

Abstract
The ability of fungal-derived β-glucan particles to induce leukocyte activation and the production of inflammatory mediators, such as tumor necrosis factor (TNF)-α, is a well characterized phenomenon. Although efforts have been made to understand how these carbohydrate polymers exert their immunomodulatory effects, the receptors involved in generating these responses are unknown. Here we show that Dectin-1 mediates the production of TNF-α in response to zymosan and live fungal pathogens, an activity that occurs at the cell surface and requires the cytoplasmic tail and immunoreceptor tyrosine activation motif of Dectin-1 as well as Toll-like receptor (TLR)-2 and Myd88. This is the first demonstration that the inflammatory response to pathogens requires recognition by a specific receptor in addition to the TLRs. Furthermore, these studies implicate Dectin-1 in the production of TNF-α in response to fungi, a critical step required for the successful control of these pathogens.
Keywords: β-glucan receptor, macrophages, inflammation, tumor necrosis factor, Candida
Introduction
β-Glucans possess anti-infective and anti-tumorigenic properties (1–3) that stem from their ability to activate leukocytes, stimulating their phagocytic activity, the production of reactive oxygen intermediates, inflammatory mediators and cytokines, including TNF-α (1, 4). The activatory properties of these fungal-derived carbohydrates, especially in particles such as zymosan (5), are widely used to examine the proinflammatory responses of phagocytes. Despite almost 50 yr of examination (6), however, the receptor(s) mediating these effects are still undefined.
We recently identified Dectin-1 (7) as a β-glucan receptor by screening a RAW264.7 cDNA library with zymosan (8). Dectin-1 is a small type II transmembrane receptor containing one lectin-like carbohydrate recognition domain, which recognizes β1,3- and/or β1,6-linked glucans and intact yeast (8), and an immunoreceptor tyrosine-based activation motif (ITAM)*in the cytoplasmic tail (7). In addition to these exogenous ligands, the receptor can also recognize an endogenous ligand on T cells (7). The receptor is expressed at high levels on macrophages (Mφ) and neutrophils, and to a lesser extent on dendritic cells and a subpopulation of T cells (9). The human homologue was found to be structurally and functionally similar to the murine receptor (10).
We have shown that Dectin-1 acts as a major receptor for zymosan and other β-glucans on Mφ (11) and wondered how this receptor contributed to the cellular responses induced by these carbohydrates. We show here that Dectin-1 mediates cellular responses to zymosan as well as to live yeast pathogens. Furthermore, we demonstrate that this response occurs at the cell surface and requires the cytoplasmic tail of this receptor and the Toll-like receptor (TLR) pathway. These results are the first evidence that the proinflammatory response requires recognition by pathogen-specific receptors in addition to the TLRs, and does not require pathogen internalization.
Materials and Methods
DNA Constructs.
Hemagglutinin (HA)-tagged Dectin-1 was generated by modifying the 3′ end of Dectin-1 cDNA (7, 11) by PCR to allow the cloning of an adaptor, encoding an HA tag and a stop codon. The fusion of the HA tag onto the carbohydrate recognition domain did not affect the ability of this molecule to recognize zymosan. PCR was used to generate the cytoplasmic mutant constructs from the full-length HA-tagged Dectin-1, including the ITAM mutant where the motif YX11YXXL (amino acids 3–18) was mutated to FX11FXXL, and the truncation mutant where the 5′ end of the gene encoding the cytoplasmic tail (amino acids 1–39) was replaced with a start codon (AUG) and Kozak sequence. All constructs were cloned into the pFB-neo retroviral vector (Stratagene), packaged into virions using 293T φNX ecotropic cells (provided by G. Nolan, Stanford University, Stanford, CA), and used to transduce RAW264.7 (no. TIB-71; American Type Culture Collection [ATCC]) Mφ. Vector-only transduced cells (RAW-FB) were used as controls. All cell lines were used as uncloned populations to reduce any founder effects and were generated and tested at least twice to confirm their phenotype. V5-tagged TLR-2 was provided by A. Ozinsky (University of Washington, Seattle, WA; 12).
Mφ.
Thioglycollate-elicited peritoneal Mφ were isolated from C57BL/6 mice by standard procedures. Animals were kept and handled according to institutional guidelines. TLR-2−/−, MyD88−/−, and control bone marrow–derived Mφ (BMDM; 13, 14) were provided by A. Edwards and C. Reiss e Sousa (Cancer Research UK, London, United Kingdom) and T. Kaisho and S. Akira (Osaka University, Osaka, Japan), respectively. Cells were maintained in RPMI with 10% heat-inactivated FCS, 50 IU/ml penicillin G, 50 μg/ml streptomycin, and 2 mM glutamine (RPMI medium), except for the BMDM, which were cultured in RPMI medium supplemented with 15% (vol/vol) L cell–conditioned medium as a source of M-CSF (15). Experiments with BMDM were performed in the absence of M-CSF 5–7 d after isolation and culture in L cell–conditioned medium. Transduced RAW264.7 Mφ were cultured in RPMI medium supplemented with 600 μg ml-1 G418 (GIBCO BRL).
Fluorescent Zymosan Binding and TNF-α Assays.
Cold fluorescence-based binding assays using FITC-labeled zymosan (Molecular Probes) were performed as previously described (11). For all other assays, Mφ were plated at 2.5 × 105 cells/well in 24-well plates in RPMI medium the day before each experiment. At the start of the assay, the cells were cooled to 4°C and washed three times with prechilled culture medium. When appropriate, glucan phosphate (GluP; reference 16) was added to 100 μg/ml and the cells were incubated for 20 min at 4°C to allow inhibition of Dectin-1 (11). After the addition of FITC-labeled zymosan (20–25 particles/cell) or unlabeled zymosan (100 μg/ml; Sigma-Aldrich), the cells were incubated at 37°C in 5% CO2 for 30 min unless stated otherwise. After washes to remove unbound particles, the cells were then incubated for an additional 3 h at 37°C after which samples of supernatant were taken for TNF-α measurements. The amount of FITC zymosan bound by the cells was quantified after lysis with 3% Triton X-100 using a Titretek Fluoroskan II (Labsystems Group) as previously described (11).
To obtain opsonized particles, FITC-labeled zymosan was incubated with 20% normal mouse serum in HBSS for 30 min at 37°C and then washed extensively in RPMI medium before use. Binding assays of the opsonized FITC zymosan were performed as described above for the unopsonized particles. Similar methodology was used to assess the effects of LPS (5 ng/ml; Sigma-Aldrich) and live yeast particles. For these experiments, Saccharomyces cerevisiae (BY4741; ATCC no. 201388) and Candida albicans (ATCC no. 18804) were grown overnight in YPD (CLONTECH Laboratories, Inc.) at 30°C, washed extensively in PBS before use, enumerated, and added to the Mφ in RPMI medium. For experiments requiring inhibition of actin polymerization, cytochalasin D in 2 μM DMSO (Sigma-Aldrich) or DMSO alone was added to the cells 40 min before the start of the assay and then maintained throughout the experiment. This concentration of cytochalasin D was sufficient to inhibit zymosan phagocytosis in RAW Mφ (unpublished data).
TNF-α was measured using the OptEIA murine TNF-α ELISA kit (BD Biosciences) as described by the manufacturers.
All experiments were repeated in duplicate or triplicate at least three times. All data are presented as the mean and standard deviation from a representative experiment.
Flow Cytometry and Immunofluorescence Microscopy.
Flow cytometry was performed according to conventional protocols. Cells were examined by two-color FACS® analysis using anti–Dectin-1 mAb (2A11; reference 11) and phycoerythrin-conjugated goat anti–rat IgG (Leinco Technologies). A nonspecific rat IgG2b was used as an isotype control. For immunofluorescence microscopy, RAW-Dectin cells were electroporated with the V5-tagged TLR-2 as previously described (17), plated on acid-treated glass coverslips, and cultured overnight as described above. After incubation with zymosan, the cells were fixed, permeabilized, and blocked in 5% goat serum and stained with anti-V5 mAb (Invitrogen) and Cy3 goat anti–mouse IgG (Jackson ImmunoResearch Laboratories). After a second blocking step in 5% mouse serum, the cells were stained with Alexaflor 488 anti-HA (HA11; Covance) to detect Dectin-1. After mounting, the cells were observed by confocal laser scanning microscopy (Biorad 1024) on a Nikon Diaphot 200 inverted microscope. Images were processed using Adobe Photoshop.
Results and Discussion
The Response to Zymosan Can Be Blocked by Soluble β-glucans in Primary Mφ.
To explore the possibility that Dectin-1 was involved in mediating the proinflammatory responses to β-glucans, we examined the effect of GluP, a potent soluble β-glucan inhibitor of Dectin-1 (8), on the ability of zymosan to induce TNF-α production in primary Mφ (Fig. 1 a). The addition of zymosan induced the release of TNF-α into the Mφ culture supernatants within 3 h as previously described (18). However, this response could be completely inhibited by the addition of GluP and was specific for zymosan as LPS-induced TNF-α production was unaffected by the presence of the soluble β-glucan. The effect of GluP was consistent with the inhibitory activities described for other soluble β-glucans on zymosan-mediated proinflammatory responses (1, 19) and suggested a role for Dectin-1 in this process.
Figure 1.

Zymosan-induced TNF-α production is β-glucan dependent and mediated by Dectin-1. (a) GluP specifically inhibits the production of TNF-α in response to unlabeled zymosan in thioglycollate-elicited Mφ. (b) RAW264.7 Mφ express low levels of endogenous Dectin-1 as measured by FACS® staining using mAb 2A11. Increased levels of this receptor are obtained by stably transducing the Mφ with HA-tagged Dectin-1, including two cytoplasmic tail mutants. (c) RAW264.7 Mφ overexpressing Dectin-1 (RAW-Dectin; ▴) have an enhanced ability to bind FITC-labeled zymosan compared with the vector-transduced control (RAW-FB; ▪) and (d) show a dose-dependent increase in TNF-α production.
Expression of Dectin-1 in RAW264.7 Mφ Correlates with TNF-α Production in Response to Zymosan.
To define the role of Dectin-1, the cellular response to zymosan was examined using RAW264.7 Mφ. This Mφ cell line expressed low levels of endogenous Dectin-1, allowing us to examine the effects of zymosan on cells retrovirally transduced to overexpress a HA-tagged version of this receptor (Fig. 1 b). When compared with control cells (RAW-FB), increased expression of Dectin-1 (RAW-Dectin) correlated with an increased ability of these cells to bind zymosan (Fig. 1 c), as expected (11), but also led to an enhanced, dose-dependent TNF-α response in these cells (Fig. 1 d). As we observed in the primary Mφ, the enhanced TNF-α production could be completely inhibited by the addition of GluP (see Fig. 4 a and b). Thus, the expression of Dectin-1 not only correlates with the ability of these cells to bind zymosan, but also their ability to produce TNF-α in response to these particles.
Figure 4.

The β-glucan–dependent TNF-α response to zymosan is mediated by Dectin-1. Although opsonization increases the amount of FITC-labeled zymosan bound by RAW-FB Mφ (a), TNF-α production is only marginally increased and is no longer β-glucan dependent (b). In contrast, Mφ overexpressing Dectin-1 (RAW-Dectin) still possess an enhanced β-glucan–dependent response to opsonized zymosan.
Dectin-1 Mediates the Production of TNF-α in Response to Live Yeast Pathogens.
As Dectin-1 can recognize intact yeast particles (8), and as TNF-α is an essential cytokine required for the successful control of many fungal pathogens (20–23), we examined the possibility that this receptor could also mediate a proinflammatory response to live organisms (Fig. 2) . When live C. albicans (Fig. 2 a) and S. cerevisiae (Fig. 2 b) were added to the Mφ, they induced enhanced levels of TNF-α in RAW-Dectin Mφ compared with the control (RAW-FB) cells. Significantly, these responses could be inhibited by the addition of GluP. Thus, this is the first description of a receptor involved in generating a proinflammatory response to fungal pathogens and it suggests that Dectin-1 may have an important role in the innate response to these organisms.
Figure 2.

Dectin-1 induces the production of TNF-α in response to live yeasts. RAW-Dectin Mφ display an enhanced ability to induce TNF-α in response to live (a) C. albicans and (b) S. cerevisiae. Live yeasts were added at a multiplicity of infection of 5:1. Note that the response to S. cerevisiae is significantly greater than to C. albicans and that these responses are β-glucan dependent.
β-Glucan is traditionally thought to be buried within the fungal cell wall (24), but the ability of Dectin-1 to recognize these organisms implies that β-glucan is present on the yeast cell surface. In addition, S. cerevisiae induced far greater levels of TNF-α than C. albicans (Fig. 2) and were also bound more efficiently to the Mφ (unpublished data), indicating differences in the amount of exposed β-glucan in these yeasts. This difference and the ability of other pathogens, such as Paracoccidioides brasiliensis (25), to modify their cell wall β-glucan content during infection is suggestive that fungal pathogens may mask their cell wall β-glucan to avoid immune recognition through Dectin-1.
The TLR Pathway Is Also Required to Trigger the Proinflammatory Response to Zymosan.
TLRs are thought to sample the phagosomal contents, triggering a response appropriate to the pathogen, and have been shown to be involved in the recognition of fungal pathogens (26), zymosan (27), and other β-glucans (28). Indeed, BMDM from MyD88 and TLR-2 knockout mice were unable to induce TNF-α production in response to zymosan (Fig. 3 a), despite being capable of binding and internalizing this particle (unpublished data). We also observed colocalization of TLR-2 and Dectin-1 in membrane areas in contact with zymosan in RAW-Dectin Mφ (Fig. 3 b–e). These results, in conjunction with the data presented in Fig. 1, indicated that the enhanced β-glucan–dependent TNF-α response observed in the transduced RAW264.7 Mφ may have been a result of increased, Dectin-1–mediated zymosan availability to which the TLRs were subsequently recognizing and responding.
Figure 3.
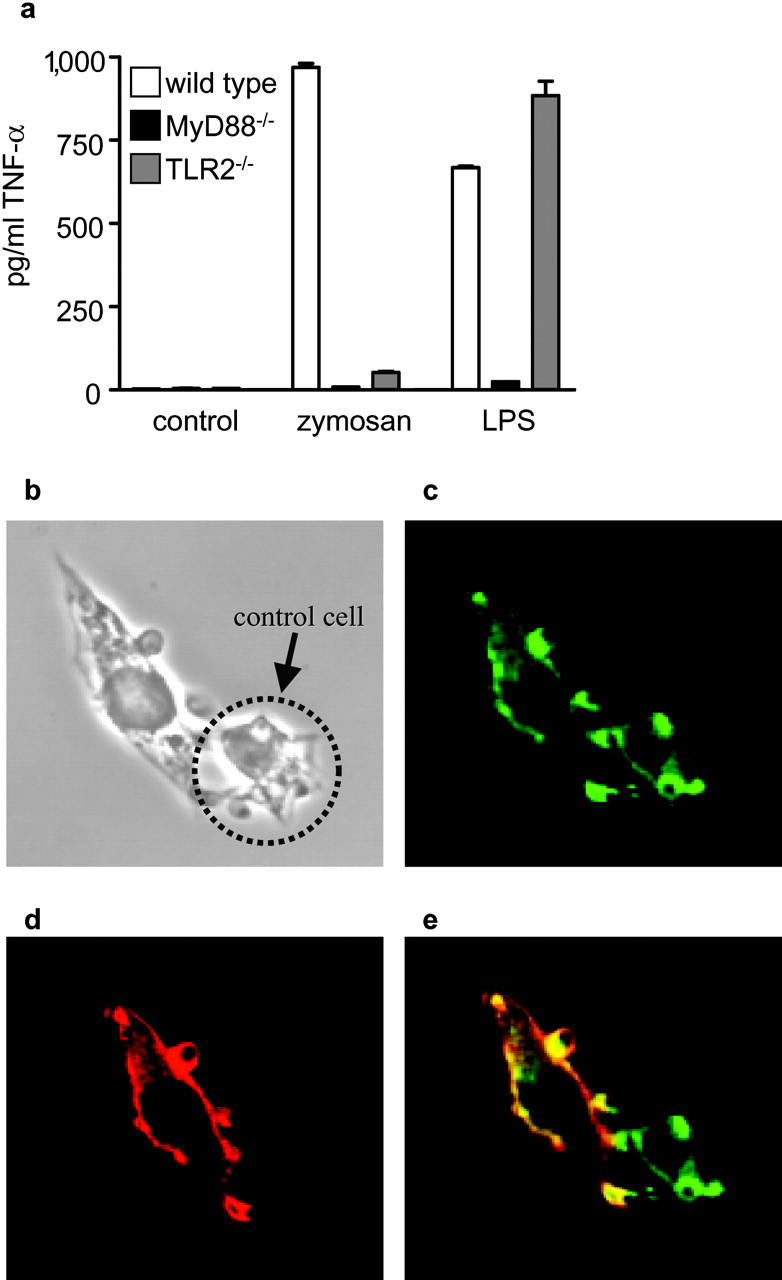
TLRs are required for TNF-α production in response to zymosan. (a) BMDM from TLR-2– or MyD88-deficient mice do not produce TNF-α after overnight incubation with unlabeled zymosan. The response to overnight incubation with 5 ng/ml LPS is shown as a control. (b–e) V5-Tagged TLR-2 (red) and HA-tagged Dectin-1 (green) colocalize within 5 min after the addition of zymosan. (b) Light micrograph showing RAW-Dectin cells binding zymosan. Anti-HA (c) and anti-V5 (d) staining showing colocalization (e) of Dectin-1 and TLR-2, respectively, at the zymosan phagocytic cup. The cell indicated with an arrow was not transfected with V5-tagged TLR-2 and is presented as a staining control. Colocalization of these receptors at the cell surface in contact with zymosan were also observed after 30-min incubation at 37°C with Cytochalasin D (unpublished data).
To investigate this hypothesis, we used complement opsonized zymosan for Dectin-1–independent delivery of zymosan to the Mφ cell surface and examined the TNF-α response in the presence or absence of GluP (Fig. 4) . Opsonized zymosan is recognized by both Dectin-1 and CR3 (11), the latter of which is expressed at equivalent levels in RAW-FB and RAW-Dectin Mφ (unpublished data). Although complement opsonization led to greatly increased levels of zymosan binding in RAW-FB Mφ (Fig. 4 a) as expected, the levels of TNF-α were only marginally increased and were no longer β-glucan dependent (Fig. 4 b). Mφ overexpressing Dectin-1, however, still possessed an enhanced β-glucan–dependent TNF-α response to opsonized zymosan. Thus, these data suggested that it was not the increased availability of zymosan that was leading to the enhanced, β-glucan–dependent production of TNF-α, but was rather a direct result of the higher levels of Dectin-1 expressed in these cells.
As Dectin-1 appeared to contribute to the TNF-α response to zymosan, we next looked at the possibility that the cytoplasmic tail of this receptor was involved in this process. For this analysis we generated RAW264.7 cell lines expressing mutant forms of Dectin-1, including a cell line expressing receptors lacking a functional ITAM motif (RAW-ITAM) as well as a cell line expressing a truncated receptor lacking the entire cytoplasmic tail (RAW-Truncation). Both cell lines expressed the mutated forms of Dectin-1 at the cell surface (Fig. 1 b) and mediated enhanced β-glucan–dependent zymosan binding to these cells, comparable to that obtained with the wild-type receptor (Fig. 5 a). In contrast, however, Mφ expressing the mutated receptors did not show elevated levels of TNF-α in response to zymosan (Fig. 5 b).
Figure 5.

The cytoplasmic tail of Dectin-1 is involved in mediating the TNF-α response to zymosan. The cell lines expressing the Dectin-1 cytoplasmic tail mutants display an enhanced ability to bind FITC-labeled zymosan (a), but they do not transduce an intracellular signal inducing the TNF-α response to this particle (b). This effect is not due to lack of internalization of the zymosan as signaling still occurs, and is even enhanced, when phagocytosis is inhibited with cytochalasin D (c; cytoD).
Because the cytoplasmic tail and ITAM motif of Dectin-1 are required for phagocytosis of zymosan (unpublished data and reference 8), it was possible that the failure to induce TNF-α was due to the lack of particle internalization. We explored this possibility by inhibiting zymosan phagocytosis using cytochalasin D (29) and measuring the effects on TNF-α production (Fig. 5 c). Although the ability of these Mφ to bind zymosan was unaffected by the presence of cytochalasin D (unpublished data), greatly elevated levels of β-glucan–dependent TNF-α were obtained in both the control (RAW-FB) and RAW-Dectin cells. Furthermore, colocalization of TLR-2 and Dectin-1 with zymosan at the cell surface was still observed after cytochalasin D treatment (unpublished data). Thus, these data suggest that the TNF-α response to zymosan does not require particle internalization but does require the cytoplasmic tail of Dectin-1.
Overall, our data suggest that Dectin-1 is a key receptor involved in mediating the biological effects of β-glucans. Furthermore, through its ability to recognize these carbohydrates, Dectin-1 is likely to be centrally involved in the innate response to fungal pathogens. We have also shown that pathogen-specific receptors, in addition to the TLR receptors, are required to generate proinflammatory responses to pathogens. Given the multitude of non-TLR receptors involved in pathogen recognition, future studies will undoubtedly show that this is a common paradigm in innate immunity.
Acknowledgments
We thank Dr. Philip Taylor for critically reading this manuscript.
This work was supported by the Wellcome Trust.
Footnotes
Abbreviations used in this paper: BMDM, bone marrow–derived macrophages; GluP, glucan phosphate; HA, hemagglutinin; ITAM, immunoreceptor tyrosine-based activation motif; Mφ, macrophages; TLR, Toll-like receptor.
References
- 1.Czop, J.K. 1986. The role of beta-glucan receptors on blood and tissue leukocytes in phagocytosis and metabolic activation. Pathol. Immunopathol. Res. 5:286–296. [DOI] [PubMed] [Google Scholar]
- 2.Williams, D.L. 1997. Overview of (1,3)-beta-D-glucan immunobiology. Mediators Inflamm. 6:247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross, G.D., V. Vetvicka, J. Yan, Y. Xia, and J. Vetvickova. 1999. Therapeutic intervention with complement and beta-glucan in cancer. Immunopharmacology. 42:61–74. [DOI] [PubMed] [Google Scholar]
- 4.Williams, D.L., A. Mueller, and W. Browder. 1996. Glucan-based macrophage stimulators. Clinical Immunotherapy. 5:392–399. [Google Scholar]
- 5.Riggi, S.J., and N.R. Di Luzio. 1961. Identification of a reticuloendothelial stimulating agent in zymosan. Am. J. Physiol. 200:297–300. [DOI] [PubMed] [Google Scholar]
- 6.Benacerraf, B., and M.M. Sebestyen. 1957. Effect of bacterial endotoxins on the reticuloendothelial system. Fed. Proc. 16:860–867. [PubMed] [Google Scholar]
- 7.Ariizumi, K., G.L. Shen, S. Shikano, S. Xu, R. Ritter III, T. Kumamoto, D. Edelbaum, A. Morita, P.R. Bergstresser, and A. Takashima. 2000. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 275:20157–20167. [DOI] [PubMed] [Google Scholar]
- 8.Brown, G.D., and S. Gordon. 2001. Immune recognition: a new receptor for beta-glucans. Nature. 413:36–37. [DOI] [PubMed] [Google Scholar]
- 9.Taylor, P.R., G.D. Brown, D.M. Reid, J.A. Willment, L. Martinez-Pomares, S. Gordon, and S.Y.C. Wong. 2002. The beta-glucan receptor, Dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 269:3876–3882. [DOI] [PubMed] [Google Scholar]
- 10.Willment, J.A., S. Gordon, and G.D. Brown. 2001. Characterisation of the human beta-glucan receptor and its alternatively spliced isoforms. J. Biol. Chem. 276:43818–43823. [DOI] [PubMed] [Google Scholar]
- 11.Brown, G.D., P.R. Taylor, D.M. Reid, J.A. Willment, D.L. Williams, L. Martinez-Pomares, S.Y.C. Wong, and S. Gordon. 2002. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 296:407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozinsky, A., D.M. Underhill, J.D. Fontenot, A.M. Hajjar, K.D. Smith, C.B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA. 97:13766–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11:443–451. [DOI] [PubMed] [Google Scholar]
- 14.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 9:143–150. [DOI] [PubMed] [Google Scholar]
- 15.Hume, D.A., and S. Gordon. 1983. Optimal conditions for proliferation of bone marrow-derived mouse macrophages in culture: the roles of CSF-1, serum, Ca2+, and adherence. J. Cell. Physiol. 117:189–194. [DOI] [PubMed] [Google Scholar]
- 16.Muller, A., P.J. Rice, H.E. Ensley, P.S. Coogan, J.H. Kalbfleish, J.L. Kelley, E.J. Love, C.A. Portera, T. Ha, I.W. Browder, et al. 1996. Receptor binding and internalization of a water-soluble (1→3)-beta-D-glucan biologic response modifier in two monocyte/macrophage cell lines. J. Immunol. 156:3418–3425. [PubMed] [Google Scholar]
- 17.Underhill, D.M., A. Ozinsky, K.D. Smith, and A. Aderem. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA. 96:14459–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stein, M., and S. Gordon. 1991. Regulation of tumor necrosis factor (TNF) release by murine peritoneal macrophages: role of cell stimulation and specific phagocytic plasma membrane receptors. Eur. J. Immunol. 21:431–437. [DOI] [PubMed] [Google Scholar]
- 19.Tapper, H., and R. Sundler. 1995. Glucan receptor and zymosan-induced lysosomal enzyme secretion in macrophages. Biochem. J. 306:829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawakami, K., X. Qifeng, M. Tohyama, M.H. Qureshi, and A. Saito. 1996. Contribution of tumour necrosis factor-alpha (TNF-alpha) in host defence mechanism against Cryptococcus neoformans. Clin. Exp. Immunol. 106:468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Netea, M.G., L.J. van Tits, J.H. Curfs, F. Amiot, J.F. Meis, J.W. van der Meer, and B.J. Kullberg. 1999. Increased susceptibility of TNF-alpha lymphotoxin-alpha double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J. Immunol. 163:1498–1505. [PubMed] [Google Scholar]
- 22.Mehrad, B., R.M. Strieter, and T.J. Standiford. 1999. Role of TNF-alpha in pulmonary host defense in murine invasive aspergillosis. J. Immunol. 162:1633–1640. [PubMed] [Google Scholar]
- 23.Allendoerfer, R., and G.S. Deepe, Jr. 1998. Blockade of endogenous TNF-alpha exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J. Immunol. 160:6072–6082. [PubMed] [Google Scholar]
- 24.Klis, F.M., P. de Groot, and K. Hellingwerf. 2001. Molecular organization of the cell wall of Candida albicans. Med. Mycol. 39:1–8. [PubMed] [Google Scholar]
- 25.Borges-Walmsley, M.I., D. Chen, X. Shu, and A.R. Walmsley. 2002. The pathobiology of Paracoccidioides brasiliensis. Trends Microbiol. 10:80–87. [DOI] [PubMed] [Google Scholar]
- 26.Netea, M.G., C.A. Van Der Graaf, A.G. Vonk, I. Verschueren, J.W. Van Der Meer, and B.J. Kullberg. 2002. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 185:1483–1489. [DOI] [PubMed] [Google Scholar]
- 27.Underhill, D.M., A. Ozinsky, A.M. Hajjar, A. Stevens, C.B. Wilson, M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 401:811–815. [DOI] [PubMed] [Google Scholar]
- 28.Kataoka, K., T. Muta, S. Yamazaki, and K. Takeshige. 2002. Activation of macrophages by linear (1,3)-beta-D-glucans. J. Biol. Chem. 277:36825–36831. [DOI] [PubMed] [Google Scholar]
- 29.Lombard, Y., J. Giaimis, M. Makaya-Kumba, P. Fonteneau, and P. Poindron. 1994. A new method for studying the binding and ingestion of zymosan particles by macrophages. J. Immunol. Methods. 174:155–165. [DOI] [PubMed] [Google Scholar]