

Abstract
Genome stability is regulated by the balance between efficiencies of the repair machinery and genetic alterations such as mutations and chromosomal rearrangements. It has been postulated that deregulation of class switch recombination (CSR) and somatic hypermutation (SHM), which modify the immunoglobulin (Ig) genes in activated B cells, may be responsible for aberrant chromosomal translocations and mutations of non-Ig genes that lead to lymphocyte malignancy. However, the molecular basis for these genetic instabilities is not clearly understood. Activation-induced cytidine deaminase (AID) is shown to be essential and sufficient to induce both CSR and SHM in artificial substrates in fibroblasts as well as B cells. Here we show that constitutive and ubiquitous expression of AID in transgenic mice caused both T cell lymphomas and dysgenetic lesions of epithelium of respiratory bronchioles (micro-adenomas) in all individual mice. Point mutations, but not translocations, were massively introduced in expressed T cell receptor (TCR) and c-myc genes in T lymphoma cells. The results indicate that AID can mutate non-Ig genes including oncogenes, implying that aberrant AID expression could be a cause of human malignancy.
Keywords: class switch recombination, somatic hypermutation, genomic instability, lymphoma, T cell receptor gene
Introduction
Antigen stimulation of mature B lymphocytes induces somatic hypermutation (SHM)* and/or class switch recombination (CSR) that diversify the variable (V) region and the heavy-chain constant (CH) region genes of Ig, respectively (1, 2). SHM introduces point mutations in rearranged V region genes, generating antibodies with higher affinity to antigens when coupled with selection by limited amounts of antigens (2). In parallel, the Ig heavy-chain genes also undergo another genetic alteration, CSR, to produce antibodies of various isotypes (IgG, IgA, or IgE) that carry different effector functions with the same antigen specificity. CSR changes the Ig CH region gene to be expressed from Cμ to one of the other CH genes by looping-out deletion of CH genes by recombination between μ switch (Sμ) region and one of downstream switch (S) regions located 5′ to each CH gene (1).
These frequent genetic alterations in the Ig loci have been suspected to be responsible for the genome instability. In fact, chromosomal translocations involving Ig loci are one of hallmarks of mature B cell lymphomas (3) and multiple proto-oncogenes are hypermutated in B cell diffuse large-cell lymphomas (DLCLs) in a similar way to SHM of Ig genes (4). As a large number of B cell lymphomas originate in germinal centers (GCs) where CSR and SHM take place, these chromosomal translocations and mutations are supposed to be mediated by aberrant hypermutation and class switching activity in GC B cells (3). However, the molecular basis for such deregulation of CSR and SHM activity has not been clearly described.
Activation-induced cytidine deaminase (AID) is specifically expressed in germinal center B cells (5) and has been shown to be required for both SHM and CSR (6, 7). Furthermore, expression of AID alone is sufficient to induce CSR and SHM in transcribed artificial constructs in fibroblasts as well as B cells (8–10). Thus, we speculated that deregulation of AID could lead to aberrant CSR and SHM activity and play a critical role in lymphomagenesis. In the present study, we generated transgenic (Tg) mice that express AID constitutively and ubiquitously to test this possibility. We found that AID Tg mice develop malignant T cell lymphomas and micro-adenomas in lung, and that the T lymphoma cells had frequent point mutations in expressed TCR and c-myc genes, which appeared to be introduced by AID activity. Our results suggest that ectopic or deregulated expression of AID might be responsible in part to human malignancy.
Materials and Methods
Tg Mice.
Murine AID cDNA (0.6 kb) were cloned into the EcoRI site of the pCXN2 vector (11) containing the chick β actin (CAG) promoter. The purified SalI-HindIII fragment containing the AID transgene was microinjected into either C57BL/6 or (C57BL/6 × C3H/He) F2 fertilized eggs to generate the B1, 2, and C1–3 lines of AID Tg mice, respectively. All Tg lines were maintained, in specific pathogen-free condition in the animal facility of the Department of Medical Chemistry, as hemizygotes by backcrossing male Tg mice with wild-type C57BL/6 females. Control mice were littermates carrying no transgene. Expression of AID in Tg tissues was analyzed by RT-PCR using following primers: AID-S1 (5′-CGGCATGAGACCTACCTCTGCTAC-3′) and AID-R1 (5′-CCAGGCTTTGAAAGTTCTTTCACG-3′). AID transcripts were amplified 30 cycles for Wt and 20 cycles for Tg mice. As an internal control, GAPDH transcripts were amplified by 20 cycles of PCR as described (12).
Histology.
Tissues were fixed with 10% formalin in PBS and embedded in paraffin. Sections were stained with hematoxylin and eosin by standard method. For immunofluorescence studies, tissue samples were frozen in tissue-tek O.C.T. compound (Sakura Finetechnical). Sections (8 μm thick) were prepared and fixed in CytoFix/CytoPerm reagent (BD Biosciences) for 10 min. Biotinylated anti-IgM (Cappel), FITC-conjugated anti-CD4 (clone RM4–5; eBioscience), streptavidin-Texas Red (Vector) were used for staining. The sections were mounted in Slow Fade (Molecular Probes), and slides were analyzed with a Bio-Rad confocal laser scanning microscope (Model MCR-1024).
Flow Cytometry.
Single suspensions from spleens, thymi, and lymph nodes were stained with following antibodies: FITC- or PE-conjugated anti-CD3 (clone 145–2C11), APC-conjugated anti-B220 (clone RA3–6B2), PE-conjugated anti-CD4 (clone RM4–5), APC-conjugated anti-CD8 (clone 53–6.7), FITC-conjugated anti-TCRβ (clone H57–597), PE-conjugated anti-PD-1 (clone J43), streptavidin-APC (eBioscience), and biotinylated-anti Vβ8 (clone F23.1; BD Biosciences). At least 10,000 live cells were analyzed on a FACSCalibur™ and CELLQuest™ software (Becton Dickinson). Dead cells were excluded from the analysis by forward and side scatter intensity and propidium iodide gating.
Southern Blot Analysis.
Genomic DNA was isolated by standard procedure. 2 μg of genomic DNA was digested with EcoRI and electrophoresed on a 0.7% agarose gel. Southern blots were hybridized with a 32P-labeled 2.3 kb TCR Jβ2 probe containing Jβ2.1–2.6 segments (13).
Spectral Karyotyping.
Lymphoma cells derived from primary mice or SCID mice were grown in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 86.4 μM 2-mercaptoethanol, and antibiotics. Cells were arrested at mitosis by treatment with colcemid (KaryoMAX Colcemid solution; GIBCO BRL) at 200 ng/ml for 2–3 h. Mitotic chromosome spreads were prepared by standard procedures, and spectral karyotyping (SKY) analysis was performed as described (14). A SkyPaint probe mixture (Applied Spectral Imaging), which contains all 21 combinatorially labeled painting probes specific for each mouse chromosome, was used for hybridization.
Sequencing.
Rearranged TCR Vβ8 genes and the c-myc gene were amplified by genomic PCR using following primers: Vβ8F1 (5′-CATGGAGGCTGCAGTCACCCAAAG-3′) and Jβ2.7R1 (5′-GGACCGAAGTACTGTTCATAGGAGC-3′) for TCR Vβ8, and myc575F (5′-GCGTTTTTTTCTGACTCGCTGTAG-3′) and myc1556R (5′-GCGGGGGTCAGGCTAAATTTTACT-3′) for c-myc. Genomic PCR was performed 35 cycles using Pyrobest polymerase (TaKaRa). The PCR products were A-tailed using rTaq polymerase (TaKaRa), subcloned into pGEM-Teasy vector (Promega), and sequenced. Vβ8-Cβ2 transcripts from rearranged alleles were amplified by RT-PCR using the following primers: Vβ8F1 and Cβ2R2 (5′-AGGGTGAAGAACGGCTCAGGATGC-3′). RT-PCR was performed 35 cycles and the products were sequenced, as described for genomic PCR.
Results
AID Transgenic Mice Die Rapidly with Enlarged Lymphoid Organs.
To elucidate whether deregulated expression of AID is involved in genetic instability, we generated five independent lines of Tg mice expressing AID under the control of CAG promoter (11). Although in normal mice, AID is expressed specifically in activated B lymphocytes (5), previous in vitro studies showed that AID could function not only in activated B cells but also in T cells and fibroblasts (8–10). Expression of AID in Tg tissues was confirmed by RT-PCR using young and healthy mice (Fig. 1 a). In Tg mice, AID was expressed ubiquitously and the expression level was not dramatically different among tissues including T and B cells in spleen. In wild-type mice, endogenous AID was expressed at low levels in mesenteric lymph nodes and spleen consistent with our previous result (5). AID Tg mice were born healthy except that they tended to be slightly smaller than their wild-type littermates (unpublished data). Flow cytometric analysis using surface markers showed that the total numbers and compositions of B and T lymphocytes including pre-B, pro-B, and mature B cells in spleen, lymph nodes, thymus, and bone marrow of young Tg mice were indistinguishable from those of their wild-type littermates (unpublished data), suggesting that premature, continuous expression of AID does not disrupt the general development of B and T cells.
Figure 1.
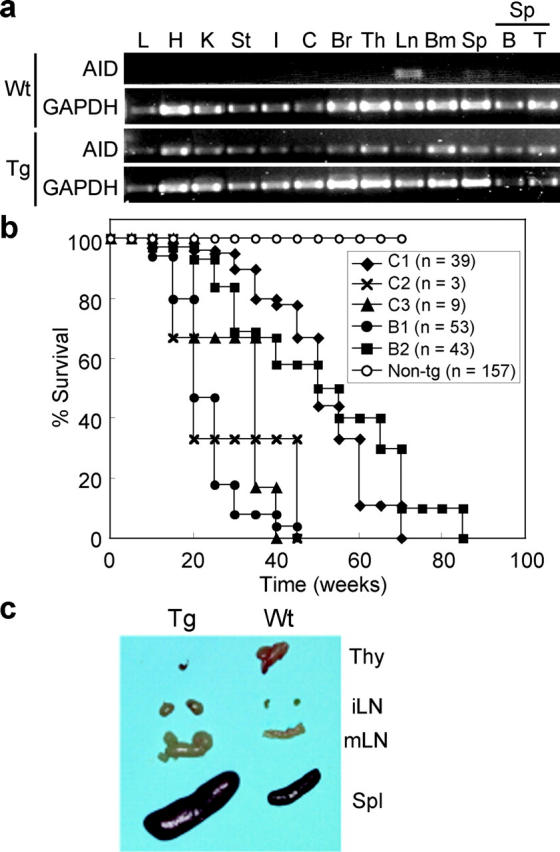
Early death and enlargements of lymphoid organs in AID Tg mice. (a) RT-PCR analysis of AID expression in wild-type and Tg tissues. Young (12 wk old) and healthy mice were used for analysis. GAPDH is an internal control. L, lung; H, heart; K, kidney; St, stomach; I, small intestine; C, colon; Br, brain; Th, thymus; Ln, mesenteric lymph nodes; Bm, bone marrow; Sp, spleen; B, purified splenic B cells; T, purified splenic T cells. No amplification was observed in RT(−) samples. (b) Survival curves of five independent lines of AID Tg mice and wild-type control. C1–3, Tg lines on C57BL/6 × (C57BL/6 × C3H/He) background; B1 and 2, Tg lines on C57BL/6 background. (c) Thymus (Thy), inguinal (iLN), and mesenteric (mLN) lymph nodes and spleen (Spl) from a 16-wk-old AID Tg mouse and its littermate control.
However, all Tg lines had greatly shorter life spans than wild-type littermates (Fig. 1 b). Mice of all Tg lines died with enlarged lymphoid organs but with variations in terms of onset of swelling and life spans. A line designated as B1 was chosen for more detailed analyses because it developed the phenotype at youngest ages. While lymphoid organs of the B1 Tg mice were enlarged with age, there were two groups that were distinguished by enlargement or shrinkage of thymus. In 65% of AID Tg mice, spleen and systemic lymph nodes were enlarged, whereas thymi were reduced in size to almost unrecognizable levels (Fig. 1 c). In contrasts, thymi of the remaining 35% of Tg mice were extremely enlarged with frequent enlargements of peripheral lymphoid organs (unpublished data). Histological analysis of enlarged lymphoid organs revealed that spleen, lymph nodes and thymus were filled with monotonous atypical lymphocytes with cytological features of lymphoblastic lymphoma such as enlarged round nuclei, irregular nuclear contours, scant cytoplasm, and frequent mitotic figures (Fig. 2 , a–c). In affected lymph nodes, normal B cell follicles and T cell zones were destroyed and occupied by larger T cells weakly expressing CD4 (Fig. 2 b). Furthermore, lymphoma cells invaded massively in nonlymphoid organs including liver, kidney, lung (Fig. 2 c), pancreas, and occasionally heart (unpublished data).
Figure 2.
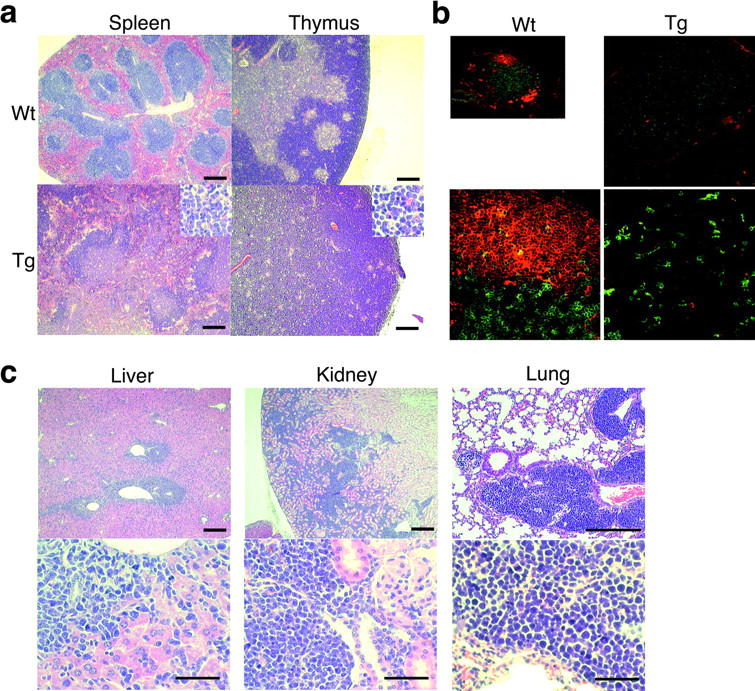
Histological analysis of lymphomas in AID Tg mice. (a) Hematoxylin and eosin staining of spleen and thymus from AID Tg and wild-type mice. Scale bar: 250 μm. (b) Immunohistochemical staining of lymph nodes from an AID Tg mouse and a wild-type control. Green, CD4; red, IgM. (c) Lymphoma infiltration into liver, kidney, and lung in AID Tg mice. Enlarged round nuclei, irregular nuclear contours, scant cytoplasm, and frequent mitotic figures of lymphoma cells are evident. Scale bars: 250 μm for top panels and 50 μm for bottom panels.
AID Transgenic Mice Develop Malignant T Cell Lymphomas and Micro-adenomas/Adenocarcinomas in Lung.
Strikingly, flow cytometric analyses revealed that lymphoma cells in all AID Tg mice were from T cells but not B cells. This is also the case in other Tg lines than the B1 line. Lymphoma cells in mice that had enlarged spleen and lymph nodes, and minute thymus were abnormally large, CD4dull single positive T cells expressing CD3 and TCRβ (Fig. 3 a), whereas CD8+ single positive T cell lymphomas were never observed in AID Tg mice. In most cases, the expression level of CD4 tended to be low and broad, and both CD3 and TCR levels were often decreased. In addition, all lymphoma cells expressed activation markers such as B220 (Fig. 3 a) and PD-1 (Fig. 3 c), and occasionally CD69 (unpublished data), implying that they derived from activated T cells in the periphery. Lymphoma cells in mice with enlarged thymus, on the other hand, were CD4CD8 double positive T cells expressing CD3 and TCRβ, suggesting that they derived from thymocytes (Fig. 3 b). In contrast to the peripheral type, they did not express B220 (Fig. 3 b), PD-1 (Fig.3 d), or CD69 (unpublished data).
Figure 3.
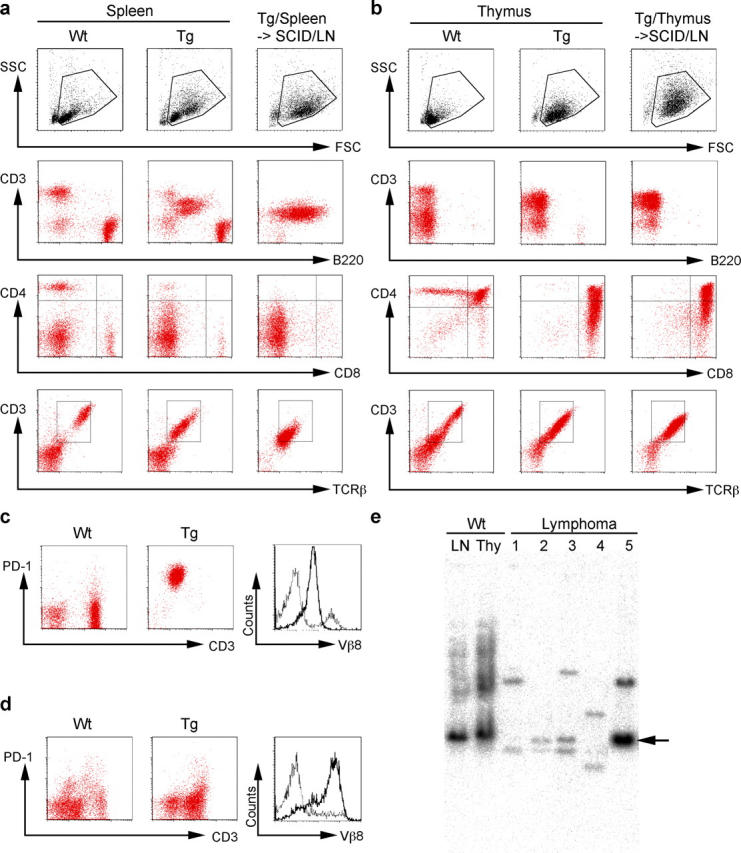
Analysis of malignant T lymphoma cells in AID Tg mice. (a) Flow cytometric analysis of lymphomas of peripheral origin. Splenocytes from wild-type control (left panels) and an AID Tg mouse (middle panels), and lymph node cells from a SCID mouse that received splenocytes from the same individual Tg mouse (right panels) are compared. Data are representative of nine affected AID Tg mice. (b) Flow cytometric analysis of lymphomas of thymic origin. Thymocytes from a wild-type control (left panels) and an AID Tg mouse (middle panels), and lymph node cells from a SCID mouse that received thymocytes from the same individual Tg mouse (right panels) are compared. Data are representative of five affected AID Tg mice. (c and d) Flow cytometric analysis of lymphoma cells of peripheral origin (c) and thymic origin (d) showing PD-1/CD3 and Vβ8 profiles. Lymph node cells (c) and thymocytes (d) from wild-type littermates were used as control. Broken lines, wild-type controls; solid lines, lymphoma cells. (e) Southern blot analysis of lymphoma DNA. EcoRI-digested DNA from five independent lymphomas was probed with Jβ2 probe. DNA from wild-type lymph node cells and thymocytes was used as control. Arrow indicates germline band. Lymphomas 1–3 and 5 are of peripheral origin, and 4 is of thymic origin. Lymphomas 1 and 4 are shown in (a and c) and (b and d), respectively.
To verify the malignancy of these abnormal T cells, 2 × 106 cells from affected lymph nodes, spleens, or thymi of six primary animals were injected either intraperitoneally or intravenously into SCID mice or syngenic C57BL/6 mice. All of them, irrespective of strain of recipients and the route of injection, generated lymphomas in the recipients and the mice became moribund within 3 to 6 wk after injection with enlarged lymph nodes, spleens, and thymi. In addition, the lymphoma cells could be passed through mice more than three times. Although the primary spleen, thymus, and lymph nodes contained normal T and B cells in addition to the abnormal T cells, only the abnormal T cells expanded in the recipient SCID mice (Fig. 3, a and b and unpublished data). Furthermore, Vβ staining (Fig. 3, c and d) and Southern blot analysis of TCR gene rearrangements (Fig. 3 e) revealed that the lymphomas were monoclonal with both alleles rearranged in many cases. Of 9 cases examined, 5 were monoclonally Vβ8+, although the reason why Vβ8 was preferred remained unclear. The transplantability and monoclonality of the abnormal T cells indicate that they are indeed malignant T cell lymphomas.
Interestingly, all AID Tg mice from all independent lines, even young lymphoma-free mice, developed micro-adenomas or dysgenetic lesions of respiratory bronchiole (Fig. 4) . The number of such lesions was ∼80 per section of one lobe from lymphoma-developed mice and less than 30 from young (10–13 wk) and lymphoma-free mice. These micro-adenomas were preferentially located in subpleura areas of lung. Such distribution pattern is quite similar to that of lung adenocarcinomas in human. Furthermore, two out of two mice that survived more than 60 wk had adenocarcinoma of bronchilo-alveolar type in addition to T lymphoma. As the number and size of micro-adenomas appeared to be larger in older mice (unpublished data), this benign micro-adenoma might have progressed to malignant adenocarcinoma when mice lived long by surviving from T lymphoma. Notably, 2 out of 25 mice that survived more than 30 wk had benign epidermalcystes in addition to T lymphoma and micro-adenomas in lung, while epithelium of gastrointestinal tracts of those Tg mice appeared normal (unpublished data). Thus, AID could potentially affect many tissues, although the susceptibility seemed different among tissues.
Figure 4.

Development of micro-adenoma and adenocarcinoma in lung of AID Tg mice. Hematoxylin and eosin staining of lung from wild-type control and AID Tg mice. Arrows indicate micro-adenomas of respiratory bronchiole. Old AID Tg mice (84- and 67-wk-old) developed adenocarcinomas of bronchilo-alveolar type. Scale bars: 250 μm for top panels and 50 μm for bottom panels.
Point Mutations but Not Translocations Are Introduced in Expressed TCR and c-myc Genes in T Lymphoma Cells.
SKY of five lymphomas revealed that lymphomas had no apparent chromosomal translocations (Fig. 5 a) although gains and losses of random chromosomes were occasionally observed in some metaphases, indicating that the lymphomas did not arise from a specific chromosomal translocation. The fact that AID introduces hypermutations in the GFP gene, Ig genes, and the AID transgene per se in AID-overexpressing cells in vitro (8–10, 15) led us to examine whether AID mutated non-Ig genes in the lymphomas. We first sequenced the rearranged and expressed TCR genes of three lymphomas (lymphomas 1, 2, and 4) of monoclonal Vβ8+ T cells. Sequenced were products of genomic PCR using primers located in Vβ8 and Jβ2.7 segments. The rearrangements were all productive and comprised of either Vβ8.2-Jβ2.4 (lymphomas 1 and 2) or Vβ8.2-Jβ2.5 (lymphoma 4). The junctions of recombination were identical among all the plasmid clones from each lymphoma, confirming its clonality (unpublished data). This result was in marked contrast to wild-type lymph node T cells, which contained heterogeneous junctions and nonproductive rearrangements.
Figure 5.
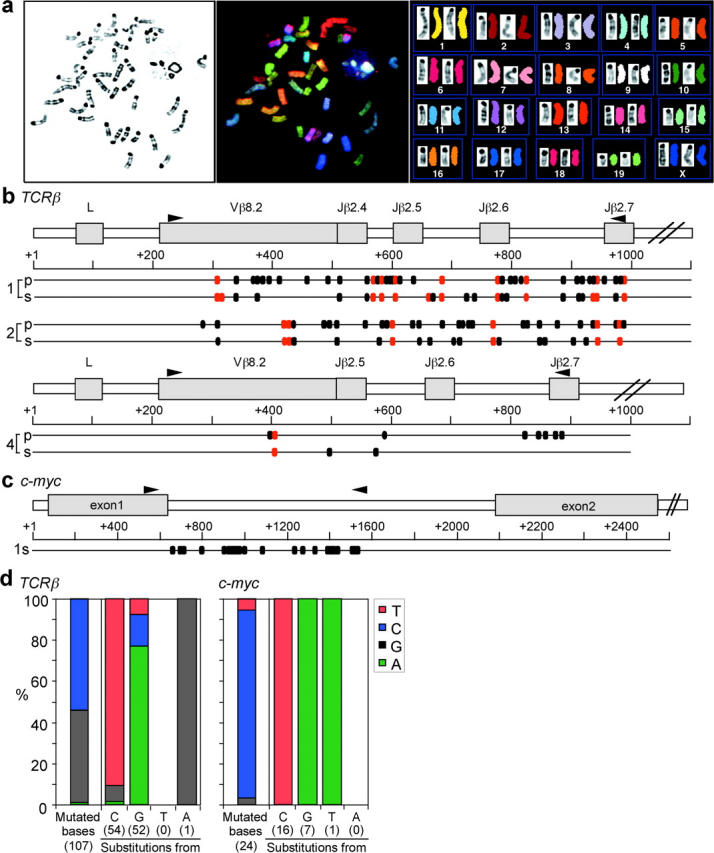
Frequent point mutations in the rearranged TCR and the c-myc genes in lymphoma cells. (a) SKY analysis of lymphoma cells from AID Tg mice. Data is representative of lymphomas from five mice. Shown are the inverted image of a 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI)-stained metaphase spread (G-band-like image) (left); spectral color image of the same metaphase spread (middle); karyotype of the spectra-based classification-colored chromosomes and their corresponding inverted DAPI images (right). (b) Distribution of mutations in the rearranged TCRβ genes of lymphomas from three independent Tg mice. Data of lymphomas 1, 2, and 4 are shown. p, primary cells; s, cells recovered from SCID mice. Red dots indicate mutations common to all plasmid clones from the same individual mice; black dots, sporadic mutations. Primers used for amplification are shown above genomic loci. (c) Mutations in the c-myc gene of lymphoma 1 from SCID mice. Primers used for amplification are shown above genomic locus. (d) Mutation patterns of the TCRβ and c-myc genes. Frequency of mutated bases is corrected for the base composition of the unmutated sequences. The number of mutations is shown in parentheses.
Massive numbers of point mutations were found in the Vβ sequences, while sequences from wild type T cells had almost no mutations (Fig. 5 b and Table I) . There were mutations common among all plasmid clones from the same lymphoma in addition to sporadic mutations, suggesting that a single T cell with some mutations expanded as lymphoma and that additional mutations continuously accumulated during subsequent proliferation. Interestingly, mutations were also found in the Cβ2 region, which was amplified by RT-PCR between Vβ8 and 3′ end of Cβ2. However, mutations in the Cβ2 region were much less than those in the Vβ8 region and located only in exon 1 and never in exon 2–4 (Table I). Considering that exon 1 is 3 kb downstream of the Jβ2.7 segment and exon2 is further 0.5 kb downstream, the distance from the promoter might explain less mutations in the C region as previously described in SHM of both Ig and non Ig loci (2, 4, 16). Although no mutations were found in the 5′ and 3′ regions of the Fas gene (unpublished data), the c-myc gene also accumulated high levels of mutations in the region encompassing the exon 1 and intron 1, the hotspot of mutations and breakpoints of translocations in B cell DLCLs (4) and Burkitt lymphomas (3, 17–19; Fig. 5 c, Table I). Point mutations in this region of the c-myc gene are known to alter myc transcription by releasing a blockade mechanism on transcriptional elongation (17, 19). Furthermore, targeted bases were strongly biased to GC bases and base substitution pattern was almost all either C to T or G to A (Fig. 5 d), consistent with the feature of mutations found in AID-overexpressing cells (9, 10) and in DLCLs (4). Therefore, we concluded that the mutations in the T lymphomas are attributable to AID activity.
Table I.
Mutations in the Rearranged TCR and the c-myc Genes in Lymphomas
| Cell source | Number of mutated clones |
10−4 × mutation frequencya |
|
|---|---|---|---|
| TCRVβ8–Jβ2 (genome) | |||
| Lymphoma 1 | Primary LN | 23/23 | 25 (41/16,330) |
| Transferred | 24/24 | 15 (25/17,040) | |
| Lymphoma 2 | Primary SPL | 24/24 | 19 (32/17,136) |
| Transferred | 24/24 | 11 (19/17,136) | |
| Lymphoma 4 | Primary THY | 24/24 | 5.4 (8/14,832) |
| Transferred | 23/23 | 2.1 (3/14,214) | |
| Wild-type | LN | 1/22 | 0.68 (1/14,799) |
| TCR Vβ8–Cβ2 (cDNA) | |||
| Lymphoma 1 | Transferred | ||
| Vβ8–Jβ2.4 | 29/29 | 12 (11/9,019) | |
| Cβ2 | Exon 1–4 | 29/29 | 3.9 (6/15,486) |
| Exon 1 | 29/29 | 5.5 (6/10,875) | |
| Exon 2–4 | 0/29 | <2.2 (0/4,611) | |
| Wild-type | T cells | ||
| Vβ8–Jβ2 | 0/24 | <1.3 (0/7,411) | |
| Cβ2 | Exon 1–4 | 0/24 | <0.78 (0/12,816) |
| c-myc (genome) | |||
| Lymphoma 1 | Transferred | 19/19 | 13 (24/18,183) |
| Wild-type | LN | 0/21 | <0.50 (0/20,097) |
Identical mutations in different plasmid clones from the same individual lymphomas were counted only once unless genealogies indicated that the mutation was unique.
Number of mutations per number of total base pairs sequenced is shown in parentheses.
Life Spans of AID Transgenic Mice Are Shortened with the Passage of Generations.
Unexpectedly, one of Tg lines, B2, which had much slower manifestation of T lymphoma in the parental mouse, showed an earlier onset of T lymphomas and a clear reduction of life spans with the passage of generations (Fig. 6) , suggesting that some effects, probably mutations, caused by AID expression may be accumulated in germ cells. The reduction of life spans appears to stop after it reached the level of the B1 line, progenies of which did not show any reduction of life spans (unpublished data).
Figure 6.

Reduction of life spans of AID Tg mice with the passage of generations. Separate survival curves of the founder (P) and F1, F2, and F3 Tg progenies of line B2 mice.
Discussion
We have shown that mice constitutively expressing AID succumbed to T cell lymphomas. Furthermore, the mice developed micro-adenomas and adenocarcinomas in lung. Strikingly, point mutations but not translocations were massively introduced in the rearranged and expressed TCR genes as well as in the c-myc proto-oncogene with similar distribution and base specificity to those observed in B cells (2), B lymphomas (4), and AID-overexpressing cells in vitro (9, 10, 15). Although a recent study showed that multiple proto-oncogenes are hypermutated in DLCLs (4) which are derived from GC B cells and express AID, whether these SHM-like mutations and expression of AID are responsible to or resultant from lymphomagenesis is unclear since most tumor cells are genetically unstable (20). However, our findings indicate that deregulated or ectopic expression of AID contributes to tumorigenesis by introducing mutations in non-Ig genes rather than generating chromosomal translocations. This is the first report of tumorigenesis by a gene product that can act as a direct, potent mutator, but not by breakdown of systems that ensure the genome stability.
The marked reduction of life spans in progenies of the B2 line, which originally showed slower onset of diseases, implies that mutations affecting tumor susceptibility accumulated in germ cells. Since generation transfer of Tg mice were mediated by three males and all Tg progenies of the same generation showed similar phenotypes, the number of mutation targets that are probably involved in tumor susceptibility or life spans may be very large. The accumulation of mutations in oncogenes or tumor-suppressor genes are thought to result in tumorigenesis (20). The genome instability is observed at both nucleotide and chromosomal levels. Defects in the repair systems such as nucleotide-excision repair and mismatch repair (MMR) systems are involved in subtle sequence instabilities, while defects in the checkpoint machinery such as mitotic spindle checkpoint and DNA-damage checkpoint systems are involved in chromosomal instabilities including gains and losses of whole chromosomes and chromosomal translocations (20). Curiously, mice lacking genes involved in either the MMR system (Msh2 [21, 22]) or the checkpoint machinery (p53 [23], Atm [24], Brca1 [25], and Brca2 [26]) develop T cell lymphomas almost exclusively or preferentially. Such exclusive occurrence of T cell lymphomas was also observed in AID Tg mice. The reason why other types of tumors than T lymphomas were rarely observed in AID Tg mice may be simply because T lymphoma cells take off and expand well in advance of generation of tumors in other cells. Although the reason why no B cell lymphoma developed is also unclear, it is possible that B cells have a protective system to prevent excessive activity of AID, making the onset of B lymphomagenesis slower than that of T lymphomagenesis. However, many types of tumors might potentially occur because in AID Tg mice mutations were introduced in the c-myc and probably other proto-oncogenes.
AID shows homology to apolipoprotein B mRNA editing catalytic subunit 1, APOBEC-1, a member of RNA-editing cytidine deaminase family (5, 27). Tumorigenesis in AID Tg mice is reminiscent of development of hepatocellular carcinomas in mice ectopically expressing APOBEC-1 in liver (28). Other hepatic mRNAs involved in cell growth and regulation than apo-B mRNA were aberrantly edited in APOBEC-1 Tg mice (28, 29). As the apo-B gene in Tg livers was not mutated (28), the mechanism for tumorigenesis in APOBEC-1 Tg mice may be attributed to aberrant editing of mRNAs, but not to direct action on DNA. On the contrary, non-Ig genes including the c-myc proto-oncogene were mutated in AID Tg mice in a similar way to the Ig gene, although whether AID acts on RNA or DNA is still controversial (1, 5, 30–32).
As constitutive expression of AID causes tumorigenesis, expression of AID has to be tightly regulated in normal mice. Otherwise, deregulatedly or ectopically expressed AID will cause enormous genomic instability. Thus, ectopic expression of AID could be partly responsible to malignancy in mouse as well as human. It is important to screen whether AID is ectopically expressed in human cancers of non-B cell origins. In addition, AID Tg mice might be a better model system to screen tumor-suppressor genes, as mutations in AID Tg mice are restricted to transcribed genes and accumulate through generations.
Acknowledgments
We are grateful to Drs. S. Fagarasan, T. Okazaki, H. Nagaoka, K. Ikuta, and S. Takeda for their useful suggestions and critical reading of the manuscript; Dr. J. Miyazaki for providing the pCXN2 vector, Dr. H. Kohda, Mses. Y. Tabuchi, T. Toyoshima, K. Yurimoto, A. Kawamura, and Y. Sasaki for their excellent technical assistance; and Ms. Y. Shiraki and T. Nishikawa for secretarial help.
This work was supported by Center of Excellence Grant from Ministry of Education, Culture, Sports, Science and Technology of Japan. I. Okazaki is a research fellow of the Japan Society for the Promotion of Science.
Footnotes
Abbreviations used in this paper: AID, activation-induced cytidine deaminase; CSR, class switch recombination; DLCL, B cell diffuse large-cell lymphoma; GC, germinal center; SHM, somatic hypermutation; SKY, spectral karyotyping; Tg, transgenic.
References
- 1.Honjo, T., K. Kinoshita, and M. Muramatsu. 2002. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 20:165–196. [DOI] [PubMed] [Google Scholar]
- 2.Neuberger, M.S., and C. Milstein. 1995. Somatic hypermutation. Curr. Opin. Immunol. 7:248–254. [DOI] [PubMed] [Google Scholar]
- 3.Kuppers, R., and R. Dalla-Favera. 2001. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 20:5580–5594. [DOI] [PubMed] [Google Scholar]
- 4.Pasqualucci, L., P. Neumeister, T. Goossens, G. Nanjangud, R.S. Chaganti, R. Kuppers, and R. Dalla-Favera. 2001. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 412:341–346. [DOI] [PubMed] [Google Scholar]
- 5.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 6.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 7.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 8.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 9.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 10.Martin, A., P.D. Bardwell, C.J. Woo, M. Fan, M.J. Shulman, and M.D. Scharff. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 415:802–806. [DOI] [PubMed] [Google Scholar]
- 11.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 108:193–199. [DOI] [PubMed] [Google Scholar]
- 12.Lee, C.G., K. Kinoshita, A. Arudchandran, S.M. Cerritelli, R.J. Crouch, and T. Honjo. 2001. Quantitative regulation of class switch recombination by switch region transcription. J. Exp. Med. 194:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikuta, K., M. Hattori, K. Wake, S. Kano, T. Honjo, J. Yo, and N. Minato. 1986. Expression and rearrangement of the alpha, beta, and gamma chain genes of the T cell receptor in cloned murine large granular lymphocyte lines. No correlation with the cytotoxic spectrum. J. Exp. Med. 164:428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liyanage, M., A. Coleman, S. du Manoir, T. Veldman, S. McCormack, R.B. Dickson, C. Barlow, A. Wynshaw-Boris, S. Janz, J. Wienberg, et al. 1996. Multicolour spectral karyotyping of mouse chromosomes. Nat. Genet. 14:312–315. [DOI] [PubMed] [Google Scholar]
- 15.Martin, A., and M.D. Scharff. 2002. Somatic hypermutation of the AID transgene in B and non-B cells. Proc. Natl. Acad. Sci. USA. 99:12304–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storb, U., A. Peters, E. Klotz, N. Kim, H.M. Shen, J. Hackett, B. Rogerson, and T.E. Martin. 1998. Cis-acting sequences that affect somatic hypermutation of Ig genes. Immunol. Rev. 162:153–160. [DOI] [PubMed] [Google Scholar]
- 17.Cesarman, E., R. Dalla-Favera, D. Bentley, and M. Groudine. 1987. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science. 238:1272–1275. [DOI] [PubMed] [Google Scholar]
- 18.Rabbitts, T.H., P.H. Hamlyn, and R. Baer. 1983. Altered nucleotide sequences of a translocated c-myc gene in Burkitt lymphoma. Nature. 306:760–765. [DOI] [PubMed] [Google Scholar]
- 19.Raffeld, M., T. Yano, A.T. Hoang, B. Lewis, H.M. Clark, T. Otsuki, and C.V. Dang. 1995. Clustered mutations in the transcriptional activation domain of Myc in 8q24 translocated lymphomas and their functional consequences. Curr. Top. Microbiol. Immunol. 194:265–272. [DOI] [PubMed] [Google Scholar]
- 20.Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1998. Genetic instabilities in human cancers. Nature. 396:643–649. [DOI] [PubMed] [Google Scholar]
- 21.Reitmair, A.H., R. Schmits, A. Ewel, B. Bapat, M. Redston, A. Mitri, P. Waterhouse, H.W. Mittrucker, A. Wakeham, B. Liu, et al. 1995. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat. Genet. 11:64–70. [DOI] [PubMed] [Google Scholar]
- 22.de Wind, N., M. Dekker, A. Berns, M. Radman, and H. te Riele. 1995. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 82:321–330. [DOI] [PubMed] [Google Scholar]
- 23.Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4:1–7. [DOI] [PubMed] [Google Scholar]
- 24.Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J.N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 86:159–171. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig, T., P. Fisher, S. Ganesan, and A. Efstratiadis. 2001. Tumorigenesis in mice carrying a truncating Brca1 mutation. Genes Dev. 15:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connor, F., D. Bertwistle, P.J. Mee, G.M. Ross, S. Swift, E. Grigorieva, V.L. Tybulewicz, and A. Ashworth. 1997. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet. 17:423–430. [DOI] [PubMed] [Google Scholar]
- 27.Teng, B., C.F. Burant, and N.O. Davidson. 1993. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 260:1816–1819. [DOI] [PubMed] [Google Scholar]
- 28.Yamanaka, S., M.E. Balestra, L.D. Ferrell, J. Fan, K.S. Arnold, S. Taylor, J.M. Taylor, and T.L. Innerarity. 1995. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc. Natl. Acad. Sci. USA. 92:8483–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamanaka, S., K.S. Poksay, K.S. Arnold, and T.L. Innerarity. 1997. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA- editing enzyme. Genes Dev. 11:321–333. [DOI] [PubMed] [Google Scholar]
- 30.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 31.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 32.Harris, R.S., S.K. Petersen-Mahrt, and M.S. Neuberger. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell. 10:1247–1253. [DOI] [PubMed] [Google Scholar]