

Abstract
Few hepatitis C virus (HCV) infections resolve spontaneously but those that do appear to afford protective immunity. Second infections are usually shorter in duration and are less likely to persist but mechanisms of virus control in immune individuals have not been identified. In this study we investigated whether memory helper and/or cytotoxic T lymphocytes provide protection in chimpanzees serially reinfected with the virus. Clearance of the first infection took 3–4 mo and coincided with the delayed onset of CD4+ and CD8+ T cell responses. High frequencies of memory T cells targeting multiple HCV proteins were stable over 7 yr of follow-up. Animals were infected for a second time to assess the protective role of memory T cells. In contrast to the prolonged course of the first infection, viremia was terminated within 14 d. Control of this second infection was kinetically linked to rapid acquisition of virus-specific cytolytic activity by liver resident CD8+ T cells and expansion of memory CD4+ and CD8+ T cells in blood. The importance of memory CD8+ T cells in control of HCV infection was confirmed by antibody-mediated depletion of this lymphocyte subset before a third infection. Virus replication was prolonged despite the presence of memory CD4+ T helper cells primed by the two prior infections and was not terminated until HCV-specific CD8+ T cells recovered in the liver. These experiments demonstrate an essential role for memory CD8+ T cells in long-term protection from chronic hepatitis C.
Keywords: liver diseases, viral hepatitis, hepatitis C, immunologic memory, T lymphocytes
Introduction
Primary hepatitis C virus (HCV)* infection resolves spontaneously in some individuals but most develop a persistent viremia that is often associated with progressive liver disease over a period of years to decades (1). A substantial body of evidence indicates that cellular immune responses are involved in control of the infection (2–6). Virus clearance in humans and chimpanzees has been linked in most studies to the onset of sustained HCV-specific CD4+ and CD8+ T cell responses (2–6) and an increase in IFN-γ mRNA levels in the liver (2). In contrast, cellular immune responses fail in those who develop persistent infections (2–6). HCV-specific CD4+ T cells are absent or transiently present in the blood (2, 4, 5, 7, 8), do not accumulate in substantial numbers in the liver (2), and may be skewed toward production of regulatory rather than antiviral cytokines (9). CD8+ T cells are sometimes detected in persistently infected humans and chimpanzees but they may lack adequate effector function (4, 5, 10–13), target a limited repertoire of viral epitopes (3, 5), and rapidly select for viruses that contain MHC class I escape mutations (14–16). Despite this circumstantial evidence linking the outcome of HCV infection with the strength of cellular immune responses, the effector mechanisms and the relative importance of CD4+ versus CD8+ T cells in prevention of persistence, are still unknown.
Individuals who successfully resolve primary HCV infections appear to have robust immunity against the virus because subsequent infections rarely persist (17). Studies in chimpanzees have revealed that the duration of viremia is also shortened after a second infection (18–22). So far this animal model has provided limited insight into the nature of protective immune responses. Resolution of a second infection was associated with a rapid type I IFN response in one study (21) and IFN-γ in another (22). Involvement of memory CD4+ and/or CD8+ T cells has not been established. Primed memory T cells were not detected in chimpanzees with resolved HCV infections (20–23). Moreover, after reinfection virus-specific lymphoproliferative responses were very weak or not consistently detected in all animals (20–22) so a temporal relationship between rapid clearance of virus and accelerated T cell responses has not been established.
Here we report that memory CD8+ T lymphocytes are required to rapidly terminate HCV replication upon reexposure to the virus. In a series of three infections in chimpanzees we demonstrate that (a) memory CD4+ and CD8+ T cell frequencies are set immediately after resolution of primary HCV infection and then do not vary over a period of several years, (b) the duration and peak of viremia are substantially diminished after a second infection and this control is temporally associated with a rapid and massive expansion of fully functional memory helper and cytotoxic T cells, and (c) antibody-mediated depletion of the cytotoxic CD8+ subset immediately before a third infection resulted in a prolonged course of viremia that was not terminated until memory CD8+ T cells recovered in the liver.
Materials and Methods
HCV Infections.
Chimpanzees (Pan troglodytes) were maintained under standard conditions for humane care and in compliance with NIH guidelines at the New Iberia Research Center, Lafayette, LA. They were infected intravenously with 100 chimpanzee infectious doses (CID) of HCV-1/910 stock (24) and then rechallenged 7 yr later with the same virus. 6 mo after clearance of the second infection, three doses of the anti-CD8 antibody cM-T807 (25, 26) were administered intravenously 14, 11, and 7 d before a third HCV challenge with 100 CID of HCV-1/910. An initial dose of 5 mg of cM-T807 per Kg body weight was followed by two reinforcing doses of 2.5 mg/Kg.
HCV RNA.
HCV RNA levels in frozen plasma collected in EDTA were analyzed by HCV 3.0 RNA branched DNA (bDNA; sensitivity 3,500 genome equivalents [GE]/ml) and HCV RNA TMA QL (sensitivity 50 GE/ml) assays by Bayer Diagnostics.
HCV Antibody Titers.
HCV-specific antibody titers in serum were measured using a multi-antigen ELISA as described previously (27, 28). Assay cut off was the mean O.D. of plasma from HCV seronegative humans + 7 standard deviations. Relative units of antibody titers were determined as O.D. of sample multiplied by the dilution factor. Serum titers are expressed as: (+++) >1,000, (++) 100–1,000, (+) 10–100, (+/−) <10 and (–) undetectable.
Isolation of Lymphocytes from Blood and Liver.
PBMCs were isolated using Ficoll density gradient. To recover intrahepatic lymphocytes (IHL), liver biopsies were gently homogenized in PBS containing 1% FCS, CD8+ T cells were enriched using anti–human CD8+ dynabeads (Dynal) and expanded using anti-human CD3 antibodies (Immunotech) as described previously (3, 29). Expanded cells were tested in either CTL or IFN-γ ELISPOT assays. For direct tetramer staining, liver homogenate was filtered through 70 μ cell strainers and stained directly without in vitro expansion as described below.
Liver CTL Assays.
CD8+ T cells expanded from liver were tested for HCV-specific cytotoxic activity in a standard 4 h 51Cr release assay as described previously (3, 29). Target cells were autologous EBV-transformed B cell lines infected with HCV-1 recombinant vaccinia vectors (a kind gift of Chiron Corporation, Emeryville, CA) that span the following regions: vv core/E1 (amino acids [aa]: 1–384), vv E2-NS3 (aa: 364–1618), vv NS4 (aa: 1590–2050), vv NS5A (aa: 2005–2396), and vv NS5B (aa: 2396–3011). Specific killing greater than 10% of prechallenge baseline at two E:T ratios was considered positive.
ELISPOT Assay.
Nine pools containing 30–40 peptides each were prepared using a set of 301 overlapping peptides (Mimotopes Pty) encompassing the entire HCV-1 polyprotein according to the published sequence by Choo et al. (24). Peptides were 20 amino acids long overlapping by 10 residues. Human IFN-γ ELISPOT kits were purchased from U-cytech. Cryopreserved PBMCs were used to assay the immune response during primary infection. Freshly isolated PBMCs were used in the secondary and tertiary infections. Comparison of T cell frequencies post primary infection using fresh and frozen PBMCs collected at the same time point yielded comparable results. 1–2 × 105 PBMCs were cocultured in duplicate in round bottom 96-well plates with peptide pools for 40–48 h at 0.5 μg/ml final concentration of each peptide in Aim-HS (Aim-V lymphocyte media [Invitrogen] supplemented with 2% human AB serum [Gemini-BioScience]). PBMCs were then transferred to precoated ELISPOT plates, incubated at 37°C for 16–18 h and then developed according to the manufacturer's protocol. The ovalbumin derived peptide “SIINFEKL” was used as a negative control. Number of specific spots was calculated by subtracting the mean number of spots in negative control duplicates from the mean number of spots in test duplicates, then normalized to number of specific spots/106 PBMCs. In some assays, CD8+ T cells were removed from PBMCs using paramagnetic beads (Dynal), and the cells incorporated in the assay as outlined above.
CD8+ IHLs were isolated and expanded from liver biopsies as described above. For the ELISPOT assay, 105 irradiated autologous PBMCs were used as antigen presenting cells with 0.5–1 × 105 expanded intrahepatic CD8+ T cells for 40–48 h then processed for spot formation as outlined above.
Flow Cytometry.
Antibodies used included anti–human CD3, CD4 (Leu 3a), CD8-PerCP (Leu-2a), CD28 (CD28.2), CD49d (9F10), IFN-γ–FITC (4S.B3), and CD69-APC (L78) all from BD Biosciences. Patr B2301/E2445 tetramer was synthesized at the NIH tetramer core facility, Emory University, Atlanta, GA. Cryopreserved PBMCs were used to assay the frequency of tetramer+ cells during primary infection. Freshly isolated PBMCs were used in the secondary and tertiary infections. 106 fresh or frozen PBMCs in AIM-HS medium were used per stain. For intracellular cytokine staining, cells were first stimulated with E2445 peptide (1 μg/ml) for 6 h at 37°C in presence of anti-CD28 and CD49d (1 μg/ml each) and addition of Brefeldin A (10 μg/ml) after the first hour. The reaction was stopped by resuspension in FACS buffer (PBS, 2% FCS, 0.1% NaN3). Cells were then stained with anti-CD8 antibody for 20 min at 4°C, followed by the tetramer for 30 min at room temperature, permeabilized using cytofix-cytoperm solution (BD Biosciences), and finally stained with anti-IFN-γ–FITC and CD69-APC for 30 min at 4°C. Cells were resuspended in 4% formaldehyde solution and analyzed on a FACSCalibur™ instrument using CELLQuest™ software. 100,000 events were acquired in the CD8+ T lymphocytes gate. For direct tetramer staining on liver homogenate, 10,000–15,000 events were acquired.
Depletion of T cell subsets in the peripheral blood after cM-T807 treatment was monitored by 4 color flow cytometry using anti-CD3 and subset-specific antibodies against CD4 (Leu 3a) and CD8 (DK25) (DakoCytomation), an antibody previously shown to bind CD8 protein in presence of cM-T807 (25).
Results
Primary Infection
Virus Replication and Immune Responses.
Chimpanzees CB0556 and CB0572 were enrolled in a vaccine study by Chiron Corporation in 1994. Recombinant envelope glycoproteins E1 and E2 produced in Chinese hamster ovary (CHO) cells were used to vaccinate the animals 3 times before challenge with 100 CID of the HCV-1/910 virus inoculum. Serum antibodies against E1 and E2 on the day of virus challenge did not protect them from infection. They exhibited a prolonged course of viremia that lasted for 3 (CB0572) or 4 (CB0556) mo (Fig. 1, A and B) . An identical pattern of viremia and clearance was observed in a mock-vaccinated control animal (unpublished data).
Figure 1.
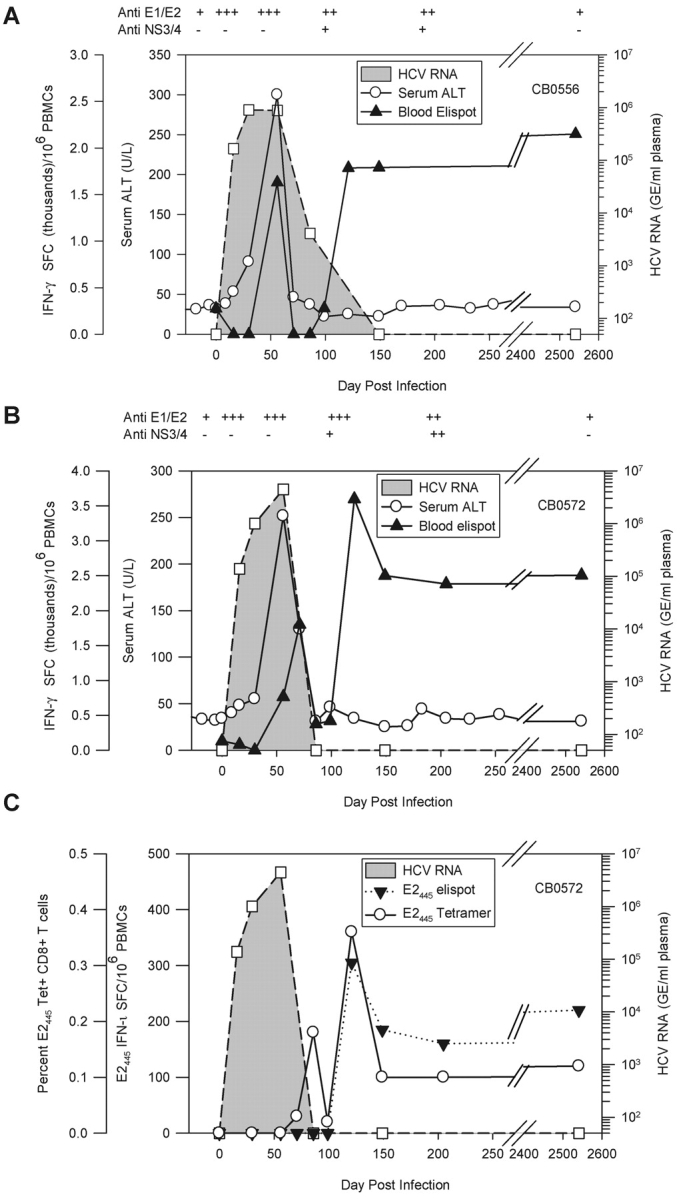
Course of acute resolving HCV-1 infection in chimpanzees CB0556 and CB0572. Virus replication and HCV-specific immune responses after primary infection of CB0556 (A) and CB0572 (B). HCV RNA genome equivalents (GE) per milliliter of plasma (GE/ml) are shown. Spot forming cells (SFCs) (×10−3) represent the sum of responses to pools of overlapping peptides. Day 2541 (7 yr) is representative of four independent time points. (C) Detection of E2445-specific CD8+ T cells using IFN-γ ELISPOT and a Patr B2301 tetramer during primary infection of CB0572.
We next examined the relationship between control of viral infection and T cell–mediated immune responses using an IFN-γ ELISPOT assay with pooled peptides that spanned the HCV polyprotein. A weak vaccine-induced IFN-γ T cell response against the envelope glycoproteins was observed in the blood of both animals before infection but it rapidly disappeared by day 16 after infection when HCV RNA was first detected in their plasma (Table I). Over the next 2 wk virus titers increased to ∼1 × 106 GE/ml of plasma and remained stable (CB0556) or increased slightly (CB0572) until day 56 after infection when a sharp alanine aminotransferase (ALT) increase and T cell responses against nonstructural proteins were detected for the first time (Table I, and Fig. 1, A and B). These experiments provided additional support for an infection model where HCV replicates for weeks or months in the absence of overt liver damage and clearance is associated with the onset of a sustained T cell response that can cause immunopathology (2–4).
Table I.
Diversity of the HCV-specific Response During Primary Infection
| CB0556
|
CB0572
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γ SFC/104 PBMCs
|
IFN-γ SFC/104 PBMCs
|
|||||||||
| Day after infection |
HCV RNAa(GE/ml plasma) | Core-NS2 | NS3 | NS4 | NS5 | HCV RNAa(GE/ml plasma) | Core-NS2 | NS3 | NS4 | NS5 |
| 0 | <2,500 | 275 | 0 | 0 | 0 | <2,500 | 130 | 0 | 0 | 0 |
| 16 | 1.6 × 105 | 0 | 0 | 0 | 0 | 1.4 × 105 | 0 | 0 | 0 | 0 |
| 30 | 9 × 105 | 0 | 0 | 0 | 0 | 1 × 106 | 0 | 0 | 0 | 0 |
| 56 | 8.8 × 105 | 250 | 735 | 450 | 195 | 4.4 × 106 | 95 | 570 | 100 | 0 |
| 71 | ND | 0 | 0 | 0 | 0 | ND | 100 | 1,130 | 405 | 165 |
| 86 | 4 × 103 | 0 | 0 | 0 | 0 | <2,500 | 0 | 375 | 0 | 0 |
| 99 | ND | 55 | 225 | 0 | 0 | ND | 95 | 195 | 130 | 0 |
| 121 | ND | 510 | 585 | 485 | 205 | ND | 800 | 1,385 | 475 | 935 |
| 149 | <2,500 | 560 | 595 | 570 | 0 | <2,500 | 625 | 905 | 380 | 590 |
| 205 | ND | ND | ND | ND | ND | ND | 530 | 970 | 470 | 410 |
| 7 yrb | <2,500 | 660 | 705 | 665 | 119 | <2,500 | 580 | 915 | 550 | 460 |
Determined by HCV-RNA 3.0 assay (sensitivity <2,500 GE/ml plasma).
Representative data from four independent time points before secondary infection at 7 yr.
Evolution and Stability of Memory T Cell Populations.
We observed an unexpected reduction in HCV-specific T cell frequencies in the blood of both animals just as HCV infection was controlled. Strong cytotoxic T cell activity was present in the liver at these time points (unpublished data), suggesting that effector lymphocytes were localizing to the liver. HCV-specific T cell frequencies rebounded ∼1 mo later to form a memory pool targeting multiple viral proteins that did not wane over 7 yr of follow-up (Table I). In contrast to the stability of memory T cells, serum antibodies against the envelope glycoproteins induced by vaccination or infection were undetectable or present at very low titers 7 yr later (Fig. 1, A and B).
Similar results were obtained by monitoring memory CD8+ T cell frequencies against a Patr-B2301 restricted epitope designated E2445 targeted by chimpanzee CB0572 (Table II). This epitope was identified in the liver and blood of CB0572 at 6 yr after infection. Tetramer analysis of cryopreserved PBMCs collected during the primary infection demonstrated that this population first appeared in blood at day 86 after infection and stabilized at a frequency of ∼0.14% of CD8+ PBMCs by month 5 after infection that did not change over the next 7 yr (Fig. 1 C). Interestingly, E2445-specific T cells visualized in blood with the tetramer starting on day 86 after infection were not consistently detected in the IFN-γ ELISPOT assay until day 121 after infection (Fig. 1 C), implying that this population transiently lacked effector function as previously suggested by others (4, 5, 10–13).
Table II.
MHC Class I–restricted Epitopes Identified at 6 yr After Primary Infection
| HCV protein | Epitope designation | Patr MHC restriction | Amino acid coordinates | Peptide sequence | |
|---|---|---|---|---|---|
| CB0556 | P7 | P7756 a | A0701 | 756–770 | AASLAGTHGLVSFL |
| CB0572 | E2 | E2445 a , b | B2301 | 445–457 | HKFNSSGCPERL |
| NS5B | NS52663 b | B2401 | 2663–2673 | CDLDPQARVAI |
Identified in blood at 6 yr after primary infection.
Isolated by limit dilution cloning from the liver at 6 yr after primary infection.
Secondary Infection
Virus Replication and Immune Responses.
Chimpanzees CB0556 and CB0572 were rechallenged seven years after the first infection with the identical strain and dose of HCV to determine if these long-lived memory T cell responses could blunt viral infection. HCV RNA peaked at 105 GE/ml of plasma in both chimpanzees by day 7 after infection and declined thereafter (Fig. 2, A and B) . ALT increases were mild compared with the first infection (unpublished data). Antibody titers were boosted by the second infection but only after the virus was permanently cleared from plasma (unpublished data), so it is unlikely that they exerted antiviral activity. Memory T cell frequencies detected by an IFN-γ ELISPOT assay did not increase through the first 10 d of infection. However, a 3–6 fold expansion was detected at the next sampling point 4 d later when HCV RNA levels were very low in CB0556 (64 GE/ml) and undetectable in CB0572 (Fig. 2, A and B). This response peaked on day 21 and was mediated by CD4+ and CD8+ T cells because frequencies were reduced by 50% if CD8+ T cells were removed before the assay (unpublished data). Involvement of HCV-specific CD8+ T cells was confirmed by monitoring responses against MHC class I–restricted epitopes identified in the liver and/or the blood of each chimpanzee before the secondary infection (Table II). Memory CD8+ T cells from CB0556 that recognized the Patr-A0701 restricted epitope designated P7756 (Table II) expanded more than 20-fold in blood between days 10 and 14 after infection (Fig. 2 A). T cells from CB0572 targeting the E2445 epitope remained stable in blood at a frequency of 0.1% by IFN-γ ELISPOT and MHC class I tetramer assays through the first 10 d of the second infection. Remarkably, they expanded 45-fold (to 4.5% by tetramer analysis) by day 14 (Fig. 2, B and C) and were fully functional because they expressed the activation marker CD69 and produced IFN-γ after in vitro peptide stimulation (Fig. 2 D). Frequencies of HCV-specific cells in both chimpanzees declined gradually to baseline values by day 94 after infection (Fig. 2, A and B).
Figure 2.

Virus replication and HCV-specific immune responses after rechallenge with HCV-1 at 7 yr after primary infection IFN-γ SFC after a second infection of CB0556 (A) and CB0572 (B) represent the sum of responses to pools of overlapping peptides or the individual CD8+ T cell epitopes P7756 (A) and E2445 (B). Prechallenge ELISPOT represents the mean SFC/106 PBMC at four independent time points. Frequency of E2445 specific T cells measured by tetramer staining is represented as percent of CD8+ T cells. (C) Detection of E2445-specific T cells using a Patr B2301 tetramer. PBMCs from CB0556 (Patr B2301−) (left panel) and from CB0572 (Patr B2301+) at ∼7 yr after the first infection (middle panel) and at day 14 after the second infection (right panel) were stained with E2445/Patr B2301 tetramer. A total of 100,000 events were acquired in the CD8+ lymphocyte gate and the frequency is presented as percent of CD8+ tetramer+ T cells. (D) Peptide activation of E2445-specific T cells from blood of CB0572 collected on day 14 after infection.
Intrahepatic CD8+ T Cell Responses.
Too few CD8+ T cells were recovered from typical liver specimens (<104/gram of tissue) to directly measure virus-specific populations so they were first expanded with anti-CD3 antibodies. HCV-specific T cells were not detected in the liver of either animal before reinfection using tetramer staining (Fig. 3 A) or cytotoxicity assays (Fig. 3, B and C), probably because they were present at a frequency too low to be measured in uncloned, anti-CD3 expanded T cell populations. However, HCV-specific cytolytic activity was detected in the liver of CB0572 and CB0556 by day 10 after infection against autologous target cells expressing the E2-P7-NS2-NS3 proteins (vv E2-NS3; Fig. 3, B and C). In the case of CB0572, at least some of this cytolytic activity on days 14 and 21 was directed against the E2445 epitope because targets pulsed with the peptide epitope were killed (Fig. 3 B) and a high percentage of the expanded CD8+ T cells stained with the Patr-B2301/E2445 tetramer (Fig. 3 A). Furthermore, the response broadened in both animals on day 14 after infection to include targets expressing the NS5B protein and returned to baseline values by day 44 after infection (Fig. 3, B and C). These results suggested that intrahepatic CD8+ T cells that gain cytolytic function within a few days of infection contribute to rapid decline in viremia.
Figure 3.

Intrahepatic HCV-specific responses upon rechallenge with HCV-1. (A) Detection of E2445 specific T cells using a Patr B2301 tetramer on in vitro expanded CD8+ T cells (IHL). CD8+ lymphocytes isolated from the liver of CB0572 at the indicated time points were expanded with anti-CD3 antibodies for 2–3 wk and stained with the Patr B2301/E2445 tetramer. Frequency is presented as percent of CD8+ tetramer+ T cells. Acquisition of HCV-specific CTL activity by intrahepatic CD8+ T cells (IHL) isolated from CB0572 (B) and CB0556 (C). Intrahepatic CD8+ T cells expanded with anti-CD3 antibodies were tested for cytolytic activity against autologous B lymphoblastoid cell lines infected with recombinant vaccinia viruses expressing all regions of the HCV polyprotein. Killing was detected only against targets that expressed E2-P7-NS2-NS3 (vvE2-NS3; aa 364–1618) and NS5B (vvNS5B; aa 2396–3011) and targets pulsed with the E2445 peptide. Cytolytic activity was scored as positive (*) when it exceeded baseline (preinfection) values by 10% at two effector to target (E:T) cell ratios. Data shown are for a 25:1 E:T ratio.
Tissue Distribution of HCV-specific T Cells.
Evidence for selective retention of memory CD8+ T cells in the liver after clearance of the second infection was obtained by comparing decay of E2445-specific CD8+ T cells in blood and liver without anti-CD3 antibody expansion. Two weeks after clearance of infection (i.e., at day 28 after infection) ∼4.5% of CD8+ T cells in liver stained with the tetramer compared with 1% in blood (Fig. 4 A). E2445-specific T cells declined in both tissue compartments by 6 mo after resolution of the second infection but were still ∼10 times more concentrated in liver (1.8%) than blood (0.2%; Fig. 4 A). Interestingly, memory CD8+ T cells in the liver but not blood expressed the activation marker CD69 at multiple time points during the six months follow-up period (Fig. 4 B). This constitutively activated phenotype is identical to that of tissue-resident effector memory CD8+ T cells previously described in mice (30, 31).
Figure 4.
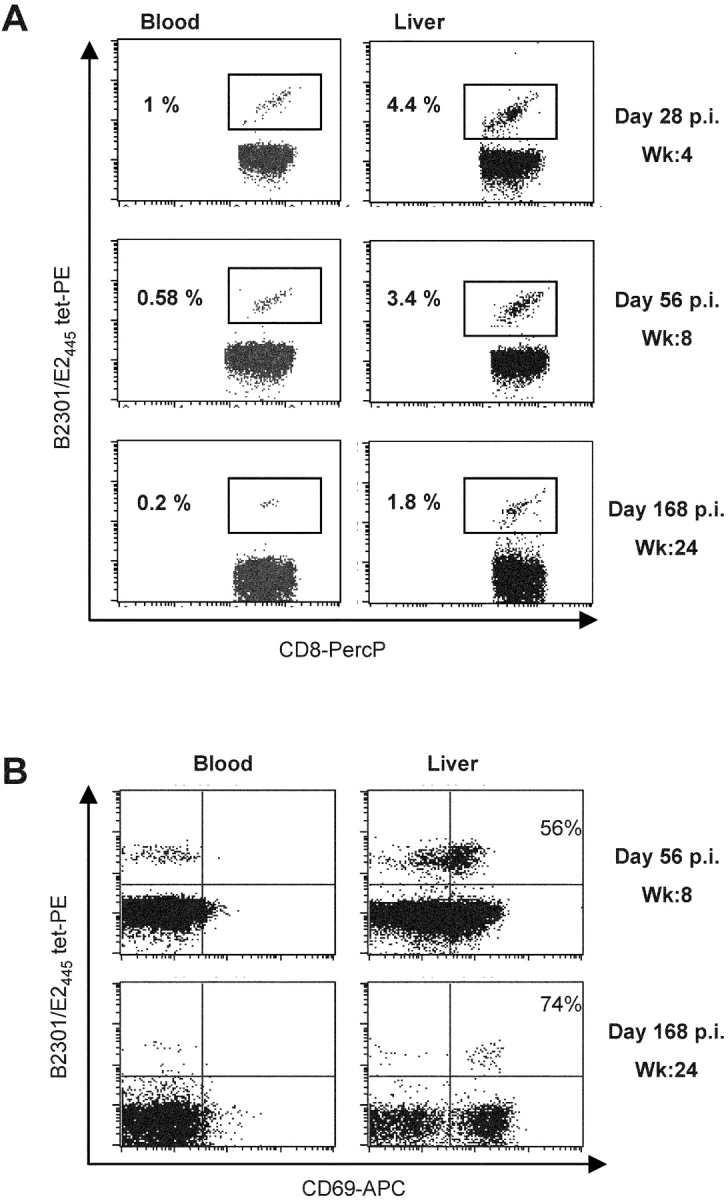
Tissue distribution and phenotype of Patr B2301/E2445 tetramer-reactive CD8+ T cells. Lymphocytes were isolated directly from blood and liver of CB0572 at the indicated time points and stained (without anti-CD3 antibody expansion) with anti-CD8 antibodies and Patr B2301/E2445 tetramer (A). Cells were also stained with anti-CD69 antibodies (B). 10,000–15,000 events were acquired in the CD8+ T lymphocytes gate and frequency is represented as percent of CD8+ tetramer+ cells (A) or percent of tetramer+ CD69+ cells (B).
Tertiary Infection
Antibody-mediated CD8+ T Cell Depletion and the Course of HCV Infection.
We next assessed the importance of memory CD8+ T cells in control of HCV infection by treating both animals with the anti-CD8 antibody cM-T807 6 mo after resolution of the second infection. This antibody was used successfully in rhesus monkeys (25) and chimpanzees (26) to asses the role of CD8+ T cells in control of simian immunodeficiency virus (SIV; reference 25) and hepatitis B virus (HBV; reference 26) infections, respectively. Three doses of cM-T807 were administered intravenously 14, 11, and 7 d before a third HCV challenge. Prior to administration of the third antibody dose (day −7) CD8+ T cells were already undetectable in blood using a monoclonal antibody (DK25) that does not compete with cM-T807 for CD8 binding (25) and they remained at less than 15% of baseline values through 6 mo of follow-up (Fig. 5 A, Table III). CD4+ T cell numbers were stable in the blood of both animals (Fig. 5 A, Table III). 7 d after the third dose of cM-T807 both animals were challenged for a third time with 100 CID of HCV-1/910. HCV RNA was first detected in the plasma of CB0556 at day 7 after infection and CB0572 at day 10 after infection (Table III, Fig. 5, B and C), and peak titers were not substantially different from those observed after the secondary infection when CD8+ T cell compartments were intact (Fig. 6) . Importantly, HCV infection was prolonged by at least 3 wk in both animals (Fig. 6). HCV RNA was consistently detected in the plasma of CB0556 and CB0572 for 35 d after the third infection (Table III, Fig. 5, B and C), and contrasts with the second infection where HCV RNA was detected in CB0556 through day 14 (at less than 100 GE/ml) and in CB0572 through day 10 (Fig. 2, A and B).
Figure 5.

Virus replication and intrahepatic immune responses following antibody-mediated depletion of CD8+ T cells. (A) CD4+ and CD8+ T cell subsets were monitored in the blood of CB0556 (top panel) and CB0572 (bottom panel) after three doses of cMT-807 (vertical dashed lines). Anti-CD3 expanded CD8+ T cells from the liver of CB0556 (B) and CB0572 (C) were tested for IFN-γ production in an ELISPOT assay using HCV-1 peptide pools and individual epitopes. * Indicates inability to recover and expand CD8+ T cells from liver tissue until days 42 and 28 after infection from CB0556 (B) and CB0572 (C), respectively. (NS) indicates that no liver sample was obtained from either chimpanzee on day 35 after infection.
Table III.
Virus Replication and Immune Responses in Blood After CD8 Depletion
| CB0556
|
CB0572
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ELISPOT
|
ELISPOT
|
|||||||||
| Day (after infection) |
HCV RNAa(GE/ml) |
CD8+
T cells/mm3 blood |
CD8b | CD4c | Totald | HCV RNAa(GE/ml) |
CD8+
T cells/mm3 blood |
CD8b | CD4c | Totald |
| −28 | <50 | 1,328 | 125 | 1,130 | 4,635 | <50 | 1,073 | 220 | 145 | 655 |
| −14 | Antibody-mediated depletion of CD8 + T cellse | Antibody-mediated depletion of CD8 + T cellse | ||||||||
| −7 | <50 | 9 | 0 | 1,930 | 1,110 | <50 | 10 | 0 | 120 | 400 |
| 0 | HCV-1 infection | HCV-1 infection | ||||||||
| 7 | 48,356 | 2 | 0 | 0 | 135 | <50 | 6 | 0 | 980 | 980 |
| 10 | 41,175 | 1 | 0 | 185 | 185 | 543 | 1 | 0 | 0 | 240 |
| 15 | 31,641 | 3 | 0 | 1,090 | 835 | 35,384 | 8 | 0 | 0 | 0 |
| 21 | 19,373 | 3 | 0 | 875 | 1,215 | 24,623 | 4 | 0 | 390 | 615 |
| 28 | 26,133 | 5 | 0 | 2,225 | 2,040 | 760 | 12 | 0 | 1,050 | 965 |
| 35 | 5,987 | 4 | 0 | 1,685 | 1,470 | 234 | 67 | 0 | 420 | 440 |
| 42 | <50 | 47 | 135 | 1,790 | 2,330 | <50 | 146 | 0 | 3,210 | 3,885 |
| 49 | <50 | 98 | 0 | 3,955 | 4,900 | <50 | 166 | 0 | 2,895 | 3,060 |
| 56 | <50 | 60 | 0 | 2,445 | 1,670 | <50 | 67 | 0 | 765 | 1,085 |
| 70 | <50 | 61 | 0 | 795 | 915 | <50 | 95 | 0 | 890 | 1,545 |
| 84 | <50 | 70 | 0 | 605 | 685 | <50 | 70 | 0 | 390 | 595 |
| 112 | <50 | 204 | 0 | 1,185 | 1,645 | <50 | 69 | 0 | 590 | 410 |
| 126 | <50 | 139 | 0 | 2,040 | 1,220 | <50 | 158 | 0 | 705 | 795 |
| 140 | <50 | 236 | 0 | 2,270 | 2,390 | <50 | 155 | 0 | 1,990 | 3,810 |
HCV RNA genome equivalents/ml plasma determined by quantitative TMA assay. Sensitivity of the assay is 50 GE/ml plasma.
Data represent total number of SFCs/106 PBMCs (SFC/106) in response to the dominant CD8 CTL epitopes P7756 and E2445 targeted by CB0556 and CB0572, respectively.
Data represent total number of SFCs/106 PBMCs (depleted of CD8+ T cells) in response to all HCV peptide pools.
Data represent total number of SFCs/106 PBMCs (SFC/106) in response to all HCV peptide pools.
Three doses of the anti-CD8 antibody (cM-T807) were administered on days −14, −11, and −7.
Figure 6.

Comparison of virus replication patterns after three consecutive infections of CB0556 and CB0572 with HCV-1.
Memory T cell frequencies were partially reduced in both animals immediately after cM-T807 treatment. Importantly, responses to the defined MHC class I–restricted epitopes were lost entirely, reflecting the effectiveness of in vivo CD8+ T cell depletion (Table III). CD4+ T cell frequencies were unaffected by cM-T807 treatment because they remained identical in both animals before (day –28) and immediately after (day –7) antibody administration (Table III). HCV-specific CD4+ T cell frequencies declined transiently in the blood of both animals when viremia was first detected but they recovered to baseline values by day 21 after infection, well before HCV RNA began to decline in plasma (Table III), suggesting that this T cell subset alone was not sufficient to rapidly clear the infection.
Resolution of HCV Infection Requires Intrahepatic CD8+ T Cells.
After cM-T807 treatment and infection, the few CD8+ T cells that remained in blood failed to produce IFN-γ in response to HCV peptide pools and only transiently against the dominant MHC class I–restricted epitope P7756 in CB0556 on day 42 after infection (Table III). We therefore postulated that HCV-specific CD8+ T lymphocytes recovered in the liver before the blood and were responsible for control of plasma viremia.
CD8+ T cells could not be expanded from the liver of CB0556 at any time through the first 35 d of infection when virus titer stabilized at ∼4 × 104 GE/ml of plasma (Fig. 5 B). CD8+ T cells were first recovered from a liver sample collected at day 42 after infection when plasma viremia abruptly declined to undetectable levels (Fig. 5 B). These CD8+ lymphocytes produced IFN-γ in response to multiple HCV peptide pools and class I epitopes (unpublished data), including the Patr-A0701 restricted epitope P7756 contained in the core-NS2 peptide pool (Fig. 5 B). A similar outcome was observed in CB0572 (Fig. 5 C). CD8+ T cells were not recovered from liver until day 28 after infection when a 30-fold reduction in plasma viremia was observed. They were also HCV-specific, producing IFN-γ in response to the Patr-B2401 restricted epitope NS52663 (Table II) contained in the NS5 peptide pool (Fig. 5 C). Several HCV peptide pools were recognized at the next sampling point (day 42 after infection) when virus was finally cleared from plasma (unpublished data). The predominant target was a peptide pool containing all structural proteins (Core-E1-E2-NS2; Fig. 5 C). At least part of this response targeted the Patr-B2301–restricted epitope E2445 (Fig. 5 C). HCV-specific CD8+ T cells remained in the liver but were undetectable in the blood of either animal throughout the entire 6 mo of follow-up (Table III, Fig. 5 C).
Discussion
Three key features of the memory T cell response against HCV were revealed by serial infection of chimpanzees CB0556 and CB0572. First, the frequency of memory HCV-specific T cells was fixed immediately after resolution of primary infection. This memory cell set-point did not vary over a period of several years despite an apparent absence of virus replication. Second, the response of memory CD4+ and CD8+ T cells to reinfection was accelerated compared with the first infection and coincided with control of viremia in less than 2 wk. Third and perhaps most importantly, this rapid antiviral response was lost in the absence of memory CD8+ T cells. The infection was not terminated until this subset recovered in the liver.
Memory CD8+ T Cells Alter the Outcome of HCV Infection.
Chronic hepatitis C is uncommon in humans and chimpanzees who resolved earlier infections with the virus (17–22). Our studies indicate that CD8+ T cells are a critical component of this protective immunity. HCV-specific CTL activity detected in the liver of both chimpanzees as early as day 10 after infection was kinetically associated with initial control of viremia. Antibody-mediated depletion of CD8+ T cells before a third infection provides direct proof that this lymphocyte subset is important for termination of HCV replication. Prolonged viral infection in both CD8+ T cell–depleted animals would not have been predicted from reinfection studies involving animals with intact immune systems. Indeed, in contrast to the results reported here, the duration and peak of viremia are usually reduced after successive infections even with different HCV genotypes (18–22). The importance of CD8+ T cells is further reinforced by the precise temporal relationship between their recovery in the liver at 4–5 wk and simultaneous clearance of HCV RNA from plasma. Our experiments were not designed to address the mechanism by which CD8+ T cells control HCV replication. Nevertheless, it is noteworthy that liver transaminases did not increase substantially when CD8+ T cells finally recovered in the liver of anti-CD8–treated animals. It is possible that very few hepatocytes were infected compared with the first infection so the scale of liver damage by recovering cytotoxic T lymphocytes was minimized. On the other hand we cannot rule out noncytolytic control by IFN-γ or other mechanisms that might silence expression of the HCV RNA genome in hepatocytes (32, 33). Indeed, clearance of HCV without an elevation in serum transaminases (2, 4) but with an increase in liver IFN-γ mRNA (2) has been reported. Furthermore, CD8+ T cells were recently shown to exert noncytolytic control of another hepatotropic virus, the HBV (26).
It should be emphasized that primed memory CD4+ T cells were present in the blood and liver of both animals after treatment with anti-CD8 antibodies. Their involvement in control of the third infection is unknown. CD4+ T cells needed to sustain CD8+ T cell responses (34) and helper function(s) might have been especially critical after anti-CD8 antibody treatment. Effector CD8+ T cells probably expanded from a small memory pool that survived antibody treatment in the liver, and CD4+ T cells might have facilitated recovery by producing growth factors such as IL-2 or by inducing the maturation of antigen presenting cells. It is also possible that memory CD4+ T cells exerted a direct antiviral effect. We did observe a plateau in virus replication at levels ∼10 to 50-fold lower than those measured after the primary infection of both animals. As noted above, IFN-γ has the potential to inhibit HCV replication and it is produced in the liver after primary (2) and secondary (22) HCV infections. Whether IFN-γ gene expression is temporally linked to the presence of memory CD4+ or CD8+ T cells in the liver of animals treated with anti-CD8 antibodies is being investigated.
Tissue Localization and Phenotypic Markers of Memory T Cells.
Studies in mice revealed that memory T cells can reside in tissues for long periods of time where they could provide a rapid first line of defense upon reinfection (30, 31, 35, 36). This may also be true for HCV infection because our results, although preliminary, indicate that memory virus-specific CD8+ T cells reside in the liver for months and possibly years after apparent control of viral infection. Anecdotally we were able to establish clonal HCV-specific CD8+ T cells (described in Table II) from the liver of CB0572 several years after resolution of the primary infection at a time when they were undetectable by either tetramer staining or CTL killing. At least some of these populations were localized preferentially to the liver because they could not be expanded from blood even after several rounds of stimulation with the cognate peptide (unpublished data).
Corroborative evidence for maintenance of memory cells in the liver was obtained after the second infection. Dominant CD8+ T cell populations were detected in the liver by tetramer staining within 1–2 wk of reinfection and persisted for at least 6 mo after resolution. Memory cell frequencies appeared to be higher in liver than blood and they were also phenotypically distinct because they constitutively expressed the activation marker CD69 found on tissue resident effector memory T cells in murine models of infection (30, 31). HCV RNA was not detected in plasma or liver at multiple time points after resolution of the first or second infections. We cannot exclude the possibility of low level virus persistence that contributed to the maintenance of memory T cells. However, we favor the hypothesis that most HCV-specific memory T cells are maintained in tissues for long periods of time by periodic exposure to cross-reactive antigens (37) or cytokine-driven homeostatic proliferation (38).
Functional Memory T Cells Respond Rapidly to Reinfection.
It is important to emphasize that there was no delay in the response of memory T cells to the second infection. Memory CD8+ T cells with antiviral activity were detected on day 10, only 3 d after HCV RNA was first detected in plasma. Infection was controlled over the next 4 d and T cells present in blood at the peak of the response were fully capable of producing IFN-γ when stimulated with antigen. This clearly differs from the prolonged course of primary infection observed in these animals and reported for other primary infections of humans and chimpanzees (2, 4). A delay in T cell immunity might be explained, at least in part, by infection with a hepatotropic virus that is thought to be noncytopathic. The apparent lack of widespread tissue destruction could delay effective antigen transfer to professional antigen-presenting cells and/or signals for T cell homing to the site of infection. Effector memory CD8+ T cells residing in the liver could contribute to accelerated immunity upon reinfection by mediating an early inflammatory response and destruction of infected hepatocytes. In support of this mechanism we observed infiltration of CD8+ T cells into the liver parenchyma immediately after the second infection (unpublished data) and they exhibited virus-specific CTL activity in vitro. We also cannot exclude the possibility that HCV selectively interferes with the efficient generation of primary but not secondary T cell responses. For instance viral subversion of innate immune responses, or dendritic cell function or maturation, could have a greater impact on generation of primary but not secondary T cell responses.
Our data also suggest that virus clearance can be slow even after induction of HCV-specific T cells during acute primary hepatitis C. After the first infection of these animals HCV T cells targeting multiple HCV proteins were detected in blood on day 56 but viremia was not terminated until 4 to 8 wk later. At least part of this delay in the effector phase of acute hepatitis C can be attributed to homing and expansion of T cells in the liver until they reach a threshold for virus control in this large solid organ. However, it should also be noted that E2445-specific T cells failed to make IFN-γ when they first appeared in the blood of CB0572 during the primary infection (Fig. 1 C) but this effect was transient. In contrast, memory T cells targeting the same epitope were fully functional through the second infection and rapidly responded to antigenic stimulation by up-regulation of the activation marker CD69 and production of IFN-γ (Fig. 2, B and D). This suggests that HCV can transiently interfere with the activation or differentiation of naive (4, 5, 10–13) but not memory T cells into functional effector populations.
Features of Protective Immune Responses and Implications for HCV Vaccines.
Both animals in this study were vaccinated with recombinant envelope glycoproteins before the first challenge with HCV. We cannot exclude the possibility that vaccine-induced antibodies and T cells subtly influenced the outcome of primary infection. Nevertheless, any protection provided by the vaccine did not match that afforded by successful resolution of acute hepatitis C. The CD4+ T cells primed by this vaccine disappeared from the circulation of both animals after infection, a common feature of helper cell responses when the virus persists (2, 4, 5, 7, 8). In this instance HCV infection was eventually contained several weeks later when a sustained CD4+ and CD8+ T cell response targeting structural and nonstructural proteins was detected in blood. Memory T cell responses were maintained in blood and possibly also liver for several years even though serum antibody titers against HCV proteins were low or undetectable, a profile that has also been described in humans studied ∼2 decades after resolution of infection (39). Rapid control of the second infection in chimpanzees before a boost in serum anti-envelope antibodies provides evidence that cellular immune responses alone are sufficient for protection from HCV persistence. Vaccine approaches focused solely on induction of neutralizing antibodies may not be adequate to protect from chronic infection with viruses that display remarkable genetic diversity in envelope genes. Moreover, the critical role of CD8+ T cells in this process revealed by antibody-mediated depletion suggests that the same vaccine strategies for priming CD4+ and CD8+ T cells to control HIV infection (40) should also be considered for prevention of HCV persistence. Where memory CD8+ T cells reside might also be a key factor influencing the rate of HCV clearance and emergence of class I MHC escape variants (14, 16, 29). Establishing a repository of memory cells in the liver that respond rapidly upon reinfection might also enhance vaccine-mediated protection against chronic hepatitis C.
Acknowledgments
We thank Dr. Dana Hasselschwert and Neal Smith of the New Iberia Research Center, New Iberia, LA, for outstanding veterinary and technical support.
This work was supported by Public Health Service grants (RO1A147367 and U19AI48231) to C.M. Walker. N.H. Shoukry is supported by postdoctoral fellowships from the Canadian Institute for Health Research (CIHR) and the American Liver Foundation (ALF). A. Grakoui is supported by a postdoctoral fellowship from the Cancer Research Institute, New York, NY. Patr-B2301 tetramers were synthesized at the NIH tetramer core facility, Emory University, Atlanta, GA. Production of the cM-T807 antibody was supported by a Public Health Service resource award (R24RR16001) to K.A. Reimann.
Footnotes
Abbreviations used in this paper: CID, chimpanzee infectious doses; GE, genome equivalents; HCV, hepatitis C virus; IHL, intrahepatic lymphocyte; SFC, spot forming cell.
References
- 1.Lauer, G.M., and B.D. Walker. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52. [DOI] [PubMed] [Google Scholar]
- 2.Thimme, R., J. Bukh, H.C. Spangenberg, S. Wieland, J. Pemberton, C. Steiger, S. Govindarajan, R.H. Purcell, and F.V. Chisari. 2002. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA. 99:15661–15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper, S., A.L. Erickson, E.J. Adams, J. Kansopon, A.J. Weiner, D.Y. Chien, M. Houghton, P. Parham, and C.M. Walker. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity. 10:439–449. [DOI] [PubMed] [Google Scholar]
- 4.Thimme, R., D. Oldach, K.M. Chang, C. Steiger, S.C. Ray, and F.V. Chisari. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lechner, F., D.K. Wong, P.R. Dunbar, R. Chapman, R.T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B.D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191:1499–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruener, N.H., T.J. Gerlach, M.C. Jung, H.M. Diepolder, C.A. Schirren, W.W. Schraut, R. Hoffmann, R. Zachoval, T. Santantonio, M. Cucchiarini, et al. 2000. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J. Infect. Dis. 181:1528–1536. [DOI] [PubMed] [Google Scholar]
- 7.Diepolder, H.M., R. Zachoval, R.M. Hoffmann, E.A. Wierenga, T. Santantonio, M.C. Jung, D. Eichenlaub, and G.R. Pape. 1995. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet. 346:1006–1007. [DOI] [PubMed] [Google Scholar]
- 8.Gerlach, J.T., H.M. Diepolder, M.C. Jung, N.H. Gruener, W.W. Schraut, R. Zachoval, R. Hoffmann, C.A. Schirren, T. Santantonio, and G.R. Pape. 1999. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 117:933–941. [DOI] [PubMed] [Google Scholar]
- 9.Wang, H., and D.D. Eckels. 1999. Mutations in immunodominant T cell epitopes derived from the nonstructural 3 protein of hepatitis C virus have the potential for generating escape variants that may have important consequences for T cell recognition. J. Immunol. 162:4177–4183. [PubMed] [Google Scholar]
- 10.Lechner, F., N.H. Gruener, S. Urbani, J. Uggeri, T. Santantonio, A.R. Kammer, A. Cerny, R. Phillips, C. Ferrari, G.R. Pape, and P. Klenerman. 2000. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur. J. Immunol. 30:2479–2487. [DOI] [PubMed] [Google Scholar]
- 11.Gruener, N.H., F. Lechner, M.C. Jung, H. Diepolder, T. Gerlach, G. Lauer, B. Walker, J. Sullivan, R. Phillips, G.R. Pape, and P. Klenerman. 2001. Sustained dysfunction of antiviral CD8(+) T lymphocytes after infection with hepatitis C virus. J. Virol. 75:5550–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wedemeyer, H., X.S. He, M. Nascimbeni, A.R. Davis, H.B. Greenberg, J.H. Hoofnagle, T.J. Liang, H. Alter, and B. Rehermann. 2002. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169:3447–3458. [DOI] [PubMed] [Google Scholar]
- 13.Urbani, S., C. Boni, G. Missale, G. Elia, C. Cavallo, M. Massari, G. Raimondo, and C. Ferrari. 2002. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J. Virol. 76:12423–12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson, A.L., Y. Kimura, S. Igarashi, J. Eichelberger, M. Houghton, J. Sidney, D. McKinney, A. Sette, A.L. Hughes, and C.M. Walker. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 15:883–895. [DOI] [PubMed] [Google Scholar]
- 15.Weiner, A., A.L. Erickson, J. Kansopon, K. Crawford, E. Muchmore, A.L. Hughes, M. Houghton, and C.M. Walker. 1995. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. USA. 92:2755–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, K.M., B. Rehermann, J.G. McHutchison, C. Pasquinelli, S. Southwood, A. Sette, and F.V. Chisari. 1997. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J. Clin. Invest. 100:2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta, S.H., A. Cox, D.R. Hoover, X.H. Wang, Q. Mao, S. Ray, S.A. Strathdee, D. Vlahov, and D.L. Thomas. 2002. Protection against persistence of hepatitis C. Lancet. 359:1478–1483. [DOI] [PubMed] [Google Scholar]
- 18.Prince, A.M., B. Brotman, T. Huima, D. Pascual, M. Jaffery, and G. Inchauspe. 1992. Immunity in hepatitis C infection. J. Infect. Dis. 165:438–443. [DOI] [PubMed] [Google Scholar]
- 19.Farci, P., H.J. Alter, S. Govindarajan, D.C. Wong, R. Engle, R.R. Lesniewski, I.K. Mushahwar, S.M. Desai, R.H. Miller, and N. Ogata. 1992. Lack of protective immunity against reinfection with hepatitis C virus. Science. 258:135–140. [DOI] [PubMed] [Google Scholar]
- 20.Bassett, S.E., B. Guerra, K. Brasky, E. Miskovsky, M. Houghton, G.R. Klimpel, and R.E. Lanford. 2001. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology. 33:1479–1487. [DOI] [PubMed] [Google Scholar]
- 21.Weiner, A.J., X. Paliard, M.J. Selby, A. Medina-Selby, D. Coit, S. Nguyen, J. Kansopon, C.L. Arian, P. Ng, J. Tucker, et al. 2001. Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J. Virol. 75:7142–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Major, M.E., K. Mihalik, M. Puig, B. Rehermann, M. Nascimbeni, C.M. Rice, and S.M. Feinstone. 2002. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J. Virol. 76:6586–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomson, M., M. Nascimbeni, M.B. Havert, M. Major, S. Gonzales, H. Alter, S.M. Feinstone, K.K. Murthy, B. Rehermann, and T.J. Liang. 2003. The clearance of hepatitis C virus infection in chimpanzees may not necessarily correlate with the appearance of acquired immunity. J. Virol. 77:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choo, Q.L., G. Kuo, A.J. Weiner, L.R. Overby, D.W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 244:359–362. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz, J.E., M.J. Kuroda, S. Santra, V.G. Sasseville, M.A. Simon, M.A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B.J. Scallon, et al. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 283:857–860. [DOI] [PubMed] [Google Scholar]
- 26.Thimme, R., S. Wieland, C. Steiger, J. Ghrayeb, K.A. Reimann, R.H. Purcell, and F.V. Chisari. 2003. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 77:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chien, D.Y., Q.L. Choo, A. Tabrizi, C. Kuo, J. McFarland, K. Berger, C. Lee, J.R. Shuster, T. Nguyen, and D.L. Moyer. 1992. Diagnosis of hepatitis C virus (HCV) infection using an immunodominant chimeric polyprotein to capture circulating antibodies: reevaluation of the role of HCV in liver disease. Proc. Natl. Acad. Sci. USA. 89:10011–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chien, D.Y., Q.L. Choo, A. Tabrizi, C. Kuo, J. McFarland, K. Berger, C. Lee, J.R. Shuster, T. Nguyen, D.L. Moyer, et al. 1993. Use of recombinant HCV antigen in the serodiagnosis of hepatitis C virus infection: significant improvement in HCV antibody detection as compared with the first generation HCV C100-3 ELISA and the synthetic peptide EIA tests. J. Gastroenterol. Hepatol. S33-S39. [Google Scholar]
- 29.Erickson, A.L., M. Houghton, Q.L. Choo, A.J. Weiner, R. Ralston, E. Muchmore, and C.M. Walker. 1993. Hepatitis C virus-specific CTL responses in the liver of chimpanzees with acute and chronic hepatitis C. J. Immunol. 151:4189–4199. [PubMed] [Google Scholar]
- 30.Hogan, R.J., W. Zhong, E.J. Usherwood, T. Cookenham, A.D. Roberts, and D.L. Woodland. 2001. Protection from respiratory virus infections can be mediated by antigen-specific CD4(+) T cells that persist in the lungs. J. Exp. Med. 193:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogan, R.J., E.J. Usherwood, W. Zhong, A.A. Roberts, R.W. Dutton, A.G. Harmsen, and D.L. Woodland. 2001. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 166:1813–1822. [DOI] [PubMed] [Google Scholar]
- 32.Frese, M., V. Schwarzle, K. Barth, N. Krieger, V. Lohmann, S. Mihm, O. Haller, and R. Bartenschlager. 2002. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology. 35:694–703. [DOI] [PubMed] [Google Scholar]
- 33.Lanford, R.E., B. Guerra, H. Lee, D.R. Averett, B. Pfeiffer, D. Chavez, L. Notvall, and C. Bigger. 2003. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(i)-poly(c), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J. Virol. 77:1092–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalams, S.A., and B.D. Walker. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188:2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413–2417. [DOI] [PubMed] [Google Scholar]
- 36.Reinhardt, R.L., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 37.Welsh, R.M., and L.K. Selin. 2002. No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2:417–426. [DOI] [PubMed] [Google Scholar]
- 38.Prlic, M., L. Lefrancois, and S.C. Jameson. 2002. Multiple choices: regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J. Exp. Med. 195:F49–F52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takaki, A., M. Wiese, G. Maertens, E. Depla, U. Seifert, A. Liebetrau, J.L. Miller, M.P. Manns, and B. Rehermann. 2000. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 6:578–582. [DOI] [PubMed] [Google Scholar]
- 40.McMichael, A., and T. Hanke. 2002. The quest for an AIDS vaccine: is the CD8+ T-cell approach feasible? Nat. Rev. Immunol. 2:283–291. [DOI] [PubMed] [Google Scholar]