JNK, anti-JNK, and anti-phospho-GSK-3 were purchased from Cell Signaling. The rabbit anti-murine Rlk antiserum used to immunoprecipitate Rlk was provided by Dr. Pamela Schwartzberg (National Institutes of Health, Bethesda, MD) and its specificity described previously (17). Rabbit anti–mouse antibody was obtained from Sigma-Aldrich and goat anti–mouse from Jackson ImmunoResearch Laboratories. Anti-T-bet antibody was provided by Dr. Laurie Glimcher (Harvard School of Public Health, Boston, MA).
Cytokine Assays.
Cytokine concentrations were measured by ELISA using commercially available kits: IL-2, and IL-5 (Endogen), IL-13, IL-4, and IFN-γ (R&D Systems). For intracellular cytokine staining, purified spleen CD4+ T cells were stimulated with plate-bound anti-CD3 and anti-CD28 for 72 h. After blocking of Fc receptors with excess mouse Fc Block™ (2.4G2), 106 CD4+ T cells were cell surface stained with Peridinin Chlorophyll-a Protein-anti–mouse CD4 (RM4–5). Fixation and permeabilization was done using reagents from the Cytofix-Cytoperm Plus kit with the Golgi-plug protocol. Allophycocyanin-anti–mouse IL-4 (11B11) and Phycoerythrin-anti–mouse IL-5 (TRFK-5) were added to permeabilized cells (30 min on ice) for the staining of cytoplasmic IL-4 and IL-5, respectively. Samples were analyzed by flow cytometry using FACSCalibur™ and CELLQuest™ software (BD Biosciences) and the frequency of cytokine-positive cells was obtained by subtracting the value obtained with the respective isotype controls. All antibodies and reagents were purchased from BD Biosciences.
Preparation of Nuclear Extracts, Immunoblot Analysis, and Electrophoretic Mobility Shift Assay.
CD4+ T cells were stimulated with immobilized anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) and IL-2 (50 U/ml) in the presence of IL-4 (200 U/ml; Becton Dickinson) and anti–IFN-γ (XMG1.2) for Th2 differentiation or IL-12 (5 ng/ml; R&D Systems) and anti–IL-4 (4 μg/ml; Becton Dickinson) for Th1 differentiation with or without of TGF-β (50 pM). Nuclear extracts were prepared at the indicated time points and used for electrophoretic mobility shift assay (EMSA) or Western blot analysis essentially as described previously (12, 18–20).
Retroviral Infections of Primary Murine CD4+ T Cells.
The retroviral plasmid construct pMSCV-caNFATC1-H-2Kk (caNFATc1), and methods for retrovirus production and infection have been described previously (21). CD4+ T cells were stimulated under neutral conditions with anti-CD3 and anti-CD28 and infected at 24 and 48 h after activation by centrifugation at 2,000 rpm for 1.5 h at room temperature with 1.5 ml viral supernatants (containing 6 μg/ml polybrene [Sigma-Aldrich] and 10 μg/ml mIL-2). The viral supernatant was removed after the spin infection and replaced with medium containing 10 μg/ml murine IL-2. The expression of green fluorescent protein (GFP) in infected CD4+ T cells was determined by FACS® analysis and over 40% of the cells were found to be infected. The infected CD4+ T cells were then restimulated under Th2 conditions in the presence or absence of TGF-β. Nuclear extracts were prepared after 5 d of culture as described previously and culture supernatants were used for cytokine estimation (12, 18, 19).
In Vitro Kinase Assays.
The Itk kinase assay was performed as described previously (22, 23) with minor modifications. Itk immunocomplexes were washed twice in kinase buffer (Cell Signaling) and then incubated in 45 μl of kinase buffer containing 10 μCi of [γ-32P] ATP (NEN Life Science Products), 10 μM ATP and 5 μg of a peptide (Arg-Arg-Leu-Ile-Glu-Asp-Ala-Glu-Tyr-Ala-Ala-Arg-Gly; Sigma-Aldrich) derived from the sequence surrounding the SRC tyrosine kinase autophosphorylation site. This mixture was incubated for 15 min at room temperature and the reaction was stopped by adding 30% glacial acetic acid. The mixture was then blotted onto phosphocellulose paper and washed 6 times with 1% phosphoric acid. The amount of γ32P incorporated into the peptide was determined by scintillation counting. To immunoprecipitate Rlk, equivalent amounts of whole cell protein were incubated with the rabbit anti-rlk serum for 2 to 4 h at 4°C. The complexes were recovered with protein A-Sepharose (Amersham Biosciences), washed two times with ice cold lysis buffer, and analyzed by Western blotting with the goat anti-rlk antibody (Santa Cruz Biotechnology, Inc.). Rlk kinase assay was performed as described previously using 20 μCi of [γ-32P] ATP (NEN Life Science Products) and 10 μM ATP and incubating for 15 min at room temperature.(17).
Measurement of Intracellular Calcium Concentration by Microscopy.
Chambered glass coverslips were washed and first coated with poly l-lysine and next with anti-CD3 and anti-CD28 (1 μg/ml). The coverslips were extensively washed, covered with a thin layer of PBS and stored at 4°C for up to 1 wk before use. Purified CD4+ T cells were loaded with 2 μM Fura2-AM (Molecular Probes) for 30 min at 37°C in serum free medium. The cells were next washed and suspended in HEPES buffered media for imaging. Coverslips coated with antibodies were mounted in a Harvard Systems culture chamber and heated to 37°C. Using a 40× dry High NA objective (0.95), and an Olympus automated microscope, driven by Simple PCI, the upper surface of the coverslip was found and focused. Cells were added to the chamber, and images collected every 10 s using motorized excitation filters at 340 and 380 nm, emission at 510 nm and an Orca 100 camera. The total imaging period for each experiment was 10 min. Using Simple PCI, ratios were calculated for the 340/380 spectra and corrected for background fluorescence.
Measurement of Intracellular Ca2+ Concentration ([Ca2+]i) by Flow Cytometry.
The Ca2+ flux in CD4+ T cells was examined using Fluo 3-AM essentially as described previously (24). Purified CD4+ T cells were loaded with Fluo 3-AM by incubation for 30 min at room temperature with 3 μM Fluo 3-AM in the presence of 0.01% pluronic F-127 in HBSS supplemented with 1% FCS. Loaded CD4+ T cells were washed 4 times and resuspended in a Hepes-buffered saline solution containing 137 mM NaCl, 5 mM KCl, 1 mM Na2HPO4, 5 mM glucose, 1 mM CaCl2, 0.5 mM MgCl2, BSA 1 mg/ml, Hepes 10 mM, pH 7.4. The cells were then activated with 5 μg each of anti-CD3 and anti-CD28 in HEPES-buffered saline solution for 15 min on ice. The antibody-bound cells were washed and subjected to Ca2+ flux analysis by flow cytometry for 90 s at 37°C. Next prewarmed HEPES-buffered saline solution containing 20 μg/ml rabbit anti-mouse was added with or without TGF-β. [Ca2+]i was monitored for the next 600 s at 37°C and results were analyzed by CELLQuest™.
Online Supplemental Material.
Videos included as online supplemental material show Ca2+ flux in real time in CD4+ T cells maintained with or without TGF-β. Images were collected every 10 s for a total of 10 min. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20021170/DC1.
Results
Impaired Th2 Polarization in the Presence of TGF-β.
We first investigated whether we could set up conditions in which the effect of TGF-β on cell proliferation could be dissociated from its effects on T cell differentiation. Toward this end, we stimulated CD4+ T cells from OVA TCR transgenic mice with antigen (Ag; pOVA323–339) in the presence of various doses of TGF-β. Cell proliferation was measured by [3H]thymidine incorporation and IL-2 production was examined. Fig. 1 A shows dose-dependent inhibition of cell proliferation by TGF-β. TGF-β affected IL-2 production even at the lowest dose of 10 pM (Fig. 1 A, top panel). However, when IL-2 (50 U/ml) was provided in culture, T cell proliferation was sustained even at a dose of 100 pM of TGF-β (Fig. 1 A, bottom panel). As a dose of 50 pM TGF-β did not interfere with cell proliferation in the presence of exogenously added IL-2, we used this dose in subsequent experiments to investigate its effects on T cell differentiation.
Figure 1.
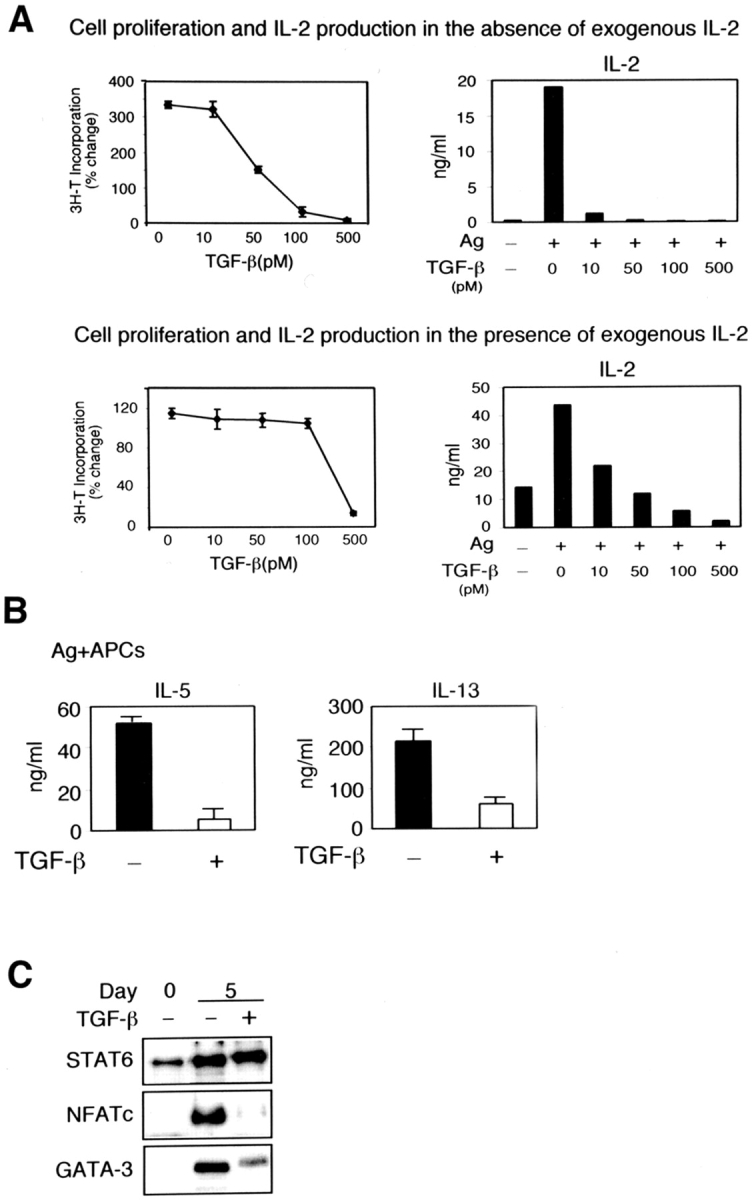
Inhibition of Th2 differentiation by TGF-β. (A) CD4+ T cells from DO11.10 TCR transgenic mice were stimulated with pOVA323–339 (5 μg/ml) and splenic APCs under Th2 differentiating conditions with or without rIL-2 (50 U/ml) ± TGF-β at different concentrations. On day 3 after stimulation, cells were pulsed with [3H]thymidine and incorporation was measured. The background was subtracted from the results and results were expressed as percent change from baseline. (B) CD4+ T cells from DO11.10 mice were stimulated with pOVA323–339 (5 μg/ml) and splenic APCs under Th2 differentiating conditions with or without pOVA323–339 (5 μg/ml) and ± TGF-β (50 pM). After 5 d, cells were harvested and restimulated at equivalent cell numbers. Supernatants were harvested after 48 h and assayed for the production of IL-5 and IL-13. (C) Nuclear extracts prepared from CD4+ T cells stimulated in the presence or absence of TGF-β during Th2 differentiation were analyzed by immunoblotting with antibodies to STAT6, NFATc, and GATA-3.
We next investigated the effects of TGF-β on Th2 differentiation to identify proximal targets during T cell differentiation. CD4+ T cells were primed under Th2 conditions with Ag and APCs in the presence or absence of TGF-β for 5 d. Cells were then harvested and restimulated for 48 h and Th2 cytokine production was measured. A significant decrease in Th2 cytokine production was observed in the presence of TGF-β (Fig. 1 B). These results indicate that TGF-β not only exerts antiproliferative effects on T cells, but even when conditions are conducive to proliferation in the presence of IL-2 in the microenvironment, TGF-β is able to inhibit T cell differentiation. When CD4+ T cells are stimulated to differentiate, several transcription factors important for Th2 differentiation are activated through phosphorylation or dephosphorylation events which ultimately result in the presence of transcriptionally competent factors inside the nucleus (11, 25, 26). We next examined whether TGF-β affected the nuclear presence of these factors. Consistent with the finding of other groups (27, 28), TGF-β did not affect nuclear translocation of STAT6 even after incubation with TGF-β for 5 d. However, the transcription factors NFATc and GATA-3 could be barely detected in the nuclear fraction prepared from TGF-β–treated cells (Fig. 1 C). These results suggested that TGF-β spares IL-4 receptor-mediated signaling and probably targets TCR/CD28-signaling.
TGF-β Does Not Inhibit NF-κB Activation but Impairs NFATc Translocation and GATA-3 Expression.
The series of experiments described next were performed to determine the effects of TGF-β during primary stimulation of CD4+ T cells under Th2-differentiating conditions. As shown in Fig. 2 A, TGF-β inhibited Th2-type cytokine production during primary stimulation of cells using a combination of anti-CD3/anti-CD28. Recently, we have shown that TCR-induced NF-κB activation is essential for GATA-3 expression and in turn Th2 differentiation (12). Because our data suggested that TGF-β may interfere with TCR signaling, we asked whether TGF-β interfered with the activation of NF-κB. As shown in Fig. 2 B, there was essentially no effect of TGF-β on NF-κB activation as measured by EMSA.
Figure 2.
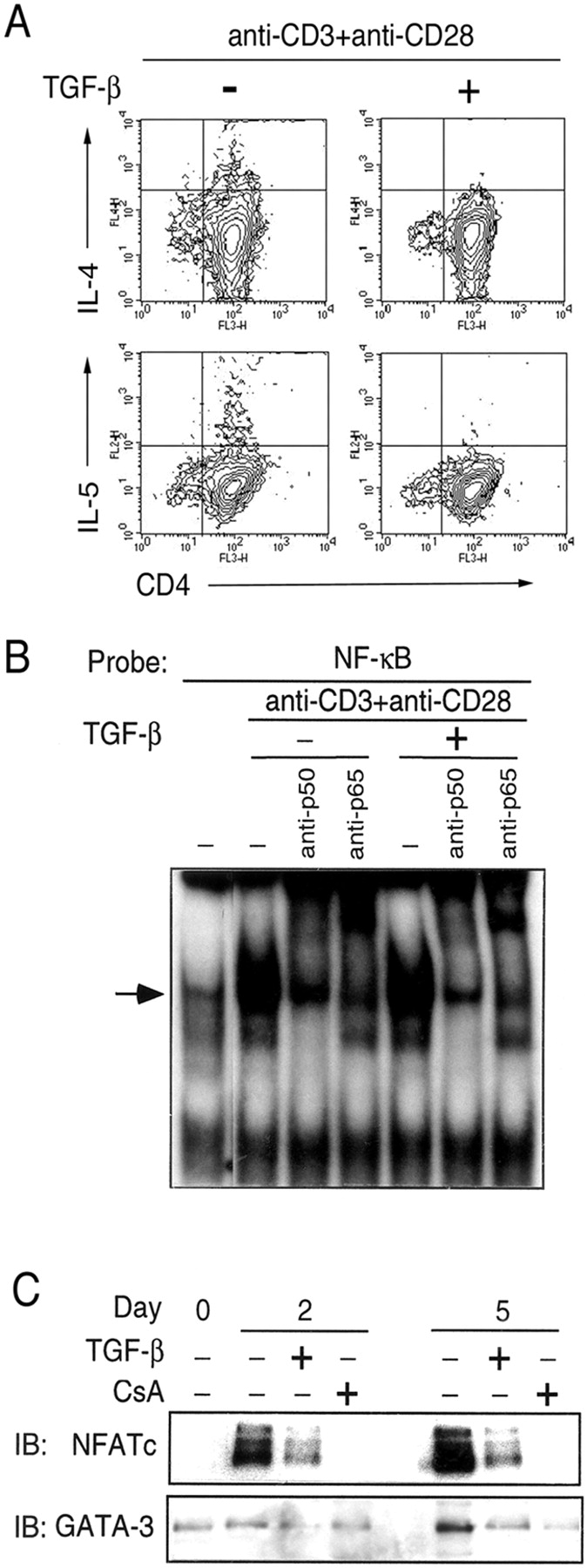
NFATc, and not NF-κB, is the target of TGF-β during Th2 differentiation. (A) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th2 differentiating conditions with or without the addition of TGF-β (50 pM). After 72 h incubation, the cells were harvested and stained for the simultaneous expression of cell-surface CD4, and intracellular IL-4 and IL-5. Flow cytometry was performed with 50,000 gated events collected for analysis of each sample. Appropriate isotype controls were included for each antibody. Cells in the lymphoid gate consisted of >98% CD4+ cells in all experimental groups. Compared with controls stimulated with anti-CD3 + anti-CD28 alone, TGF-β inhibited intracellular expression of both cytokines: 0.76% versus 0.11% of CD4+ cells for IL-4, and 0.38% versus 0.04% of CD4+ cells for IL-5, respectively. (B) To determine the effects on NF-κB activation, cells were stimulated in the presence or absence of TGF-β for 3 h. Nuclear extracts were prepared and analyzed for NF-κB activation by EMSA. (C) TGF-β and CsA both inhibit NFATc translocation as well as up-regulation of GATA-3 expression during Th2 differentiation. CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th2- differentiating conditions with or without TGF-β (50 pM) or cyclosporin A (100 ng/ml). Nuclear extracts were prepared and analyzed for the presence of NFATc and GATA-3. It is to be noted that induced, but not basal, expression of GATA-3 is inhibited by CsA and TGF-β.
NFATc is known to be an important regulator of IL-4 production and thus of Th2 differentiation (29, 30). In resting cells, NFATc resides in the cytoplasm as a phosphorylated protein. Upon stimulation, NFATc undergoes dephosphorylation by the calcium-dependent phosphatase calcineurin, which allows it to translocate to the nucleus to induce gene transcription (31, 32). The dephosphorylation of NFATc by calcineurin is inhibited by cyclosporin A (CsA). As shown in Fig. 2 C, treating cells with CsA not only inhibited the nuclear translocation of NFATc, but the up-regulation of GATA-3 expression in stimulated cells was also compromised despite the presence of exogenously added IL-4. It is to be noted that the basal low level of GATA-3 expression that is typically observed in CD4+ T cells was not affected by either CsA or TGF-β but only the induced expression was inhibited by both reagents. A direct effect of calcineurin on GATA-3 is unlikely as, unlike NFATc, GATA-3 resides in the nucleus in nuclear bodies in unstimulated cells (33) and undergoes phosphorylation and not dephosphorylation upon stimulation of cells, as we recently demonstrated (34). These results suggest a potential role for NFATc in the expression of GATA-3 which may be direct or indirect through regulation of other NFATc-dependent transcription factors. The early induction of NFATc during Th2 differentiation and its essential role in IL-4 gene expression together with the inhibition of GATA-3 expression by CsA prompted us to explore effects of TGF-β on TCR/CD28-induced proximal pathways that are linked to NFATc activation.
The effects of TGF-β on NFATc could be either on suppression of NFATc expression or on inhibition of NFATc translocation. As shown in Fig. 3 A, CD4+ T cells stimulated by a combination of anti-CD3 and anti-CD28 under Th2-differentiating conditions displayed a net increase in NFATc levels which was not inhibited by either TGF-β or CsA as determined by immunoblotting of whole cell lysates. There are three splice variants of NFATc and the slightly slower migration pattern of the bands in TGF-β– and CsA-treated cultures reflects inhibition of dephosphorylation of NFATc that is normally effected by calcineurin. These observations showed that although TGF-β, like CsA, inhibits processing of NFATc, it does not interfere with net NFATc accumulation in the cell. In a recent study, Feske et al. showed that the duration of nuclear presence of NFAT influences the pattern of expression of several cytokines in human T cells (35). This observation led us to suspect that TGF-β may interfere with the nuclear residence of NFATc either by inhibiting its import (by suppressing dephosphorylation) or by expediting its export (by enhancing rephosphorylation). To investigate this, we used the drug leptomycin B (LMB), which inhibits Crm-1–mediated export of NFATc causing accumulation of NFATc in the nucleus (36). Thus, if TGF-β interfered with NFATc import, LMB would be unable to cause nuclear accumulation of NFATc. On the other hand, if TGF-β did not interfere with import of NFATc but only expedited its export, similar levels of NFATc should be observed in control and TGF-β-treated cells in the presence of LMB. As shown in Fig. 3 B, cells stimulated under Th2-differentiating conditions in the presence of both TGF-β and LMB showed very little evidence of nuclear trapping by LMB compared with control stimulated cells that were treated with LMB suggesting that TGF-β interfered with a signaling event that controls NFATc import (dephosphorylation).
Figure 3.

TGF-β interferes with signals essential for NFATc translocation but not gene expression. In all experiments (A) whole cell lysates were prepared from CD4+ T cells stimulated under Th2 differentiating conditions in the presence or absence of TGF-β or CsA. The expression of NFATc was assessed by immunoblotting. (B) CD4+ T cells were incubated with or without leptomycin B (20 ng/ml) for 30 min and then stimulated with anti-CD3 and anti-CD28 under Th2 differentiating conditions ± TGF-β (50 pM). Nuclear extracts were prepared after 4 h of stimulation and analyzed for the presence of NFATc. (C) CD4+ T cells were stimulated with PMA and ionomycin under Th2 differentiating conditions with or without the addition of TGF-β (50 pM). Nuclear extracts were prepared and analyzed for the presence of NFATc and GATA-3. It is to be noted that while stimulation by Ag + APC or anti-CD3 + anti-CD28 maintains nuclear presence of NFATc up to 5 d, very little active NFATc is detected at 5 d after stimulation with a combination of PMA and ionomycin. (D) Expression of constitutively active NFATc (caNFATc1) in CD4+ T cells restores the expression of GATA-3 and IL-13 inhibited by TGF-β. Activated CD4+ T cells were infected with recombinant retrovirus expressing caNFATc1 and or control virus. Infected CD4+ T cells were subsequently incubated under Th2-differentiating condition with or without TGF-β (50 pM). Nuclear extracts were prepared at 5 d of culture and analyzed by immunoblotting for the presence of GATA-3. The same blot was then stripped and reprobed with anti-Oct-1 antibody. Culture supernatants were analyzed for the presence of IL-13.
One key check point in the initiation and maintenance of nuclear presence of NFATc is calcium (Ca2+) influx in cells (37, 38). We reasoned that if Ca2+ influx into cells was affected by the presence of TGF-β, then we should be able to bypass the effects of TGF-β by addition of a calcium ionophore such as ionomycin to the cells. CD4+ T cells were stimulated with phorbol myristate acetate (PMA) and ionomycin under Th2-stimulatory conditions in the presence or absence of TGF-β and the expression of NFATc and GATA-3 was analyzed. As illustrated in Fig. 3 C, TGF-β did not inhibit the nuclear presence of NFATc or of GATA-3 when cells were treated with PMA and ionomycin indicating that TGF-β targeted pathways that regulated [Ca2+]i levels.
To assess the importance of inhibited NFATc1 on T cell differentiation by TGF-β, a constitutively active mutant form of NFATc1 or control virus was introduced into CD4+ T cells by retroviral-mediated gene transfer techniques (21). As shown in Fig. 3 D, infection of cells with virus expressing the mutant but not with the control virus restored GATA-3 expression in the cells. It is interesting that this mutant was found to increase IFN-γ expression and actually decrease IL-4 expression in recent studies (21). However, as shown in Fig. 3 D, the levels of the cytokine IL-13 in the culture supernatant was restored to levels in the absence of TGF-β when caNFATc1 was expressed in the presence of TGF-β. Similarly, we have also found significant restoration of IL-5 levels (unpublished data). It should be noted that since TGF-β was added to the cultures after infection with retrovirus to allow efficient infection with the virus, the inhibition of IL-13 production by TGF-β was not as drastic as shown in Figs. 1 and 2 where TGF-β was present since the beginning. Given its ability to restore GATA-3 expression and maintain IL-5 and IL-13 expression, the effect of caNFATc1 on IL-4 could be specific and suggests that while NFATc1 is undoubtedly important for IL-4 expression as previous studies have shown (29, 30), unregulated expression of NFATc1 may ultimately inhibit IL-4 expression by conflicting with the expression of or antagonizing other transcription factors such as c-maf that are important for IL-4 gene transcription but not for IL-5 or IL-13 expression (39).
TGF-β Does Not Prevent Th2 Responses in Committed Th2 Cells.
To investigate whether TGF-β was also able to influence Th2 responses in already committed Th2 cells which have high levels of GATA-3 expression, we allowed CD4+ T cells to differentiate along the Th2 lineage without TGF-β and added it only during restimulation. As shown in Fig. 4 , addition of TGF-β during secondary stimulation did not impair either GATA-3 or NFATc expression or Th2 cytokine production. This shows that TGF-β can prevent the development of Th2 cells but does not affect activation of already committed Th2 cells. Our results are not unexpected since it has been previously shown that T cells developing along the Th2 lineage undergo loss of calcium signaling pathways (40) and thus these cells can be expected to be less susceptible to inhibition by TGF-β.
Figure 4.
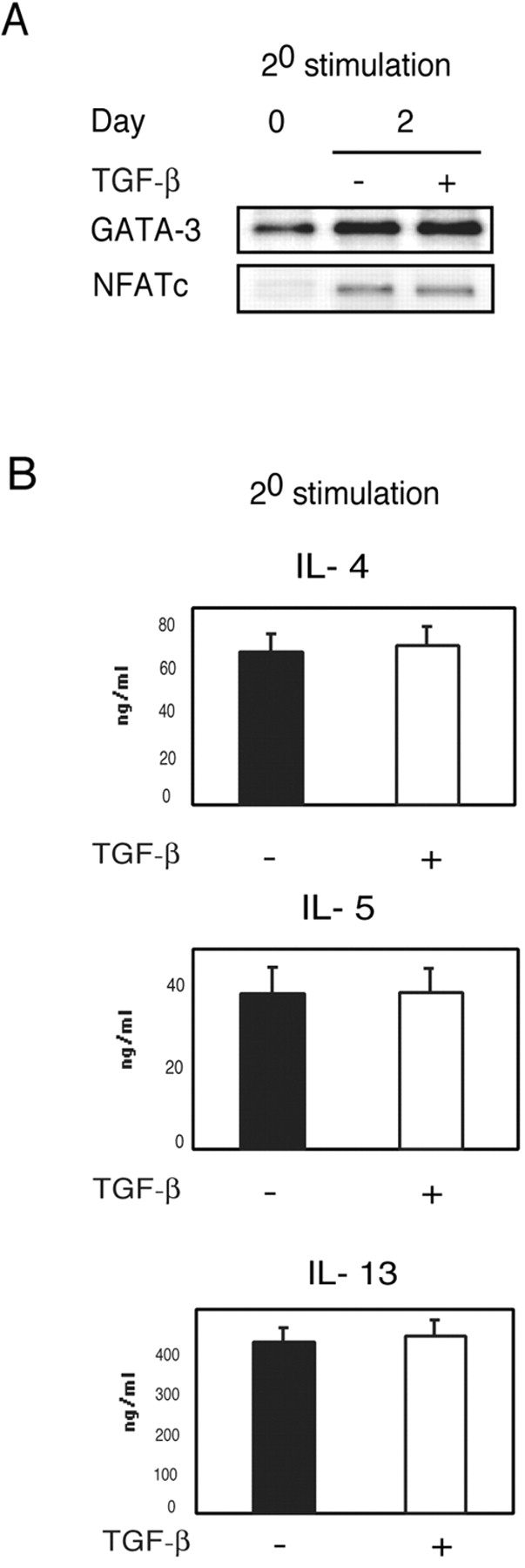
TGF-β does not inhibit Th2 cytokine production when added during secondary stimulation of already committed Th2 cells. (A) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th2 differentiating conditions. After 5 d of culture, cells were harvested and restimulated with anti-CD3 and anti-CD28 (IL-2, 50 U/ml) in the presence or absence of TGF-β (50 pM). Nuclear extracts were prepared 2 d after restimulation and analyzed by immuno-blotting with antibodies to GATA-3 and NFATc. (B) Supernatants were harvested after 48 h and assayed for the production of IL-4, IL-5, and IL-13 by ELISA.
TGF-β Affects Th1 Development by Inhibiting Both T-bet and NFATc Expression.
Although TGF-β has been shown to affect Th1 development (41, 42), given the IFN-γ–stimulating effect of the constitutively active NFATc and the demonstrated role of NFATc in IFN-γ gene transcription (43, 44), we were interested to determine whether TGF-β was able to inhibit NFATc activation in Th1-developing cells. As shown in Fig. 5 A, while TGF-β did inhibit T-bet expression as shown previously, the effect on NFATc was more pronounced. These inhibitory effects were reflected in inhibition of IFN-γ production by the cells (Fig. 5 B).
Figure 5.

Inhibition of Th1 differentiation by TGF-β. (A) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th1 differentiating conditions ± TGF-β (50 pM). Nuclear extracts were prepared at 3 d and 5 d of culture and analyzed for the presence of T-bet. The same blot was then stripped and sequentially reprobed with anti-NFATc antibody and anti-CREB-1 antibody. (B) IFN-γ concentrations in the culture supernatants collected at days 2 and 5 after stimulation were determined by ELISA.
TGF-β Interferes with Activation of the Tec Kinase Itk.
Stimulation of the TCR/CD28 signaling pathways results in a complex series of sequential phosphorylation/dephosphorylation events that control cell proliferation and differentiation. The Tec family of nonreceptor tyrosine kinases has an important role in integrating multiple signals from TCR/CD28 and transmitting them to molecules downstream in the signaling cascade. In T cells, three Tec-family members have been well studied: Itk, Rlk, and Tec (45). Both TCR signaling and costimulatory signals contribute to tyrosine phosphorylation of these kinases resulting in their activation (45). Tec kinases such as Itk and Tec have been demonstrated to play a role in TCR signaling for IL-2 production, Ca2+ mobilization and in turn NFATc translocation (46–48). To determine if TGF- β could interfere with Itk activity, Itk was immunoprecipitated with anti-Itk antibody after stimulation of CD4+ T cells in the presence or absence of TGF-β and Itk phosphorylation was analyzed by immunoblotting with anti-phosphotyrosine antibody. As shown in Fig. 6 A, Itk phosphorylation was rapidly induced when cells were stimulated by cross-linking of cell surface-bound anti-CD3 and anti-CD28. The phosphorylation peaked at 2 min and was detected until 20 min after stimulation (unpublished data). When cells were stimulated with plate-bound antibodies, the increased phosphorylation was sustained until 60 min after TCR/CD28 stimulation in the absence of TGF-β. Itk phosphorylation, however, did not increase in the presence of TGF-β using either method of cell stimulation. We also examined Itk kinase activity in Th2-differentiating cells in the presence or absence of TGF-β and found an inhibition of Itk kinase activity in the presence of TGF-β (Fig. 6 B). Recently, unlike Itk−/− mice which are defective in Th2 responses in vivo, Itk−/− Rlk−/− mice were found to maintain Th2 responses (49). To investigate Rlk phosphorylation in Th2 developing cells, Rlk was immunoprecipitated and an immunecomplex kinase assay was performed to detect autophosphorylation of Rlk as described previously (17). As shown in Fig. 6 C, we detected basal levels of phsophorylation of Rlk which did not change upon stimulation of cells. In other experiments, this protein band was not detected using control rabbit serum confirming the specificity of the antiserum (unpublished data). Because Itk has been shown to regulate Ca2+ mobilization, which is critical for NFATc translocation, we next examined the Ca2+ response in the presence of TGF-β.
Figure 6.
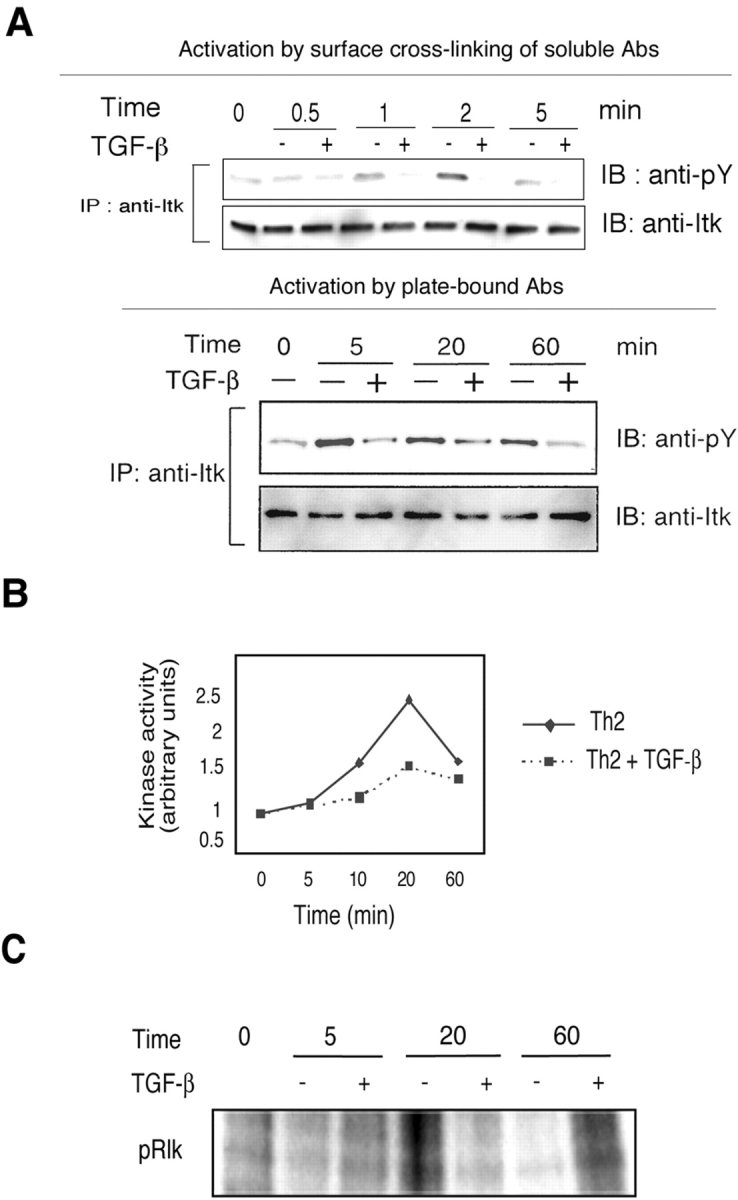
Impaired Itk phosphorylation in the presence of TGF-β. (A) CD4+ T cells were stimulated either by cross-linking of cell surface-bound anti-CD3 and anti-CD28 with rabbit anti–mouse antibody or with plate-bound anti-CD3 and anti-CD28 antibodies ± TGF-β (50 pM). Cells were harvested and lysed at the indicated time points. Cell lysates were immunoprecipitated with anti-Itk antibody. The immunoprecipitates were analyzed by immunoblotting using anti-phosphotyrosine antibody (4G10) to detect the level of phosphorylation. The same blot was then stripped and reprobed with anti-Itk antibody. (B) TGF-β decreases Itk kinase activity in stimulated CD4+ T cells. CD4+ T cells were stimulated by anti-CD3 and anti-CD28 under Th2 differentiating conditions ± TGF-β (50 pM). Equal amounts of cell lysate proteins were immunoprecipitated with anti-Itk antibody. The kinase reaction was performed during 15 min with [γ-32P] ATP, cold ATP, and the SRC peptide. The reaction was stopped, blotted onto phosphocellulose paper, and incorporation was measured. (C) Effect of TGF-β on Rlk autophosphorylation. CD4+ T cells were stimulated by anti-CD3 and anti-CD28 under Th2 differentiating conditions with or without TGF-β (50 pM). Equal amounts of cell lysate proteins were immunoprecipitated with anti-Rlk antiserum. The kinase reaction was performed with [γ-32P] ATP and cold ATP for 15 min. The reaction was stopped by adding SDS-sample buffer and the samples were subjected to SDS-PAGE. Protein phosphorylation was assessed by autoradiography.
To determine the effects of TGF-β on Ca2+ flux in activated CD4+ T cells, we used both flow cytometry and real-time imaging techniques using the Ca2+-binding ratiometric dye Fura-2M. In the flow cytometry method, cells were loaded with fluo-3 AM and incubated with anti-CD3 and anti-CD28. Cells were activated by cross-linking the bound antibodies and the intracellular Ca2+ level was monitored by flow cytometry. As shown in Fig. 7 A, in the absence of TGF-β, the typical biphasic response was noted with an initial rapid increase in [Ca2+]i that was followed by a sustained low level of [Ca2+]i. Addition of TGF-β caused the initial increase in [Ca2+]i which dropped to basal levels by 4 min. We have also monitored [Ca2+]i in the cells using a ratiometric technique that is being increasingly used to track T cell dynamics at several levels in vitro (50). In this technique, Fura-2M–loaded CD4+ T cells were added to anti-CD3 and anti-CD28–coated chambered glass coverslips mounted in a Harvard Systems culture chamber maintained at 37oC and containing medium with or without TGF-β. Images were collected every 10 s using motorized excitation filters at 340 and 380 nm and emission at 515 nm. Images at two time points are shown in Fig. 7 B. As shown in Fig. 7 C, there was an initial small change in the ratio in both control and TGF-β–treated cells which returned to baseline by ∼150 s. However, the subsequent steady, dramatic increase in the ratio observed in the control cells due to increased calcium levels in the cells was not observed in the presence of TGF-β.
Figure 7.


TGF-β blocks Ca2+ influx in activated CD4+ T cells. (A) Fluo-3 AM loaded CD4+ T cells were incubated with anti-CD3 and anti-CD28 for 15 min on ice. After measuring the basal mean fluorescence intensity (mean F0) for 80 s, cells were stimulated by cross-linking with rabbit anti–mouse antibody in a Hepes-buffered saline solution containing IL-2 (50 U/ml) with or without TGF-β. Ca2+ flux was monitored by measuring mean fluorescence intensity (F) for the next 10 min. (B) CD4+ T cells were added to the warmed chambered anti-CD3– and anti-CD28–coated cover-slip containing culture medium and images were collected every 10 s. The cells are colored to show the relative intensities of the 340 nm (green) and 380 nm (red) signal. At early time points (for example in the 1 min and 40 s time point shown here), most cells appear red/orange reflecting the higher 380 nm component to the signal. However, at later time points (in this example the 8 min 30 s images are shown), while there is little change in the ratio in cells treated with TGF-β, the control cells show a clear change in the ratio, and most of the cells have a significant increase in the 340 component (bar = 50 microns). (C) Graphical representation of the results. The measured values represent the ratio for 15 cells in each experimental condition.
TGF-β Does Not Affect Lck or ZAP-70 Phosphorylations but Inhibits ERK Phosphorylation.
To determine whether inhibition of Itk activation was a consequence of inhibition of Lck and/or ZAP-70 phosphorylation, which are known to regulate Itk activation (22, 51), cells were stimulated in the presence or absence of TGF-β and cell extracts were prepared to examine protein phosphorylations. As shown in Fig. 8 A, TGF-β did not inhibit either Lck or ZAP-70 phosphorylations. We also investigated whether TGF-β could alter other TCR-induced signaling events that have been suggested to regulate NFATc function. T cells deficient in Itk show an inability to activate the mitogen-activated protein (MAP) kinases ERK1 and ERK2 (52). ERK phosphorylation induced upon TCR/CD28 ligation was inhibited by TGF-β (Fig. 8 B). Interestingly, as observed with Itk phosphorylation, use of plate-bound antibodies for cell activation resulted in a delayed profile of ERK phosphorylation (at ∼60 min; unpublished data) as has been previously observed in other studies (53). The JNK MAP kinase pathway has been shown to inhibit Th2 responses through phosphorylation of NFATc (54, 55). We therefore examined whether TGF-β treatment led to increase in JNK phosphorylation. However, no increase in JNK phosphorylation was observed in the presence of TGF-β. Yet another mechanism that regulates shuttling of NFATc between the cytoplasm and the nucleus is glycogen synthase kinase-3 (GSK-3). By phosphorylating serine residues within serine-rich regions and serine-proline repeats in NFATc, GSK-3 enhances export of NFATc from the nucleus. Thus while a kinase-inactive mutant of GSK-3 has been shown to slow down nuclear export of NFATc, overexpression of GSK-3 expedites nuclear export of this factor (56). As TCR engagement leads to phosphorylation of GSK-3, which inhibits its activity thereby allowing nuclear localization of NFATc, we investigated whether TGF-β interfered with GSK-3 phosphorylation which would prevent normal activation of NFATc. Our results show that TGF-β does not interfere with the phosphorylation status of GSK-3 in stimulated cells (Fig. 8 B).
Figure 8.


TGF-β does not inhibit Lck or ZAP-70 phosphorylations but inhibits ERK phosphorylation. Whole cell lysates were prepared from CD4+ T cells stimulated by cross-linking anti-CD3 and anti-CD28 bound to cells under neutral conditions (IL-2, 50 U/ml) in the presence or absence of TGF-β (50 pM). Total cell lysates were prepared and analyzed for the phosphorylation of (A) Lck and ZAP-70 or (B) ERK, JNK, and GSK-3.
Discussion
The immunosuppressive functions of TGF-β are well recognized. TGF-β has been found to inhibit primary responses in CD4+ T cells during both Th1 and Th2 differentiation and memory Th2 cells were found to resist TGF-β–mediated suppression (57). In Th1 cells, the inhibitory effect of TGF-β was initially associated with the attenuation of IL-12Rβ2 expression and, more recently, with inhibition of T-bet expression (41, 42). Consistent with the reports of two other groups (27, 28), we show inhibition of GATA-3 expression in Th2 cells in the presence of TGF-β and absence of any effects on STAT6 activation. In developing Th1 cells, TGF-β caused inhibition of IFN-γ production and T-bet expression as shown previously (42). Addition of TGF-β during differentiation to either lineage inhibited NFATc activation. A critical requirement for NFATc activation is sustained levels of elevated [Ca2+]i. We show that TGF-β prevents Ca2+ influx in CD4+ T cells that is important for both Th1 and Th2 differentiation. The phosphorylation of the Tec kinase, Itk, which regulates Ca2+ mobilization and NFATc translocation (45, 47, 52, 58) was impaired in the presence of TGF-β.
We and others have demonstrated an essential role for GATA-3 in Th2 differentiation (18, 59). Inhibition of GATA-3 function in mice limits Th2 responses in a model of allergic airway inflammation (19). We have also shown that the p50 subunit of NF-κB plays a nonredundant role in GATA-3 gene expression in developing Th2 cells (12), most likely involving the classic NF-κB complex, the p50:p65 heterodimer, that is activated by TCR signaling. NF-κB, however, does not appear to be a target of TGF-β. Instead our data suggest that the inhibition of GATA-3 expression by TGF-β may be secondary to inhibition of NFATc activation by TGF-β since CsA inhibited not only NFATc translocation but also GATA-3 expression and inhibition of a constitutively active mutant of NFATc restored GATA-3 expression and also Th2 cytokine production. The precise mechanism by which NFATc controls GATA-3 expression remains to be determined. It is to be noted that multiple NFAT-binding sequences can be identified in the 5′-flanking region of the GATA-3 gene.
Activation of transcription factors such as NF-κB and NFATc, that play crucial roles in orchestrating multiple responses in T cells including proliferation, differentiation, and apoptosis, is dependent on Ca2+ mobilization. There are two stages of Ca2+ mobilization in T and B cells which are coupled to differential transcription factor activation through various signaling intermediates. TCR stimulation leads to an initial rapid but transient peak of Ca2+ influx. This is triggered by TCR-induced activation of PLC-γ which cleaves phosphatidylinositol 4,5 biphospahate (PIP2) to inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 then binds to IP3 receptors on the endoplasmic reticulum which triggers release of intracellular stores of Ca2+ into the cytoplasm. This initial rise in [Ca2+]i then signals influx of Ca2+ across the plasma membrane which is mediated by specialized depletion-activated channels called calcium-release-activated Ca2+ channels (CRAC; reference 60). The initial [Ca2+]i spike is sufficient to activate NF-κB (38). However, other transcription factors such as NFAT require more sustained [Ca2+]i levels (37, 38). Our results show that while TGF-β does not affect the initial rapid increase in [Ca2+]i, it compromises the duration of the Ca2+ signal which is consistent with our observation that the activation of NFAT, but not of NF-κB, is affected by TGF-β.
After TCR ligation or engagement of IgM on B cells, nonreceptor tyrosine kinases including members of the Src and Syk family are activated by phosphorylation, which in turn activate another family of kinases, the Tec kinases, which include Itk/Tsk/Emt, TxK/Rlk, and Tec in T cells and Btk in B cells which play an important role in transmitting signals that ultimately result in Ca2+ mobilization and appropriate responses in T cells and B cells (17, 47, 52, 61). It is believed that the Tec kinases are involved in sustained Ca2+ influx through capacitative calcium entry which requires prolonged activation of PLC-γ in stimulated lymphocytes (46). Gene targeting studies showed that Btk- or Itk- or Itk/Rlk-deficiency affects PLC-γ activation as well as Ca2+ mobilization and MAPK activation after TCR ligation despite preservation of normal patterns of tyrosine phosphorylation and maintenance of the initial rise in [Ca2+]i due to release from intracellular stores (46, 52, 58). The phosphorylation of Itk has been shown to depend on the activation of both Lck and ZAP70 although Itk does not appear to be a direct substrate of ZAP-70. As our studies did not reveal any inhibition of Lck or ZAP70 phosphorylation by TGF-β, lack of Itk phosphorylation in the presence of TGF-β may be due to blockade by TGF-β of Itk recruitment to the membrane complex that is necessary for Itk phosphorylation and activation. It is also possible that the kinase that phosphorylates Itk is a more proximal target of TGF-β.
Itk has been shown to be essential for Th2 responses in vivo (62). However, stimulated CD4+ T cells from Itk-deficient mice are unable to mount Ca2+ flux across the plasma membrane that is essential for NFATc activation (58). As an increase in [Ca2+]i is important for both Th1 and Th2 differentiation and the subsequent translocation of NFATc also appears to be important for both Th1 and Th2 responses, it stands to reason that Itk is important for both Th1 and Th2 differentiation. By establishing Itk as a target of TGF-β–mediated immunosuppression, our studies strengthen the importance of Tec kinases in controlling critical downstream events in T cell differentiation such as the induction of sustained [Ca2+]i levels and the nuclear localization of NFAT. It will be interesting to determine the precise relationship between proximal signaling events such as Tec kinase phosphorylation, Ca2+ influx and NFATc and the expression of the transcription factors T-bet in Th1 cells and GATA-3 in Th2 cells.
Acknowledgments
We thank P. Ray for helpful discussions, L. Glimcher and P. Schwartzberg for anti-T-bet and anti-Rlk antiserum, respectively, and M. Yoshida for leptomycin B and A. Gambotto for retrovirus production.
Supported by grants AI 48927, HL 60995, and P50-HL56389 from the National Institutes of Health (to A. Ray).
The online version of this article contains supplemental material.
C.-H. Chen and C. Seguin-Devaux contributed equally to this work.
Footnotes
Abbreviations used in this paper: EMSA, electrophoretic mobility shift assay; ERK, extracellular signal–regulated kinase; IP3, inositol 1,4,5-triphosphate; LMB, leptomycin B; MAP, mitogen-activated protein; NF, nuclear factor; PMA, phorbol myristate acetate.
References
- 1.Cerwenka, A., and S.L. Swain. 1999. TGF-beta1: immunosuppressant and viability factor for T lymphocytes. Microbes Infect. 1:1291–1296. [DOI] [PubMed] [Google Scholar]
- 2.Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, et al. 1992. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorelik, L., and R.A. Flavell. 2000. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 12:171–181. [DOI] [PubMed] [Google Scholar]
- 4.Kehrl, J.H., L.M. Wakefield, A.B. Roberts, S. Jakowlew, M. Alvarez-Mon, R. Derynck, M.B. Sporn, and A.S. Fauci. 1986. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 163:1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sad, S., and T.R. Mosmann. 1994. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J. Immunol. 153:3514–3522. [PubMed] [Google Scholar]
- 6.Haneda, K., K. Sano, G. Tamura, T. Sato, S. Habu, and K. Shirato. 1997. TGF-beta induced by oral tolerance ameliorates experimental tracheal eosinophilia. J. Immunol. 159:4484–4490. [PubMed] [Google Scholar]
- 7.Hansen, G., J.J. McIntire, V.P. Yeung, G. Berry, G.J. Thorbecke, L. Chen, R.H. DeKruyff, and D.T. Umetsu. 2000. CD4(+) T helper cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. J. Clin. Invest. 105:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakao, A., S. Miike, M. Hatano, K. Okumura, T. Tokuhisa, C. Ra, and I. Iwamoto. 2000. Blockade of transforming growth factor beta/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J. Exp. Med. 192:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray, A., and L. Cohn. 1999. Th2 cells and GATA-3 in asthma: new insights into the regulation of airway inflammation. J. Clin. Invest. 104:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glimcher, L.H., and K.M. Murphy. 2000. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 14:1693–1711. [PubMed] [Google Scholar]
- 11.Kuo, C.T., and J.M. Leiden. 1999. Transcriptional regulation of T lymphocyte development and function. Annu. Rev. Immunol. 17:149–187. [DOI] [PubMed] [Google Scholar]
- 12.Das, J., C.-H. Chen, L. Yang, L. Cohn, P. Ray, and A. Ray. 2001. A critical role for NF-kB in Gata3 expression and Th2 differentiation in allergic airway inflammation. Nat. Immunol. 2:45–50. [DOI] [PubMed] [Google Scholar]
- 13.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 14.Murphy, K.M., W. Ouyang, J.D. Farrar, J. Yang, S. Ranganath, H. Asnagli, M. Afkarian, and T.L. Murphy. 2000. Signaling and transcription in T helper development. Annu. Rev. Immunol. 18:451–494. [DOI] [PubMed] [Google Scholar]
- 15.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 16.Szabo, S.J., B.M. Sullivan, C. Stemmann, A.R. Satoskar, B.P. Sleckman, and L.H. Glimcher. 2002. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 295:338–342. [DOI] [PubMed] [Google Scholar]
- 17.Debnath, J., M. Chamorro, M.J. Czar, E.M. Schaeffer, M.J. Lenardo, H.E. Varmus, and P.L. Schwartzberg. 1999. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol. Cell. Biol. 19:1498–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang, D.-H., L. Cohn, P. Ray, K. Bottomly, and A. Ray. 1997. Transcription factor GATA-3 is differentially expressed in Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272:21597–21603. [DOI] [PubMed] [Google Scholar]
- 19.Zhang, D.H., L. Yang, L. Cohn, L. Parkyn, R. Homer, P. Ray, and A. Ray. 1999. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 11:473–482. [DOI] [PubMed] [Google Scholar]
- 20.Yang, L., L. Cohn, D.H. Zhang, R. Homer, A. Ray, and P. Ray. 1998. Essential role of nuclear factor kappaB in the induction of eosinophilia in allergic airway inflammation. J. Exp. Med. 188:1739–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter, C.M., and N.A. Clipstone. 2002. Sustained NFAT signaling promotes a Th1-like pattern of gene expression in primary murine CD4+ T cells. J. Immunol. 168:4936–4945. [DOI] [PubMed] [Google Scholar]
- 22.Gibson, S., A. August, Y. Kawakami, T. Kawakami, B. Dupont, and G.B. Mills. 1996. The EMT/ITK/TSK (EMT) tyrosine kinase is activated during TCR signaling. J. Immunol. 156:2716–2722. [PubMed] [Google Scholar]
- 23.Gibson, S., K. Truitt, Y. Lu, R. Lapushin, H. Khan, J.B. Imboden, and G.B. Mills. 1998. Efficient CD28 signaling leads to increases in kinase activities of the TEC family tyrosine kinase EMT/ITK/TSK and the SRC family tyrosine kinase LCK. Biochem. J. 330:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandenberghe, P.A., and J.L. Ceuppens. 1990. Flow cytometric measurement of cytoplasmic free calcium in human peripheral blood T lymphocytes with fluo-3, a new fluorescent calcium indicator. J. Immunol. Methods. 127:197–205. [DOI] [PubMed] [Google Scholar]
- 25.Jain, J., P.G. McCaffrey, V.E. Valge-Archer, and A. Rao. 1992. Nuclear factor of activated T cells contains Fos and Jun. Nature. 356:801–804. [DOI] [PubMed] [Google Scholar]
- 26.Rengarajan, J., S.J. Szabo, and L.H. Glimcher. 2000. Transcriptional regulation of Th1/Th2 polarization. Immunol. Today. 21:479–483. [DOI] [PubMed] [Google Scholar]
- 27.Heath, V.L., E.E. Murphy, C. Crain, M.G. Tomlinson, and A. O'Garra. 2000. TGF-beta1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur. J. Immunol. 30:2639–2649. [DOI] [PubMed] [Google Scholar]
- 28.Gorelik, L., P.E. Fields, and R.A. Flavell. 2000. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 165:4773–4777. [DOI] [PubMed] [Google Scholar]
- 29.Ranger, A.M., M.R. Hodge, E.M. Gravallese, M. Oukka, L. Davidson, F.W. Alt, F.C. de la Brousse, T. Hoey, M. Grusby, and L.H. Glimcher. 1998. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity. 8:125–134. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida, H., H. Nishina, H. Takimoto, L.E. Marengere, A.C. Wakeham, D. Bouchard, Y.Y. Kong, T. Ohteki, A. Shahinian, M. Bachmann, et al. 1998. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity. 8:115–124. [DOI] [PubMed] [Google Scholar]
- 31.Loh, C., K.T. Shaw, J. Carew, J.P. Viola, C. Luo, B.A. Perrino, and A. Rao. 1996. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J. Biol. Chem. 271:10884–10891. [DOI] [PubMed] [Google Scholar]
- 32.Shibasaki, F., E.R. Price, D. Milan, and F. McKeon. 1996. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature. 382:370–373. [DOI] [PubMed] [Google Scholar]
- 33.Elefanty, A.G., M. Antoniou, N. Custodio, M. Carmo-Fonseca, and F.G. Grosveld. 1996. GATA transcription factors associate with a novel class of nuclear bodies in erythroblasts and megakaryocytes. EMBO J. 15:319–333. [PMC free article] [PubMed] [Google Scholar]
- 34.Chen, C.H., D.H. Zhang, J.M. LaPorte, and A. Ray. 2000. Cyclic AMP activates p38 mitogen-activated protein kinase in Th2 cells: phosphorylation of GATA-3 and stimulation of Th2 cytokine gene expression. J. Immunol. 165:5597–5605. [DOI] [PubMed] [Google Scholar]
- 35.Feske, S., R. Draeger, H.H. Peter, K. Eichmann, and A. Rao. 2000. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J. Immunol. 165:297–305. [DOI] [PubMed] [Google Scholar]
- 36.Nishi, K., M. Yoshida, D. Fujiwara, M. Nishikawa, S. Horinouchi, and T. Beppu. 1994. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem. 269:6320–6324. [PubMed] [Google Scholar]
- 37.Timmerman, L.A., N.A. Clipstone, S.N. Ho, J.P. Northrop, and G.R. Crabtree. 1996. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 383:837–840. [DOI] [PubMed] [Google Scholar]
- 38.Dolmetsch, R.E., R.S. Lewis, C.C. Goodnow, and J.I. Healy. 1997. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 386:855–858. [DOI] [PubMed] [Google Scholar]
- 39.Kim, J.I., I.-C. Ho, M.J. Grusby, and L.H. Glimcher. 1999. The transcription factor c-Maf controls the production of IL-4 but not other Th2 cytokines. Immunity. 10:745–751. [DOI] [PubMed] [Google Scholar]
- 40.Sloan-Lancaster, J., T.H. Steinberg, and P.A. Allen. 1997. Selective loss of the calcium ion signaling pathway in T cells maturing toward a T helper 2 phenotype. J. Immunol. 159:1160–1168. [PubMed] [Google Scholar]
- 41.Gorham, J.D., M.L. Guler, D. Fenoglio, U. Gubler, and K.M. Murphy. 1998. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J. Immunol. 161:1664–1670. [PubMed] [Google Scholar]
- 42.Gorelik, L., S. Constant, and R.A. Flavell. 2002. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 195:1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sica, A., L. Dorman, V. Viggiano, M. Cippitelli, P. Ghosh, N. Rice, and H.A. Young. 1997. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J. Biol. Chem. 272:30412–30420. [DOI] [PubMed] [Google Scholar]
- 44.Sweetser, M.T., T. Hoey, Y.-L. Sun, W.M. Weaver, G.A. Price, and C.B. Wilson. 1998. The roles of nuclear factor of activated T cells and ying-yang 1 in activation-induced expression of interferon-γ promoter in T cells. J. Biol. Chem. 273:34775. [DOI] [PubMed] [Google Scholar]
- 45.Schaeffer, E.M., and P.L. Schwartzberg. 2000. Tec family kinases in lymphocyte signaling and function. Curr. Opin. Immunol. 12:282–288. [DOI] [PubMed] [Google Scholar]
- 46.Scharenberg, A.M., and J.P. Kinet. 1998. PtdIns-3,4,5-P3: a regulatory nexus between tyrosine kinases and sustained calcium signals. Cell. 94:5–8. [DOI] [PubMed] [Google Scholar]
- 47.Bunnell, S.C., M. Diehn, M.B. Yaffe, P.R. Findell, L.C. Cantley, and L.J. Berg. 2000. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 275:2219–2230. [DOI] [PubMed] [Google Scholar]
- 48.Yang, W.C., M. Ghiotto, R. Castellano, Y. Collette, N. Auphan, J.A. Nunes, and D. Olive. 2000. Role of tec kinase in nuclear factor of activated T cells signaling. Int. Immunol. 12:1547–1552. [DOI] [PubMed] [Google Scholar]
- 49.Schaeffer, E.M., G.S. Yap, C.M. Lewis, M.J. Cza, D.W. McVicar, A.W. Cheever, A. Sher, and P.L. Schwartzberg. 2001. Mutation of Tec family kinases alters T helper cell differentiation. Nat. Immunol. 2:1183–1188. [DOI] [PubMed] [Google Scholar]
- 50.Cahalan, M.D., I. Parker, S.H. Wei, and M.J. Miller. 2002. Two-photon tissue imaging:seeing the immune system in a fresh light. Nat. Rev. Immunol. 2:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan, X., and R.L. Wange. 1999. Itk/Emt/Tsk activation in response to CD3 cross-linking in Jurkat T cells requires ZAP-70 and Lat and is independent of membrane recruitment. J. Biol. Chem. 274:29323–29330. [DOI] [PubMed] [Google Scholar]
- 52.Schaeffer, E.M., J. Debnath, G. Yap, D. McVicar, X.C. Liao, D.R. Littman, A. Sher, H.E. Varmus, M.J. Lenardo, and P.L. Schwartzberg. 1999. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 284:638–641. [DOI] [PubMed] [Google Scholar]
- 53.Zhu, J., H. Huang, L. Guo, T. Stonehouse, C.J. Watson, J. Hu-Li, and W.E. Paul. 2000. Transient inhibition of interleukin 4 signaling by T cell receptor ligation. J. Exp. Med. 192:1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong, C., D.D. Yang, C. Tournier, A.J. Whitmarsh, J. Xu, R.J. Davis, and R.A. Flavell. 2000. JNK is required for effector T-cell function but not for T-cell activation. Nature. 405:91–94. [DOI] [PubMed] [Google Scholar]
- 55.Porter, C.M., M.A. Havens, and N.A. Clipstone. 2000. Identification of amino acid residues and protein kinases involved in the regulation of NFATc subcellular localization. J. Biol. Chem. 275:3543–3551. [DOI] [PubMed] [Google Scholar]
- 56.Beals, C.R., C.M. Sheridan, C.W. Turck, P. Gardner, and G.R. Crabtree. 1997. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 275:1930–1934. [DOI] [PubMed] [Google Scholar]
- 57.Ludviksson, B.R., D. Seegers, A.S. Resnick, and W. Strober. 2000. The effect of TGF-beta1 on immune responses of naive versus memory CD4+ Th1/Th2 T cells. Eur. J. Immunol. 30:2101–2111. [DOI] [PubMed] [Google Scholar]
- 58.Liu, K.Q., S.C. Bunnell, C.B. Gurniak, and L.J. Berg. 1998. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med. 187:1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng, W., and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 60.Putney, J.W., Jr. 1986. A model for receptor-regulated calcium entry. Cell Calcium. 7:1–12. [DOI] [PubMed] [Google Scholar]
- 61.Scharenberg, A.M., O. El-Hillal, D.A. Fruman, L.O. Beitz, Z. Li, S. Lin, I. Gout, L.C. Cantley, D.J. Rawlings, and J.P. Kinet. 1998. Phoasphatidylinositol-3,4,5-triphosphate (PtdIns-3,4,5-P3)/Tec kinase-dependent calcium signaling pathway: a target for SHIP-mediated inhibitory signals. EMBO J. 17:1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fowell, D.J., K. Shinkai, X.C. Liao, A.M. Beebe, R.L. Coffman, D.R. Littman, and R.M. Locksley. 1999. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 11:399–409. [DOI] [PubMed] [Google Scholar]