

Abstract
Changes in chromatin structure induced by posttranslational modifications of histones are important regulators of genomic function. Phosphorylation of histone H2AX promotes DNA repair and helps maintain genomic stability. Although B cells lacking H2AX show impaired class switch recombination (CSR), the precise role of H2AX in CSR and somatic hypermutation (SHM) has not been defined. We show that H2AX is not required for SHM, suggesting that the processing of DNA lesions leading to SHM is fundamentally different from CSR. Impaired CSR in H2AX−/− B cells is not due to alterations in switch region transcription, accessibility, or aberrant joining. In the absence of H2AX, short-range intra-switch region recombination proceeds normally while long-range inter-switch region recombination is impaired. Our results suggest a role for H2AX in regulating the higher order chromatin remodeling that facilitates switch region synapsis.
Keywords: class switch recombination, somatic hypermutation, activation-induced cytidine deaminase, H2AX, non-homologous end joining
Introduction
During development in the bone marrow, B cells rearrange their immunoglobulin genes to assemble a functional B cell receptor through site specific V(D)J recombination (1, 2). Upon antigen encounter in secondary lymphoid organs the antibody repertoire is further diversified by somatic hypermutation (SHM)* and class switch recombination (CSR). SHM introduces point mutations in the variable region of rearranged Ig V genes to generate families of related B cell clones bearing receptors with different antigen binding affinities which are selected by antigen in germinal centers (3). Transcription dependent mutations occur within a 2 kb region downstream of the Ig promoter (4–6). Although a consensus for initiation of mutation is not defined, ∼50–60% of the mutations are found in RGYW hotspots (7, 8).
CSR is a region-specific recombination event that replaces the IgM constant region (Cμ) with a downstream CH gene (γ, ɛ, or α), thereby switching the antibody isotype expressed while retaining receptor specificity (9). CSR involves large repetitive switch (S) region sequences located upstream of each CH gene. This deletional recombination reaction is dependent on components of the nonhomologous end joining pathway of DNA repair (NHEJ; references 10–12) and results in the loss of intervening DNA as a circular episome (13–15). Like SHM, CSR is transcription dependent and recombination is targeted to individual switch regions by T cell–derived cytokines that induce transcription from intronic (I) promoters located upstream of each switch region. These promoters support “sterile” transcription through the switch region and they are essential for CSR (16–21), possibly because transcription promotes switch region accessibility (9).
A major advance in understanding SHM and CSR was achieved when activation-induced cytidine deaminase (AID) was shown to be required for both reactions (22, 23). AID functions upstream of the DNA lesions that trigger CSR and SHM (24, 25). Based on sequence similarity with APOBEC-1, an RNA editing cytidine deaminase, AID was proposed to initiate CSR and SHM directly by deaminating cytidine residues in DNA (25–27) or alternatively by modifying a yet unknown ubiquitous mRNA to create an endonuclease (22, 28). The DNA deamination model is supported by experiments in mice, chicken DT40 cells, and E. coli showing a role for uracyl-DNA glycosylase (UNG) downstream of AID (25–27). Furthermore, biochemical and genetic experiments suggest that single-stranded DNA exposed during the transcription reaction is the target for AID (29, 30, 30a, 30b). AID-mediated DNA deamination produces U:G mismatches in DNA that might be replicated to generate transition mutations (from C to T and G to A). Alternatively, recognition and removal of the uracyl by UNG would result in an abasic site that might be replicated to produce transition or transversion mutations. The abasic site could also be cleaved by an apurinic endonuclease, processed by additional nucleases, and repaired by error prone polymerases to introduce mutations at locations other than the original lesion (31–35). Finally, the U:G mismatch might be recognized by the mismatch repair pathway components MSH2/MSH6 and processed to produce distant mutations or double strand DNA (dsDNA) breaks. In this model CSR would proceed through dsDNA breaks produced by the UNG (major) or MSH2 (minor) pathways (26).
We have shown that in cells undergoing CSR phosphorylated histone H2AX (γ-H2AX) and the Nijmegen breakage syndrome protein (Nbs1) form nuclear foci at the IgH locus in the G1 phase of the cell cycle (24). These foci are AID dependent, suggesting that γ-H2AX acts downstream of AID during CSR (24). In addition, H2AX−/− lymphocytes show impaired CSR (24, 36). Histone H2AX is one of the three H2A subfamily members that participate in packaging eukaryotic DNA into nucleosomes. It is unique in being posttranslationally modified by phosphorylation of serine residues in the COOH-terminal domain by the PI3-kinases ataxia-telangiectasia mutated (ATM) and ATM-related (ATR; references 37–39) in response to dsDNA breaks (40–42). Although the precise role of γ-H2AX in DNA repair is still to be defined, γ-H2AX forms foci at dsDNA breaks and has been implicated both in homologous recombination and nonhomologous end joining DNA repair pathways (24, 36, 42, 43). In the absence of H2AX eukaryotic cells show multiple chromosomal abnormalities consistent with a role for H2AX in maintaining genomic stability (36, 43). Here we report on the role of histone H2AX in CSR and SHM.
Materials and Methods
Mice and Immunizations.
Wild-type (C57BL/6), AID−/− (22), H2AX−/− (36), Ku80−/− with a Bcl2 transgene carrying prerearranged heavy and light chains (12, 44), and mice carrying a prerearranged VHB1–8 gene (45) were bred and maintained under specific pathogen free conditions. Mutant mice were maintained by intercrossing. Age-matched 8–10-wk-old mice were immunized by footpad injection with 50 μg of NP-CGG (Biosearch Technologies) in complete Freund's adjuvant.
Lymphocyte Cultures and Cell Sorting.
B lymphocytes were isolated from spleen using CD43 microbeads (Miltenyi Biotech), labeled with CFDA-SE for 10 min at 37°C (5 μM; Molecular Probes), and cultured (106 cells/ml) with LPS (25 μg/ml) and IL-4 (5 ng/ml) for 1–4 d. Peyer's patches (PPs) and lymph nodes were dissected before or after immunization. Germinal center B cells were stained with APC-anti-B220, FITC-anti-GL7, and PE-anti-FAS monoclonal antibodies (BD Biosciences). In all cell-sorting experiments propidium iodide (PI: 0.5 μg/ml) was added immediately before laser excitation to exclude dead cells. Cell sorting was performed on a FACSVantage™ (Becton Dickinson), and an aliquot of each of the sorted fractions was reanalyzed for purity on a FACSCalibur™ (Becton Dickinson).
Hybridoma Analysis.
B cells were stimulated with LPS and IL-4 for 72 h and fused to the SP2/0Ag-14 myeloma cell line. IgM secreting clones were selected by ELISA for further analysis. Genomic DNA was prepared and Southern blot analysis performed using standard techniques.
PCR and Mutation Analysis.
Genomic DNA was amplified by PCR using Pfu Turbo DNA polymerase (Stratagene) from 5,000 sorted cell equivalents in four independent reactions that were pooled for cloning experiments. For the Sμ, Sγ1, Iγ1, Sγ3, JH4-intron, VHB1–8, and Eμ regions amplification conditions were 25 cycles 94°C (30 s), 60°C (30 s), 72°C (40 s). Sμ-Sγ1 junctions were amplified using Expand long template PCR system (Roche). Amplification conditions were 10 cycles at 94°C (10 s), 60°C (30 s), 68°C (1 min), and 20 cycles at 94°C (10 s), 60°C (30 s), 68°C (1 min and 20 s/cycle). PCR products were cloned using TOPO-TA cloning kit (Invitrogen) and sequenced using M13 universal primers. Sequence analysis was performed using Sequence Manager II software (DNASTAR). Primers were: μ Switch region: 5μ3 (5′-AATGGATACCTCAGTGGTTTTTAATGGTGGGTTTA-3′) and SμR5 (5′-GCGGCCCGGCTCATTCCAGTTCATTACAG-3′); μ intronic enhancer: 5μE2 (5′-ATTTTTAAATGAATTGAGCAATGTTGAGTTGGAGT-3′) and μE-R (5′-AAAGATTTGTGAAGCCGTTTTGACCAGAATGTC-3′); γ3 Switch region: γ3-F (5′-AGGAGAGCATAGGGGACCTGGATAAGC-3′) and γ3-R (5′-GTCCCCACACCCCACATACC-3′); CHμ probe: CHμ (5′- AGCCCCTCCCCCACCTCCACCTACCTATTAC-3′) and CH2 (5′-CTGTGATCGGTTTTGGAGTGAAGTTCGTG-3′); Sγ1 probe: Sγ1-F (5′-GAAGCTGGAGCTGATGGGTATAAAAGGAAC-3′) and Sγ1-R (5′-GGT-TCTCCACAATTCTTTCCCCCAGTTCTT-3′); Eμ probe: 5μE2 and 3μEA (5′-GGCAACTTCAAATTCATTAAACCACAT-3′); JH4 probe: JH4-F (5′-TATGCTATGGACTACTGG-3′)and μE-R; Sμ-Sγ1 junctions: 5μ3 and γ1-R (5′-CAATTAGCTCCTGCTCTTCTGTGG-3′); JH4-intron: VHJ588/FR3 (46) (5′-GGAATTCGCCTGACATCTGAGGACTCTGC-3′) and μE-R.
Quantitative Real Time RT-PCR.
Total RNA was extracted with TRIzol (Invitrogen), reverse transcribed with random hexamers and superscript II reverse transcriptase (Invitrogen). 1st strand cDNA was used for SYBR Green fluorogenic dye real time PCR (Applied Biosystems). Specificity of the μ, γ1, γ1-CT, and GAPDH primers was determined using negative controls and by the analysis of dissociation curves. Serially diluted cDNA samples were used to estimate the efficiency (Ef) of each PCR, which was 0.96, 0.95, 0.98, and 0.97 for μ, γ1, γ1-CT, and GAPDH, respectively. Calculation of the fold differences was based on equation (1 + EfGAPDH)Ct GAPDH x − CtGAPDH cal (1 + Efμ, γ1, or γ1-CT)Ctμ (or γ1 or γ1-CT) cal − Ct μ (or γ1 or γ1-CT) x, where Ct is the number of cycles at which the threshold of fluorescence is reached, x is a given sample, and cal is the calibrator used to normalize all results (resting B cells). Primers were: GAPDH: (5′-TGAAGCAGGCATCTGAGGG-3′) and (5′-CGAAGGTGGAAGAGTGGGAG-3′); μ germline transcript: (5′-CTCGGTGGCTTTGAAGGAAC-3′) and (5′-TGGTGCTGGGCAGGAAGT-3′); γ1 germline transcript: (5′-TCGAGAAGCCT-GAGGAATGTG-3′) and (5′-ATGGAGTTAGTTTGGGCA-GCA-3′); γ1 circle transcript: (5′-TCGAGAAGCCTGA-GGAATGTG-3′) and (5′-GAAGACATTTGGGAAGGACTGACT-3′).
Results
Hypermutation in Sμ Requires Ku80 and Is Linked to CSR.
DNA upstream of the Sμ core is mutated by an AID-dependent mechanism in B cells undergoing class switch recombination (24, 47). To further characterize this type of mutation we performed time course experiments on wild-type B cells stimulated with LPS and IL-4. A significant increase in mutation frequency in the Sμ region was found after 48 h. Mutations increased with time in culture accumulating at a rate of 1.6 × 10−4 mutations/bp/day (Fig. 1 a). This increase in mutation frequency was accompanied by a concomitant increase in the proportion of mutated sequences reaching a maximum of 23% at 96 h of stimulation (Fig. 1 a). Mutation was specific for the Sμ region (24, 47), as no significant increase in mutation frequencies in the μ intronic enhancer, VH, or the IgG3 switch region (Sγ3) was observed (Fig. 1 b). 141 mutations were analyzed and their distribution in the Sμ sequence is shown in Fig. 1 c. Consistent with previous reports (24, 47), 60% of the mutations were found within RGYW hotspot motifs (7, 8) and the nucleotide substitution preference (data not depicted) was similar to that reported for SHM (48). Thus, mutations induced by LPS and IL-4 resemble SHM (24, 47), are specific to Sμ and these mutations accumulate over time in culture.
Figure 1.
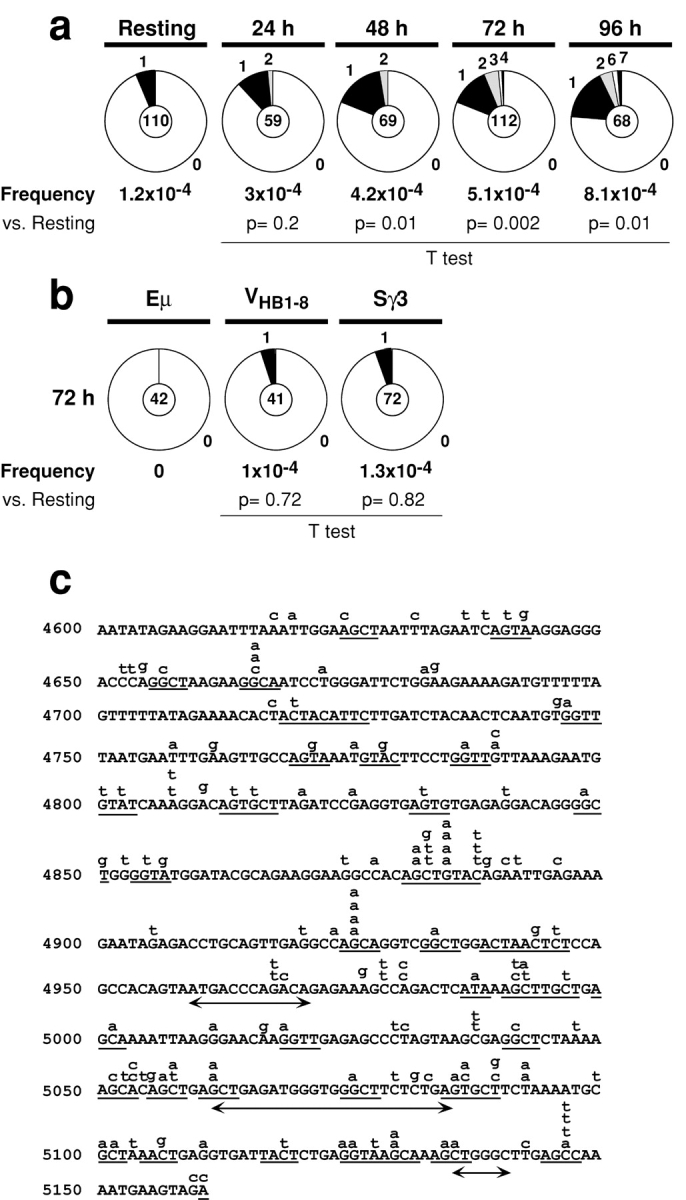
Mutations in Sμ. (a) Time course of Sμ mutation in wild-type B cells stimulated with LPS and IL-4. The number of mutations was: resting B cells: 7 mutations/59,301 bp; 24 h: 9 mutations/30,060 bp; 48 h: 15 mutations/36,000 bp; 72 h: 32 mutations/62,720 bp; 96 h: 30 mutations/37,066 bp. (b) Proportion of μ intronic enhancer (Eμ), VHB1–8, and Sγ3 sequences carrying mutations after 72 h of stimulation with LPS and IL-4. The number of mutations was: μ intronic enhancer: 0 mutations/22124 bp; Sγ3: 4 mutations/31812 bp and VHB1–8: 2 mutations/20,427 bp. Analysis of VHB1–8 sequences was performed on B cells obtained from mice carrying a prerearranged VHB1–8 gene (reference 45). Segment sizes in the pie charts are proportional to the number of sequences carrying the number of mutations indicated in the periphery of the charts. The frequency of mutations per bp sequenced and the total number of independent sequences analyzed is indicated underneath and in the center of each chart respectively. Statistical significance was determined by a two-tailed t test assuming unequal variance and comparing to background (Resting B cells). P values are indicated below each pie chart. (c) Distribution of point mutations in Sμ. The region sequenced is indicated with the first base corresponding to position 4600 in the Sμ germline sequence (GenBank/EMBL/DDBJ accession no. J0040). Lower-case letters above the line indicate independent mutations (141 total). Mutations occurring at any base within the RGYW motif (references 7 and 8) were considered to be hotspot mutations. RGYW motifs containing mutations are underlined. A double head arrow underneath the line indicates deletions.
To determine whether there is a relationship between mutations in Sμ, cell division, and CSR we labeled B cells with CFSE and analyzed mutation in cells sorted on the basis of surface expression of IgM or IgG1 and cell division (as determined by dye dilution). As described by others (49), the proportion of IgG1+ cells increased proportionally with the number of cell divisions (Fig. 2 a). We found that increased cell division was also associated with increased mutations in Sμ (average of 1.7 × 10−4 mutations/bp/cell division; Fig. 2 b). The increase in mutation frequency with cell division is the result of an increase in the proportion of mutated clones as well as the number of mutations per clone (Fig. 2 b). Finally, mutations in Sμ accumulate at slightly higher rates in IgG1+ cells (51 mutations/47,040 bp) than in IgM+ B cells (17 mutations/45,356 bp; Fig. 2 c, P = 0.005). AID−/− B cells proliferate as efficiently as wild-type but are unable to switch (22, 23) and their Sμ sequences remain un-mutated (24, 47) even after multiple rounds of cell division (Fig. 2, a and d). We conclude that mutations in the Sμ region are coupled to cell division, that they occur in IgM+ cells that failed to undergo CSR, but are found more frequently in cells that have undergone productive CSR.
Figure 2.

Analysis of CSR and mutations in Sμ of wild-type, AID− / −, Ku80− / −, Ku80− / −Bcl2+, and H2AX− / − B cells. (a) Flow cytometric analysis of IgG1 expression on CFSE-labeled wild-type (WT), AID−/−, H2AX−/−, Ku80−/−, and Ku80−/−Bcl2+ B cells stimulated with LPS plus IL-4 for 4 d. Cell division as measured by CFSE dye dilution is shown in the upper panel. The percentage of cells expressing IgG1 after a specific number of cell divisions is indicated on the lower panel. (b) Sμ mutations in wild-type B cells sorted for 1, 3, or 5 cell divisions. The number of mutations was: 1 division, 7 mutations/33,306 bp; 3 divisions, 14 mutations /30,818 bp; 5 divisions, 33 mutations/32,460 bp. (c) Sμ mutations in wild-type B cells sorted for 5 cell divisions and IgM or IgG1 cell surface expression. The number of mutations was: IgM+: 17 mutations/45,356 bp; IgG1+: 51 mutations/47,040 bp. (d) Sμ mutations in AID−/−, Ku80−/−, and Ku80−/−Bcl2+ B cells sorted for 5 cell divisions and expressing IgM. The number of mutations was: AID−/−, 2 mutations/34,124 bp; Ku80−/−, 7 mutations/31,360 bp; Ku80−/−Bcl2+, 4 mutations/27,740 bp. (e) Sμ mutations in H2AX−/− B cells sorted for 5 cell divisions and IgM or IgG1 cell surface expression. The number of mutations was: IgM+: 12 mutations/35,810 bp; IgG1+: 20 mutations/33,946 bp. Pie charts and statistics are as in Fig. 1.
To determine whether NHEJ is required for Sμ mutation we reconstituted the B cell compartment in Ku80 deficient (Ku80−/−) or Ku80−/ − Bcl2 transgenic mice (Ku80−/−Bcl2+) with prerecombined heavy and light chain genes (12, 44). LPS and IL-4 treatment caused proliferation and cell death in Ku80−/− B cells and Ku80−/− Bcl2+ B cells and CSR was severely impaired (11, 12) even in cells that had undergone several rounds of cell division (Fig. 2 a). Live Ku80−/− and Ku80−/−Bcl2+ B cells that had completed 5 cell divisions and were positive for cell surface IgM expression were sorted for Sμ sequence analysis. In contrast to wild-type, Ku80−/− and Ku80−/−Bcl2+ B cells showed no increase in mutation in Sμ with cell division (Fig. 2 d). We conclude that Sμ mutation is Ku80 dependent.
Hypermutation Does Not Require H2AX.
To determine whether H2AX is required for Sμ mutation we analyzed H2AX−/− B cells stimulated with LPS and IL-4. We found similar levels of Sμ mutation in H2AX−/− and control B cells that had completed 5 cell divisions (Fig. 2 e). To examine the role of H2AX in IgV gene somatic hypermutation we compared germinal center B cells (B220+Fas+GL-7+) from H2AX−/− and control mice. Analysis of the JH4-intron sequences (46) revealed no differences between H2AX−/− and wild-type PPs germinal center cells in mutation frequencies or in the proportion of mutated clones (Fig. 3 a). A similar result was also obtained by analyzing VHB1–8 gene sequences from sorted germinal center B cells obtained from the lymph nodes of H2AX−/− and control mice immunized with NP-CGG (Fig. 3 b), and there was no significant bias in the nucleotide substitution patterns found in VHB1–8 genes cloned from wild-type and H2AX−/− germinal center B cells (Fig. 3 c). We conclude that H2AX is not required for IgV gene SHM or Sμ mutation and that H2AX deficiency results in a CSR specific defect.
Figure 3.

Somatic hypermutation is unaffected in the absence of H2AX. (a) Wild-type and H2AX−/− germinal center B cells (B220+Fas+GL-7+) obtained from the PPs of unimmunized mice. Pie charts show proportion of JH4-intron sequences carrying different number of mutations. (b) Wild-type and H2AX−/− GC B cells (B220+Fas+GL-7+) from lymph nodes of NP-immunized mice. Pie charts show proportion of VHB1–8 sequences carrying different number of mutations. (c) Percent nucleotide substitutions in VHB1–8 from GC B cells adjusted for base composition. Percentage of mutations within hotspot motifs is indicated underneath each panel. Total number of mutations analyzed was: wild-type = 653 mutations; H2AX−/−= 475 mutations. Pie charts and statistics are as in Fig. 1.
Switch Region Accessibility Is Normal in the Absence of H2AX.
CSR is reduced in the absence of H2AX but the nature of the defect has not been determined (24, 36). We used real time PCR to measure the μ and γ1 preswitch sterile transcripts and the post-switch γ1 circle transcript (50) before and after stimulation with LPS and IL-4 over a 72 h time course (Fig. 4) . Induction of μ and γ1 sterile transcripts was similar in H2AX−/− and control B cells (Fig. 4, a and b). In wild-type B cells the γ1 sterile transcript level dropped off while the γ1 circle transcript increased with time in culture, presumably due to recombinational deletion of the I promoter and exon and concomitant switch circle formation during CSR (Fig. 4, b and c). In contrast, the γ1 sterile transcription remained elevated in H2AX−/− B cells and the γ1 circle transcript was only modestly increased, consistent with decreased CSR in the absence of H2AX (24, 36; Fig. 4, b and c). We conclude that sterile transcription of the IgM and IgG1switch regions and its effect on switch region accessibility is not altered in the absence of H2AX (24). Furthermore, the target switch regions continue to be transcribed in H2AX−/− B cells in culture despite inefficient CSR.
Figure 4.

Switch region accessibility is normal in the absence of H2AX. Real time RT-PCR for (a) μ sterile transcript, (b) γ1 sterile transcript, and (c) γ1 circle transcript in wild-type (solid circles) and H2AX−/− (open circles) B cells stimulated with LPS and IL-4 over 3 d. Mean results from two independent cultures are expressed as fold induction relative to resting B cells. (d) Proportion of mutations in the Sγ1 5′ region in wild-type, AID−/−, and H2AX−/− B cells stimulated with LPS and IL-4 and sorted for IgM surface expression and 5 divisions. The number of mutations was: Resting B cells, 5 mutations/59,301 bp; wild-type, 26 mutations/53,624 bp; AID−/−, 5 mutations/52,164 bp; H2AX−/−, 21 mutations/53,838 bp. Pie charts and statistics are as in Fig. 1.
To determine whether H2AX plays a direct role in regulating accessibility of switch regions to AID we analyzed mutations in Sγ1 in the presence or absence of H2AX. Wild-type, H2AX−/−, and AID−/− IgM+ B cells that had completed 5 cell divisions after stimulation with LPS and IL-4 were isolated by cell sorting and assayed for Sγ1 mutation (Fig. 4 d). We found a significant accumulation of point mutations in sequences from wild-type B cells (26 mutations/53624 bp, P = 0.02 vs. background) and H2AX−/− B cells (21 mutations/53,838 bp, P = 0.01 vs. background) but not AID−/− B cells (5 mutations/52,164 bp, P = 0.7 vs. background). We conclude that AID-mediated lesions are induced in Sγ1 during CSR and that these can occur in the absence of H2AX. Therefore, impaired switching in H2AX−/− B cells is not due to impaired access of AID to Sγ1.
CSR Junctions Are Normal in the Absence of H2AX.
In the absence of NHEJ, large deletions and significant increases in the length of microhomology of recombination junctions are found (51, 52). Although CSR is inefficient in the absence of H2AX some cells do succeed in producing IgG1 in response to LPS and IL-4. To determine whether CSR junctions were normal in the absence of H2AX we compared IgG1 CSR junctions from H2AX−/− (n = 30 junctions) and wild-type (n = 38 junctions) B cells. We found no significant differences in the amount of donor/acceptor homology at the junctions (the average length of overlap was 1.8 bp in H2AX−/− and 1.7 bp in controls; Fig. 5 , a–c). Mutation frequency in the vicinity of the junctions (±50 bp) was lower in H2AX−/− (1.6 × 10−2) than in wild-type (2.6 × 10−2); however, this difference was not statistically significant as determined by a two-tailed t test (P = 0.69). Furthermore, the breakpoint distribution in Sμ and Sγ1 measured by scatter analysis was similar in H2AX−/− and control cells (Fig. 5 d). We conclude that switch recombination junctions are unaffected in H2AX−/− B cells, suggesting that DNA ends are processed normally by NHEJ during CSR in the absence of H2AX.
Figure 5.
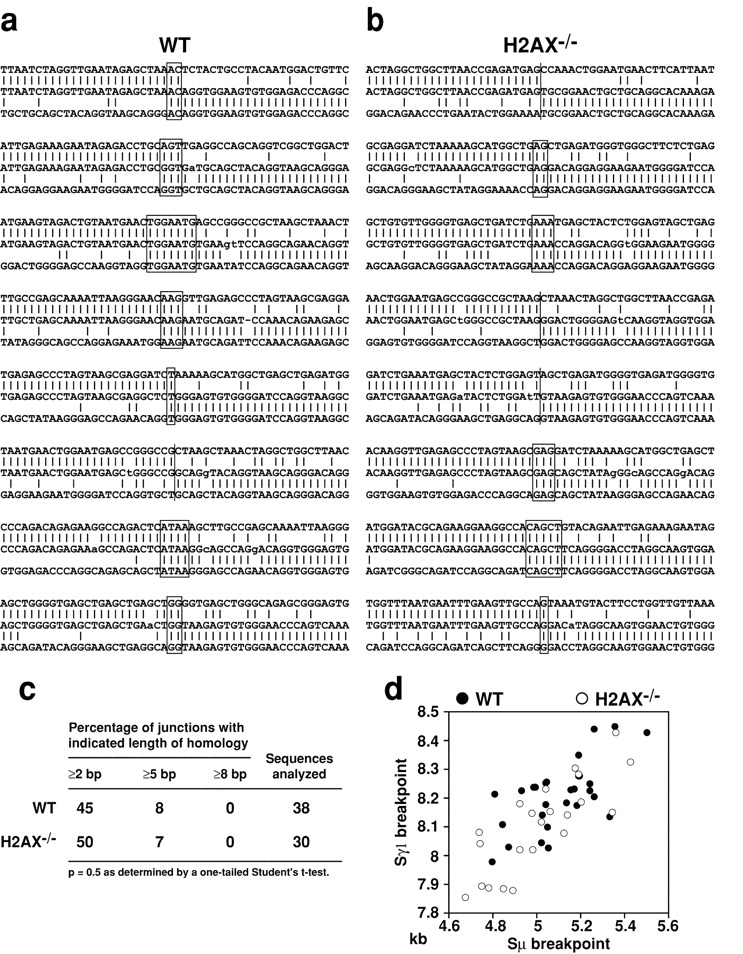
Switch recombination junctions are normal in the absence of H2AX. Sμ-Sγ1 switch recombination junctions from wild-type (a) and H2AX−/− (b) B cells. Overlap was determined by identifying the longest region at the switch junction of perfect uninterrupted donor/acceptor identity. The Sμ and Sγ1 germline sequences are shown above and below each junction sequence respectively. Lower-case letters indicate mutations, (|) indicates identity between nucleotides and (−) indicates a deletion. Homology at the junctions is boxed. (c) Length of microhomologies at Sμ/Sγ1 junctions in wild-type and H2AX−/− B cells. (d) Scatter analysis of the μ/γ1 breakpoints derived from in vitro–stimulated B cells. The axes indicate the position relative to GenBank/EMBL/DDBJ sequences J00440 (Sμ) and D78340 (Sγ1). Open circles denote breakpoints from H2AX−/−, filled circles from wild-type controls.
CSR-induced Sμ Internal Deletions Are Dependent on AID and Ku80 but Not H2AX.
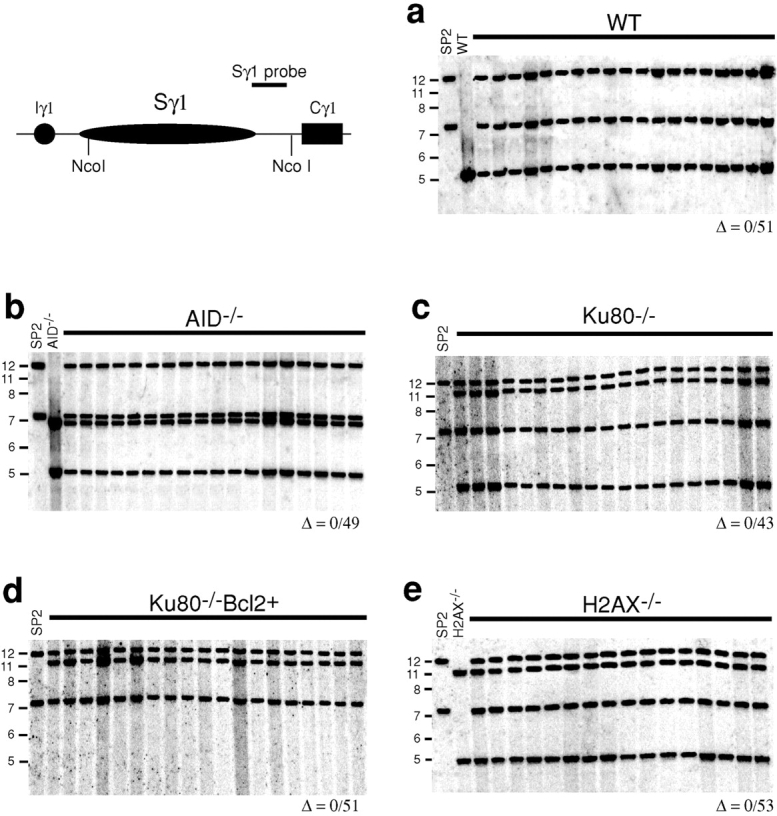
B cells stimulated to undergo CSR suffer frequent AID dependent internal Sμ deletions (17, 53–56). To determine whether these are H2AX dependent and repaired by NHEJ we examined the switch regions in hybridomas from wild-type, AID−/−, H2AX−/−, Ku80−/−, and Ku80−/−Bcl2+ B cells. Hybridomas were produced from B cells stimulated with LPS and IL-4 to divide in vitro and IgM secretors were selected by ELISA for analysis by Southern blotting (Figs. 6 and 7) . Consistent with published reports (56), structural alterations in Sμ were detected in 9 out of 51 wild-type hybridomas (18%; Fig. 6 a). The changes in Sμ were AID dependent (56), as all of the 49 AID−/−-derived hybridomas were in germline configuration as assessed by 5′ and 3′ probes (Fig. 6 b, data not depicted). A similar defect in intra-switch region recombination was observed in the analysis of the hybridomas derived from Ku80-deficient B cells. Only 1 of the 43 Ku80−/− hybridomas (Fig. 6 c) and none of the 51 Ku80−/− Bcl2+ hybridomas showed structural alterations in Sμ (Fig. 6 d). In contrast, the rate of Sμ alteration in H2AX−/− hybridomas was similar to wild-type, 11 out of 53 hybridomas showed deletions or insertions (21%; Fig. 6 e). Sγ1 differed from Sμ in that Sγ1 was always in the germline configuration (Fig. 7), suggesting that targeting of Sγ1 is either less frequent or occurs as part of a cleavage coupled repair reaction that leads to authentic CSR. Although no large deletions were observed in Sγ1, it is possible that smaller deletions are induced upon stimulation; however, this is difficult to assess by PCR and sequencing due to the repetitive nature of the Sγ1 sequence. We conclude that recombination within Sμ induced by LPS and IL-4 resembles authentic CSR in that it is induced by AID and repaired by a Ku80-dependent pathway. Like SHM, intra-switch region recombination differs from CSR in that it proceeds normally in the absence of H2AX.
Figure 6.

Resolution of Sμ internal deletions is independent of H2AX but requires AID and Ku80. Southern blot analysis of the Sμ region in IgM secreting hybridomas derived from (a) wild-type (WT), (b) AID−/−, (c) Ku80−/−, (d) Ku80−/−Bcl2+, and (e) H2AX−/− B cells. Restriction enzymes and probes used are indicated in the top left panel. Δ indicates deletions in Sμ. The SP2/0Ag-14 (SP2) cell line has a deletion in Cμ and shows no hybridization. The same deletions in Sμ were found using an Eμ probe (unpublished data). Number of deletions over hybridomas screened is indicated below each panel. Molecular weight markers in kilobase pairs are indicated on the left side of each panel.
Figure 7.
The Sγ1 locus is intact in IgM secreting hybridomas stimulated for CSR. Southern blot analysis of the Sγ1 region in IgM secreting hybridomas from (a) wild-type (WT), (b) AID−/−, (c) Ku80−/−, (d) Ku80−/−Bcl2+, and (e) H2AX−/− mice. Restriction enzymes and probes used are indicated in the top left panel. Molecular weight markers in kilobase pairs are indicated on the left side of each panel. Number of deletions over hybridomas screened is indicated below each panel.
Discussion
Somatic hypermutation and class switch recombination are linked by a requirement for AID (22, 23), which is believed to initiate these DNA modification reactions by deaminating cytidine residues in DNA (25–27). Transcription facilitates the targeting step of the CSR and SHM reactions by exposing single stranded DNA, which is the substrate for AID (29, 30, 30a, 30b) and the efficiency of both CSR and SHM correlates directly with the rate of transcription (57–60). Histone modifications such as acetylation and methylation alter transcription by modifying chromatin structure (61, 62). Similarly, histone H2AX phosphorylation is thought to alter chromatin structure (63, 64) and this modification could alter transcription. However, H2AX deficiency does not have an impact on transcription of switch regions (24), or the accessibility of switch regions to AID as measured by mutation, or Sμ intra-switch region recombination. Finally, neither the rate nor distribution of SHM is affected by the absence of H2AX. Thus, H2AX is entirely dispensable for the targeting stage and initial lesion formation in the CSR and SHM reactions.
The AID mediated U:G mismatches that trigger the CSR reaction are thought to be recognized and processed by uracyl-DNA glycosylase and components of the mismatch repair pathway including MSH2, PMS2, and MLH1 (26, 65–68). In the absence of any of these factors CSR is impaired. In addition, MLH1 or PMS2 deficiency leads to an increase in the length of homology of CSR junctions whereas the absence of MSH2 affects local sequence specificity and focusing of CSR junctions to consensus motifs (65–68). These DNA repair factors are also thought to play a role in the SHM reaction because in their absence there is an altered spectrum of mutation in Ig V genes (69–73). In contrast, we found no alteration in CSR junctional profiles or SHM in H2AX−/− B cells. Therefore, H2AX alters the efficiency of the CSR reaction but it does not have a role in processing of DNA ends before joining.
CSR is a deletional recombination reaction and therefore dsDNA breaks must be intermediates in the reaction (26, 74, 75). As isotype switching is impaired in mice that lack Ku80, or Ku70, or DNA-PKcs these breaks are thought to be repaired by the NHEJ pathway (10–12). However, the requirement for DNA-PKcs is not absolute (76, 77) and the potential impact of altered cell division in absence of Ku was never assessed (11, 12). Our experiments support the idea that NHEJ is required for CSR independently of a possible role for Ku in cell division because Ku80−/− B cells, which divide in response to LPS and IL-4 are unable to complete the CSR reaction as measured by IgG1 expression or intra-switch region recombination. We propose that B cells that produce dsDNA breaks in the absence of Ku80 are unable to repair these lesions and die by apoptosis. However, not all cells undergo a CSR reaction in response to LPS and IL-4 and those cells that divide but fail to produce dsDNA breaks retain their Sμ DNA in the germline configuration.
Finding that Sμ mutations are absent in Ku80−/− B cells exposed to LPS and IL-4 was surprising because DNA-PKcs and Ku70 are not required for SHM in chicken DT40 cells (78). Indeed, it has been proposed that NHEJ is not involved in repairing the DNA lesions that lead to SHM (79). We have not been able to measure Ig V region mutation in Ku80−/− mice because they lack T cells and therefore cannot produce germinal centers, which are required for efficient SHM. Thus, it is a distinct possibility that Ku80 is required for Sμ mutation but not Ig V region SHM. The dependence of Sμ mutations on Ku80 could be explained if these mutations were associated with repair of dsDNA breaks that occur in CSR. This explanation would be consistent with the increased number of such mutations found at CSR junctions (80–83) and in the switch regions of B cells that have undergone internal Sμ deletions (56).
H2AX phosphorylation promotes the assembly of repair factors into nuclear foci localized at sites of DNA damage (36, 42, 43, 84). One potential explanation for impaired CSR in H2AX−/− B cells would be the inefficient assembly of DNA repair factors. However, AID induced internal deletions in Sμ occur at wild-type frequencies in H2AX−/− B cells and we found no aberrant recombination by Southern blot (5′ and 3′ probes hybridize with the same DNA fragments; Fig. 6, and data not depicted). In addition, fluorescence in situ hybridization using a combination of IgH variable and constant region specific probes and whole painting of chromosome 12 showed no evidence of translocations (unpublished data). Chromosomal translocations are frequent in H2AX−/− fibroblasts but such translocations have not been detected in B cells possibly because they lead predominantly to cell death. Finally, switch region mutation was found at wild-type frequencies in both Sμ and Sγ1. Thus, the DNA lesions that trigger the CSR reaction are resolved efficiently in the absence of H2AX. Consistent with these results, we have found that although higher order assembly into nuclear foci is dependent on γ-H2AX, absence of H2AX does not affect the initial recruitment of factors to dsDNA breaks (84a).
If AID targeting and repair factor recruitment to switch regions are both normal in H2AX−/− B cells, why is CSR impaired in the absence of H2AX? Histone H2AX phosphorylation is propagated over large distances in the genome (41) and γ-H2AX foci are found at sites of DNA damage in eukaryotic cells after exposure to ionizing radiation (40, 41), during meiotic recombination (85), V(D)J recombination (86), and class switch recombination (24, 36). The role of focus formation in DNA repair has yet to be determined but absence of H2AX leads to genomic instability and widespread chromosomal abnormalities (36, 43). Phosphorylation of H2A in yeast induces a change in chromatin structure that facilitates DNA repair (63). Moreover, H2AX phosphorylation is essential for the condensation and synapsis of the sex chromosomes during male meiosis, as well as for preventing the premature separation of broken ends during replication and in response to γ-irradiation (64). Thus, modification of the H2AX tail by phosphorylation may change the overall structure of the chromosomal domain at the site of a dsDNA break. We propose that during CSR, modification of the COOH-terminal tail of H2AX by phosphorylation alters the overall structure of the nucleosome at the site of a dsDNA break and throughout the constant region of the Ig heavy chain locus. In this model, chromatin conformational changes induced by H2AX phosphorylation would facilitate switch region synapsis and therefore class switch recombination.
Acknowledgments
We thank members of the Nussenzweig laboratories, N. Yannoutsos for advice on Southern blotting, K. Velinzon for cell sorting, A. Celeste for generating the mice, F. Weiss-Garcia for hybridoma fusions, and E. Besmer for help with the manuscript.
This work was supported in part by grants from National Institutes of Health to M.C. Nussenzweig. M.C. Nussenzweig is a Howard Hughes Medical Institute investigator.
Footnotes
Abbreviations used in this paper: AID, activation-induced cytidine deaminase; CSR, class switch recombination; NHEJ, nonhomologous end joining pathway of DNA repair; PP, Peyer's patch; SHM, somatic hypermutation.
References
- 1.Tonegawa, S. 1983. Somatic generation of antibody diversity. Nature. 302:575–581. [DOI] [PubMed] [Google Scholar]
- 2.Schatz, D.G., M.A. Oettinger, and D. Baltimore. 1989. The V(D)J recombination activating gene, RAG-1. Cell. 59:1035–1048. [DOI] [PubMed] [Google Scholar]
- 3.Papavasiliou, F.N., and D.G. Schatz. 2002. Somatic hypermutation of immunoglobulin genes: merging mechanisms for genetic diversity. Cell. 109(Suppl):S35–S44. [DOI] [PubMed] [Google Scholar]
- 4.Peters, A., and U. Storb. 1996. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 4:57–65. [DOI] [PubMed] [Google Scholar]
- 5.Goyenechea, B., N. Klix, J. Yelamos, G.T. Williams, A. Riddell, M.S. Neuberger, and C. Milstein. 1997. Cells strongly expressing Ig(kappa) transgenes show clonal recruitment of hypermutation: a role for both MAR and the enhancers. EMBO J. 16:3987–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukita, Y., H. Jacobs, and K. Rajewsky. 1998. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 9:105–114. [DOI] [PubMed] [Google Scholar]
- 7.Rogozin, I.B., and N.A. Kolchanov. 1992. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim. Biophys. Acta. 1171:11–18. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro, G.S., K. Aviszus, D. Ikle, and L.J. Wysocki. 1999. Predicting regional mutability in antibody V genes based solely on di- and trinucleotide sequence composition. J. Immunol. 163:259–268. [PubMed] [Google Scholar]
- 9.Stavnezer, J. 1996. Immunoglobulin class switching. Curr. Opin. Immunol. 8:199–205. [DOI] [PubMed] [Google Scholar]
- 10.Rolink, A., F. Melchers, and J. Andersson. 1996. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 5:319–330. [DOI] [PubMed] [Google Scholar]
- 11.Manis, J.P., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F.W. Alt. 1998. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwasato, T., A. Shimizu, T. Honjo, and H. Yamagishi. 1990. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 62:143–149. [DOI] [PubMed] [Google Scholar]
- 14.Matsuoka, M., K. Yoshida, T. Maeda, S. Usuda, and H. Sakano. 1990. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 62:135–142. [DOI] [PubMed] [Google Scholar]
- 15.von Schwedler, U., H.M. Jack, and M. Wabl. 1990. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 345:452–456. [DOI] [PubMed] [Google Scholar]
- 16.Stavnezer-Nordgren, J., and S. Sirlin. 1986. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. EMBO J. 5:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu, H., Y.R. Zou, and K. Rajewsky. 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 73:1155–1164. [DOI] [PubMed] [Google Scholar]
- 18.Jung, S., K. Rajewsky, and A. Radbruch. 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science. 259:984–987. [DOI] [PubMed] [Google Scholar]
- 19.Seidl, K.J., J.P. Manis, A. Bottaro, J. Zhang, L. Davidson, A. Kisselgof, H. Oettgen, and F.W. Alt. 1999. Position-dependent inhibition of class-switch recombination by PGK-neor cassettes inserted into the immunoglobulin heavy chain constant region locus. Proc. Natl. Acad. Sci. USA. 96:3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu, L., B. Gorham, S.C. Li, A. Bottaro, F.W. Alt, and P. Rothman. 1993. Replacement of germ-line epsilon promoter by gene targeting alters control of immunoglobulin heavy chain class switching. Proc. Natl. Acad. Sci. USA. 90:3705–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, J., A. Bottaro, S. Li, V. Stewart, and F.W. Alt. 1993. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. EMBO J. 12:3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 23.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 24.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 26.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 27.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 28.Doi, T., K. Kinoshita, M. Ikegawa, M. Muramatsu, and T. Honjo. 2003. Inaugural article: de novo protein synthesis is required for the activation-induced cytidine deaminase function in class-switch recombination. Proc. Natl. Acad. Sci. USA. 100:2634–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID- mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 30.Bransteitter, R., P. Pham, M.D. Scharff, and M.F. Goodman. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 30b.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zan, H., A. Komori, Z. Li, A. Cerutti, A. Schaffer, M.F. Flajnik, M. Diaz, and P. Casali. 2001. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 14:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogozin, I.B., Y.I. Pavlov, K. Bebenek, T. Matsuda, and T.A. Kunkel. 2001. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2:530–536. [DOI] [PubMed] [Google Scholar]
- 33.Zeng, X., D.B. Winter, C. Kasmer, K.H. Kraemer, A.R. Lehmann, and P.J. Gearhart. 2001. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2:537–541. [DOI] [PubMed] [Google Scholar]
- 34.Faili, A., S. Aoufouchi, E. Flatter, Q. Gueranger, C.A. Reynaud, and J.C. Weill. 2002. Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymerase iota. Nature. 419:944–947. [DOI] [PubMed] [Google Scholar]
- 35.Pavlov, Y.I., I.B. Rogozin, A.P. Galkin, A.Y. Aksenova, F. Hanaoka, C. Rada, and T.A. Kunkel. 2002. Correlation of somatic hypermutation specificity and A-T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc. Natl. Acad. Sci. USA. 99:9954–9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward, I.M., and J. Chen. 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276:47759–47762. [DOI] [PubMed] [Google Scholar]
- 38.Burma, S., B.P. Chen, M. Murphy, A. Kurimasa, and D.J. Chen. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- 39.Andegeko, Y., L. Moyal, L. Mittelman, I. Tsarfaty, Y. Shiloh, and G. Rotman. 2001. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 276:38224–38230. [DOI] [PubMed] [Google Scholar]
- 40.Rogakou, E.P., D.R. Pilch, A.H. Orr, V.S. Ivanova, and W.M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 41.Rogakou, E.P., C. Boon, C. Redon, and W.M. Bonner. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paull, T.T., E.P. Rogakou, V. Yamazaki, C.U. Kirchgessner, M. Gellert, and W.M. Bonner. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10:886–895. [DOI] [PubMed] [Google Scholar]
- 43.Bassing, C.H., K.F. Chua, J. Sekiguchi, H. Suh, S.R. Whitlow, J.C. Fleming, B.C. Monroe, D.N. Ciccone, C. Yan, K. Vlasakova, et al. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA. 99:8173–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaux, D.L., S. Cory, and J.M. Adams. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 335:440–442. [DOI] [PubMed] [Google Scholar]
- 45.Shih, T.A., E. Meffre, M. Roederer, and M.C. Nussenzweig. 2002. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat. Immunol. 3:570–575. [DOI] [PubMed] [Google Scholar]
- 46.Jolly, C.J., N. Klix, and M.S. Neuberger. 1997. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res. 25:1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagaoka, H., M. Muramatsu, N. Yamamura, K. Kinoshita, and T. Honjo. 2002. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J. Exp. Med. 195:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sale, J.E., M. Bemark, G.T. Williams, C.J. Jolly, M.R. Ehrenstein, C. Rada, C. Milstein, and M.S. Neuberger. 2001. In vivo and in vitro studies of immunoglobulin gene somatic hypermutation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCall, M.N., and P.D. Hodgkin. 1999. Switch recombination and germ-line transcription are division-regulated events in B lymphocytes. Biochim. Biophys. Acta. 1447:43–50. [DOI] [PubMed] [Google Scholar]
- 50.Kinoshita, K., M. Harigai, S. Fagarasan, M. Muramatsu, and T. Honjo. 2001. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc. Natl. Acad. Sci. USA. 98:12620–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu, Y., K.J. Seidl, G.A. Rathbun, C. Zhu, J.P. Manis, N. van der Stoep, L. Davidson, H.L. Cheng, J.M. Sekiguchi, K. Frank, et al. 1997. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 7:653–665. [DOI] [PubMed] [Google Scholar]
- 52.Bogue, M.A., C. Wang, C. Zhu, and D.B. Roth. 1997. V(D)J recombination in Ku86-deficient mice: distinct effects on coding, signal, and hybrid joint formation. Immunity. 7:37–47. [DOI] [PubMed] [Google Scholar]
- 53.Hummel, M., J.K. Berry, and W. Dunnick. 1987. Switch region content of hybridomas: the two spleen cell Igh loci tend to rearrange to the same isotype. J. Immunol. 138:3539–3548. [PubMed] [Google Scholar]
- 54.Winter, E., U. Krawinkel, and A. Radbruch. 1987. Directed Ig class switch recombination in activated murine B cells. EMBO J. 6:1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bottaro, A., F. Young, J. Chen, M. Serwe, F. Sablitzky, and F.W. Alt. 1998. Deletion of the IgH intronic enhancer and associated matrix-attachment regions decreases, but does not abolish, class switching at the mu locus. Int. Immunol. 10:799–806. [DOI] [PubMed] [Google Scholar]
- 56.Dudley, D.D., J.P. Manis, A.A. Zarrin, L. Kaylor, M. Tian, and F.W. Alt. 2002. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc. Natl. Acad. Sci. USA. 99:9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bachl, J., C. Carlson, V. Gray-Schopfer, M. Dessing, and C. Olsson. 2001. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol. 166:5051–5057. [DOI] [PubMed] [Google Scholar]
- 58.Lee, C.G., K. Kinoshita, A. Arudchandran, S.M. Cerritelli, R.J. Crouch, and T. Honjo. 2001. Quantitative regulation of class switch recombination by switch region transcription. J. Exp. Med. 194:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 60.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 61.Strahl, B.D., and C.D. Allis. 2000. The language of covalent histone modifications. Nature. 403:41–45. [DOI] [PubMed] [Google Scholar]
- 62.Jenuwein, T., and C.D. Allis. 2001. Translating the histone code. Science. 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 63.Downs, J.A., N.F. Lowndes, and S.P. Jackson. 2000. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 408:1001–1004. [DOI] [PubMed] [Google Scholar]
- 64.Fernandez-Capetillo, O., S.K. Mahadevaiah, A. Celeste, P.J. Romanienko, D.R. Camerini-Otero, W. Bonner, K. Manova, P.S. Burgoyne, and A. Nussenzweig. 2003. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell. 4:497–508. [DOI] [PubMed] [Google Scholar]
- 65.Ehrenstein, M.R., and M.S. Neuberger. 1999. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 18:3484–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ehrenstein, M.R., C. Rada, A.M. Jones, C. Milstein, and M.S. Neuberger. 2001. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 98:14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schrader, C.E., J. Vardo, and J. Stavnezer. 2002. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J. Exp. Med. 195:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rada, C., M.R. Ehrenstein, M.S. Neuberger, and C. Milstein. 1998. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 9:135–141. [DOI] [PubMed] [Google Scholar]
- 70.Winter, D.B., Q.H. Phung, A. Umar, S.M. Baker, R.E. Tarone, K. Tanaka, R.M. Liskay, T.A. Kunkel, V.A. Bohr, and P.J. Gearhart. 1998. Altered spectra of hypermutation in antibodies from mice deficient for the DNA mismatch repair protein PMS2. Proc. Natl. Acad. Sci. USA. 95:6953–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Phung, Q.H., D.B. Winter, R. Alrefai, and P.J. Gearhart. 1999. Hypermutation in Ig V genes from mice deficient in the MLH1 mismatch repair protein. J. Immunol. 162:3121–3124. [PubMed] [Google Scholar]
- 72.Kim, N., G. Bozek, J.C. Lo, and U. Storb. 1999. Different mismatch repair deficiencies all have the same effects on somatic hypermutation: intact primary mechanism accompanied by secondary modifications. J. Exp. Med. 190:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiesendanger, M., B. Kneitz, W. Edelmann, and M.D. Scharff. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wuerffel, R.A., J. Du, R.J. Thompson, and A.L. Kenter. 1997. Ig Sgamma3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 159:4139–4144. [PubMed] [Google Scholar]
- 75.Chen, X., K. Kinoshita, and T. Honjo. 2001. Variable deletion and duplication at recombination junction ends: implication for staggered double-strand cleavage in class-switch recombination. Proc. Natl. Acad. Sci. USA. 98:13860–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bosma, G.C., J. Kim, T. Urich, D.M. Fath, M.G. Cotticelli, N.R. Ruetsch, M.Z. Radic, and M.J. Bosma. 2002. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J. Exp. Med. 196:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 78.Sale, J.E., D.M. Calandrini, M. Takata, S. Takeda, and M.S. Neuberger. 2001. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature. 412:921–926. [DOI] [PubMed] [Google Scholar]
- 79.Bemark, M., J.E. Sale, H.J. Kim, C. Berek, R.A. Cosgrove, and M.S. Neuberger. 2000. Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PK(cs)) or recombination-activating gene (RAG)1 activity. J. Exp. Med. 192:1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dunnick, W., M. Wilson, and J. Stavnezer. 1989. Mutations, duplication, and deletion of recombined switch regions suggest a role for DNA replication in the immunoglobulin heavy-chain switch. Mol. Cell. Biol. 9:1850–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li, J., G.A. Daniels, and M.R. Lieber. 1996. Asymmetric mutation around the recombination break point of immunoglobulin class switch sequences on extrachromosomal substrates. Nucleic Acids Res. 24:2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pan, Q., and L. Hammarstrom. 1999. Targeting of human switch recombination breakpoints: implications for the mechanism of mu-gamma isotype switching. Eur. J. Immunol. 29:2779–2787. [DOI] [PubMed] [Google Scholar]
- 84.Fernandez-Capetillo, O., H.T. Chen, A. Celeste, I. Ward, P.J. Romanienko, J.C. Morales, K. Naka, Z. Xia, R.D. Camerini-Otero, N. Motoyama, et al. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993–997. [DOI] [PubMed] [Google Scholar]
- 84a.Celeste, A. O. Fernandez-Capetillo, M. Kruhlak, D.R. Pilch, D.W. Staudt, A. Lee, R.F. Bonner, W.M. Bonner, and A. Nussenzweig. 2003. Histone H2AX phosphorylation is dispensible for the initial recognition of DNA breaks. Nat. Cell Biol. In press. [DOI] [PubMed] [Google Scholar]
- 85.Mahadevaiah, S.K., J.M. Turner, F. Baudat, E.P. Rogakou, P. de Boer, J. Blanco-Rodriguez, M. Jasin, S. Keeney, W.M. Bonner, and P.S. Burgoyne. 2001. Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet. 27:271–276. [DOI] [PubMed] [Google Scholar]
- 86.Chen, H.T., A. Bhandoola, M.J. Difilippantonio, J. Zhu, M.J. Brown, X. Tai, E.P. Rogakou, T.M. Brotz, W.M. Bonner, T. Ried, and A. Nussenzweig. 2000. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 290:1962–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]