

Abstract
A defect in RelB, a member of the Rel/nuclear factor (NF)-κB family of transcription factors, affects antigen presenting cells and the formation of lymphoid organs, but its role in T lymphocyte differentiation is not well characterized. Here, we show that RelB deficiency in mice leads to a selective decrease of NKT cells. RelB must be expressed in an irradiation-resistant host cell that can be CD1d negative, indicating that the RelB expressing cell does not contribute directly to the positive selection of CD1d-dependent NKT cells. Like RelB-deficient mice, aly/aly mice with a mutation for the NF-κB–inducing kinase (NIK), have reduced NKT cell numbers. An analysis of NK1.1 and CD44 expression on NKT cells in the thymus of aly/aly mice reveals a late block in development. In vitro, we show that NIK is necessary for RelB activation upon triggering of surface receptors. This link between NIK and RelB was further demonstrated in vivo by analyzing RelB+/− × aly/+ compound heterozygous mice. After stimulation with α-GalCer, an antigen recognized by NKT cells, these compound heterozygotes had reduced responses compared with either RelB+/− or aly/+ mice. These data illustrate the complex interplay between hemopoietic and nonhemopoietic cell types for the development of NKT cells, and they demonstrate the unique requirement of NKT cells for a signaling pathway mediated by NIK activation of RelB in a thymic stromal cell.
Keywords: T lymphocytes, NF-κB, lymphocyte development, lipid antigens, CD1
Introduction
RelB is a member of the Rel/nuclear factor (NF)*-κB family of transcription factors, of which five family members have been described in mammalian cells: RelA (p65), c-Rel, RelB, NF-κB1 (p50), and NF-κB2 (p52; reference 1). The Rel/NF-κB family controls gene expression essential for cytokine expression, developmental processes, and cell differentiation and survival, particularly in the immune system (2). Rel proteins have distinct biologic roles as evidenced by the variety of phenotypes of gene-knockouts of Rel family members. For example, RelA-deficient mice exhibit embryonic lethality due to a defect in liver development, whereas mice lacking functional c-Rel are viable, but their mature B and T cells are unresponsive to most mitogenic stimuli (3). RelB forms heterodimers with p50 or p52 to activate gene transcription (2). RelB-deficient mice have a complex phenotype with a variety of defects in their hemopoietic and immune systems. They lack a normal thymic medullary epithelium (4) and have a profound defect in dendritic cells, particularly in the myeloid related CD8α− dendritic cells (DCs; reference 5). The development of lymphoid organs and splenic germinal centers also is impaired (6, 7). In addition, RelB mutant mice display a late onset, severe and fatal multi-organ inflammation, including splenomegaly and myeloid hyperplasia in the bone marrow, as well as skin lesions similar to atopic dermatitis (4, 8, 9). The inflammation observed in RelB−/− mice is T cell dependent, but no gross abnormalities were found in conventional T or B lymphocyte differentiation before the development of inflammation (10). However, the mice were not analyzed for potential defects in the subsets of specialized T cells that could have regulatory function. NKT cells are one such specialized population of mature lymphocytes. They rapidly secrete a variety of cytokines after activation, which may give them the ability to regulate a variety of immune responses. NKT cells coexpress NK receptors and TCRs, and they can be subdivided into at least two major categories. First, the majority in mice expresses a Vα14-Jα18 rearrangement with an invariant CDR3 region (11–14) and they are positively selected by CD1d, a nonclassical class I antigen-presenting molecule. These cells are CD4+ or CD4−CD8− (double negative, DN), and they recognize the glycolipid α-galactosylceramide (α-GalCer) presented by CD1d (15, 16). The other NK1.1+ T cell subpopulation is more heterogeneous, including both CD1d-dependent and independent cells, a greater percentage of CD8+ lymphocytes, and a more diverse TCR repertoire (17). CD1d-dependent NKT cells with the invariant Vα14 TCR can be readily distinguished from other populations by staining with α-GalCer loaded CD1d tetramers (18, 19), because nearly all the cells with this α rearrangement recognize α-GalCer presented by CD1d. There are several circumstances in which these cells do not express NK1.1, however, including when they are immature and after activation (17). Therefore, we refer to them here as Vα14i T cells or Vα14i NKT cells.
The developmental pathway followed by Vα14i NKT cells is now beginning to be elucidated. A number of gene deficiencies that disrupt Vα14i NKT cell development leave conventional T cells unaffected (17), providing evidence that Vα14i NKT cell differentiation is divergent from conventional T cells. It is believed, however, that Vα14i NKT cells branch off from the conventional T cell developmental pathway after random TCR rearrangement in the thymus and subsequent positive selection by CD1d expressing double positive thymocytes (17, 19–21).
Here we report that the development of Vα14i NKT cells requires RelB expression in a radiation resistant host cell, and that RelB activation requires a functional NF-κB–inducing kinase (NIK). The differentiation of Vα14i NKT cells is blocked at a relatively late stage, after acquisition of the canonical TCR. These data therefore define a signaling pathway in stromal cells that has a specific effect on Vα14i NKT cells.
Materials and Methods
Mice and Immunizations.
RelB + / − and RelB − / − mice, all on an inbred C57BL/6 background, have been described previously (4). C57BL/6, RAG2−/−, and β2m−/− mice were obtained from The Jackson Laboratory. CD45.1+ congenic mice were purchased from Taconic. The aly/aly homozygous mice and the control aly/+ heterozygous mice (22) were purchased from CLEA Japan. The aly/+ × RelB + / − compound heterozygotes were obtained by crossing aly/aly homozygous with RelB + / − mice and typing for the RelB-allele. Experiments were initiated with 5 to 8-wk-old mice. RAG2−/− mice were used at 8–12 wk of age as recipients in the bone marrow chimera experiments. All mice were housed and bred under specific pathogen free conditions in the La Jolla Institute for Allergy and Immunology Vivarium. For in vivo immunizations, α-GalCer was dissolved in 0.5% polysorbate 20 (Nikko Chemicals) in a 0.9% NaCl solution. Mice of both sexes were immunized both intraperitoneally and intravenously with either vehicle alone or 2 μg of α-GalCer as described previously (23). At the indicated time points, blood was obtained from the retro-orbital plexus.
Reagents and Antibodies.
α-GalCer was synthesized at the Pharmaceutical Research Laboratories, Kirin Brewery Co., LTD. The following mAbs were used in cytokine ELISAs: anti–IFN-γ mAbs R4–6A2 and biotinylated XMG1.2 and anti–IL-4 mAbs BVD4–1D11 and biotinylated BVD6–24G2. The cytokine standards consisted of the corresponding recombinant cytokines IFN-γ (108 U/mg) and IL-4 (107 U/mg). The anti–mouse LTβR mAb (3C8, IgG1) was produced from a Sprague-Dawley rat immunized with mouse LTβR-Fc decoy protein (24). This antibody is agonistic for induction of VCAM1 expression on mouse fibroblasts and does not cross react with other TNFR family proteins. The following mAbs were used for phenotypic analysis: anti-CD16/32 (2.4G2) for blocking Fc receptors, FITC or allophycocyanin (APC)-labeled anti-TCRβ (H57–597), PE-labeled anti-CD1d (1B1), PE or PerCP-labeled anti-NK1.1 (PK136), Cy-Chrome or FITC-labeled anti-CD4 (H129.19), APC or PerCP-labeled anti-CD8α (53–6.7), APC-labeled anti-CD44 (IM7), and fluorochrome-labeled isotype-matched controls. Adhesion molecule expression on fibroblasts was determined using FITC-labeled anti–VCAM-1 (429) and PE-labeled anti–ICAM-1 (3E2). Unless otherwise mentioned, all antibodies and recombinant cytokines were purchased from BD Biosciences. α-GalCer/CD1d tetramers were produced as described previously (18).
Cell Preparation and Flow Cytometry.
Liver mononuclear cells were prepared as described previously (25). Cells from thymus, spleen, and bone marrow were prepared by conventional methods. Red blood cells were removed from spleen cell suspensions using a standard Ficoll gradient (Accurate Chemical & Scientific Corporation). For surface staining, cells were suspended in buffer comprised of PBS (pH 7.4) containing 2% BSA (wt/vol) and 0.02% NaN3 (wt/vol). After blocking with 2.4G2 anti-FcγR mAb, the cells were stained at 4°C for 20 min with the labeled mAbs, then washed and analyzed on a FACSCalibur™ (Becton Dickinson) flow cytometer. Lymphocytes were enumerated out of the heterogeneous cell population by electronic gating, as determined by forward angle and side angle light scatter. Stainings with α-GalCer/CD1d tetramers were performed as described previously (18). Unloaded CD1d tetramers were used as controls.
Cytokine Assays.
Cytokine levels were detected using standard sandwich ELISAs, according to the manufacturer's protocol (BD Biosciences). Cytokine levels are expressed as mean ± SD of culture triplicates.
Preparation of APCs and Primary Cultures of Responder Cells.
For DC enrichment, spleen cell suspensions were incubated for 1 h with 400 U/ml of type III collagenase (Sigma-Aldrich), washed, and allowed to adhere onto plastic of tissue culture flasks at 107 cells/ml in culture medium for 1 h, 30 min at 37°C, 5% CO2 as described previously (15). Nonadherent cells were then removed, and adherent cells were reincubated overnight with 80 ng/ml of α-GalCer or with the vehicle only. The cells that became nonadherent during the secondary culture period were recovered and centrifuged over a 50% Percoll gradient (Amersham Biosciences). Such a purified population contained more than 80% of DCs, as identified by morphology and flow cytometry (CD16/32low, CD11c+, CD11blow, MHC class IIhigh). Pulsed DCs were washed extensively before being added to the cultures. Responder cells were prepared by depleting APCs from freshly isolated splenocytes of C57BL/6 mice with I-Ab and CD19 mAbs using magnetic beads (Dynal). Obtained purity was >98%. The responders were seeded at 2.5 × 105 cells/well in 96-well plates. Primary APCs were pulsed for 2 to 3 h at 37°C with glycolipids, then irradiated (7,000 rad) and extensively washed before being added at 6 × 104 cells/well, as indicated in the figure legends. Supernatants were harvested at indicated time points, and cytokine levels were assessed by ELISA.
Generation of Bone Marrow Chimeras.
T cell–depleted (using anti-Thy1.1 mAbs) bone marrow cells (5–10 × 106) from RelB − / −, RelB + / −, or C57BL/6 mice were intravenously transferred into γ-irradiated (1,300 rad) RAG2−/− or β2m−/− recipient mice. Thymus, liver, and spleen of each recipient were analyzed 8–12 wk later for NKT cells.
Fibroblast Cultures.
Fibroblasts isolated from the kidneys of wild-type or RelB − / − mice were isolated and maintained as described previously (26, 27). Embryonic fibroblasts from aly/aly and aly/+ mice were established as described previously (28). Cultures were maintained in Dulbecco's minimal essential medium containing 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete medium). Stimulation of cells was performed by incubation with an agonistic anti-LTβR mAb (2 μg/ml) or isotype control.
Cell Extracts.
Cells were harvested, and whole cell and nuclear extracts were prepared as described previously (29, 30). Briefly, cell pellets were resuspended and lysed at 4°C for 25 min in whole cell extract lysis buffer (20 mM HEPES, 0.4 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, and 1 mM DTT) containing phosphatase inhibitors (40 mM fl-glycerophosphate, 20 mM NaF, 1 mM Na3VO4, 20 mM p-nitrophenyl phosphate [Calbiochem]); and protease inhibitors (aprotinin 10 μg/ml, leupeptin 10 μg/ml, bestatin 10 μg/ml, and pepstatin 10 μg/ml; Calbiochem) and 1 mM phenylmethylsulfonyl chloride (Sigma-Aldrich). Lysates were centrifuged at 13,000 g for 10 min at 4°C, and the resulting supernatants were transferred to fresh tubes. Protein concentrations in the supernatants were determined by the Bradford assay (Bio-Rad Laboratories).
Electrophoretic Mobility Shift Assays.
Electrophoretic mobility shift assays (EMSAs) were conducted as described previously with a γ32P labeled oligonucleotide probe corresponding to a consensus NF-κB binding site (29, 30). The composition of the activated NF-κB complex was examined by supershift analysis with antisera to Rel family members (Santa Cruz Biotechnology, Inc.).
Homeostasis of CFSE-labeled Thymocytes.
Thymuses were isolated from CD45.1+ congenic mice and single cell suspensions were made. CD8+ thymocytes were depleted using MACs beads (Miltenyi Biotech) according to the manufacturer's protocol. The cells were suspended at a concentration of 10 × 106 cells/ml in PBS/0.1%BSA for labeling with CFSE (Molecular Probes). 1 μl of a 5 mM CFSE stock (prepared in DMSO) was added per 1 ml of cell suspension. The cells were then incubated for 10 min at 37°C, and the labeling reaction was stopped by adding cold PBS. The cells were then washed and resuspended in PBS. 7 × 107 cells were injected into γ-irradiated (700 rad) aly/aly or aly/+ mice. Recipient mice were analyzed 1 wk after transfer.
Statistical Analysis.
Quantitative differences between two samples were compared with the Mann Whitney U (rank sum) test. When three groups were being compared, a Kruskal-Wallis test was used, followed by a Dunn's post-hoc test to determine which mouse strain was different.
Results
Vα14i NKT Cell Deficiency in RelB−/− Mice.
Mononuclear cells were isolated from the principal sites where Vα14i NKT cells are found, including thymus, spleen, liver, and bone marrow, from both RelB + / + and RelB − / − mice, and the fraction of α-GalCer/CD1d-tetramer+ TCRβ+ cells was determined by flow cytometry (Fig. 1, A and B) . The mice analyzed were between 5 and 8 wk old, before the full development of multi-organ inflammatory disease. The percentage and absolute number of Vα14i NKT cells were greatly and consistently reduced in RelB − / − versus RelB + / + mice in the thymus, spleen, liver (Fig. 1), and bone marrow (unpublished data). Expression of other membrane proteins characteristic for Vα14i NKT cells, such as IL-2Rβ and members of the Ly49 killer-inhibitory receptor family, also were severely diminished in RelB − / − mice (unpublished data). Interestingly, the proportion of Vα14i NKT cells in heterozygous RelB + / − mice, which do not have inflammation, was also significantly reduced compared with RelB + / + mice (Fig. 1, A and B), indicating that the observed defect in RelB − / − mice is not secondary to inflammation. The number of conventional T cells was not affected by RelB deficiency in any organ examined (unpublished data). Consistent with this decrease, IL-4 could not be detected in serum of RelB − / − mice upon in vivo administration of α-GalCer (Fig. 1 C). The absence of cytokine in the serum suggests a systemic defect in Vα14i NKT cell numbers, in all organs of RelB − / − mice, rather than an altered tissue distribution.
Figure 1.

Vα14i NKT cell deficiency in RelB− / − mice. (A) Representative dot plots showing TCRβ versus α-GalCer/CD1d tetramer binding in the thymus and liver of RelB+/+, RelB+/−, or RelB−/− mice. The average percentage of Vα14i NKT lymphocytes is indicated. Numbers are mean ± SEM of 4 to 17 mice analyzed in each group. (B) Total number of Vα14i NKT cells. Thymus, liver, and spleen mononuclear cells of the indicated mice were labeled with mAbs against TCRβ and α-GalCer/CD1d tetramers. Using the total cell count obtained from each organ, absolute numbers of NKT cells (gated as shown in A) were determined. Numbers are mean ± SEM of 4 to 17 mice analyzed in each group. *Significantly different from RelB+/+ (P < 0.05, Kruskal-Wallis; Dunn's post-hoc test), †RelB+/− versus RelB−/− (P < 0.05, Kruskal-Wallis; Dunn's post-hoc test). (C) Measurement of serum IL-4 upon in vivo administration of α-GalCer. RelB+/+ (n = 7), RelB+/− (n = 3), and RelB−/− (n=2) mice on the C57BL/6 background were immunized with α-GalCer (2 μg/mouse) and analyzed 4 h after immunization for serum levels of IL-4 as determined by ELISA. (D) α-GalCer presentation by splenic DCs. (Top panel) Purified DCs from the indicated mice were isolated as described and stained with anti-CD1d mAb or isotype control and analyzed by flow cytometry. Representative histograms for CD1d (black) or controls (open lines) are shown. (Bottom panel) α-GalCer-pulsed DCs were seeded at 6 × 104 cells/well with responder spleen cells from C57BL/6 mice at 2.5 × 105 cells/well. After 3 d of culture, IFN-γ levels were assayed by ELISA. Data represent mean ± SEM of triplicate cultures. One representative experiment of three is shown.
Despite the reduced number of Vα14i NKT cells in RelB − / − mice, CD1d levels on antigen presenting cells were found to be unaffected in all organs analyzed (Fig. 1 D, top panel). Furthermore, RelB − / − splenocytes were able to present α-GalCer to mouse Vα14/Vβ8+ hybridomas as efficiently as RelB + / + splenocytes (unpublished data). This indicates that the reduced α-GalCer responses in RelB − / − mice are not due to a reduced ability to form stimulating lipid-CD1d complexes at the surface of antigen presenting cells.
RelB − / − mice have been reported to have reduced numbers of myeloid related CD8α− DCs. To assess the antigen presenting ability of DC from RelB−/− mice, we purified total DCs from the spleen and tested their ability to stimulate responder Vα14i NKT cells from the spleen of C57BL/6 mice after stimulation with α-GalCer. Interestingly, no differences in the levels of IFN-γ were measured in the supernatants of those in vitro cultures with DCs from RelB−/−, heterozygotes or wild-type mice (Fig. 1 D, bottom panel). This indicates that there is no intrinsic defect in the ability of RelB-deficient DCs to present α-GalCer.
The Requirement of Vα14i NKT Cells for RelB Is Not Cell Autonomous.
To determine whether the observed defect in Vα14i NKT cell number is cell autonomous, bone marrow chimeric mice were constructed by transfer of T cell-depleted bone marrow cells from RelB − / − or RelB + / + mice into lethally irradiated RAG2−/− recipients. 8 to 10 wk after transfer, chimeric mice were analyzed for the presence of Vα14i NKT cells. Vα14i NKT cells were present in the liver of RAG2−/− mice repopulated with RelB − / − bone marrow (Fig. 2 A). The fraction of Vα14i NKT cells among the total intrahepatic lymphocytes (Fig. 2 A) or among the gated, TCRβ+ lymphocytes in the liver, spleen, and thymus tissue (Fig. 2 B) was similar when chimeric mice reconstituted with bone marrow from RelB − / − were compared with those reconstituted with bone marrow from wild-type mice. These findings indicate that the generation of Vα14i NKT cells requires the presence of a radiation-resistant host cell expressing RelB, and that RelB expression is not required in the Vα14i NKT cell lineage or in the double-positive thymocyte that selects these cells.
Figure 2.

Development of CD1d-dependent Vα14i NKT cells is restored in bone marrow chimeras. (A) Representative staining with α-GalCer/CD1d tetramers. Bone marrow chimeric mice were made as described in Materials and Methods, and liver mononuclear cells of the indicated recipient mice were stained with mAb against TCRβ and with α-GalCer/CD1d tetramers. The fraction of total intrahepatic lymphocytes staining with α-GalCer/CD1d tetramers in RAG2−/− recipients reconstituted with either RelB + / + or RelB − / − bone marrow is indicated. The numbers represent mean ± SEM of at least six individual mice in each group. (B) Mononuclear cells of liver, spleen, and thymus were stained with anti-TCRβ and α-GalCer/CD1d tetramers, and the fraction of CD1d tetramer+ cells among the gated, TCRβ+ lymphocytes was determined. Numbers are the mean ± SEM of at least six mice analyzed in each group.
RelB Does Not Need to be Expressed by the β2m-positive Cell Required for Positive Selection.
If the RelB expressing cell type(s) required for Vα14i NKT cell development were directly responsible for the positive selection of these lymphocytes, they should also express β2-microglobulin (β2m), which is required for expression of CD1d on the cell surface. Such a result, however, would contradict the well-established finding that bone marrow–derived cells select Vα14i NKT cells (31–35). Bone marrow cells from either RelB − / − or RelB + / + donor mice therefore were transferred to lethally irradiated β2m−/− mice, and recipients were analyzed 8 to 12 wk later. In this separate series of chimeras, the percentage of Vα14i NKT cells was reduced approximately twofold compared with the previous set (Figs. 2 A and 3), perhaps reflecting the use of irradiated immune competent rather than irradiated RAG-deficient recipients. Nevertheless, Vα14i NKT cell development was restored to comparable levels in liver, spleen, and thymus of β2m−/− recipients of either RelB − / − or RelB + / + bone marrow (Fig. 3 , and unpublished data). These observations are consistent with a model in which the RelB+ stromal cell required for the full development of Vα14i NKT cells is not directly responsible for their positive selection.
Figure 3.

The RelB expressing cell required for Vα14i NKT cell development does not need to express β2m. Liver mononuclear cells of the indicated recipient mice were stained with mAb against TCRβ and with α-GalCer/CD1d tetramers. The fraction of TCRβ+ cells staining with α-GalCer/CD1d tetramers in β2m−/− recipients reconstituted with either RelB + / + or RelB − / − bone marrow was determined. Numbers represent mean ± SEM of two to three individual mice analyzed in each group.
NIK Is Required for the Generation of Vα14i NKT Cells.
To examine the role of molecules potentially upstream of RelB and involved in its activation, we analyzed cells from the alymphoplasia (aly/aly) mouse, which has a defect in the organogenesis of lymph nodes and Peyer's patches (22). This strain has a spontaneous point mutation in the NIK, although the mutant enzyme retains catalytic potential (36). Previously, the aly/aly mouse also was reported to have a deficiency in NKT cells, as determined by staining for NK1.1 and TCRβ expression as opposed to the use of α-GalCer/CD1d tetramers (29, 30). As for RelB, NIK expression was found to be required in a radiation resistant host cell that does not express CD1d, rather than in the double positive thymocyte responsible for NKT cell positive selection, the NKT cell precursor or NKT cell itself (37, 38). We confirmed this NIK requirement using α-GalCer/CD1d tetramers to directly enumerate the Vα14i expressing, CD1d reactive, NKT cells. With this reagent, NIK mutant mice were found to have a reduced number and proportion of Vα14i NKT cells in spleen, thymus, liver (Fig. 4, A and B) , and bone marrow (unpublished data). Consistent with this finding, aly/aly mice did not respond to α-GalCer in vivo as assessed by cytokine release into the serum (Fig. 4 C). The decrease in Vα14i NKT cells in aly/aly mice, however, was less severe than the RelB defect. Furthermore, aly/+ mice have normal numbers and function of their Vα14i NKT cells (compare Figs. 1 and 4), indicating that a single copy of the wild-type NIK gene is sufficient to support the generation of these cells.
Figure 4.
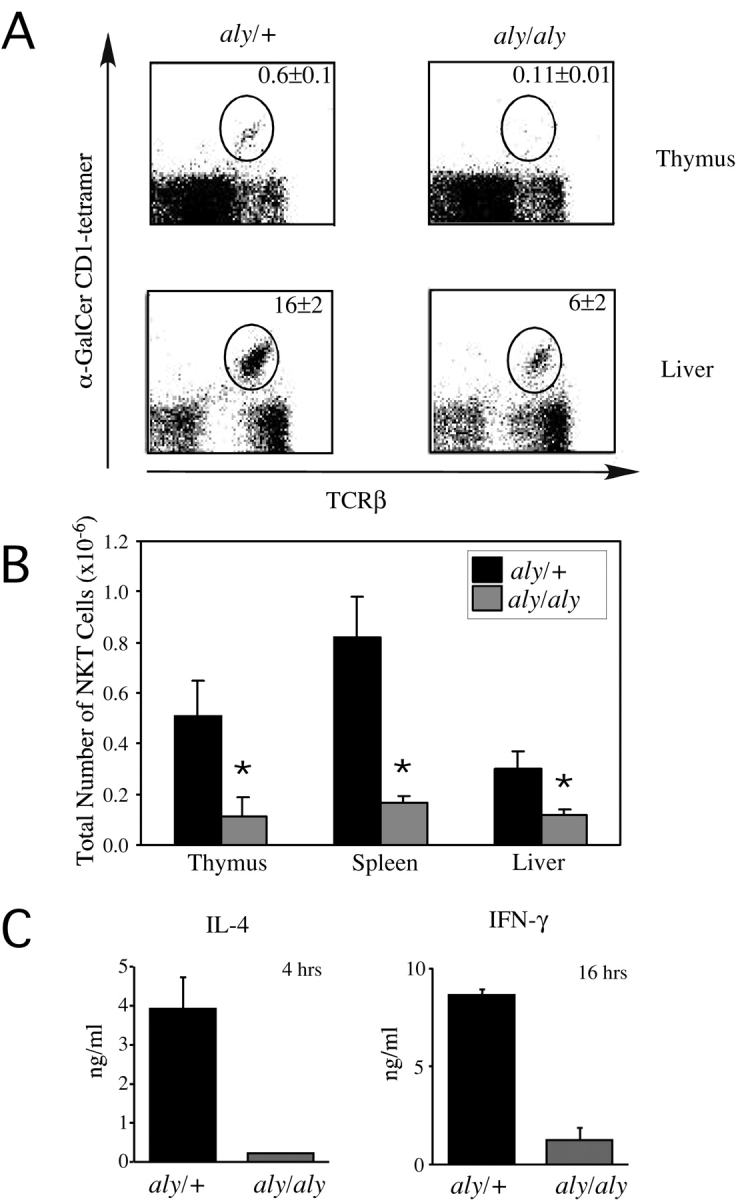
Vα14i NKT cell deficiency in aly/aly mice. (A) Representative dot plots showing TCRβ versus α-GalCer/CD1d tetramer staining in thymus and liver from aly/+ and aly/aly mice. Percentage of Vα14i NKT lymphocytes is indicated. Numbers are mean ± SEM of four mice analyzed in each group. (B) Total number of Vα14i NKT cells. Thymus, liver, and spleen mononuclear cells of the indicated mice were labeled with mAbs against TCRβ and α-GalCer/CD1d tetramers. Using the total cell count obtained from each organ, absolute numbers of Vα14i NKT lymphocytes (gated as shown in A) were determined. Numbers are mean ± SEM of four mice analyzed in each group. *P < 0.05 Mann Whitney U (rank sum) test. This data is representative of two separate experiments in which four mice of each strain were analyzed. (C) Measurement of IFN-γ and IL-4 release upon in vivo administration of α-GalCer. aly/+ or aly/aly mice were immunized with α-GalCer (2 μg/mouse) and analyzed four (IL-4) and 16 h (IFN-γ) after immunization. Serum levels of IL-4 and IFN-γ were tested by ELISA. Numbers are the mean ± SEM of four mice analyzed in each group.
RelB Acts Downstream of NIK.
In addition to the Vα14i NKT cell defect, NIK and RelB mutant mice share defects in secondary lymphoid organ development (22). These observations suggested that RelB may act downstream of NIK to govern the development of Vα14i NKT cells as well as secondary lymphoid organs, although NIK also can activate other NF-κB family transcription factors (39). We therefore performed experiments in vitro to provide evidence for NIK activation of RelB. Adhesion molecules are induced on primary fibroblasts after LTβR stimulation with an agonistic antibody (28). This induction is defective, however, in NIK mutant aly/aly fibroblasts (28). To determine if RelB might act downstream of NIK, we analyzed the induction of adhesion molecules on primary fibroblasts from wild-type and RelB mutant mice after LTβR stimulation. Interestingly, similar to aly/aly mice, up-regulation of both ICAM-1 (Fig. 5 A) and VCAM-1 (unpublished data) were absent in RelB − / − fibroblasts after stimulation with an agonistic anti-LTβR mAb. Upon LTβR triggering, however, RelB − / − fibroblasts displayed a similar degree of IκBα degradation as RelB + / + cells (unpublished data). To directly assess RelB activation, NF-κB DNA-binding activity was determined by EMSA using a consensus NF-κB DNA binding sequence as a probe. Stimulation of mouse embryonic fibroblasts (MEFs) from wild-type or aly/aly mice with an agonistic LTβR antibody resulted in a profound activation of NF-κB (Fig. 5 B), which lasted for more than 8 h. To determine the identity of the NF-κB containing complexes, supershift assays were performed. These assays revealed that RelB and p65 containing complexes were present in wild-type MEFs after LTβR signaling (Fig. 5 B). By contrast, in aly/aly MEFs, LTβR stimulation did not result in RelB activation. Taken together, these data unambiguously demonstrate that NIK is essential for RelB activation after LTβR stimulation in fibroblasts, but not for the activation of complexes containing RelA (p65).
Figure 5.

LTβR mediated activation of RelB through NIK. (A) ICAM-1 induction by LTβR stimulation. Fibroblasts from the kidneys of RelB + / + or RelB − / − mice were stimulated with an agonistic α-LTβR mAb (2 μg/ml) for 24 h. Cell surface levels of ICAM-1 were determined by flow cytometry. Histograms representing ICAM-1 levels before (thin line) and after stimulation (bold line) are shown. One representative example of three independent experiments is shown. (B) NF-κB/Rel binding activities in wild-type and aly/aly mice after LTβR ligation. MEFs from C57BL/6 and aly/aly mice were stimulated with an α-LTβR mAb (2 μg/ml) for 8 h. Nuclear extracts were prepared from unstimulated and LTβR triggered cells. Extracts were incubated with a palindromic κB-binding site as described in Materials and Methods. The results from addition of specific anti-sera against RelA (p65) and RelB are indicated at the bottom. One representative experiment of three is shown.
To determine if NIK might be required for RelB activation in vivo, we analyzed compound heterozygous mice obtained by back crossing aly/aly mice with RelB heterozygotes. As shown in Fig. 6 A, in addition to lacking peripheral lymph nodes, RelB − / − mice lack Peyer's patches. In agreement with previous results (22), we found that aly/aly mice also lack Peyer's patches. The aly/+ heterozygotes have normal numbers of Peyer's patches. Similar to the defect in NKT cells, however, there is haplo-insufficiency for RelB, as RelB + / − mice have a reduced number of Peyer's patches compared with wild-type controls. Furthermore, the size of the remaining Peyer's patches in the RelB + / − mice is reduced compared with wild-type control mice (Fig. 6 B). In the aly/+, RelB + / − compound heterozygotes, the number of Peyer's patches is even further reduced compared with RelB + / − heterozygotes. Moreover, Peyer's patches in the compound heterozygotes were dramatically reduced in size compared with either the RelB + / − or aly/+ mice (unpublished data). Therefore, these data suggest that NIK and RelB act in a pathway in vivo that is important for the genesis of Peyer's patches. Similar groups of mutant mice were analyzed for the number and function of Vα14i NKT cells. The number of Vα14i NKT cells was not significantly reduced in the compound heterozygotes over the reduction observed in RelB + / − mice (unpublished data). Vα14i NKT function was assessed by measuring cytokines in the blood 6 h after α-GalCer injection. As shown in Fig. 6 C, after injection of α-GalCer, there was a significant reduction in the amount of IFN-γ in the blood in the compound heterozygotes, even when compared with RelB + / − mice. Therefore, similar to the case for Peyer's patch formation, NIK is likely to act upon RelB in vivo in the development of Vα14i NKT cells.
Figure 6.

In vivo requirement of NIK for RelB activation. (A) The number of Peyer's patches depends upon RelB and NIK. Peyer's patches were counted in RelB − / −, aly/aly mice, and RelB/aly compound heterozygotes. Each dot represents the number of Peyer's patches in an individual mouse and the horizontal bar indicates the average value. *RelB + / − × aly/+ versus aly/+ and RelB + / − mice (P < 0.05, Student's t test). (B) Development of normal sized Peyer's patches is RelB dependent. Shown are representative Peyer's patches in adult RelB − / −, RelB + / −, and RelB + / + mice. (C) Measurement of IFN-γ release upon in vivo administration of α-GalCer. RelB + / −, RelB + / − × aly/+ compound heterozygotes and aly/+ mice were immunized with α-GalCer (2 μg/mouse) and analyzed 16 h after immunization. Serum levels of IFN-γ were tested by ELISA. Numbers represent mean ± SEM of four to six individual mice analyzed in each group. *RelB + / − × aly/+ versus aly/+ and RelB + / − mice (P < 0.05, Student's t test).
A Developmental Block Underlies the Vα14i NKT Cell Deficiency in NIK and RelB Mutant Mice.
Most recent data indicate that Vα14i NKT cells are generated predominantly in the thymus (20, 21, 35, 40), although this point remains controversial (41, 42). Despite this likely thymic origin, the great majority of Vα14i NKT cells in the thymus may be mature cells that are not dividing (21, 35), but which have the ability to secrete cytokines in vitro (12, 43, 44). Therefore, the decreased numbers of Vα14i NKT cells in the thymus tissue and elsewhere in the mutant mice could reflect either a defect in the homeostasis or expansion of mature Vα14i NKT cells or a block in development. We performed several experiments to distinguish between these possibilities. First, to determine if the observed Vα14i NKT cell defect is related to the homeostasis of mature cells, CD8-depleted thymocytes from CD45.1+ congenic mice were labeled with CFSE and injected into irradiated wild-type or NIK mutant mice. The CD8-depleted thymocyte population is enriched for Vα14i NKT cells, although the majority is CD4 single positive cells. By avoiding positive selection in the enrichment of Vα14i NKT cells, we could circumvent the activation induced cell death that this might cause (18). This protocol has been used recently to analyze the influence of cytokines and CD1d expression on the lymphopenia-induced proliferation of Vα14i NKT cells (45). aly/aly recipient mice were analyzed for this experiment, because of the potential secondary effects of RelB-mediated inflammation on Vα14i NKT cell biology or the postirradiation survival of the recipients. 7 d after transfer, the recipients were killed and the homeostatic proliferation of donor CD45.1+ Vα14i NKT cells was analyzed by flow cytometry. As shown in Fig. 7 A, Vα14i NKT cells underwent three to four cell divisions in both wild-type and aly/aly recipients. Similarly, no differences in the homeostatic proliferation of conventional T cells could be observed in the two types of recipients (unpublished data).
Figure 7.
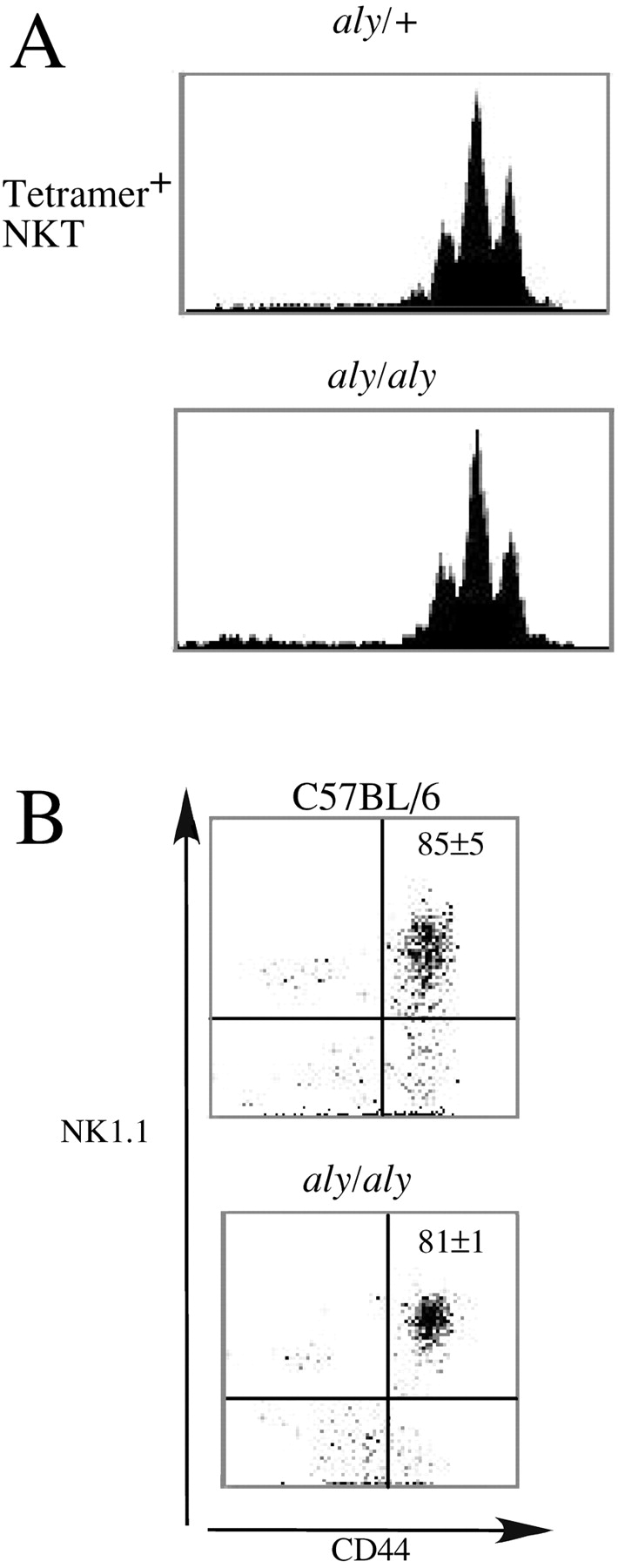
Vα14i NKT cell differentiation, not homeostasis, is affected by disrupting NIK signaling. (A) Homeostatic proliferation of α-GalCer reactive Vα14i NKT cells. CD45.1+, CD8 depleted thymocytes were labeled with CFSE and adoptively transferred to CD45.2+ aly/+ or aly/aly recipients as described. 7 d after transfer the number of cell divisions of CD45.1+ α-GalCer/CD1d-tetramer+ NKT cells in the liver of the recipient mice was analyzed by flow cytometry. Histograms representing CFSE staining in gated tetramer+ cells in aly/+ or aly/aly recipients are shown. One representative example of four independent experiments is shown. (B) Maturity of thymic NKT cells. Thymocytes of the indicated mice were stained with mAbs against TCRβ, CD44, NK1.1, and with α-GalCer/CD1d tetramers. Vα14i NKT cells were gated as shown in Fig. 1 A and Fig. 3 A and analyzed for the expression of CD44 and NK1.1. The average percentage of CD44+NK1.1+ Vα14i NKT cells is indicated. Numbers are mean ± SEM of four (aly/aly) or five (C57BL/6) mice analyzed in each group.
Cells from the thymus of the mutant mice were analyzed by multi-parameter flow cytometry to determine if a phenotypic alteration consistent with a defect in Vα14i NKT cell differentiation was evident. Development of Vα14i NKT cells is likely to proceed through a double positive (DP) intermediate (21), but these cells are so rare that a discrete population of tetramer+ DP cells in the adult thymus cannot be observed. NK1.1+CD44+ Vα14i NKT cells have been identified as the most mature thymus subset, however, and the data indicate that they differentiate from a NK1.1− and CD44− α-GalCer/CD1d tetramer+ precursor (20, 46). These markers therefore were used in order to determine the developmental stage reached by Vα14i NKT cells in aly/aly mice. Similar to C57BL/6 mice, most Vα14i NKT cells in the thymus of adult aly/aly mice had a NK1.1+CD44+ phenotype (Fig. 7 B) indicating that the block seen in NIK-deficient mice is likely to occur late in Vα14i NKT cell development. The proportion of CD4+ and DN NKT cells was also similar between the strains (unpublished data). Collectively, these data suggest that the development rather than the homeostasis of tetramer+ cells is impaired in the absence of signals mediated through NIK and RelB.
Discussion
The results from this study reveal two important new features of the biology of NF-κB family transcription factors containing RelB. First, the data demonstrate a selective requirement for RelB in the differentiation of Vα14i NKT cells. The haplo-insufficiency of RelB with respect to Vα14i NKT cell differentiation indicates that the defect is not secondary to inflammatory disease, which is not found in RelB + / − mice. Second, the results delineate an upstream pathway for the selective activation of RelB, providing a connection between NIK and RelB activity. This pathway was documented in vitro, and data also were presented indicating it operates in vivo as well to control the differentiation of Vα14i NKT cells and the formation of Peyer's patches. The results from several experiments are consistent with a block at a late stage in Vα14i NKT cell differentiation in mutant mice that cannot activate RelB through NIK. First, α-GalCer–reactive NKT cells were greatly reduced in RelB − / − and aly/aly mice in all principal sites where these cells normally are found, including the thymus (31–35). Second, Vα14i NKT cell responses could not be detected systemically in the serum after α-GalCer stimulation, suggesting that functional cells were absent, rather than having migrated to a different location. Despite this, CD1d surface expression and the ability of CD1d to present a glycolipid antigen were not affected by RelB deficiency. Third, the migration of CFSE-labeled thymic Vα14i NKT cells to the liver was unimpaired in aly/aly mice, as was their homeostatic proliferation in a lymphopenic environment, consistent with a primary defect in Vα14i NKT cell differentiation as opposed to homeostasis or homing. While the pathway for Vα14i NKT cell development remains incompletely characterized, the mature phenotype of the residual Vα14i NKT cells present in the thymus of aly/aly mice is consistent with a partial block late in differentiation. This phenotype is different, however, from that observed in IL-15−/− mice, which have a block in differentiation at an earlier stage, as well as effects on the lymphopenia induced proliferationVα14i NKT cells (45).
Previous studies have demonstrated an essential role for CD1d expressed by bone marrow–derived cells, probably cortical CD4+CD8+ thymocytes, in the positive selection of α-GalCer–reactive NKT cells (31–35). The data here demonstrate that a radiation resistant host cell that expresses RelB also is indispensable. The results from transfer of RelB − / − bone marrow cells to β2m−/− mice demonstrate that this radiation-resistant host cell does not need to express CD1d. Conversely, both the positively-selecting CD1d+ thymocyte and the NKT cell progenitor do not require RelB. Thus, rather than being directly responsible for CD1d-mediated positive selection, the RelB expressing cell has an indirect role. In aly/aly mice, a similar CD1d-independent requirement for a NIK expressing stromal cell was reported for the generation of NK1.1+ TCRβ+ cells (37, 38). The previous studies did not use CD1d tetramers to detect the affected ell population. NK1.1 expression can vary in different genetic backgrounds making NKT cell detection problematic, and the previous studies could not have detected immature NK1.1− Vα14i T cells, and therefore did not identify the stage at which the differentiation of the cells was blocked.
It remains to be determined where the required interaction between the maturing Vα14i NKT cell and the RelB expressing stromal cells occurs within the thymus. Transfer of a medullary epithelial cell line into aly/aly mice resulted in an increase in NK1.1+ TCR+ cells in the thymus (38), and RelB-deficient mice have a specific defect in medullary epithelial cells, as identified by UEA-1 staining (6). These findings suggest that normal Vα14i NKT cell development requires the activity of the NIK/RelB pathway in epithelial cells in the thymic medulla. It is noteworthy that a number of mutations alter Vα14i T cell differentiation while having only a minimal or no effect on conventional T cells (for a review, see reference 17). The complexity of the gene program required to produce Vα14i T cells probably reflects the combined genetic requirements for conventional positive selection as well as the requirements for the subsequent activation and expansion of these Vα14i T cells in the thymus.
Despite the obvious importance of RelB for the immune system, scarce data are available on its regulation and the upstream receptors that might be responsible for its activation. The basal NF-κB activity in thymus and spleen largely consists of p50-RelB and p52-RelB heterodimers, as opposed to the inducible NF-κB binding activity, which often consists of RelA (p65) and c-Rel containing complexes (47–50). It was therefore believed that RelB primarily had a role in the constitutive expression of NF-κB–dependent genes (47–49). RelB activation has been shown in vitro, however, by stimulation of primary B cells with CD40L (51, 52). This activation was found to be selective to stimulation through the TNF family receptor CD40, as opposed to other stimulatory signals, but the in vivo significance of this pathway remains to be determined. Stimulation of MEFs through the LTβ-R has been shown to lead to RelB translocation to the nucleus as well. RelB is sequestered in the cytosol by the p100 molecule. Upon stimulation through the LTβ-R, p100 is processed into p52, and the p52-RelB complex can subsequently be found in the nucleus. This translocation was found to require the action of IKKα (53–55).
Here we have demonstrated that LTβR ligation results in sustained NF-κB DNA binding with a unique composition of Rel complexes, consisting of both RelA and RelB DNA binding complexes. Interestingly, by contrast with RelA, we have shown that the activation of RelB is strictly dependent upon functional NIK in vitro. Although the analysis of compound heterozygotes is complicated by the haplo-insufficiency of RelB, enhanced defects in Peyer's patch formation and Vα14i NKT cell function in RelB and aly compound heterozygotes suggests that this pathway also is important in vivo. The role of RelB in lymphoid development, however, is independent from its proinflammatory role, as aly/aly mice do not develop inflammation. Our data are consistent with those from a recent study demonstrating that cells with a null mutation of NIK displayed normal NF-κB DNA binding activity when treated with TNF or LTβR antibodies (39). However, NIK was selectively required for MCP-1 gene transcription induced through ligation of the LTβR, but not TNF receptors (39). Altogether, the results suggest that NIK activity is induced by a subset of TNF family receptors, and that it in turn selectively regulates the transcriptional activity NF-κB family members.
RelB forms active heterodimers with two NF-κB subunits, p50 and p52, and the question arises whether the inflammatory versus developmental roles can be ascribed to the different RelB-containing heterodimers. Defects in secondary lymphoid tissue formation have been reported in p52 but not in p50-deficient mice (56, 57). Interestingly, the processing of p100 by NIK to generate p52 (58) has been recently reported using gene overexpression systems. These results, together with the data presented here, suggest that RelB-p52 activation, perhaps through NIK by p100 processing, may be essential for aspects of lymphoid development as well as Vα14i T cell differentiation. The increased inflammatory phenotype observed in RelB − / − p50−/− mice, compared with RelB − / − mice, suggests that the lack of RelB is in part compensated by other p50-containing complexes (59). However, p50 complexes that do not contain RelB have not been characterized. Furthermore, other mechanisms have been proposed by which RelB might control inflammation, such as regulation of stability of the inhibitor protein IκBα (60).
In summary, we have shown that RelB activation through NIK is an essential mediator of Vα14i NKT cell differentiation and Peyer's patch formation in vivo. In Vα14i NKT cell development, this pathway is required on a thymus stromal cell that is not directly required for CD1d-mediated positive selection. These findings demonstrate the interplay between hemopoietic and nonhemopoietic cells in the unique pathway governing the development of Vα14i NKT cells.
Acknowledgments
We thank Kirin Pharmaceutical Research for providing α-GalCer (KRN7000), Drs. L. Brossay, C.A. Benedict, F. Koning, E. Dejardin, and S. Santee for helpful suggestions and advice, D. Martin for technical assistance, C. Reilly and C. Lena for maintaining the mouse colony, and Dr. O.V. Naidenko for providing α-GalCer/CD1d tetramers.
This is manuscript no. 412 of the La Jolla Institute for Allergy and Immunology. This work was supported in part by National Institutes of Health grants RO1 CA52511 (M. Kronenberg), RO1 AI33068 and RO1 CA69381 (C.F. Ware), R29 DK54451 (H. Cheroutre), and the Crohn's and Colitis Foundation of America (CCFA; H. De Winter). D. Elewaut is a recipient of a CCFA Career Development Award.
D. Elewaut and R.B. Shaikh contributed equally to this work.
D. Elewaut's present address is Department of Rheumatology, University Hospital, Ghent, Belgium.
Footnotes
Abbreviations used in this paper: α-GalCer, α-galactosylceramide; DC, dendritic cell; EMSA, electrophoretic mobility shift assay; MEF, mouse embryonic fibroblast; NF, nuclear factor; NIK, NF-κB–inducing kinase.
References
- 1.Karin, M., and M. Delhase. 2000. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin. Immunol. 12:85–98. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh, S., M.J. May, and E.B. Kopp. 1998. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260. [DOI] [PubMed] [Google Scholar]
- 3.Beg, A.A., W.C. Sha, R.T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- 4.Burkly, L., C. Hession, L. Ogata, C. Reilly, L.A. Marconi, D. Olson, R. Tizard, R. Cate, and D. Lo. 1995. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 373:531–536. [DOI] [PubMed] [Google Scholar]
- 5.Wu, L., A. D'Amico, K.D. Winkel, M. Suter, D. Lo, and K. Shortman. 1998. RelB is essential for the development of myeloid-related CD8alpha− dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity. 9:839–847. [DOI] [PubMed] [Google Scholar]
- 6.Lo, D., H. Quill, L. Burkly, B. Scott, R.D. Palmiter, and R.L. Brinster. 1992. A recessive defect in lymphocyte or granulocyte function caused by an integrated transgene. Am. J. Pathol. 141:1237–1246. [PMC free article] [PubMed] [Google Scholar]
- 7.Weih, D.S., Z.B. Yilmaz, and F. Weih. 2001. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol. 167:1909–1919. [DOI] [PubMed] [Google Scholar]
- 8.Weih, F., D. Carrasco, S.K. Durham, D.S. Barton, C.A. Rizzo, R.P. Ryseck, S.A. Lira, and R. Bravo. 1995. Multi-organ inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 80:331–340. [DOI] [PubMed] [Google Scholar]
- 9.Barton, D., H. HogenEsch, and F. Weih. 2000. Mice lacking the transcription factor RelB develop T cell-dependent skin lesions similar to human atopic dermatitis. Eur. J. Immunol. 30:2323–2332. [DOI] [PubMed] [Google Scholar]
- 10.DeKoning, J., L. DiMolfetto, C. Reilly, Q. Wei, W.L. Havran, and D. Lo. 1997. Thymic cortical epithelium is sufficient for the development of mature T cells in relB-deficient mice. J. Immunol. 158:2558–2566. [PubMed] [Google Scholar]
- 11.Lantz, O., and A. Bendelac. 1994. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4−8− T cells in mice and humans. J. Exp. Med. 180:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arase, H., N. Arase, K. Ogasawara, R.A. Good, and K. Onoe. 1992. An NK1.1+ CD4+8− single-positive thymocyte subpopulation that expresses a highly skewed T-cell antigen receptor V beta family. Proc. Natl. Acad. Sci. USA. 89:6506–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohteki, T., and H.R. MacDonald. 1996. Stringent V beta requirement for the development of NK1.1+ T cell receptor-alpha/beta+ cells in mouse liver. J. Exp. Med. 183:1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eberl, G., R. Lees, S.T. Smiley, M. Taniguchi, M.J. Grusby, and H.R. MacDonald. 1999. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J. Immunol. 162:6410–6419. [PubMed] [Google Scholar]
- 15.Burdin, N., L. Brossay, Y. Koezuka, S.T. Smiley, M.J. Grusby, M. Gui, M. Taniguchi, K. Hayakawa, and M. Kronenberg. 1998. Selective ability of mouse CD1 to present glycolipids: alpha-galactosylceramide specifically stimulates V alpha 14+ NK T lymphocytes. J. Immunol. 161:3271–3281. [PubMed] [Google Scholar]
- 16.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, H. Koseki, and M. Taniguchi. 1997. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 17.Kronenberg, M., and L. Gapin. 2002. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2:557–568. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benlagha, K., A. Weiss, A. Beavis, L. Teyton, and A. Bendelac. 2000. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J. Exp. Med. 191:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellicci, D.G., K.J. Hammond, A.P. Uldrich, A.G. Baxter, M.J. Smyth, and D.I. Godfrey. 2002. A natural killer T (NKT) cell developmental pathway involving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J. Exp. Med. 195:835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gapin, L., J.L. Matsuda, C.D. Surh, and M. Kronenberg. 2001. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat. Immunol. 2:971–978. [DOI] [PubMed] [Google Scholar]
- 22.Miyawaki, S., Y. Nakamura, H. Suzuka, M. Koba, R. Yasumizu, S. Ikehara, and Y. Shibata. 1994. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 24:429–434. [DOI] [PubMed] [Google Scholar]
- 23.Burdin, N., L. Brossay, and M. Kronenberg. 1999. Immunization with alpha-galactosylceramide polarizes CD1-reactive NK T cells towards Th2 cytokine synthesis. Eur. J. Immunol. 29:2014–2025. [DOI] [PubMed] [Google Scholar]
- 24.Force, W.R., B.N. Walter, C. Hession, R. Tizard, C.A. Kozak, J.L. Browning, and C.F. Ware. 1995. Mouse lymphotoxin-beta receptor. Molecular genetics, ligand binding, and expression. J. Immunol. 155:5280–5288. [PubMed] [Google Scholar]
- 25.Watanabe, H., K. Ohtsuka, M. Kimura, Y. Ikarashi, K. Ohmori, A. Kusumi, T. Ohteki, S. Seki, and T. Abo. 1992. Details of an isolation method for hepatic lymphocytes in mice. J. Immunol. Methods. 146:145–154. [DOI] [PubMed] [Google Scholar]
- 26.Feng, L., Y. Xia, J.I. Kreisberg, and C.B. Wilson. 1994. Interleukin-1 alpha stimulates KC synthesis in rat mesangial cells: glucocorticoids inhibit KC induction by IL-1. Am. J. Physiol. 26:F713–F22. [DOI] [PubMed] [Google Scholar]
- 27.Xia, Y., M.E. Pauza, L. Feng, and D. Lo. 1997. RelB regulation of chemokine expression modulates local inflammation. Am. J. Pathol. 151:375–387. [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto, M., K. Iwamasa, P.D. Rennert, T. Yamada, R. Suzuki, A. Matsushima, M. Okabe, S. Fujita, and M. Yokoyama. 1999. Involvement of distinct cellular compartments in the abnormal lymphoid organogenesis in lymphotoxin-alpha-deficient mice and alymphoplasia (aly) mice defined by the chimeric analysis. J. Immunol. 163:1584–1591. [PubMed] [Google Scholar]
- 29.DiDonato, J.A., F. Mercurio, and M. Karin. 1995. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol. Cell. Biol. 15:1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elewaut, D., J.A. DiDonato, J.M. Kim, F. Truong, L. Eckmann, and M.F. Kagnoff. 1999. NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J. Immunol. 163:1457–1466. [PubMed] [Google Scholar]
- 31.Bix, M., M. Coles, and D. Raulet. 1993. Positive selection of V beta 8+ CD4−8− thymocytes by class I molecules expressed by hematopoietic cells. J. Exp. Med. 178:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bendelac, A., N. Killeen, D.R. Littman, and R.H. Schwartz. 1994. A subset of CD4+ thymocytes selected by MHC class I molecules. Science 263:1774–1778. [DOI] [PubMed] [Google Scholar]
- 33.Coles, M.C., and D.H. Raulet. 1994. Class I dependence of the development of CD4+ CD8− NK1.1+ thymocytes. J. Exp. Med. 180:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bendelac, A. 1995. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J. Exp. Med. 182:2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coles, M.C., and D.H. Raulet. 2000. NK1.1+ T cells in the liver arise in the thymus and are selected by interactions with class I molecules on CD4+CD8+ cells. J. Immunol. 164:2412–2418. [DOI] [PubMed] [Google Scholar]
- 36.Shinkura, R., K. Kitada, F. Matsuda, K. Tashiro, K. Ikuta, M. Suzuki, K. Kogishi, T. Serikawa, and T. Honjo. 1999. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 22:74–77. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa, K., K. Iwabuchi, K. Ogasawara, M. Ato, M. Kajiwara, H. Nishihori, C. Iwabuchi, H. Ishikura, R.A. Good, and K. Onoe. 1997. Generation of NK1.1+ T cell antigen receptor alpha/beta+ thymocytes associated with intact thymic structure. Proc. Natl. Acad. Sci. USA. 94:2472–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konishi, J., K. Iwabuchi, C. Iwabuchi, M. Ato, J.I. Nagata, K. Onoe, K.I. Nakagawa, M. Kasai, K. Ogasawara, and K. Kawakami. 2000. Thymic epithelial cells responsible for impaired generation of NK-T thymocytes in Alymphoplasia mutant mice. Cell. Immunol. 206:26–35. [DOI] [PubMed] [Google Scholar]
- 39.Yin, L., L. Wu, H. Wesche, C.D. Arthur, J.M. White, D.V. Goeddel, and R.D. Schreiber. 2001. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 291:2162–2165. [DOI] [PubMed] [Google Scholar]
- 40.Tilloy, F., J.P. Di Santo, A. Bendelac, and O. Lantz. 1999. Thymic dependence of invariant V alpha 14+ natural killer-T cell development. Eur. J. Immunol. 29:3313–3318. [DOI] [PubMed] [Google Scholar]
- 41.Shimamura, M., T. Ohteki, P. Launois, A.M. Garcia, and H.R. MacDonald. 1997. Thymus-independent generation of NK1+ T cells in vitro from fetal liver precursors. J. Immunol. 158:3682–3689. [PubMed] [Google Scholar]
- 42.Shimamura, M., Y.Y. Huang, Y. Suda, S. Kusumoto, K. Sato, M.J. Grusby, H. Sato, T. Nakayama, and M. Taniguchi. 1999. Positive selection of NKT cells by CD1(+), CD11c(+) non-lymphoid cells residing in the extrathymic organs. Eur. J. Immunol. 29:3962–3970. [DOI] [PubMed] [Google Scholar]
- 43.Bendelac, A., and R.H. Schwartz. 1991. CD4+ and CD8+ T cells acquire specific lymphokine secretion potentials during thymic maturation. Nature. 3539:68–71. [DOI] [PubMed] [Google Scholar]
- 44.Bendelac, A., P. Matzinger, R.A. Seder, W.E. Paul, and R.H. Schwartz. 1992. Activation events during thymic selection. J. Exp. Med. 175:731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuda, J.L., L. Gapin, S. Sidobre, W.C. Kieper, J.T. Tan, R. Ceredig, C.D. Surh, and M. Kronenberg. 2002. Homeostasis of V alpha 14i NKT cells. Nat. Immunol. 3:966–974. [DOI] [PubMed] [Google Scholar]
- 46.Benlagha, K., T. Kyin, A. Beavis, L. Teyton, and A. Bendelac. 2002. A thymic precursor to the NK T cell lineage. Science. 296:553–555. [DOI] [PubMed] [Google Scholar]
- 47.Lernbecher, T., U. Muller, and T. Wirth. 1993. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 365:767–770. [DOI] [PubMed] [Google Scholar]
- 48.Lernbecher, T., B. Kistler, and T. Wirth. 1994. Two distinct mechanisms contribute to the constitutive activation of RelB in lymphoid cells. EMBO J. 13:4060–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weih, F., D. Carrasco, and R. Bravo. 1994. Constitutive and inducible Rel/NF-kappa B activities in mouse thymus and spleen. Oncogene. 9:3289–3297. [PubMed] [Google Scholar]
- 50.Carrasco, D., R.P. Ryseck, and R. Bravo. 1993. Expression of relB transcripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 118:1221–1231. [DOI] [PubMed] [Google Scholar]
- 51.Neumann, M., G. Wohlleben, S. Chuvpilo, B. Kistler, T. Wirth, E. Serfling, and A. Schimpl. 1996. CD40, but not lipopolysaccharide and anti-IgM stimulation of primary B lymphocytes, leads to a persistent nuclear accumulation of RelB. J. Immunol. 157:4862–4869. [PubMed] [Google Scholar]
- 52.Lin, S.C., H.H. Wortis, and J. Stavnezer. 1998. The ability of CD40L, but not lipopolysaccharide, to initiate immunoglobulin switching to immunoglobulin G1 is explained by differential induction of NF-kappaB/Rel proteins. Mol. Cell. Biol. 18:5523–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dejardin, E., V. Deregowski, R. Greimers, Z. Cai, S. Chouaib, M.P. Merville, and V. Bours. 1998. Regulation of major histocompatibility complex class I expression by NF-kappaB-related proteins in breast cancer cells. Oncogene. 16:3299–3307. [DOI] [PubMed] [Google Scholar]
- 54.Dejardin, E., N.M. Droin, M. Delhase, E. Haas, Y. Cao, C. Makris, Z.W. Li, M. Karin, C.F. Ware, and D.R. Green. 2002. The Lymphotoxin-beta Receptor Induces Different Patterns of Gene Expression via Two NF-kappaB Pathways. Immunity. 17:525–530. [DOI] [PubMed] [Google Scholar]
- 55.Dobrzanski, P., R.P. Ryseck, and R. Bravo. 1995. Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100. Oncogene. 10:1003–1007. [PubMed] [Google Scholar]
- 56.Caamano, J.H., C.A. Rizzo, S.K. Durham, D.S. Barton, C. Raventos-Suarez, C.M. Snapper, and R. Bravo. 1998. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med. 187:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sha, W.C., H.C. Liou, E.I. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 80:321–330. [DOI] [PubMed] [Google Scholar]
- 58.Xiao, G., E.W. Harhaj, and S.C. Sun. 2001. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 7:401–409. [DOI] [PubMed] [Google Scholar]
- 59.Weih, F., S.K. Durham, D.S. Barton, W.C. Sha, D. Baltimore, and R. Bravo. 1997. p50-NF-kappaB complexes partially compensate for the absence of RelB: severely increased pathology in p50(−/−)relB(−/−) double-knockout mice. J. Exp. Med. 185:1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia, Y., S. Chen, Y. Wang, N. Mackman, G. Ku, D. Lo, and L. Feng. 1999. RelB modulation of IkappaBalpha stability as a mechanism of transcription suppression of interleukin-1alpha (IL-1alpha), IL-1beta, and tumor necrosis factor alpha in fibroblasts. Mol. Cell. Biol. 19:7688–7696. [DOI] [PMC free article] [PubMed] [Google Scholar]