

Abstract
We describe here a previously unrecognized property of dendritic cells (DCs), the ability to deacylate the lipid A moiety of gram-negative bacterial LPSs. Both immature DCs of the XS52 cell line and bone marrow–derived DCs produce acyloxyacyl hydrolase, an enzyme that detoxifies LPS by selectively removing the secondary acyl chains from lipid A. Acyloxyacyl hydrolase expression decreased when DCs were incubated with IL-4, IL-1β, TNFα, and an agonistic CD40 antibody (maturation cocktail), and increased after treatment with LPS, CpG oligodeoxynucleotides, or a gram-positive bacterium (Micococcus luteus). Maturation cocktail treatment also diminished, whereas LPS treatment enhanced or maintained the cells' ability to kill Escherichia coli, deacylate LPS, and degrade bacterial protein. Enzymatic deacylation of LPS is an intrinsic, regulated mechanism by which DCs may modulate host responses to this potent bacterial agonist.
Keywords: lipopolysaccharide, dendritic cell, acyoxyacyl hydrolase, gram-negative bacteria, deacylation
Introduction
Dendritic cells (DCs),* the most potent antigen-presenting cells, initiate and modulate both innate and adaptive immune responses. Immature DCs reside in nonlymphoid tissues, such as the skin, where they are poised to capture and process microbial invaders. After an encounter with bacteria or bacterial components, immature DCs migrate via lymphatic channels to the T cell areas of regional lymph nodes, where they mature, losing their antigen-capturing and -processing ability and becoming specialized for presenting antigens to T cells (1–3). DC maturation can be induced in vitro and in vivo by various stimuli, such as inflammatory cytokines (TNFα and IL-1β), CD40 ligand, and several conserved microbial molecules (LPS, peptidoglycans, bacterial lipoproteins, DNA that contains unmethylated CpG motifs, and viral double-stranded RNA; references 2, 4). Different agonists may drive distinct maturation events and act synergistically to induce the maturation of DCs (5–7).
In addition to inducing adaptive immune responses to microbial antigens, DCs also contribute to innate immunity by ingesting and killing microbes and by secreting mediators that recruit macrophages, natural killer cells, and eosinophils to sites of infection (2, 4). However, little is known about how immature DCs help control bacterial infection and/or prevent harmful host responses to bacteria or bacterial components. In the studies described here, we show that these cells are able to degrade the most potent agonist contained in the gram-negative bacterial cell wall, the lipid A moiety of LPS.
Lipid A, the conserved bioactive center of LPS, has a glucosamine disaccharide backbone. In enterobacterial lipid A, four molecules of 3-hydroxytetradecanoate (3-OH-14:0) attach directly to this backbone. The hydroxyl groups of two or three of the 3-OH-14:0 residues are substituted with secondary acyl chains (laurate and myristate) to form acyloxyacyl groups (Fig. 1 A). Animals have an enzyme, acyloxyacyl hydrolase (AOAH), that removes secondary fatty acyl chains from the lipid A regions of diverse LPSs and greatly reduces the molecules' ability to elicit toxic responses in vivo (8). Moreover, in vitro studies using human monocytes, endothelial cells, and neutrophils have shown that the partially deacylated LPS produced by AOAH can be an LPS antagonist (9–11). Mice that are unable to produce AOAH due to targeted gene disruption have exaggerated antibody responses to LPS (unpublished results).
Although AOAH had been detected previously only in neutrophils and monocyte macrophages, we found that immature DCs also produce the enzyme. We studied the ability of DCs to alter their expression of AOAH, and their ability to deacylate LPS in ingested Escherichia coli in response to host (inflammatory cytokines and CD40 ligand) or bacterial (LPS, E. coli, Micrococcus luteus [peptidoglycan], and CpG oligonucleotides) stimuli. The results indicate that DCs coordinate AOAH expression and LPS deacylation with many other antibacterial responses, increasing or decreasing their ability to process this important bacterial molecule in response to environmental cues.
Materials and Methods
Materials.
Unless otherwise indicated, reagents were obtained from Sigma-Aldrich. Phosphorothioate-modified CpG (5′-TCCATGACGTTCCTGATGCT-3′) and GpC (5′-TCCATGAGCTTCCTGATGCT-3′) oligodeoxynucleotides (ODNs) were obtained from Invitrogen.
AOAH−/− Mice.
Targeted disruption of the murine AOAH gene was accomplished in embryonic stem cells from 129S6/SvEvTac mice by inserting a neomycin resistance gene in the first AOAH exon, and by eliminating a 705-bp region that encodes untranslated mRNA, the translation start site, the leader and pro-peptide sequences, and 41 amino acids of the small subunit of the enzyme (12). Mouse DNA was screened by Southern blot analysis using an EcoRI–BamHI probe derived from the 5′ genomic sequence upstream of the long arm of the targeting vector. For the experiments described here, peritoneal macrophages and bone marrow–derived DCs (BMDCs) were isolated from AOAH knockout and wild-type 129S6/SvEvTac mice.
Cell Lines.
XS52 is an immature DC line derived from newborn BALB/c mouse epidermis (13). XS52 cells were maintained in XS medium (complete RPMI [cRPMI] supplemented with 0.5 ng/ml recombinant murine GM-CSF [rmGM-CSF; R&D Systems] and culture supernatant [5%, vol/vol] from a NS47 stromal cell line. cRPMI is RPMI 1640 (Cellgro) with 10% heat-inactivated FBS (endotoxin < 0.06 EU/ml; Hyclone), 2 mM l-glutamine (GIBCO BRL), 100 μM of nonessential amino acids (GIBCO BRL), 100 U/ml penicillin, 0.1 mg/ml streptomycin (GIBCO BRL), 10 μM of sodium pyruvate, 25 mM Hepes, pH 7.4, and 50 μM 2-mercaptoethanol. NS47 cells (13) were cultured in cRPMI medium. XS106, a mature DC line established from A/J mice (14), was also cultured in XS medium. The BALB/c-derived macrophage line RAW264.7 was maintained in DMEM (Cellgro) supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Murine macrophage cell lines P388D1 and J774 were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin.
BMDCs and Peritoneal Macrophages.
DCs were generated from bone marrow as described by Inaba et al. (15) with minor modifications. Marrow was flushed out of the femurs and tibias. Red blood cells were lysed using RBC lysing buffer and cells were cultured in 6-well plates at 6 × 106 cells per well in 3 ml cRPMI medium supplemented with 10 ng/ml rmGM-CSF. On day 3 of culture, nonadherent cells were removed, and 4 ml of fresh medium containing rmGM-CSF were added. On day 7, nonadherent CD11c+ cells were purified using anti-CD11c monoclonal antibody N418 coupled to magnetic microbeads (Miltenyi Biotec). Fc receptors were blocked with 0.5 mg/ml of normal mouse IgG (Caltag Laboratories) before the cells were incubated with the beads. Flow cytometric analysis (FACScan™; Becton Dickinson) showed that >95% of the sorted cells were CD11c+. These purified CD11c+ cells had DC morphology (veiled and dendritic processes) when viewed using light microscopy. Thioglycollate-elicited peritoneal macrophages were prepared as described previously (16). All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and air.
Induction of DC Differentiation.
We followed the method of Yamada and Katz (17). In brief, XS52 cells were cultured in cRPMI medium supplemented with rmGM-CSF and IL-4 (10 ng/ml each) for 6 d; the medium was replaced with fresh medium containing rmGM-CSF and IL-4 on day 3. The cells were cultured for three additional days in the presence of GM-CSF, IL-4, TNF-α (10 ng/ml each, all from R&D Systems), 10 ng/ml IL-1β (BD Biosciences), and 4 μg/ml anti-CD40 (clone 1C40; R&D Systems). To study the impact of LPS on XS52 cell maturation, 10 ng/ml of E. coli O9 LPS (purified using hot phenol-water extraction from E. coli) was added to the cells in XS medium. Cells were either harvested at 24 h or they were incubated for 9 d; the medium was replaced with fresh XS52 medium containing 10 ng/ml LPS on days 3 and 6. To study the impact of living E. coli on DC maturation, 105 CFU/ml of an overnight culture of E. coli O9 was added to XS52 cells (bacteria/cell ratio = 1:5) in XS medium without antibiotics and incubated at 37°C for 1 h. The cells were washed with cRPMI medium without antibiotics, cultured in XS medium that contained 50 μg/ml gentamicin, and were harvested at 24 h. When incubation was continued for 9 d, this procedure was repeated on days 3 and 6, and the cells were harvested on day 9.
To induce the maturation of BMDCs, 10 ng/ml LPS, 1 μM CpG ODN, and control GpC ODN, 40 μg/ml M. luteus or a cocktail of IL-4, TNFα, IL-1β, and agonistic anti-CD40 antibody were added to day 5 BMDC cultures. On day 7, the nonadherent cells were harvested, and CD11c+ cells were sorted by CD11c monoclonal antibody–conjugated magnetic microbeads.
Deacylation of Purified LPS.
Double-labeled LPS (14C-glucosamine backbone and 3H-fatty acyl chains; reference 18) was incubated with cell lysates for 16 h at 37°C in AOAH reaction mixture (1 mg/ml of fatty acid–free bovine serum albumin, 5 mM CaCl2, 0.05% Triton X-100, and 20 mM Tris-citrate, pH 5). The reaction was terminated by adding 2.5 volumes of 100% ethanol. After centrifugation to precipitate intact LPS and partially deacylated LPS, the 3H-fatty acids in the supernatant were quantitated by β-scintillation counting (Packard Instrument Co.). Confirmation that the deacylating activity was AOAH-like (releasing only 12:0 and 14:0 from the LPS) was obtained using TLC (19).
Deacylation of LPS in E. coli.
E. coli LCD25 is unable to produce its own acetate or use acetate as carbon or energy source (20). When LCD25 cells are cultured in minimal medium with sodium 2-[14C]acetate (NEN Life Science Products), 14C is exclusively incorporated into fatty acyl chains. The method of Katz et al. (21) was followed to 14C-label fatty acyl chains in LCD25, yielding ∼20,000 dpm /106 CFU. 14C-Labeled LCD25 bacteria were added to XS52 cells (cell/bacteria ratio = 1:50) in XS medium without antibiotics and incubated for 1 h at 37°C. The cells were washed and incubated for 6 or 24 h in XS medium with 50 μg/ml gentamicin to kill extracellular bacteria. For BMDCs, nonadherent cells were harvested on day 7 of culture, mixed with 14C-labeled LCD25 (cell/bacteria ratio = 1:50) in cRPMI medium without antibiotics, and incubated at 37°C for 1 h. The CD11c+ cells were purified by magnetic microbeads and the CD11c− cells and unbound bacteria were washed away. The sorted CD11c+ cells were cultured in cRPMI supplemented with 10 ng/ml rmGM-CSF and 50 μg/ml gentamicin for 6 or 24 h. The cells and culture media were pooled to measure LPS deacylation. At the 0-h time point, samples were harvested immediately after 14C-labeled LCD25 bacteria were added to DCs.
LPS deacylation was quantitated by calculating the loss of the fatty acyl chains from LPS (21). In brief, the interphase of a Bligh-Dyer extraction (22), which contains LPS and partially deacylated LPS, was washed three times with chloroform and dried under argon. The interphase was hydrolyzed with 4 M HCl and 4 M NaOH (16). The hydrolyzed interphase was extracted again and the chloroform phase, which contains released fatty acids, was recovered, dried, and resuspended in 100 μl methanol/chloroform 1:1 (vol/vol). The interphase-derived fatty acids were resolved by TLC. First, the primary (3-OH-14:0) and secondary (12:0 and 14:0) fatty acids were separated using TLC system 1 (silica gel G, petroleum ether/diethyl ether/acetic acid = 70:30:1). The two bands containing 3-OH-14:0 and the nonhydroxylated fatty acids were visualized by phosphorimager (Molecular Dynamics). The 3-OH-14:0 band was scraped and the radioactivity was quantitated by β-scintillation counting. The nonhydroxylated fatty acids were extracted from the silica gel and resolved on a reverse-phase KC18 plate, using acetic acid/acetonitrile (3:7) as the solvent (TLC, system 2); the plates were developed, dried, and developed again to get a better separation of fatty acids. The bands corresponding to each species of nonhydroxylated fatty acid (12:0, 14:0, and 16:0) were scraped and quantitated. The disintegrations per minute recovered from each band were normalized to the total disintegrations per minute measured for each sample before extraction. The values at t = 0 h were set as 100%, and the values at other time points were converted to percentages by comparing them to the t = 0–h value. For example, 12:0 attached to LPS at 24 h (%) = (12:0 recovered at 24 h/total dpm of 24-h sample before extraction)/(12:0 recovered at 0 h/total dpm of 0-h sample before extraction) × 100. The distribution of the radioactivity in each sample was assayed in duplicate.
At the end of the maturation experiment, there were more cells in the cytokine-treated wells, and fewer in the LPS-treated wells, than in the untreated wells. The cell mass was estimated by measuring the amount of cell protein in each well.
Bacterial Protein Degradation.
To label bacterial proteins, LCD25 cells were cultured in minimal medium plus 1 mM sodium acetate with 0.04 μM 3H-arginine (NEN Life Science Products) and 10 μM of nonradiolabeled l-arginine, yielding ∼3,500 3H dpm/106 bacterial CFUs. 3H-Arginine–labeled LCD25 were added to XS52 cells (cell/bacteria ratio = 1:50). Incubation, washing, and harvesting were performed as in the LPS deacylation protocol described in previous paragraphs. The 0-h time point samples were harvested immediately after the 3H-arginine–labeled LCD25 bacteria were added to DCs. 0.5 ml of each sample was mixed with an equal volume of 20% TCA, incubated at 4°C for 20 min, and centrifuged. The radioactivities in the supernatant and pellet were counted. The fraction of the counts that was TCA-insoluble at 0 h was considered to represent 100%, and the values at later time points were converted to percentages by comparing them to the t = 0–h value. Each experimental condition was assayed in duplicate.
Flow Cytometry.
XS52 cells were washed with PBS and harvested with PBS containing 2 mM EDTA. BMDCs were purified by magnetic cell sorting. After blocking Fc binding with 0.5 mg/ml of normal mouse IgG, cells were incubated with anti–CD14-PE (rmC5–3), anti–CD40-PE (3:23), anti–CD86-PE (GL1), anti–CD11c-FITC (HL3), and corresponding isotype controls. These antibodies were all purchased from BD Biosciences and used at 5 μg/ml. The samples were analyzed by FACScan™, gating on cells that excluded propidium iodide. Data were analyzed using CELLQuest™ (Becton Dickinson) software.
Phagocytosis.
106 DCs were washed with PBS and suspended in 0.2 ml cRPMI medium. 0.05 ml of 109 bacteria/ml Bodipy–E. coli (Molecular Probes) was added to the cells, which were incubated at 37°C in the dark for 1 h before they were chilled on ice to stop phagocytosis. To quench extracellular fluorescence, 0.25 ml of 0.2% trypan blue in PBS was added before analysis by flow cytometry. Control cells were either pretreated with 10 μM cytochalasin D for 0.5 h before adding Bodipy–E. coli.
Bactericidal Activity.
LCD25 bacteria, labeled with 3H-arginine as described in a previous paragraph (Bacterial Protein Degradation), were added to XS52 cells in XS medium without antibiotics. Bacteria were added at a cell/bacteria ratio of 1:50. After incubation at 37°C for 0.5 h, the cells were washed and incubated for 0.5 or 2 h in XS medium without antibiotics. Cells and media were harvested together and cells were lysed by adding Triton X-100 (final concentration = 0.2%). Samples were serially diluted in PBS, and 100 μl of several dilutions were spread on LB agar plates. After overnight incubation at 37°C, the colonies were counted and the total number of CFUs recovered from each well was calculated. The total number of bacteria associated with cells after the wash was determined by measuring the cell-associated 3H dpm at this time and dividing by the 3H dpm/CFU.
mRNA Analysis.
Total RNA was isolated from untreated or treated XS52 cells (RNAqueous Kit; Ambion). A region of the AOAH cDNA was amplified using primers Seq_mAOAH-ex12F (5′- CCAACTCTCTGGTGTAACTGGATTT-3′) and Seq_mAOAH-ex12R (5′-TCTCAAACGATGGTAAATGGATTTT-3′). The probe (TaqMan® MGB, FAM™ dye-labeled) 5′-ACGAGTGGAATTGAAG-3′ and primers were designed and synthesized by Applied Biosystems. Plasmid PMF612, which contains murine AOAH cDNA, was used as a reference molecule for the standard curve calculation. TaqMan® rodent GAPDH control reagents were used to measure GAPDH gene expression. All real-time PCR reactions were performed in a 25-μl mixture with TaqMan® one-step RT-PCR Master Mix Reagents Kit on a sequence detection system (model ABI PRISM® 7700).
Results
Immature (XS52) DCs and BMDCs Express AOAH.
We first measured LPS-deacylating activity in lysates of XS52 cells, immature BMDCs, a mature DC line, XS106, and several murine macrophage lines. Both XS52 cells and BMDCs had LPS-deacylating activity similar to that of murine peritoneal macrophages and greater than that of the other cell lines tested (Fig. 1 B). Mature DCs (XS106) expressed much lower activity than immature DCs (XS52).
Figure 1.

LPS deacylation by cell lysates. (A) Structure of the lipid A moiety of E. coli LPS. AOAH (arrows) cleaves the secondary fatty acyl chains (laurate, 12:0; myristate, 14:0) attached in ester (acyloxyacyl) linkage to the glucosamine-linked 3-hydroxymyristoyl residues. (B) LPS deacylation by cell lysates. Lysates of cultured cells were assayed for their ability to deacylate purified 3H/14C-LPS as described in Materials and Methods. Each bar shows the mean of three or more independent experiments. Error bars represent 1 SEM. Asterisks, significantly different from XS52 cells: **, P < 0.01; ***, P < 0.001 (Student's t test).
XS52 Cells and BMDCs Deacylate the LPS in Whole Bacteria in an AOAH-like Manner.
In the assay described in the previous paragraph, we used purified LPS as the substrate and measured its deacylation by cell lysates in the presence of detergent. Next, we studied the ability of DCs to deacylate LPS in its natural setting, the outer membrane of gram-negative bacteria. Because the fatty acids cleaved from LPS, and other bacterial lipids can be degraded by host cells and/or incorporated into cellular lipids, we measured the disappearance of individual fatty acids from the LPS backbone. We found that both XS52 cells and BMDCs deacylated the LPS in whole bacteria in an AOAH-like manner (Fig. 2, A and B) . 3-OH-14:0, the primary fatty acyl chain of lipid A, was not removed from the LPS backbone, whereas the nonhydroxylated (secondary) fatty acids (12:0 and 14:0) were cleaved over time. Confirmation that AOAH is the deacylating enzyme came from a comparison of BMDCs from AOAH wild-type (+/+) and AOAH null (−/−) 129 mice. BMDCs from AOAH−/− mice deacylated significantly less LPS than their wild-type counterparts (Fig. 2, B and C); the apparent removal of 20–30% of the 12:0 by AOAH−/− BMDCs−/− is unexplained because they were unable to deacylate purified LPS (unpublished data), and peritoneal macrophages from AOAH−/− mice did not deacylate the LPS in E. coli (Fig. 2, D and E).
Figure 2.

Deacylation of LPS in E. coli. (A) Deacylation of LPS in E. coli by XS52 cells. (B and C) Deacylation of LPS in E. coli by BMDCs from AOAH+/+ (B) and AOAH−/− (C) mice. (D and E) Deacylation of LPS in E. coli by thioglycollate-elicited peritoneal macrophages from AOAH+/+ (D) and AOAH−/− (E) mice. The data in each panel are representative of two or more experiments with similar results. FA, fatty acid.
Regulation of AOAH Activity in XS52 Cells.
In vitro, DCs can be induced to mature by inflammatory cytokines, by CD40 ligand, or by microbes or microbial molecules such as LPS and peptidoglycan. Following the protocol of Yamada and Katz (17), we treated XS52 cells with 10 ng/ml IL-4 for 6 d and for an additional 3 d with a maturation cocktail that included IL-4, TNFα, IL-1β, and an agonistic CD40 antibody. To assess the maturation state of the cells, we measured cell surface markers by using flow cytometry. Increased surface expression of CD40, a costimulatory molecule, was used to reflect maturation (17). The LPS binding receptor, CD14, which is constitutively expressed on the XS52 cell surface, was shown to be down-regulated on XS52 cells treated with IL-4 and maturation cocktail. We also measured the phagocytic activity of untreated and treated cells. Immature DCs, which are highly phagocytic, lose this capability when they mature (5, 23).
After treatment with IL-4 for 6 d and the cytokine cocktail for three more days, CD40 expression on XS52 cells had increased a little (Fig. 3 A), whereas CD14 expression (Fig. 3 A) and phagocytic activity (Fig. 3 B) had decreased. These changes were accompanied by a sixfold decrease in AOAH activity (Fig. 3 C).
Figure 3.


Stimulus-induced changes in XS52 cells. (A) Cell surface expression of CD14 and CD40 on untreated XS52 cells and on cells treated with cytokines (IL-4 and maturation cocktail), 10 ng/ml LPS, or 105 CFU/ml E. coli for 9 d (Materials and Methods). Black line, specific antibody; gray line, control antibody of the same isotype. (B) Flow cytometric analysis of phagocytosis of Bodipy-labeled E. coli. Dotted line, cells only; black line, cells with Bodipy–E. coli; gray line, after pretreatment with cytochalasin D to inhibit phagocytosis. (C) LPS-deacylating activity in cell lysates. Each experimental condition was assayed in duplicate; the results are combined from nine independent experiments. (D) AOAH mRNA abundance as assessed by real-time PCR measurements of AOAH and GAPDH mRNA. The data are combined from two independent experiments. (C and D) Bars represent means +1 SD.
In the same experiments, we asked if exposing the cells to LPS or gram-negative bacteria would alter their ability to phagocytose or to express AOAH. After XS52 cells had been incubated with LPS or whole gram-negative bacteria for 9 d, their surface expression of CD40 increased, whereas CD14 expression, phagocytic activity, and AOAH activity were maintained or slightly increased (Fig. 3, A–C).
Prolonged incubation in the presence of LPS or E. coli was associated with some loss of XS52 cell viability. When we treated XS52 with LPS or E. coli for 24 h, with no loss of cell numbers, the results were similar: maintenance of CD14 expression, phagocytic ability, and AOAH activity were accompanied by increased expression of CD40 (unpublished data).
To investigate the basis for the changes in AOAH activity, we measured AOAH mRNA abundance in XS52 cells using real-time PCR. As shown in Fig. 3 D, AOAH mRNA abundance decreased eightfold after treatment with the maturation cocktail and increased twofold after treatment with LPS.
Maturation of XS52 cells, when induced by proinflammatory cytokines and an agonistic CD40 antibody, was thus associated with decreases in CD14 expression, phagocytic activity, and AOAH activity, whereas CD40 expression was enhanced. Although treatment with LPS or gram-negative bacteria was also followed by increased expression of the costimulatory molecule, CD40, the cells maintained their ability to recognize LPS, internalize bacteria, and deacylate LPS.
Regulation of AOAH Activity in BMDCs.
We treated BMDCs with IL-4 and the same maturation cocktail for 2 d. CD40 and CD86 expression increased (Fig. 4 A), whereas phagocytic activity decreased (Fig. 4 B). Immature BMDCs expressed very little CD14, and treatment with maturation cocktail did not change its expression (unpublished data). Cytokine-induced maturation decreased AOAH activity by sixfold (Fig. 4 C).
Figure 4.

Maturation-induced changes in BMDCs. (A) Cell surface expression of CD40 and CD86 on BMDCs and on BMDCs treated with maturation cocktail (see Materials and Methods), 10 ng/ml LPS, 1 μM CpG ODN, 1 μM GpC ODN, and 40 μg/ml M. luteus for 2 d. Black line, specific antibody; shaded gray histogram, control antibody of the same isotype. (B) Phagocytosis of Bodipy-labeled E. coli. Dotted line, cells only; black line, cells with Bodipy–E. coli; gray line, after pretreatment with cytochalasin D to inhibit phagocytosis. (C) LPS-deacylating activity in cell lysates. Each experimental condition was assayed in duplicate; the results are combined from two or more independent experiments. (C) Bars show the mean +1 SE.
When BMDCs were treated with 10 ng/ml LPS for 2 d, they expressed more surface CD40 and CD86 (Fig. 4 A) and slightly more CD14 (reference 24; unpublished data), whereas their phagocytic activity decreased (Fig. 4 B). AOAH activity increased by threefold (Fig. 4 C). Up-regulation of costimulatory molecules has also been described previously in Salmonella-infected BMDCs in vitro (25) and in response to LPS in vivo (26). Thus, the maturation cocktail and LPS both augment cell surface CD40 and CD86 expression in BMDCs and decrease their phagocytic activity. The maturation cocktail decreases BMDC AOAH activity whereas LPS increases it.
Other Bacterial Agonists Also Increase AOAH Expression in BMDCs.
Next, we wanted to know if the augmentation of AOAH activity is LPS-specific because LPS is an AOAH substrate. LPS is thought to activate DCs by interacting with the toll-like receptor (TLR) 4–MD-2 signaling complex. To find out if cell activation via other TLRs can increase or decrease AOAH expression, we incubated BMDCs with bacterial CpGs (TLR9 agonist; references 27, 28) and M. luteus (gram-positive bacterium; agonist for TLR2 [29], possibly also other TLRs). When we tested the working suspension of each preparation using Limulus amebocyte lysate (Cape Cod Inc.), the endotoxin levels were <0.03 EU/ml. As shown in Fig. 4 C, CpG and M. luteus both increased AOAH activity whereas GpC did not. These results indicate that BMDC AOAH can be regulated via signals downstream of TLRs other than TLR4.
After Exposure to Maturation Cocktail or LPS, XS52 Cells Alter Their Ability to Deacylate LPS in Whole Bacteria.
We explored how different stimuli affect the ability of XS52 cells to deacylate the LPS in whole bacteria. XS52 cells treated with IL-4 for 6 d and maturation cocktail for three additional days were compared with XS52 cells treated with LPS and with control cells that had been maintained without stimulation. The ability of the cells to deacylate LPS in E. coli was abolished in the maturation cocktail–treated group (Fig. 5 B) and was increased by LPS treatment (Fig. 5 C). In parallel experiments, we studied the ability of the cells to degrade bacterial protein. Protein degradation was fourfold lower in maturation cocktail–treated XS52 cells than in untreated cells, whereas LPS treatment maintained the protein degradation rate (Fig. 5 D). Under the conditions studied here, maturation thus altered the cells' ability to degrade bacterial LPS and protein in a similar fashion.
Figure 5.
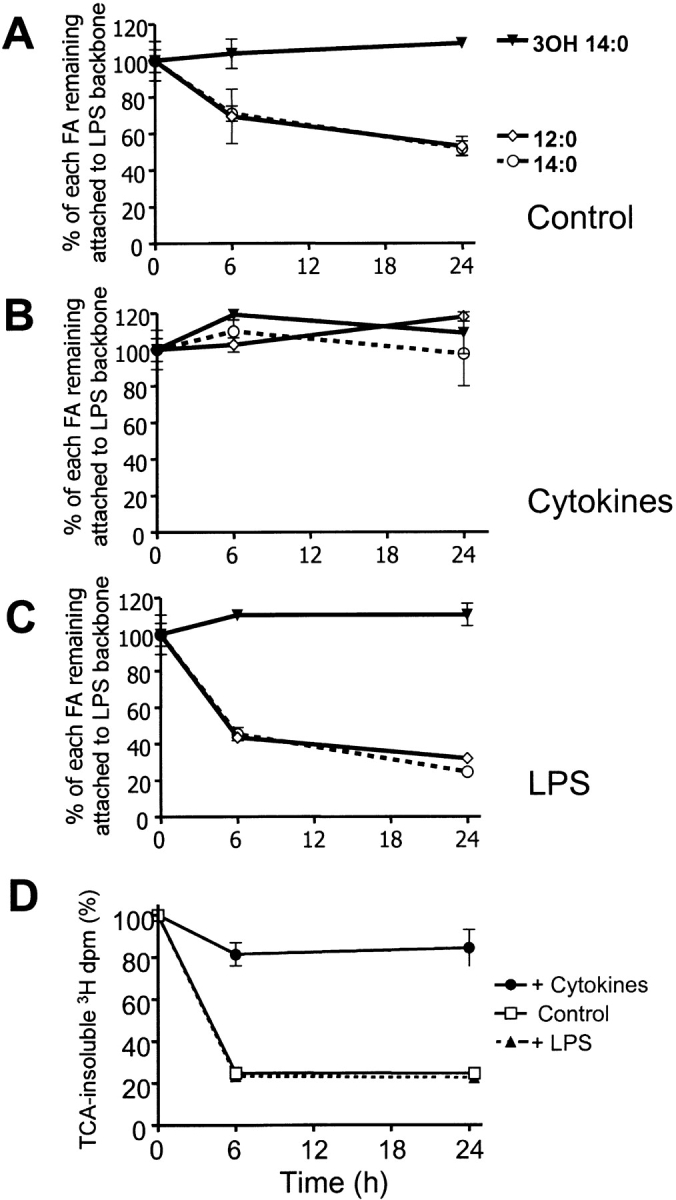
Impact of exogenous stimuli on the deacylation of LPS and degradation of bacterial protein in phagocytosed E. coli. (A–C) XS52 cells, untreated or treated for 9 d with IL-4 and maturation cocktail or LPS (Materials and Methods), were allowed to take up 14C-labeled E. coli for 1 h. After washing to remove unattached cells and further incubation for 6 h, 30% of the 12:0 and 14:0 had been removed from the LPS backbone in untreated cells (A), whereas 60–70% was removed in LPS-treated cells (C, P < 0.05, Student's t test). Cytokine treatment abolished the ability of the cells to deacylate the LPS in E. coli (B). (D) XS52 cells, untreated or treated with IL-4 and maturation cocktail or LPS, were incubated with 3H-arginine–labeled E. coli for 1 h. IL-4 and maturation cocktail treatment decreased protein degradation whereas LPS treatment maintained it. Treatment with IL-4 and maturation cocktail also reduced phagocytosis (see Fig. 3 B); this likely contributed to the decrease in LPS deacylation and protein degradation seen in these cells. The data shown in each panel are from one of three experiments with similar results. Error bars indicate ± 1 SD.
Differentiation Also Influences the Bactericidal Activity of DCs.
IL-4– and maturation cocktail–treated XS52 cells were less able (by 50%) to kill E. coli, whereas LPS treatment maintained or slightly increased killing (Fig. 6) .
Figure 6.

Impact of cell maturation on bactericidal activity Treatment for 9 d with IL-4 and maturation cocktail treatment decreased XS52 cells' ability to kill E. coli by ∼50% (*, P < 0.05, Student's t test), whereas LPS maintained bactericidal activity or increased it by ∼20% (#, P = 0.1). Data shown are mean ± 1 SD from three independent experiments.
Discussion
Deacylation of the lipid A moiety of LPS was first described (30) in Dictyostelium discoideum, a slime mold that digests internalized bacteria as a foodstuff. The discovery that human neutrophils can also carry out LPS deacylation was reported in 1983 (19), and subsequent work identified an LPS-deacylating enzyme, AOAH, in myeloid lineage cells from numerous animals. Mouse, rabbit, and human enzymes have over 70% amino acid sequence identity/similarity (31), whereas D. discoideum and mouse AOAH genes encode proteins that have ∼30% overall amino acid sequence similarity, with identity in four of the five sequence motifs that place the enzyme in the GDSL lipase family (reference 32; unpublished data). Although the enzyme has thus been highly conserved during evolution, the role(s) that it plays in modulating immune responses to LPS are not well understood.
The results of the present experiments provide strong evidence that AOAH is the major, if not the only, mammalian enzyme that deacylates the LPS contained in phagocytosed bacteria. We also identified a previously unsuspected role in LPS deacylation for DCs, key cells in the innate immune response to invading bacteria.
We first found that lysates of immature DCs, whether derived from skin (XS52 cells) or bone marrow, had AOAH activity that was equivalent to the activity found in peritoneal macrophages and considerably greater than that in several macrophage cell lines. Second, both XS52 cells and BMDCs deacylated, in an AOAH-like manner, the LPS contained in the E. coli that they ingested; this ability was greatly diminished in BMDC and macrophages from AOAH-deficient mice, indicating that this enzyme is largely, if not entirely, responsible for LPS deacylation in these cells. The absence of enzymes that remove any of the four glucosamine-linked hydroxylated fatty acids from LPS suggests that animals may have other mechanisms for digesting, and/or disposing of, partially deacylated LPS (33).
The high levels of AOAH activity found in immature DCs and in macrophages raise the possibility that these cells play important roles in regulating the body's responses to bioactive LPS. In this regard, it is intriguing that AOAH-deficient mice have exaggerated antibody responses to LPS (unpublished results); because the ability of extracellular AOAH to deacylate LPS is quite limited (34) and B cells do not produce the enzyme, partial deacylation of LPS by phagocytes may be required to limit B cell responses to gram-negative bacterial LPS in vivo.
The third significant finding from these experiments is that DCs can regulate their ability to deacylate LPS according to external cues. In response to a mixture of inflammatory cytokines and an agonistic CD40 antibody, DCs down-regulated their AOAH activity, whereas LPS treatment increased it. The enzymatic activity measured in cell lysates changed in concert with the ability of the cells to deacylate the LPS in phagocytosed bacteria. The regulation of AOAH expression was due, at least in part, to differential expression or degradation of AOAH mRNA. The finding that LPS treatment can increase AOAH mRNA abundance in murine macrophages (10–20-fold) and in vivo in mouse lung and liver (3–6-fold) was described by Cody et al. (35); whereas none of the stimuli used in their paper decreased AOAH mRNA or enzymatic activity in macrophages, we found that treatment with IL-4 and maturation cocktail greatly reduced AOAH in DCs. Thus, it appears that both up- and down-regulation of this low abundance enzyme can occur in phagocytes. The results of microarray analyses of AOAH mRNA abundance in human peripheral blood leukocytes (36) support this conclusion.
Fourth, we found that both XS52 cells and BMDCs also regulate their ability to internalize and kill E. coli. Although XS52 cells responded to IL-4 and maturation cocktail treatment by diminishing their ability to phagocytose, kill, and digest E. coli, exposure to either E. coli or to LPS maintained the cells' phagocytic ability, CD14 expression, AOAH activity, and the ability to kill and digest internalized bacteria. Expression of a costimulatory molecule (CD40), used here as a marker of maturation, increased slightly during the response to all of the stimuli studied. These results are consistent with the currently accepted paradigm in which immature DCs can internalize and process bacterial antigens (37), whereas mature DCs lose these capabilities as they gain in antigen-presenting ability (2, 4, 38). Together with the results of others (39–41), our findings suggest that immature DCs contribute not only to the processing of bacterial antigens but also to the host's innate armamentarium for killing invading bacteria and disabling their stimulatory molecules.
When BMDCs were treated with LPS, in contrast, their phagocytic activity decreased as AOAH activity increased. This finding suggests that phagocytosis (as well as endocytosis [42]) and AOAH expression may be regulated independently in BMDCs. Differential regulation downstream of the LPS signal has also been suggested by the reduced ability of LPS to induce cytokines in MyD88-deficient DCs whereas LPS induction of costimulatory molecule expression was intact (43, 44). Similarly, inhibition of p38 SAPK prevented LPS-induced up-regulation of CD80, CD83, and CD86 in monocyte-derived DCs, but did not affect changes in macropinocytosis or HLA-DR and CD40 expression (45). Furthermore, Rescigno et al. (46) found that LPS induced NF-κB translocation and that inhibition of NF-κB with a serine protease inhibitor prevents D1 (a murine DC line) maturation (CD86 and class II expression), but does not interfere with the ability of LPS to prevent DC apoptosis. In contrast, Sallusto et al. (47) found that human monocyte-derived DCs respond to LPS as well as TNF and IL-1 with a coordinated series of changes that include down-regulation of macropinocytosis and Fc receptors, disappearance of the class II compartment, and up-regulation of adhesion and costimulatory molecules.
Finally, it is noteworthy that AOAH activity increased as DCs recognized microbial agonists that activate them via several different TLRs. Thus, maintaining or increasing the ability to deacylate LPS seems to be a DC response to sensing diverse microbial molecules, including those in a gram-positive bacterium (M. luteus), which does not contain LPS. Expression of TLR4 is required for LPS, but not gram-negative bacteria, to induce the maturation of BMDCs (39).
In all phagocytes studied in vitro to date, LPS deacylation has occurred over many hours (21, 48). Thus, it is unlikely that AOAH influences the ability of the phagocytosing cell to respond to the LPS in a bacterial cell wall. Because the long-term fate of LPS within phagocytes is unknown, it is possible that LPS deacylation might either diminish a phagocyte's late responses to LPS, or even reduce the responses of other cells that encounter LPS (either on the surface of the phagocyte (49, 50), or after the LPS has been released into the phagocyte's environment; reference 51). It is also intriguing that animals should deacylate LPS so selectively: when AOAH acts on ingested E. coli, all of the hydroxylated fatty acyl chains remain attached to the glucosamine backbone of lipid A. Previous authors have raised the possibility that the partially deacylated (thus, tetra-acyl) LPS produced by AOAH could act as an LPS antagonist in vivo (9, 10, 52). The discovery of stimulus-regulated deacylation of LPS by DCs provides a new impetus for investigating the biological significance of LPS degradation by animals and the role(s) that DCs play in antibacterial host defense.
Acknowledgments
We thank J. Herz, K. Graves, J. Horton, J. Garcia, and J. Ritter for their generous advice and encouragement during the production of the AOAH knockout mouse, X.-H. Li for expert mouse husbandry, and D. Sodora for help with real-time PCR.
This work was supported by grant AI18188 from the National Institute of Allergy and Infectious Diseases and by the Jan and Henri Bromberg Chair in Internal Medicine.
Footnotes
Abbreviations used in this paper: AOAH, acyloxyacyl hydrolase; BMDC, bone marrow–derived DC; cRPMI, complete RPMI; DC, dendritic cell; ODN, oligodeoxynucleotide; TLR, toll-like receptor.
References
- 1.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 3.Steinman, R.M., K. Inaba, S. Turley, P. Pierre, and I. Mellman. 1999. Antigen capture, processing, and presentation by dendritic cells: Recent cell biological studies. Hum. Immunol. 60:562–567. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 5.Reis e Sousa, C. 2001. Dendritic cells as sensors of infection. Immunity. 14:495–498. [DOI] [PubMed] [Google Scholar]
- 6.Gatti, E., M.A. Velleca, B.C. Biedermann, W. Ma, J. Unternaehrer, M.W. Ebersold, R. Medzhitov, J.S. Pober, and I. Mellman. 2000. Large-scale culture and selective maturation of human Langerhans cells from granulocyte colony-stimulating factor-mobilized CD34+ progenitors. J. Immunol. 164:3600–3607. [DOI] [PubMed] [Google Scholar]
- 7.Lapointe, R., J.F. Toso, C. Butts, H.A. Young, and P. Hwu. 2000. Human dendritic cells require multiple activation signals for the efficient generation of tumor antigen-specific T lymphocytes. Eur. J. Immunol. 30:3291–3298. [DOI] [PubMed] [Google Scholar]
- 8.Munford, R.S., and C.L. Hall. 1989. Purification of acyloxyacyl hydrolase, a leukocyte enzyme that removes secondary acyl chains from bacterial lipopolysaccharides. J. Biol. Chem. 264:15613–15619. [PubMed] [Google Scholar]
- 9.Pohlman, T.H., R.S. Munford, and J.M. Harlan. 1987. Deacylated lipopolysaccharide inhibits neutrophil adherence to endothelium induced by lipopolysaccharide in vitro. J. Exp. Med. 165:1393–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitchens, R.L., and R.S. Munford. 1995. Enzymatically deacylated lipopolysaccharide (LPS) can antagonize LPS at multiple sites in the LPS recognition pathway. J. Biol. Chem. 270:9904–9910. [DOI] [PubMed] [Google Scholar]
- 11.Kitchens, R.L., R.J. Ulevitch, and R.S. Munford. 1992. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J. Exp. Med. 1760:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagen, F.S., F.J. Grant, J.L. Kuijper, C.A. Slaughter, C.R. Moomaw, K. Orth, P.J. O'Hara, and R.S. Munford. 1991. Expression and characterization of recombinant human acyloxyacyl hydrolase, a leukocyte enzyme that deacylates bacterial lipopolysaccharides. Biochemistry. 30:8415–8423. [DOI] [PubMed] [Google Scholar]
- 13.Xu, S., K. Ariizumi, G. Caceres-Dittmar, D. Edelbaum, K. Hashimoto, P.R. Bergstresser, and A. Takashima. 1995. Successive generation of antigen-presenting, dendritic cell lines from murine epidermis. J. Immunol. 154:2697–2705. [PubMed] [Google Scholar]
- 14.Matsue, H., K. Matsue, M. Walters, K. Okumura, H. Yagita, and A. Takashima. 1999. Induction of antigen-specific immunosuppression by CD95L cDNA-transfected ‘killer’ dendritic cells. Nat. Med. 5:930–937. [DOI] [PubMed] [Google Scholar]
- 15.Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu, and R.M. Steinman. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony–stimulating factor. J. Exp. Med. 176:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munford, R.S., and C.L. Hall. 1985. Uptake and deacylation of bacterial lipopolysaccharides by macrophages from normal and endotoxin-hyporesponsive mice. Infect. Immun. 48:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada, N., and S.I. Katz. 1999. Generation of mature dendritic cells from a CD14+ cell line (XS52) by IL-4, TNF-α, IL-1β, and agonistic anti-CD40 monoclonal antibody. J. Immunol. 163:5331–5337. [PubMed] [Google Scholar]
- 18.Munford, R.S., and A.L. Erwin. 1992. Eucaryotic lipopolysaccharide deacylating enzyme. Methods Enzymol. 209:485–492. [DOI] [PubMed] [Google Scholar]
- 19.Hall, C.L., and R.S. Munford. 1983. Enzymatic deacylation of the lipid A moiety of Salmonella typhimurium lipopolysaccharides by human neutrophils. Proc. Natl. Acad. Sci. USA. 80:6671–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munford, R.S., L.C. DeVeaux, J.E. Cronan, Jr., and P.D. Rick. 1992. Biosynthetic radiolabeling of bacterial lipopolysaccharide to high specific activity. J. Immunol. Methods. 148:115–120. [DOI] [PubMed] [Google Scholar]
- 21.Katz, S.S., Y. Weinrauch, R.S. Munford, P. Elsbach, and J. Weiss. 1999. Deacylation of LPS in E. coli during destruction by cellular and extracellular components of rabbit peritoneal inflammatory exudates. J. Biol. Chem. 274:36579–36584. [DOI] [PubMed] [Google Scholar]
- 22.Bligh, E.G., and W.J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. [DOI] [PubMed] [Google Scholar]
- 23.Svensson, M., C. Johansson, and M.J. Wick. 2000. Salmonella enterica serovar typhimurium-induced maturation of bone marrow-derived dendritic cells. Infect. Immun. 68:6311–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahnke, K., E. Becher, P. Ricciardi-Castagnoli, T.A. Luger, T. Schwarz, and S. Grabbe. 1997. CD14 is expressed by subsets of murine dendritic cells and upregulated by lipopolysaccharide. Adv. Exp. Med. Biol. 417:145–159. [DOI] [PubMed] [Google Scholar]
- 25.Yrlid, U., M. Svensson, C. Johansson, and M.J. Wick. 2000. Salmonella infection of bone marrow-derived macrophages and dendritic cells: influence on antigen presentation and initiating an immune response. FEMS Immunol. Med. Microbiol. 27:313–320. [DOI] [PubMed] [Google Scholar]
- 26.De Smedt, T., B. Pajak, E. Muraille, L. Lespagnard, E. Heinen, P. De Baetselier, J. Urbain, O. Leo, and M. Moser. 1996. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 184:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sparwasser, T., E.S. Koch, R.M. Vabulas, K. Heeg, G.B. Lipford, J.W. Ellwart, and H. Wagner. 1998. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 28:2045–2054. [DOI] [PubMed] [Google Scholar]
- 28.Askew, D., R.S. Chu, A.M. Krieg, and C.V. Harding. 2000. CpG DNA induces maturation of dendritic cells with distinct effects on nascent and recycling MHC-II antigen-processing mechanisms. J. Immunol. 165:6889–6895. [DOI] [PubMed] [Google Scholar]
- 29.Dziarski, R., Q.L. Wang, K. Miyake, C.J. Kirschning, and D. Gupta. 2001. MD-2 enables toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to gram-positive and gram-negative bacteria and their cell wall components. J. Immunol. 166:1938–1944. [DOI] [PubMed] [Google Scholar]
- 30.Nigam, V.N., D. Malchow, E. Th. Rietschel, O. Lüderitz, and O. Westphal. 1970. Die enzymatische abspaltung langkettiger fettsäuren aus bakteriellen lipopolysacchariden mittels extrakten aus der amöbe von Dictyostelium discoideum. Hoppe-Seyler's Z. Physiol. Chem. 351:1123–1132. [PubMed] [Google Scholar]
- 31.Staab, J.F., S. Fosmire, M. Zhang, A.W. Varley, and R.S. Munford. 1999. Distinctive structural features are shared by human, lapine, and murine acyloxyacyl hydrolases. J. Endotoxin Res. 5:205–208. [Google Scholar]
- 32.Upton,C., and J.T.Buckley. 1995. A new family of lipolytic enzymes? Trends Biochem. Sci. 20:178–179. [DOI] [PubMed] [Google Scholar]
- 33.Elsbach, P. 2000. Mechanisms of disposal of bacterial lipopolysaccharides by animal hosts. Microbes Infect. 2:1171–1180. [DOI] [PubMed] [Google Scholar]
- 34.Weinrauch, Y., S.S. Katz, R.S. Munford, P. Elsbach, and J. Weiss. 1999. Deacylation of purified LPS by cellular and extracellular components of a sterile rabbit peritoneal inflammatory exudate. Infect. Immun. 67:3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cody, M.J., C.A. Salkowski, B.E. Henricson, G.R. Detore, R.S. Munford, and S.N. Vogel. 1997. Effect of inflammatory and anti-inflammatory stimuli on acyloxyacyl hydrolase gene expression and enzymatic activity in murine macrophages. J. Endotoxin Res. 4:371–379. [Google Scholar]
- 36.Boldrick, J.C., A.A. Alizadeh, M. Diehn, S. Dut, C.L. Liu, C.E. Belcher, D. Botstein, L.M. Staudt, P.O. Brown, and D.A. Relman. 2002. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc. Natl. Acad. Sci. USA. 99:972–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svensson, M., B. Stockinger, and M.J. Wick. 1997. Bone marrow-derived dendritic cells can process bacteria for MHC-1 and MHC-II presentation to T cells. J. Immunol. 158:4229–4236. [PubMed] [Google Scholar]
- 38.Granucci, F., E. Ferrero, M. Foti, D. Aggujaro, K. Vettoretto, and P. Ricciardi-Castagnoli. 1999. Early events in dendritic cell maturation induced by LPS. Microbes Infect. 1:1079–1084. [DOI] [PubMed] [Google Scholar]
- 39.Rescigno, M., M. Urbano, M. Rimoldi, B. Valzasina, G. Rotta, F. Granucci, and P. Ricciardi-Castagnoli. 2002. Toll-like receptor 4 is not required for the full maturation of dendritic cells or for the degradation of Gram-negative bacteria. Eur. J. Immunol. 32:2800–2806. [DOI] [PubMed] [Google Scholar]
- 40.Unkmeir, A., U. Kämmerer, A. Stade, C. Hübner, S. Haller, A. Kolb-Mäurer, M. Frosch, and G. Dietrich. 2002. Lipooligosaccharide and polysaccharide capsule: Virulence factors of Neisseria meningitidis that determine meningococcal interaction with human dendritic cells. Infect. Immun. 70:2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouis, D.A., T.G. Popova, A. Takashima, and M.V. Norgard. 2001. Dendritic cells phagocytose and are activated by Treponema pallidum. Infect. Immun. 69:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lutz, M.B., N.A. Kukutsch, M. Menges, S. Rössner, and G. Schuler. 2000. Culture of bone marrow cells in GM-CSF plus high doses of lipopolysaccharide generates exclusively immature dendritic cells which induce alloantigen-specific CD4 T cell anergy in vitro. Eur. J. Immunol. 30:1048–1052. [DOI] [PubMed] [Google Scholar]
- 43.Kaisho, T., O. Takeuchi, T. Kawai, K. Hoshino, and S. Akira. 2001. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J. Immunol. 166:5688–5694. [DOI] [PubMed] [Google Scholar]
- 44.Pulendran, B., P. Kumar, C.W. Cutler, M. Mohamadzadeh, T. Van Dyke, and J. Banchereau. 2001. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 167:5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ardeshna, K.M., A.R. Pizzey, S. Devereaux, and A. Khwaja. 2000. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 96:1039–1046. [PubMed] [Google Scholar]
- 46.Rescigno, M., M. Martino, C.L. Sutherland, M.R. Gold, and P. Ricciardi-Castagnoli. 1998. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 188:2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sallusto, F., M. Cella, C. Danieli, and A. Lanzavecchia. 1995. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: down-regulation by cytokines and bacterial products. J. Exp. Med. 182:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luchi, M., and R.S. Munford. 1993. Binding, internalization, and deacylation of bacterial lipopolysaccharides by human neutrophils. J. Immunol. 151:959–969. [PubMed] [Google Scholar]
- 49.Wuorela, M., S. Jalkanen, P. Toivanen, and K. Granfors. 1993. Yersinia lipopolysaccharide is modified by human monocytes. Infect. Immun. 61:5261–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forestier, C., E. Moreno, J. Pizarro-Cerda, and J.P. Gorvel. 1999. Lysosomal accumulation and recycling of lipopolysaccharide to the cell surface of murine macrophages, an in vitro and in vivo study. J. Immunol. 162:6784–6791. [PubMed] [Google Scholar]
- 51.Forestier, C., E. Moreno, S. Méresse, A. Phalipon, D. Olive, P.J. Sansonetti, and J.P. Gorvel. 1999. Interaction of Brucella abortus lipopolysaccharide with major histocomopatibility complex class II molecules in B lymphocytes. Infect. Immun. 67:4048–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riedo, F.X., R.S. Munford, W.B. Campbell, J.S. Reisch, K.R. Chien, and R.D. Gerard. 1990. Deacylated lipopolysaccharide inhibits plasminogen activator inhibitor-1, prostacyclin, and prostaglandin E2 induction by lipopolysaccharide but not by tumor necrosis factor-alpha. J. Immunol. 144:3506–3512. [PubMed] [Google Scholar]