

Abstract
Activation of naive CD8 T cells to undergo clonal expansion and develop effector function requires three signals: (a) Ag, (b) costimulation, and (c) IL-12 or adjuvant. The requirement for the third signal to stimulate Ag-dependent proliferation is variable, making the greatest contribution when Ag levels are low. At high Ag levels, extensive proliferation can occur in vitro or in vivo in the absence of a third signal. However, despite having undergone the same number of divisions, cells that expand in the absence of a third signal fail to develop cytolytic effector function. Thus, proliferation and development of cytolytic function can be fully uncoupled. Furthermore, these cells are rendered functionally tolerant in vivo, in that subsequent restimulation with a potent stimulus results in limited clonal expansion, impaired IFN-γ production, and no cytolytic function. Thus, the presence or absence of the third signal appears to be a critical variable in determining whether stimulation by Ag results in tolerance versus development of effector function and establishment of a responsive memory population.
Keywords: antigens, cell division, immune tolerance, interleukin-12, lymphocytes
Introduction
In addition to central tolerance resulting from negative selection of immature T cells in the thymus, there are mechanisms that provide for the induction of peripheral T cell tolerance. Numerous studies with a diverse array of experimental models make it clear that peripheral tolerance can take several forms. “Clonal ignorance” results from a failure of potentially reactive T cells to become activated, and regulatory lymphocytes may prevent the activation of T cells in response to peripheral Ag. In addition, peripheral tolerance can be established as a result of clonal deletion and/or induction of nonresponsiveness (1–10). The use of transgenic mice, or T cells derived from TCR transgenic mice, made it possible to clearly demonstrate that tolerance of CD8 T cells as a result of challenge with Ag can result in partial or complete deletion of the Ag-specific cells (5, 7, 8, 11, 12). In most cases, establishment of the tolerant state is preceded by a vigorous response of the cells to the Ag. The extent of clonal deletion varies and is seldom complete, and the remaining Ag-specific T cells are nonresponsive and exhibit functional alterations (8–10, 13–16). It is speculated frequently that levels of Ag or costimulation influence tolerance induction; but in most cases, there is little or no direct evidence for this. Furthermore, there are few in vitro correlates of tolerance induction in CD8 T cells, making it difficult to study the mechanistic aspects of the phenomenon.
Over the past several years, the central role of dendritic cells (DC)* as the “professional APCs” responsible for activating T cells has become apparent. Cross-presentation of exogenous Ag by DC (17) appears to be the primary mode of presentation to naive CD8 T cells, but can lead to either full activation and development of effector function, or the induction of tolerance (18, 19). How cross-presentation can lead to either activation or tolerance, and the factors that determine the outcome, are not well-defined. The subset of DC presenting Ag may influence the outcome (20, 21), but the activation state of the DC is more likely to be critical (22, 23).
DC become activated in response to engagement of toll-like receptors by adjuvants such as bacterial cell wall components (24, 25) or by ligation of CD40 by CD40L expressed on CD4 helper T cells (26). One consequence of activation is increased expression of costimulatory ligands that can provide signal 2 to T cells recognizing the Ag presented by DC. In particular, B7 ligands are expressed on resting DC, but expression is up-regulated severalfold upon activation (27, 28). This is often viewed as being important for avoiding the tolerance that can occur when T cells recognize Ag but do not receive signal 2, thus rendering them anergic (29). However, there is little direct evidence that increasing B7 expression levels above the basal resting levels is critical for CD8 T cell activation. In fact, there is some evidence that it may not always be necessary (30) or sufficient (31, 32) to prevent tolerance.
Activation of DC also stimulates them to produce a variety of cytokines, and recent studies suggest that some of these cytokines may provide an essential third signal that is required to fully activate T cells and avoid tolerance induction (30, 32–34). This has been studied most extensively for CD8 T cells, where IL-12 has been shown to provide the third signal needed for strong proliferation and development of cytolytic function in response to Ag and B7 ligand in vitro (33) and in vivo (30, 34), and to shift the outcome of in vivo Ag stimulation from tolerance to full activation (30, 32, 34) and establishment of a long-lived memory population (34). Consistent with this, the CD8α+ DC subset most effective for priming CD8 T cells (35, 36) produces high amounts of IL-12 in response to stimulation, whereas CD8α− DC produce little (37).
Development of expanded Ag-specific CD8 T cell populations that lack function, and are thus tolerant, has been observed in a variety of experimental systems (32, 38–42), but the causes of this have not been defined. We show here that stimulation of naive CD8 T cells with a high level of Ag (signal 1) in the presence of costimulation can lead to strong in vitro proliferation and in vivo clonal expansion, but the cells fail to develop effector function unless signal 3 in the form of IL-12 is also provided. Furthermore, cells that initially expand in the absence of signal 3 are rendered tolerant, in that they fail to develop effector function upon rechallenge with a stimulus that is normally fully activating for naive cells. Thus, CD8 T cells can undergo substantial clonal expansion in response to Ag and costimulation, but have profoundly different fates depending on whether or not they receive the third signal.
Materials and Methods
Mice, Cell Lines, and Reagents.
OT-I mice having a transgenic TCR specific for H-2Kb and OVA257–264 were a gift from Dr. F. Carbone (University of Melbourne, Melbourne, Australia). OT-I mice were also crossed with Thy1-congenic B6.PL-Thy1a/Cy (Thy1.1) mice (Jackson ImmunoResearch Laboratories) and bred to homozygosity. The OT-I and OT-I/PL breeding colonies were maintained under specific pathogen-free conditions at the University of Minnesota. C57BL/6NCr mice were purchased from the National Cancer Institute. E.G7 tumor cells (EL-4 thymoma transfected with ovalbumin) were used as targets in in vitro cytolysis assays, and EL-4 cells were used as controls for specificity. All directly conjugated fluorescent antibodies were purchased from BD Biosciences or eBioscience. 25-D1.16 mAb that recognizes the complex of H-2Kb with OVA257–264 (43) was a gift from Dr. R. Germain (National Institutes of Health, Bethesda, MD). Binding of biotinylated 25-D1.16 was detected with streptavidin-PE (Jackson ImmunoResearch Laboratories).
Naive T Cell Purification.
Inguinal, axillary, brachial, cervical, and mesenteric LN were harvested from OT-I or OT-I/PL mice, pooled, and disrupted to obtain a single cell suspension, and passed over Cellect CD8 enrichment columns (Cedarlane Laboratories Limited). Cells were stained with anti–CD44-FITC and anti–CD8-PE mAbs and sorted using a FACSVantage™ flow cytometer (BD Biosciences). This yielded a population of naive CD8+ cells that were >99% CD8+ and >98% CD44low. Alternatively, OT-I/PL LN cells were enriched for CD8+ CD44low cells by negative selection using MACS magnetic cells sorting (Miltenyi Biotec). In brief, cells were coated with FITC-labeled antibodies specific for CD4, B220, I-Ab, CD11c, and CD44. Anti-FITC magnetic MicroBeads (Miltenyi Biotech) were added to the cells, which were passed over separation columns attached to the MACS magnet. The cells that did not bind to the column were collected and were >95% CD8+ and <0.5% CD44high.
In Vitro Proliferation and Cytotoxicity Assays.
A total of 5 × 104 purified CD8+ T cells and 2 × 105 Ag-coated latex microspheres were placed in flat-bottom microtiter wells in 200 μl RPMI 1640 medium supplemented with 10% FCS, 4 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin and streptomycin, 10 mM HEPES, and 5 μM 2-ME (RP-10). Where indicated, cultures were supplemented with 2.5 U/ml human rIL-2 (TECIN™; National Cancer Institute, Biological Resources Branch) and 2 U/ml murine rIL-12 (Genetics Institute). Proliferation was measured after 2 d by the addition of 1 μCi [3H]TdR per well for the last 8 h of culture. Triplicate determinations were done, and SDs are shown. Alternatively, cell recovery after 3 d of culture was used as the measure of proliferation, and is expressed as the fold increase in cells over the input number of cells. Cytolytic activity was determined in triplicate in a standard 51Cr release assay using E.G7 cells as targets, with El-4 cells included as a control for specificity. Results are expressed as percent-specific release, and error bars show standard deviations of triplicate values. Effectors and targets were incubated together for 4 h when effectors were generated in in vitro cultures; effectors were incubated with targets for 6 h when effectors were generated by in vivo stimulation of adoptively transferred OT-I/PL cells. In all figures, the effector to target ratio is expressed as the number of OT-I T cells in the effector population relative to the number of targets.
Antigens and B7-1 on Microspheres.
Methods for immobilizing MHC antigens and costimulatory ligands on 5-μM-diameter latex microspheres have been described previously in detail (44). Peptide–MHC complexes were a gift from M. Daniels and S. Jameson (University of Minnesota, Minneapolis, MN). Complexes were prepared using recombinant H-2Kb with the BirA recognition sequence at the carboxy terminus and human β2m that were refolded in the presence of OVA257–264 and biotinylated (45–47). Avidin was immobilized on 5-μM latex microspheres using 0.25 μg/107 beads, and the biotinylated H-2Kb–OVA257–264 complexes were allowed to bind for 1 h at 4°C. In some experiments, microspheres were coated with DimerX H-2Kb:Ig fusion protein (BD Biosciences) using 2.5 μg DimerX H-2Kb:Ig/107 latex microspheres. Peptide was loaded onto the H-2Kb portion of the fusion protein by incubating the coated microspheres with 0.1 μM OVA257–264 for 2 h at 37°C, followed by extensive washing to remove free peptide. When used, B7-1 in the form of a recombinant mouse B7-1/Fc chimera (R&D Systems) was coimmobilized on the microspheres along with Ag using 0.6 μg/107 microspheres (high B7) or 0.15 μg/107 microspheres (low B7). For all microsphere preparations, immobilization of proteins was verified by staining with fluorescent antibodies and analysis with flow cytometry.
Adoptive Transfer and Immunization of OT-I/PL Transgenic Cells.
Pooled LNs from OT-I/PL mice were disrupted to yield single cell suspensions and washed with PBS. Before transfer, the cells were analyzed by flow cytometry to determine the percentage of CD8+ cells. Their CD25, CD69, and CD44 phenotypes were determined to confirm that the cells that were transferred were not activated. A total of 1.5–3 × 106 CD8+ cells in 0.3 ml PBS were transferred via tail vein injection into age- and sex-matched naive 6–8-wk-old C57BL/6 recipients. Recipient mice were rested for 24 h before immunization. In some experiments, OT-I/PL LN cells were labeled with CFSE before transfer to C57BL/6 mice. LN cells were resuspended to 20 × 106/ml in HBSS and warmed in a 37°C water bath for 10 min before adding an equal volume of 6 μM CFSE in HBSS and incubating for an additional 5 min at 37°C. Cells were washed with ice-cold RP-10 (to stop the reaction) and washed 2× with ice-cold PBS before adoptive transfer. For immunization, the OVA257–264 synthetic peptide (SIINFEKL; Research Genetics) was dissolved in PBS and injected via the tail vein in a volume of 0.2 ml. Where indicated, animals received 1 μg of recombinant murine IL-12 (Genetics Institute) or 50 μg LPS in the same injection.
Flow Cytometric Analysis of Transferred Cells.
Mice were killed at the indicated times after adoptive transfer and in vivo challenge. Spleen cells and LN cells (pooled from axillary, brachial, cervical, inguinal, and mesenteric nodes) were counted by trypan blue dye exclusion to determine total viable cell counts, and were stained with the antibodies CD8 and Thy 1.1 to detect the transferred OT-I/PL cells. Stained cells were analyzed on a FACSCalibur™ flow cytometer using CELLQuest™ software (BD Biosciences) to determine the percent and total OT-I/PL cells in the transferred mice. When OT-I/PL cells were labeled with CFSE before adoptive transfer, analysis of cell division as indicated by dilution of CFSE fluorescence was done using ModFit software (BD Biosciences).
Intracellular Cytokine Staining After In Vitro Rechallenge.
Spleen and LN cells harvested from adoptively transferred mice were incubated at a concentration of 2 × 106 cells/ml in RP-10 with 0.2 μM OVA257–264. After 1 h of incubation at 37°C, 0.6 μl/ml of the monensin-containing solution GolgiStop (BD Biosciences) was added, and cultures were incubated for an additional 4–5 h at 37°C. Cells were washed and stained with the antibodies CD8 and Thy 1.1 to mark the OT-I/PL cells. Cells were fixed in Cytofix buffer (BD Biosciences) for 15 min at 4°C, and permeabilized in saponin-containing Perm/Wash buffer (BD Biosciences) for 15 min at 4°C before staining with APC-conjugated antibody to IFN-γ for 30 min at 4°C. Cells were washed once with Perm/Wash buffer and once with PBS containing 2% FBS. Stained cells were analyzed on a FACSCalibur™ flow cytometer using CELLQuest™ software.
In Vivo Killing Assay.
The assay was performed essentially as described previously by Mueller et al. (48). In brief, a single cell suspension of C57BL/6 spleen cells was divided into two. One sample was pulsed with 0.2 μM OVA257–264 for 45 min at 37°C, washed extensively, and incubated with a high concentration of CFSE (final concentration of 5 μM). The other population was incubated without peptide for 45 min at 37°C, washed, and labeled with a low concentration of CFSE (final concentration of 0.5 μM). Cells were resuspended at 108/ml and equal volumes of the peptide-pulsed, CFSEhigh cells and unpulsed, CFSElow cells were mixed together, and a total of 2 × 107 CFSE-labeled spleen cells were transferred by tail vein injection into mice previously injected with OT-I/PL cells and immunized as indicated. After 3 h, mice were killed and spleen cells were analyzed by flow cytometry to detect the CFSE-labeled target cells. The two target populations were distinguished by their different CFSE fluorescence intensities. The percent-specific lysis was determined by the following formula: Ratio = (percentage CFSElow/percentage CFSEhigh); Percent-specific lysis = [1 − (ratio unprimed/ratio primed) × 100]. The spleen cells from the in vivo killing assays were also stained with antibodies to CD8 and Thy 1.1 to determine the number of OT-I/PL cells in each animal.
Results
In Vitro Activation with High Signal 1 and Signal 2 Stimulates Proliferation but Not Lytic Function.
Artificial APCs in the form of microspheres having Ag and other ligands on the surface provide a means of defining the minimal requirements for activating T cells. We had shown previously that stimulation of naive CD8 T cells with native class I Ag on microspheres at physiological densities required addition of both IL-2 and IL-12 for proliferation and development of cytotoxic function to occur (33). When the B7-1 ligand was included on the microspheres to provide costimulation, the responses still depended on the addition of IL-12. We have now found that the requirement for a third signal for proliferation can be overcome when TCR engagement is very high (i.e., at high levels of signal 1), but that development of effector function remains dependent on the third signal. This was first observed in experiments using immobilized anti-TCR mAb to provide signal 1 (unpublished data), but can also be seen when very high Ag surface densities are achieved by using immobilized recombinant H-2Kb that has been refolded in the presence of OVA257–264 peptide so that the majority of the class I proteins have the relevant peptide Ag bound.
When microspheres were coated at a low density, using the refolded complex at 0.001 μg/106 beads, and used to stimulate naive OT-I CD8 T cells specific for the complex (49), proliferation (Fig. 1 A) and killing (Fig. 1 B) only occurred if both IL-2 and IL-12 were added. This low density is similar to the density of the Ag complex present on OVA-transfected E.G7 cells, based on staining with the 25-D1.6 mAb (43) that recognizes the Kb–OVA257–264 complex (unpublished data). When a 100-fold higher density of Ag complex was used, naive OT-I cells proliferated vigorously when just IL-2 was added, and proliferation was not further increased upon addition of IL-12 (Fig. 1 A). However, despite this vigorous proliferative response in the absence of IL-12, the cells failed to develop lytic effector function unless IL-12 was added (Fig. 1 C). This was the case at all times examined; stimulation with Ag and IL-12 results in no lytic activity at 24 h, weak activity at 48 h, maximal activity at 72 h, and declining activity at 96 h (unpublished data), whereas cells resulting from stimulation with just Ag and IL-2 have no cytolytic activity at any of these times (unpublished data). Thus, at high levels of TCR engagement, optimal proliferation requires only IL-2, but development of cytolytic function requires IL-12 additionally; whereas at low TCR engagement levels, both cytokines are required for proliferation as well as differentiation.
Figure 1.
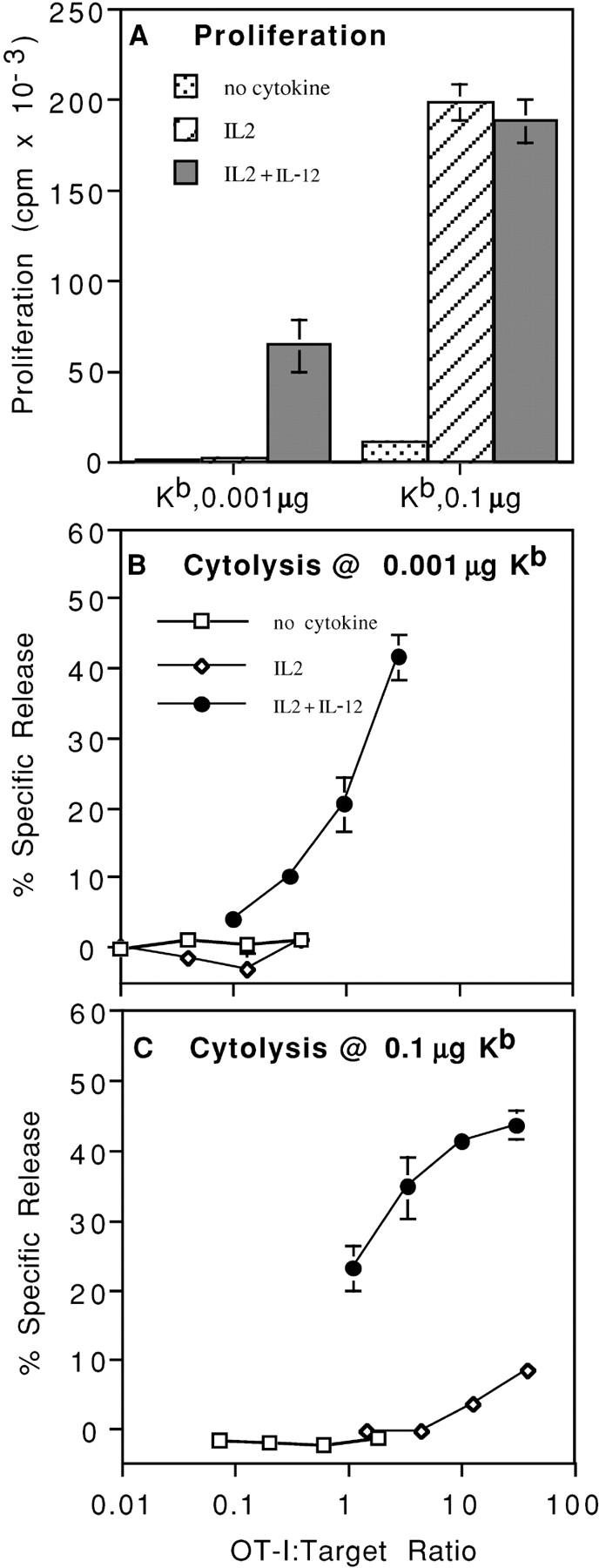
IL-12 is required for both proliferation and development of effector function at low Ag levels, but only for development of effector function at high Ag levels. Microspheres were coated with either 0.1 or 0.001 μg Kb/OVA/106 microspheres. The Kb–OVA complex was prepared by refolding recombinant heavy chain in the presence of peptide and β2m, as described in Materials and Methods. (A) CD44low CD8+ OT-I cells were stimulated with microspheres in the presence of the indicated cytokines and proliferation was determined on day 2 by measuring incorporation of [3H]TdR. (B) CD44low CD8+ OT-I cells were stimulated with microspheres made using 0.001 μg Kb/OVA per 106 microspheres in cultures with the indicated cytokines. Cells were harvested on day 3 and a 4-h 51Cr release assay was done using E.G7 cells as targets. (C) As described in B, but using 0.1 μg Kb/OVA per 106 microspheres.
TCR engagement by Ag on APCs is normally accompanied by costimulation provided by B7 ligands on the APCs. To determine whether CD28 engagement might circumvent the requirement for IL-12 in induction of cytolysis, microspheres having Kb–OVA257–264 complex coimmobilized with B7-1 were prepared (50) and used to stimulate naive OT-I T cells. Effective costimulation was apparent in that proliferation and clonal expansion occurred without addition of exogenous IL-2 when B7-1 was included on the microspheres (Fig. 2 A). Nevertheless, development of cytolytic function only occurred when IL-12 was available, regardless of the amount of IL-2 added (Fig. 2 B). Thus, development of cytolytic function remains dependent on the third signal provided by IL-12 even when signal 2 is provided by CD28 binding to B7-1.
Figure 2.

Costimulation does not replace a requirement for IL-12 for the induction of cytolytic function. Microspheres were coated with DimerX H-2Kb:Ig either alone or along with B7.1-Ig at low or high concentrations, and pulsed with OVA257–264 as described in Materials and Methods. These microspheres were used to stimulate MACS column-purified CD44low CD8+ OT-I cells in the presence of the indicated cyto-kines. (A) Proliferation was determined by counting the number of viable cells after 3 d of culture and expressed as the fold increase over input cell number. (B) Cells were assayed for lytic activity on day 3 using a 4-h 51Cr release assay using E.G7 cells as targets. Results are expressed as percent-specific lysis at various effector/target cell ratios.
In Vivo Activation with High Ag Dose Stimulates Proliferation but Not Lytic Effector Function.
There are a number of works describing CD8 T cell populations that have expanded in vivo but lack effector function (32, 38–42). To determine if a high level of signal 1 might stimulate in vivo clonal expansion without leading to development of effector function, experiments were done using adoptive transfer of OT-I T cells into normal C57BL/6 recipients to allow quantitation of the Ag-specific T cell response in a relatively normal immune system (51). OT-I cells were adoptively transferred by intravenous (tail vein) injection and allowed to equilibrate in the recipient's immune system for 1 d. Mice were challenged with varying doses of OVA257–264 peptide alone, or along with IL-12. 3 d later, at the peak of the response (34), the numbers of OT-I cells in the LN and spleen were determined and cytotoxicity was measured for the populations. At low peptide concentrations, clonal expansion was minimal in the absence of IL-12, but was at maximal levels when IL-12 was coadministered (Fig. 3 A). Clonal expansion in response to peptide in the absence of IL-12 increased in a dose-dependent manner, whereas increasing the peptide dose above 2 μg in the presence of IL-12 did not increase the number of OT-I T cells. Very similar results were obtained when peptide was administered along with LPS as an adjuvant (Fig. 3 B).
Figure 3.

In vivo activation of CD8+ cells with high Ag dose stimulates proliferation but not lytic effector function. (A) C57BL/6 mice received OT-I/PL LN cells by adoptive transfer on day −1 and were challenged on day 0 with the indicated amounts of OVA257–264 peptide either alone or with 1 μg/ mouse of IL-12. Spleens were harvested on day 4, and the number of OT-I/PL cells was determined by flow cytometry analysis as described in Materials and Methods. The values shown are averages of duplicate mice and the error bars represent the ranges. (B) C57BL/6 mice received OT-I/PL LN cells by adoptive transfer on day −1 and were challenged on day 0 with the indicated amounts of OVA257–264 peptide either alone or with 50 μg/mouse of LPS. Spleens were harvested on day 3, and the number of OT-I/PL cells was determined by flow cytometry. The values shown are averages of duplicate mice and the error bars represent the ranges. (C) Spleen cells from the animals described in A that received 10 μg of peptide, with and without IL-12, were assayed for lytic activity against 51Cr-labeled E.G7 targets at spleen cell/target ratios of 200, 100, 50, and 25:1. These ratios were converted to OT-I/PL target ratios by multiplying by the percent OT-I/PL cells in each spleen cell population. The results shown are for one of the two animals in each treatment group; splenocytes from the other animal in each group showed essentially identical lytic activity. (D) C57BL/6 mice received OT-I/PL LN cells by adoptive transfer on day −1 and were left unchallenged (transfer only) or challenged with 10 μg OVA257–264/mouse alone (peptide only) or with 1 μg/mouse of IL-12 (peptide + IL-12). On day 3, mice were injected with equal numbers of unpulsed and OVA257–264-pulsed C57BL/6 spleen cells that were labeled with low and high concentrations of CFSE, respectively. Spleens were harvested after 3 h and the cells were analyzed for the preferential loss of peptide-pulsed, CFSEhigh versus unpulsed, CFSElow target cells. Histograms show the CFSEhigh and CFSElow cells from one of two identically treated mice; the percent lysis, calculated as described in Materials and Methods, is indicated.
In the same experiment, cytolytic activity of CD8 T cells from spleens of adoptive transfer recipients challenged with peptide Ag and IL-12 could be detected directly ex vivo in a standard 51Cr release assay (Fig. 3 C). In the experiment shown in Fig. 3 C, the effector/target ratio is expressed as the number of OT-I cells present in the population. Despite large clonal expansion of OT-I cells in response to just peptide Ag, no cytolytic activity could be measured for these populations (Fig. 3 C). The cytolytic activities induced in OT-I cells by peptide or peptide and IL-12 were more directly determined using an in vivo assay of specific killing. C57BL/6 spleen cells, unpulsed or pulsed with OVA257–264 peptide, were labeled with different levels of CFSE fluorescent dye and injected into mice 3 d after challenge. 3 h later, spleen cells were harvested and flow cytometry was done to determine the numbers of cells having low (unpulsed) and high (pulsed) levels of CFSE. The ratio of these two populations in mice not challenged with Ag defines the 0% lysis level, and the ratios in the challenged mice can be converted to percent-specific lysis based on this. Consistent with the results of the ex vivo 51Cr release assay, minimal specific lysis of peptide-pulsed targets occurred in mice that received just peptide, whereas the majority of the peptide-pulsed targets were specifically eliminated in the mice that received peptide and IL-12 (Fig. 3 D). In this experiment, the total number of OT-I cells in the spleens of the unchallenged mice (transfer only) was 0.3 × 105, 2.4 × 105 in mice that received just peptide, and 10.2 × 105 in the mice challenged with peptide and IL-12. Thus, as in the in vitro experiments, high signal 1 levels in vivo can stimulate extensive clonal expansion, but the cells fail to develop lytic effector function.
Although high doses of peptide alone stimulate a large increase in the number of Ag-specific CD8 T cells, the extent of clonal expansion in the spleen and LN was not as great as when IL-12 or LPS adjuvant were administered (Fig. 3, A and B), at least over the peptide dose range that was examined. This raised the possibility that cells might undergo fewer rounds of division when stimulated with just Ag, and that failure to develop cytotoxic activity might be related to this. This was examined by labeling OT-I cells with the fluorescent dye CFSE before adoptive transfer and challenge. As the cells divide in vivo the dye is diluted twofold with each division, allowing a determination of the number of rounds of division that the cells in the population have undergone.
When CFSE-labeled OT-I cells were adoptively transferred and the recipients left unchallenged, CFSE levels remained high 3 d later (Fig. 4 A), which is consistent with an absence of clonal expansion of the cells. In contrast, challenge with either peptide alone (Fig. 4 B) or peptide and IL-12 (Fig. 4 C) resulted in extensive dilution of the dye 3 d later as a result of the clonal expansion that occurs in response to Ag. The extent of CFSE dilution was essentially the same in response to peptide in the presence or absence of IL-12, indicating that the cells had undergone a comparable number of rounds of division, even though challenge with peptide alone resulted in somewhat less clonal expansion and no cytolytic function (unpublished data).
Figure 4.

OT-I/PL cells from mice immunized with peptide alone or peptide and IL-12 undergo comparable numbers of divisions. C57BL/6 mice received CFSE-labeled OT-I/PL LN cells by adoptive transfer on day −1 and were left unchallenged or challenged with 2 μg OVA257–264/mouse either alone or with 1 μg/mouse of IL-12 on day 0. On days 2 and 3, LN cells were analyzed by flow cytometry to determine the number of OT-I/PL cells in each animal, and the amount of cell division by OT-I/PL cells was determined by analyzing CFSE fluorescence using ModFit software. (A–C) CFSE fluorescence of LN cells from mice immunized as indicated, gated on CD8+ Thy1.1+ populations. The open histograms are the CFSE profiles, and the shaded histograms represent the subpopulations of cells that have undergone the same numbers of divisions as determined by deconvolution of the CFSE profiles using ModFit software. The fluorescence intensity of the endogenous cells was about twofold lower than that of the CFSE-labeled cells (not depicted), demonstrating that the OT-I cells being examined have not become CFSE negative. (D) Comparison of the number of OT-I/PL cells recovered on days 2 and 3. (E) Comparison of the average number of cell divisions, calculated using ModFit software, on days 2 and 3 after immunization.
This was examined in more detail by comparing the numbers of OT-I cells present on days 2 and 3 of the response, and the average number of divisions that the cells had undergone. 2 d after challenge, the OT-I cells had expanded comparably to peptide with or without IL-12 (Fig. 4 D). By day 3, there was little further increase in OT-I cell numbers in the mice that received just peptide, whereas OT-I cells numbers increased substantially between days 2 and 3 in mice that received peptide and IL-12 (Fig. 4 D). Despite the difference in numbers of cells, CFSE dye dilution indicated that cells in both populations had undergone the same average number of divisions on both days 2 and 3 (Fig. 4 E). Thus, although the OT-I cells in mice that received just peptide continued to proliferate between days 2 and 3, this was not reflected in an increased number of cells. These results suggest that IL-12 may provide a survival advantage to the responding cells to allow continued clonal expansion between days 2 and 3. The results also demonstrate that the difference in cytolytic activity between the two populations (Fig. 3 and unpublished data) is not due to the cells that expand in response to just peptide having undergone fewer rounds of division.
Production of IFN-γ by CD8 T Cells Stimulated In Vivo in the Absence and Presence of IL-12.
Production of IFN-γ is an important effector function of CD8 T cells. Therefore, we examined the ability of OT-I cells stimulated by in vivo administration of peptide in the absence or presence of IL-12 to perform this function. Mice bearing adoptively transferred OT-I cells were challenged; 3 d later, cells in the LN and spleen were examined to determine the number of OT-I cells present, their cytolytic activity, and their ability to produce IFN-γ upon in vitro restimulation with peptide. IFN-γ production was determined by intracellular staining with an IFN-γ–specific mAb 5–6 h after in vitro restimulation with peptide.
A small number of OT-I cells from unchallenged mice produced IFN-γ (Fig. 5 A) consistent with a recent study demonstrating that naive human CD8 T cells are capable of rapid IFN-γ production in response to Ag (52). Challenge with peptide alone resulted in about a twofold increase in the fraction of OT-I cells that produced IFN-γ, whereas challenge with peptide and IL-12 caused a three- to fivefold increase (Fig. 5 A). As expected, the number of OT-I cells increased substantially in mice challenged with peptide, and clonal expansion was greater when the mice also received IL-12 (Fig. 5 B). The clonal expansion that occurs in response to just peptide, together with the increase in the fraction of cells that can make IFN-γ (Fig. 5 A), results in a substantial increase in the number of Ag-specific T cells capable of carrying out this function, in comparison to mice that have not been challenged (Fig. 5 B). Thus, although Ag in the absence of a third signal does not induce cytolytic effector function (Fig. 5 C), it does result in a significant increase in the number of Ag-specific cells that appear to be functionally competent with respect to IFN-γ production.
Figure 5.
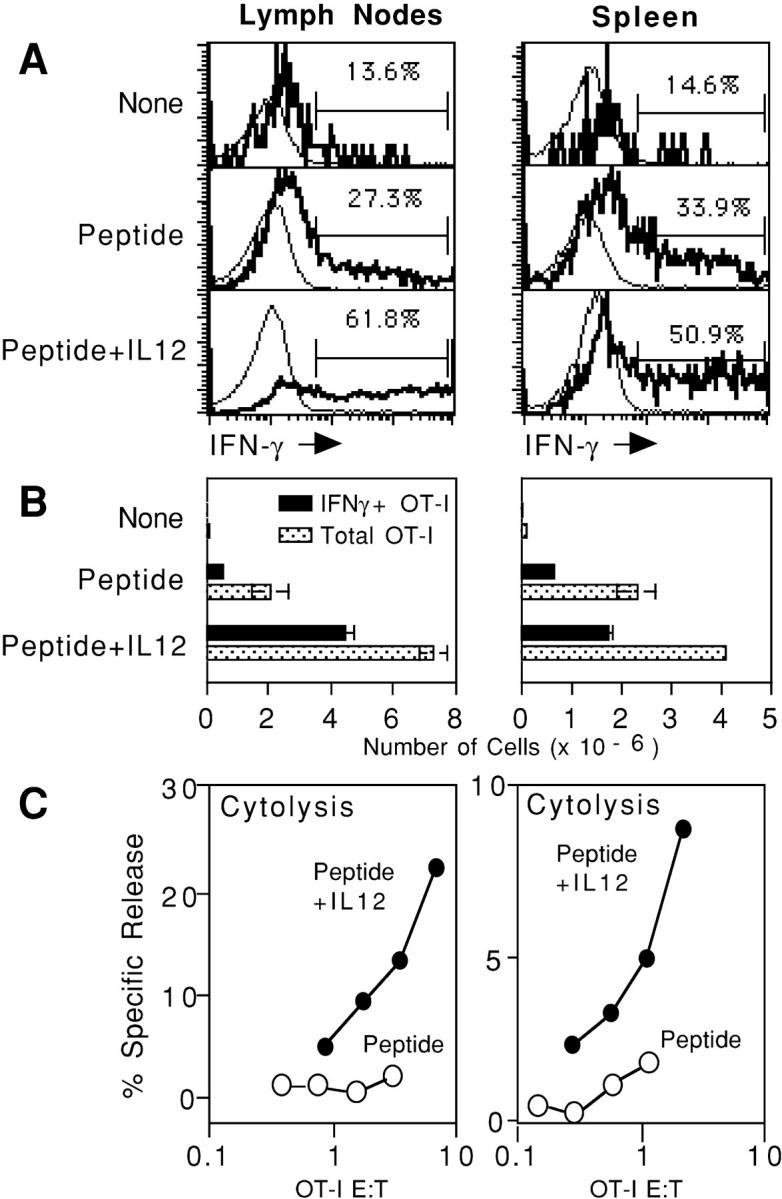
CD8+ T cells stimulated by peptide in the absence or presence of IL-12 develop the ability to produce the effector cytokine IFN-γ. C57BL/6 mice received OT-I/PL LN cells by adoptive transfer on day −1 and were left unchallenged or challenged with 10 μg OVA257–264/mouse either alone or with 1 μg/mouse of IL-12 on day 0. (A) On day 3, LN and spleen cells were harvested and incubated in vitro with peptide; cells were fixed, permeabilized, and stained to detect IFN-γ production as described previously in Materials and Methods. Histograms are gated on endogenous CD8+ (Thy1.1-negative lymphocytes [thin line]) or gated on transgenic CD8+ (Thy1.1-positive lymphocytes [bold line]). The percentage of OT-I/PL cells expressing IFN-γ is shown. Results shown are for one of two animals in each group; results were essentially the same for the other animal in each group. (B) On day 3, LN and spleen cells were analyzed by flow cytometry to determine the number of OT-I/PL cells in each animal, and the total number of IFN-γ–producing OT-I/PL cells was calculated by multiplying the percent IFN-γ+ by the total number of OT-I/PL cells. Results shown are the average from two animals; error bars represent the range. (C) On day 3, LN and spleen cells were assayed for lytic activity against 51Cr-labeled E.G7 targets at LN or spleen cell:target cell ratios of 100, 50, 25, and 12.5:1. These ratios were converted to OT-I/PL target ratios by multiplying by the percent OT-I/PL cells in each LN or spleen cell population. The results shown are for one of the two animals in each treatment group, the other animal in each group showed essentially identical lytic activity.
CD8 T Cells That Expand In Vivo in Response to High Signal 1 without Signal 3 Are Tolerant.
CD8 T cells that respond to high levels of Ag alone undergo substantial clonal expansion but do not develop cytolytic effector function (Fig. 3). To determine if these cells could be subsequently restimulated to develop effector function, mice adoptively transferred with OT-I cells were challenged with either peptide alone or peptide and IL-12. 28 d later, the mice were rechallenged with peptide and LPS adjuvant, and the number and cytolytic activity of OT-I cells in the spleen was determined 3 d later. OT-I cells were detectable in the previously primed mice at day 28 without rechallenge, and expressed high CD44 levels indicating that they had responded to the initial challenge. Clonal expansion in the LN and spleen occurred upon rechallenge with peptide and LPS when the initial challenge was with either peptide alone or peptide and IL-12, but was greater when IL-12 was present during the primary stimulation (Fig. 6, A and B) . In a total of four experiments, cells present in LN at ∼30 d after priming with peptide alone expanded 8.6 ± 5.8-fold upon rechallenge. In every experiment, greater expansion occurred for the population resulting from priming with peptide and IL-12, averaging 58.7 ± 25.6-fold expansion. Similar results were obtained when OT-I cells from the spleens were examined. Thus, cells that respond initially to Ag in the presence of IL-12 have greater capacity to expand upon secondary challenge.
Figure 6.

CD8+ T cells that expand in vivo in response to peptide in the absence of IL-12 are unable to develop lytic effector function after secondary challenge with peptide and adjuvant. C57BL/6 mice received 3 × 106 OT-I/PL LN cells by adoptive transfer on day −1 and were challenged on day 0 with 10 μg OVA257–264 either alone or with 1 μg/mouse of IL-12. After 40 d, mice were left unchallenged, or were rechallenged with 10 μg OVA257–264 with 50 μg LPS. (A and B) After an additional 3 d, LN and spleen cells were analyzed by flow cytometry to determine the number of OT-I/PL cells in each mouse. Results shown for mice that did not receive a secondary challenge are the average of two animals; error bars represent the range. Results for mice that were rechallenged with peptide and LPS represent the average of four (primary with peptide) or three (primary with peptide + IL-12) animals; SDs are shown. (C and D) LN and spleen cells from animals that were rechallenged with peptide and LPS were assayed for lytic activity against 51Cr-labeled E.G7 targets at total viable cell:target ratios of 100, 50, 25, and 12.5:1. These ratios were converted to OT-I/PL ratios based on the percentage of OT-I/PL cells in each population. Results for the individual mice in each group are shown. LN and spleen cells from mice that did not receive a secondary challenge with peptide and LPS had no lytic activity at this time, whether the primary challenge was with peptide alone or peptide and IL-12 (not depicted). The results shown are from one of four essentially identical experiments.
Even greater differences in these populations are seen with respect to the ability of the cells to acquire functional activity. Cells in the LN and spleen from mice primed initially in the presence of Ag and IL-12 developed potent cytolytic activity in the secondary response, whereas those that had responded initially to peptide only did not (Fig. 6, C and D). In three additional independent experiments examining cytolytic activity after initial priming with 10 μg of peptide, all mice (n = 8) primed with peptide alone had no lytic activity, and in one experiment where initial priming was with 2 μg of peptide, the mice (n = 4) primed with peptide alone had no lytic activity. Cells primed with peptide alone also had a reduced capacity to produce the effector cytokine IFN-γ; fewer of the cells produced IFN-γ upon restimulation (Fig. 7, A and B) and those that did, produced less (Fig. 7, C and D). Thus, cells that make an initial response to Ag in the absence of a third signal undergo only weak clonal expansion upon strong secondary stimulation, are defective in IFN-γ production, and fail to develop cytolytic activity; i.e., they are functionally tolerant.
Figure 7.

CD8+ T cells that expand in vivo in response to peptide in the absence of IL-12 have reduced ability to produce IFN-γ after secondary challenge with peptide and adjuvant. LN and spleen cells from the mice described in Fig. 6 that were rechallenged with peptide and LPS were incubated in vitro with OVA257-264 peptide for 5 h; cells were fixed, permeabilized, and stained to detect IFN-γ production as described in Materials and Methods. (A and B) The percentage of OT-I cells in the LN and spleen that were producing IFN-γ is shown. (C and D) The geometric mean fluorescence intensity of IFN-γ staining in the IFN-γ + OT-I cells is shown. Results in A–D are the averages of four (primary with peptide) and three (primary with peptide + IL-12) mice; SDs are shown.
Discussion
The initial suggestion that naive CD8 T cells require a “third signal” in addition to Ag (signal 1) and costimulation (signal 2) came from studies using artificial APC, microspheres coated with class I MHC–peptide complexes, and B7 ligand. Memory CD8 T cells from TCR transgenic mice proliferated vigorously and developed cytolytic effector function in response to the artificial APC, whereas naive cells expressing the same TCR failed to respond unless IL-12 was provided (33). Consistent with these in vitro results, in vivo studies examining responses of adoptively transferred TCR transgenic CD8 T cells have shown that IL-12 could act as a potent adjuvant (34). Administration of peptide Ag alone resulted in only weak clonal expansion and failure to develop a memory population. In contrast, peptide Ag administered with either complete Freund's adjuvant or IL-12 stimulated strong clonal expansion, development of lytic effector function, and establishment of a responsive memory population. IL-12 had this effect even when the adoptive transfer recipients lacked the IL-12 receptor, indicating that IL-12 was acting directly on the transferred CD8 T cells (30).
These initial studies used relatively low levels of Ag; i.e., levels in vitro that were similar to those on Ag-expressing cell lines and levels in vivo that gave optimal responses when delivered with adjuvant. However, as shown here, high levels of Ag, as might be reached during a virus infection, can stimulate strong proliferation in the absence of IL-12 both in vitro (Figs. 1 and 2) and in vivo (Figs. 3 and 4). In other work, we have found that IL-12 stimulates higher and more prolonged expression of CD25 on the cells than can be achieved with just Ag and B7 costimulation, thus rendering the cells more responsive to low levels of IL-2 (53). This contribution to the proliferative response becomes less important at high signal 1 levels (Figs. 1–4), perhaps as a result of increased IL-2 production under these conditions. IL-12 also appears to increase clonal expansion by enhancing survival of the proliferating cells. At high Ag levels in vivo, clonal expansion in the absence or presence of IL-12 is comparable on day 2 as measured by either the number of cells recovered or the number of divisions the cells have undergone (Fig. 4, D and E). By day 3, the cells have divided further, and the average number of divisions remains the same in the absence or presence of IL-12 (Fig. 4 E). However, by day 3, the number of OT-I T cells recovered is markedly less in the absence of IL-12 (Fig. 4 D), suggesting that survival is impaired. Marrack and colleagues have shown that adjuvants increase the survival of activated T cells (54), and increase the expression of Bcl-3 (55), a member of IkB family that can promote T cell survival. Thus, although high levels of Ag can stimulate comparable cell division in the presence or absence of a third signal, it appears that the extent of clonal expansion remains somewhat limited unless a third signal is available to promote survival (Figs. 3 and 4).
Although high levels of Ag can largely overcome the need for a third signal for proliferation of naive CD8 T cells, development of cytolytic effector function remains completely dependent on the third signal both in vitro (Figs. 1 and 2) and in vivo (Fig. 3, C and D). Development of cytolytic function does not require multiple rounds of division; CD8 cells that have undergone as few as one or two divisions can become cytolytic (56). In the experiments shown here, failure to develop cytolytic function in the absence of signal 3 is clearly not due to the cells having undergone fewer divisions. Thus, proliferation and cytolytic function are completely uncoupled in the absence of a third signal.
Although naive CD8 T cells that are stimulated to divide in the absence of signal 3 fail to develop cytolytic effector function, some of the cells do acquire the ability to produce IFN-γ. A small fraction of naive CD8 T cells rapidly produce small amounts of IFN-γ upon stimulation with Ag (Fig. 5 A), as has recently been reported for naive human CD8 T cells (52). After expansion, in response to just peptide, a substantially greater fraction produces IFN-γ upon Ag restimulation, and this is further increased when the primary stimulation is done in the presence of IL-12 (Fig. 5 A). Thus, unlike cytolytic function, production of IFN-γ is not completely dependent on a third signal. Using tetramer staining, HIV-specific CD8 cells that are able to produce cytokines, including IFN-γ, but that lack cytolytic function have been characterized in peripheral blood in chronic HIV infection (38).
The substantial clonal expansion that can occur in the absence of adjuvant/signal 3, together with the significant increase in the fraction of cells that can produce IFN-γ, results in a large increase in the number of cells that can carry out this effector function. Thus, CD8 T cells that have responded to Ag in the absence of a third signal, although lacking cytolytic effector function, have the potential to make some contribution to development of an immune response. Whether this is important will likely depend on the response being studied. These observations raise an important caution with respect to studies that examine IFN-γ production by CD8 T cells as a measure of their functional activation; it cannot be assumed that a responding population that is capable of IFN-γ production has also developed cytolytic activity.
CD8 T cells that expand in response to high Ag levels in the absence of signal 3 persist in low but detectable numbers. However, these persisting cells are not responsive to even a potent stimulation with Ag and adjuvant. Thus, upon restimulation with peptide and adjuvant, they undergo less clonal expansion and fail to acquire lytic effector function (Fig. 6). In contrast, cells that were previously stimulated in the presence of signal 3 undergo strong secondary expansion and reacquire cytolytic function in response to the same secondary stimulus. Thus, it appears that cells that make an initial response to Ag in the absence of signal 3 are rendered permanently tolerant.
Expanded Ag-specific CD8 T cell populations that lack function, and are thus tolerant, have been described previously in a number of experimental systems (32, 38–42), but in most cases the basis for the acquisition of this phenotype has not been defined. We would suggest that in many instances this results from cross-presentation of Ag to naive CD8 T cells in a noninflammatory environment so that they proliferate in response to signals 1 and 2, but fail to receive the third signal needed to develop function and avoid tolerance. In fact, Sherman and colleagues have recently reported results (32, 57) that complement our previous results (30, 33, 34) and the findings discussed here. Their studies examined the responses of Clone 4 CD8 T cells with a transgenic TCR specific for hemagglutinin upon adoptive transfer into mice that express hemagglutinin in the β cells of the pancreatic islets. In this model, Ag is cross-presented by APC in the LN draining the pancreas, and the Clone 4 cells respond by proliferating at this site, but fail to develop effector functions. Treatment of the mice with anti-CD40 mAb to activate DC resulted in enhanced proliferation of the Clone 4 cells, as well as an increase in the levels of B7.1 and B7.2 on DC in the draining LN. However, despite extensive proliferation, the Clone 4 cells still failed to develop cytolytic activity or produce IFN-γ. When mice were treated with both anti-CD40 mAb and IL-12, the Clone 4 cells developed both effector functions. Thus, their results also support a model in which three signals are required to support clonal expansion and development of effector function, and avoidance of tolerance. Our results differ somewhat from theirs because we find that a substantial fraction of the cells proliferating in the absence of signal 3 are able to make IFN-γ upon restimulation (Fig. 5).
The evidence available thus far strongly supports a model in which productive activation of CD8 T cells leading to cytolytic effector function, and avoidance of tolerance, requires that three distinct signals be delivered to the T cell, and that increasing the level of any one of them cannot bypass the need for the other two. This is the case when artificial APC are used in vitro to stimulate the cells (33), and appears to be the case in vivo as well. As described here, even very high levels of Ag fail to support development of lytic effector function, even though the cells proliferate (Fig. 3). Similarly, high levels of B7 also fail to overcome the requirement for the third signal (Fig. 2). This also appears to be the case in vivo because Sherman and colleagues, in the experiments described previously, showed that treatment of mice with anti-CD40 mAb increased levels of B7.1 and B7.2 on DC in the draining LN, but Clone 4 cells still failed to respond unless IL-12 was provided (32). Albert et al. (31) also concluded that a third signal was needed for human CD8 T cells activation when stimulated by DC cross-presentation of Ag in an in vitro system. When mature DC expressing high levels of B7.1 and B7.2 were used, the CD8 T cells failed to develop cytolytic function and were tolerized, but the addition of IL-12 or IL-1β to the cultures resulted in development of effector function. Relatively high concentrations of the cytokines were required, leading the authors to suggest that some alternative, undefined mechanism allows DC to activate the CD8 T cells. However, it would seem very likely that IL-12 and other cytokines produced by DC that are presenting Ag may reach high local concentrations at the surface of the bound T cell. Although IL-12 can clearly provide the third signal, it is not the only thing that can, because peptide Ag and adjuvant elicit strong, productive responses by CD8 T cells adoptively transferred into IL-12–deficient mice (34). In vitro experiments have implicated another soluble factor, in addition to IL-12, that is produced by splenic APCs and can provide the third signal, and we are working to identify this (unpublished data). We have tested a variety of cytokines in vitro that have failed to support development of cytolytic effector function, including IL-1, 2, 4, 7, 10, 15, and 18, and IFN-γ.
Thus, Ag and costimulation-dependent expansion of CD8 T cells that occurs in the absence of an inflammatory response appears to lead to the induction of functional tolerance of the cells, rather than development of effector function and a responsive memory population. This suggests the possibility that whether cross-presentation of Ag by CD8α+ DC will lead to full activation or tolerance (18, 19, 58) will depend on whether the DC have been activated to produce IL-12 and/or an alternate signal 3. The findings discussed here have important implications for vaccine development, and potentially for understanding many situations where CD8 T cells have clearly made a response to Ag but have failed to control the virus or tumor growth.
Acknowledgments
We thank Dr. Marc Jenkins for critical reading of the manuscript.
This work was supported by the National Institutes of Health grants AI 34824 and AI 35296 to M.F. Mescher.
Footnotes
Abbreviation used in this paper: DC, dendritic cells.
References
- 1.Aichele, P., K. Brduscha-Reim, R. Zinkernagel, H. Hengartner, and H. Pircher. 1995. T cell priming versus T cell tolerance induced by synthetic peptides. J. Exp. Med. 182:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, J., I.L. Campbell, G. Morahan, T.E. Mandel, L.C. Harrison, and J.F. Miller. 1988. Diabetes in transgenic mice resulting from over-expression of class I histocompatibility molecules in pancreatic beta cells. Nature. 333:529–533. [DOI] [PubMed] [Google Scholar]
- 3.Burkly, L.C., D. Lo, O. Kanagawa, R.L. Brinster, and R.A. Flavell. 1989. T-cell tolerance by clonal anergy in transgenic mice with nonlymphoid expression of MHC class II I-E. Nature. 342:564–566. [DOI] [PubMed] [Google Scholar]
- 4.Ferber, I., G. Schonrich, J. Schenkel, A. Mellor, G. Hammerling, and B. Arnold. 1994. Levels of peripheral T cell tolerance induced by different doses of tolerogen. Science. 263:674–676. [DOI] [PubMed] [Google Scholar]
- 5.Kyburz, D., P. Aichele, D. Speiser, H. Hengartner, R. Zinkernagel, and H. Pircher. 1993. T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur. J. Immunol. 23:1956–1962. [DOI] [PubMed] [Google Scholar]
- 6.Morahan, G., J. Allison, and J.F.A.P. Miller. 1989. Tolerance of class I histocompatibility antigens expressed extrathymically. Nature. 339:622–624. [DOI] [PubMed] [Google Scholar]
- 7.Moskophidis, D., F. Lechner, H. Pircher, and R. Zinkernagel. 1993. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 362:758–761. [DOI] [PubMed] [Google Scholar]
- 8.Rocha, B., and H. von Boehmer. 1991. Peripheral selection of the T cell repertoire. Science. 251:1225–1228. [DOI] [PubMed] [Google Scholar]
- 9.Webb, S., C. Morris, and J. Sprent. 1990. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 63:1249–1256. [DOI] [PubMed] [Google Scholar]
- 10.Wieties, K., R. Hammer, S. Jones-Youngblood, and J. Forman. 1990. Peripheral tolerance in mice expressing a liver-specific class I molecule: inactivation/deletion of a T cell population. Proc. Natl. Acad. Sci. USA. 87:6604–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirberg, J., L. Bruno, and H. vonBoehmer. 1993. CD4+8− help prevents rapid deletion of CD8+ cells after a transient response to antigen. Eur. J. Immunol. 23:1963–1967. [DOI] [PubMed] [Google Scholar]
- 12.Rocha, B., C. Tanchot, and H. vonBoehmer. 1993. Clonal anergy blocks in vivo growth of mature T cells and can be reversed in the absence of antigen. J. Exp. Med. 177:1517–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubois, P.M., M. Pihlgren, M. Tomkowiak, M. Van Mechelen, and J. Marvel. 1998. Tolerant CD8 T cells induced by multiple injections of peptide antigen show impaired TCR signaling and altered proliferative responses in vitro and in vivo. J. Immunol. 161:5260–5267. [PubMed] [Google Scholar]
- 14.Kearney, E., K. Pape, D. Loh, and M. Jenkins. 1994. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1:327–339. [DOI] [PubMed] [Google Scholar]
- 15.Rellahan, B.L., L.A. Jones, A.M. Kruisbeek, A.M. Fry, and L.A. Matis. 1990. In vivo induction of anergy in peripheral Vβ8+ T cells by staphylococcal enterotoxin B. J. Exp. Med. 172:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanchot, C., S. Guillaume, J. Delon, C. Bourgeois, A. Franzke, A. Sarukhan, A. Trautmann, and B. Rocha. 1998. Modifications of CD8+ T cell function during in vivo memory or tolerance induction. Immunity. 8:581–590. [DOI] [PubMed] [Google Scholar]
- 17.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 19.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 1:126–134. [DOI] [PubMed] [Google Scholar]
- 20.Fazekas de St Groth, B. 1998. The evolution of self-tolerance: a new cell arises to meet the challenge of self-reactivity. Immunol. Today. 19:448–454. [DOI] [PubMed] [Google Scholar]
- 21.Suss, G., and K. Shortman. 1996. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand–induced apoptosis. J. Exp. Med. 183:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 23.Sallusto, F., and A. Lanzavecchia. 1999. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J. Exp. Med. 189:611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aderem, A., and R.J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature. 406:782–787. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 26.Grewal, I., and R. Flavell. 1998. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 16:111–135. [DOI] [PubMed] [Google Scholar]
- 27.De Smedt, T., B. Pajak, E. Muraille, L. Lespagnard, E. Heinen, P. De Baetselier, J. Urbain, O. Leo, and M. Moser. 1996. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 184:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamath, A.T., J. Pooley, M.A. O'Keeffe, D. Vremec, Y. Zhan, A.M. Lew, A. D'Amico, L. Wu, D.F. Tough, and K. Shortman. 2000. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165:6762–6770. [DOI] [PubMed] [Google Scholar]
- 29.Mueller, D., and M. Jenkins. 1995. Molecular mechanisms underlying function T-cell unresponsiveness. Curr. Opin. Immunol. 7:375–381. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt, C.S., and M.F. Mescher. 2002. Peptide Ag priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12. J. Immunol. 168:5521–5529. [DOI] [PubMed] [Google Scholar]
- 31.Albert, M.L., M. Jegathesan, and R.B. Darnell. 2001. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2:1010–1017. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez, J., S. Aung, K. Marquardt, and L.A. Sherman. 2002. Uncoupling of proliferative potential and gain of effector function by CD8+ T cells responding to self-antigens. J. Exp. Med. 196:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtsinger, J.M., C.S. Schmidt, A. Mondino, D.C. Lins, R.M. Kedl, M.K. Jenkins, and M.F. Mescher. 1999. Inflammatory cytokines provide third signals for activation of naive CD4+ and CD8+ T cells. J. Immunol. 162:3256–3262. [PubMed] [Google Scholar]
- 34.Schmidt, C.S., and M.F. Mescher. 1999. Adjuvant effect of IL-12: conversion of peptide antigen administration from tolerizing to immunizing for CD8+ T cells in vivo. J. Immunol. 163:2561–2567. [PubMed] [Google Scholar]
- 35.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 37.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8α+ and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appay, V., D.F. Nixon, S.M. Donahoe, G.M.A. Gillespie, T. Dong, A. King, G.S. Ogg, H.M.L. Spiegel, C. Conlon, C.A. Spina, et al. 2000. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauvau, G., S. Vijh, P. Kong, T. Horng, K. Kerksiek, N. Serbina, R.A. Tuma, and E.G. Pamer. 2001. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science. 294:1735–1739. [DOI] [PubMed] [Google Scholar]
- 40.Lee, P.P., C. Yee, P.A. Savage, L. Fong, D. Brockstedt, J.S. Weber, D. Johnson, S. Swetter, J. Thompson, P.D. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 41.Moser, J.M., J.D. Altman, and A.E. Lukacher. 2001. Antiviral CD8+ T cell responses in neonatal mice: susceptibility to polyoma virus–induced tumors is associated with lack of cytotoxic function by viral antigen–specific T cells. J. Exp. Med. 193:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zajac, A.J., J.N. Blattman, K. Murali-Krishna, D.J.D. Sourdive, M. Suresh, J.D. Altman, and R. Ahmed. 1999. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porgador, A., J.W. Yewdell, Y. Deng, J.R. Bennink, and R.N. Germain. 1997. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 6:715–726. [DOI] [PubMed] [Google Scholar]
- 44.Tham, E.L., P.L. Jensen, and M.F. Mescher. 2001. Activation of antigen-specific T cells by artificial cell constructs having immobilized multimeric peptide-class I complexes and recombinant B7-Fc proteins. J. Immunol. Methods. 249:111–119. [DOI] [PubMed] [Google Scholar]
- 45.Altman, J.D., P.A.H. Moss, P.J.R. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]
- 46.Busch, D.H., I.M. Pilip, S. Vijh, and E.G. Pamer. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 8:353–362. [DOI] [PubMed] [Google Scholar]
- 47.Daniels, M.A., and S.C. Jameson. 2000. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J. Exp. Med. 191:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mueller, S.N., C.M. Jones, C.M. Smith, W.R. Heath, and F.R. Carbone. 2002. Rapid cytotoxic T lymphocyte activation occurs in the draining lymph nodes after cutaneous herpes simplex virus infection as a result of early antigen presentation and not the presence of virus. J. Exp. Med. 195:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hogquist, K., S. Jameson, W. Heath, J. Howard, M. Bevan, and F. Carbone. 1994. T cell receptor antagonist peptides induce positive selection. Cell. 76:17–27. [DOI] [PubMed] [Google Scholar]
- 50.Tham, E.L., and M.F. Mescher. 2001. Signaling alterations in activation-induced non-responsive (AINR) CD8 T cells. J. Immunol. 167:2040–2048. [DOI] [PubMed] [Google Scholar]
- 51.Pape, K., E. Kearney, A. Khoruts, A. Mondino, R. Merica, Z. Chen, E. Ingulli, J. White, J. Johnson, and M. Jenkins. 1997. Use of adoptive transfer of T-cell antigen-receptor-transgenic T cell for the study of T-cell activation in vivo. Immunol. Rev. 156:67–78. [DOI] [PubMed] [Google Scholar]
- 52.Mailliard, R.B., S. Egawa, Q. Cai, A. Kalinska, S.N. Bykovskaya, M.T. Lotze, M.L. Kapsenberg, W.J. Storkus, and P. Kalinski. 2002. Complementary dendritic cell–activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J. Exp. Med. 195:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valenzuela, J., C.S. Schmidt, and M.F. Mescher. 2002. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J. Immunol. 169:6842–6849. [DOI] [PubMed] [Google Scholar]
- 54.Vella, A.T., J.E. McCormack, P.S. Linsley, J.W. Kappler, and P. Marrack. 1995. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 2:261–270. [DOI] [PubMed] [Google Scholar]
- 55.Mitchell, T.C., D. Hildeman, R.M. Kedl, T.K. Teague, B.C. Schaefer, J. White, Y. Zhu, J. Kappler, and P. Marrack. 2001. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat. Immunol. 2:397–402. [DOI] [PubMed] [Google Scholar]
- 56.Oehen, S., and K. Brduscha-Riem. 1998. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J. Immunol. 161:5338–5346. [PubMed] [Google Scholar]
- 57.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belz, G.T., G.M. Behrens, C.M. Smith, J.F. Miller, C. Jones, K. Lejon, C.G. Fathman, S.N. Mueller, K. Shortman, F.R. Carbone, and W.R. Heath. 2002. The CD8α+ dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 196:1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]