

Abstract
Amongst the many ploys used by microbial pathogens to interfere with host immune responses is the production of proteins with the properties of superantigens. These properties enable superantigens to interact with conserved variable region framework subdomains of the antigen receptors of lymphocytes rather than the complementarity determining region involved in the binding of conventional antigens. To understand how a B cell superantigen affects the host immune system, we infused protein A of Staphylococcus aureus (SpA) and followed the fate of peripheral B cells expressing B cell receptors (BCRs) with VH regions capable of binding SpA. Within hours, a sequence of events was initiated in SpA-binding splenic B cells, with rapid down-regulation of BCRs and coreceptors, CD19 and CD21, the induction of an activation phenotype, and limited rounds of proliferation. Apoptosis followed through a process heralded by the dissipation of mitochondrial membrane potential, the induction of the caspase pathway, and DNA fragmentation. After exposure, B cell apoptotic bodies were deposited in the spleen, lymph nodes, and Peyer's patches. Although in vivo apoptosis did not require the Fas death receptor, B cells were protected by interleukin (IL)-4 or CD40L, or overexpression of Bcl-2. These studies define a pathway for BCR-mediated programmed cell death that is VH region targeted by a superantigen.
Keywords: tolerance, repertoire, clonal selection, Ig genes, host immunity
Introduction
The fate of a B lymphocyte clone is largely determined by signals received through the Ag receptor (i.e., B cell receptor [BCR]*). The developmental and maturational stage of the lymphocyte, the availability of costimulatory signals, and the qualitative features of the BCR signal influence the cellular outcome of these ligand exposures. Antigenic challenge can result, therefore, in either positive selection due to proliferation and pro-survival affects or negative selection due to functional inactivation, deletion, or induction of BCR editing. However, a B cell can be affected by encounters with more than one type of Ag, as the BCR of a clone might be cross-reactive with unrelated exogenous and self-ligands. Such cross-reactivity has been said to contribute to the primary mode of action of natural viral and bacterial proteins that have been termed superantigens for their capacity to interact with conserved sites in the variable regions. Consequently, interactions with superantigens are orders of magnitude more frequent in the lymphocyte repertoire than those for a conventional Ag that typically involve CDRs (for review see 1).
In recent studies, we have shown that protein A of Staphylococcus aureus (SpA) has the properties of a B cell superantigen by virtue of interactions with a large supraclonal B cell set via high affinity framework-mediated interactions with soluble and cell-associated BCR (1). Crystallographic studies have demonstrated that a domain of SpA forms a complex with human Fab via a conformational surface on BCR that involves side chains from four β strands present in framework subdomains of the clan III gene-encoded VH region (2). These binding interactions and the B cell targeting properties of SpA do not involve the site by which SpA binds Fcγ (2). Moreover, the VH surface on BCR targeted by SpA has been conserved in the B cell repertoires of amphibian, avian, and mammalian species (3), and is highly represented in the human and murine immune systems.
In the mouse, Fab-mediated SpA-binding interactions are commonly displayed by 5–10% of mature B cells, which express genes from the clan III set of related VH families, the murine homologues of human VH3 genes (4, 5). BCR encoded by clan III/S107 VH genes convey high affinity binding activity for SpA (for review see 1). Our studies of the consequences of in vivo exposure have shown that SpA can deplete VH-targeted B cells in both neonates and adult mice by a process that is unimpaired in TCRβδ−/− mice, indicating that T cells are not required (6). Exposure also results in an associated proportional loss of S107-μ transcripts and clan III VH-encoded circulating IgM and Ab responses whereas other responses are unaffected (5, 6).
Among the SpA-targeted clan III/S107-encoded clones are B cells bearing the T15 idiotype (6). T15 lymphocytes, which reside within the B-1 pool, display high affinity SpA-binding activity and are depleted after in vivo exposure (6). Because these T15 B-1 cells dominate responses to phosphorylcholine-containing immunogens, their loss after in vivo exposure to SpA explained the finding of induced phosphorylcholine-specific immune tolerance. In fact, this long-term immunologic impairment in SpA-treated mice persists despite the fact that central neo-lymphogenesis reestablishes the level of newly formed S107-expressing B cells in the periphery within 1–2 wk after the last SpA treatment (5). In explanation, the effect of SpA treatment on Ig transcript levels was found to be complex. The persistent selective loss of S107 transcripts in the spleen correlated with an induced persistent “hole” in the B-1 repertoire and resulting depletion of splenic spontaneous S107-IgM–secreting cells that subsequently cannot be replenished from the central compartment (6, 7).
Although we have postulated that SpA exposure leads to BCR-mediated cell death, the underlying process has not been examined. We could not formally rule out alternative mechanisms that include the induction of a “suppressive” regulatory circuit, nondeletional functional inactivation (i.e., anergy) accompanied by sIg down-regulation that impairs in vivo detection, or a trafficking event that removes the affected clan III VH gene–expressing B cells from the sites of investigation.
In these studies, to investigate the mechanism(s) responsible for the supraclonal lymphocyte loss, we have evaluated the outcome of in vivo exposure to this B cell toxin on affected lymphocytes. These studies elucidate the sequence of events that follows SpA exposure from the surface phenotypic changes induced on VH-restricted BCR-expressing B cells to the induction of apoptosis.
Materials and Methods
Mice and Immunogens.
Mice were obtained from The Jackson Laboratory and bred under specific pathogen-free conditions under the supervision of the University California San Diego Animal Subjects Program. Adult mice, at least 6 wk of age, received either recombinant 0.5 mg SpA (RepliGen), 0.5 mg of a control protein, OVA (Sigma-Aldrich), or 2 mg anti-Ig (H and L chain specific; Jackson ImmunoResearch Laboratories), or as indicated, with removal of contaminating endotoxin (<0.5 EU/ml). Mice received protein in 125 μl of pyrogen-free saline instilled into the peritoneal cavity. After mice were killed, tissues were harvested for immediate ex vivo analysis and aliquots were placed in culture. Mice were age and sex matched in all experiments.
Genomic PCR Southern Surveys of VHDJ Family Expression.
To evaluate the effect of treatment on B cell representation, 6-wk-old C57BL/6J mice received a dose of 1 mg protein in saline, which was repeated after 3 d, and were killed 6 d after the first dose. Freshly isolated splenocytes were purified on a ficoll gradient and genomic DNA was extracted using a DNeasy Tissue Kit (QIAGEN) with an RNase incubation step. For amplification of genomic rearrangement, DNA aliquots of 90, 30, or 10 ng were added to separate tubes containing 5 μl 10× PCR buffer (PerkinElmer), 2 μl of 10 mM dNTP (PerkinElmer), 1 μl of each 50-pmol oligonucleotide primer solution (MWG Biotech), and 1 μl Amplitaq gold (PerkinElmer), with nuclease-free H2O added to a final volume of 50 μl. Samples in triplicate were amplified using a denaturing step of 95°C for 12 min, 32 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 40 s, followed by a final 72°C for 5 min. To amplify VH family–specific rearrangement, reactions were performed with a framework 1–derived oligonucleotide primer specific for members of the J558 family (clan I) or S107 family (clan III) primer paired with an antisense JH-derived primer as previously described (5, 8). Products were electrophoretically separated, transferred to membranes, and hybridized with digoxigenin-labeled (Roche), VH family–specific 150–250-bp exon-derived DNA probes, and later reacted with peroxidase-tagged antidigoxigenin Ab before visualization of bands with ECF substrate (Amersham Biosciences) with images analyzed as previously described (5). To independently confirm the VH family specificity of the amplification reactions, aliquots were also subcloned into the TA system (Invitrogen) and sequence was determined (6 and unpublished data).
Flow Cytometry Analysis.
Adapting previously reported methods (3, 5, 6), mononuclear cells were stained with FITC-labeled anti-CD21 (clone 7G6), CD40 (clone HHM40-3), CD54 (clone 3E2), CD69 (clone H1.2F3), CD80 (clone 16-10A1), CD86 (clone GL-1), CD95 (clone JO-2), TRI-COLOR®-labeled anti-CD19 (clone 6D5; Caltag), biotin-labeled I-Ab (clone KH74), PE- or biotin-labeled mSpA, or isotype controls used with streptavidin-labeled Per CP® or APC (BD Biosciences) in the presence of Fc block (BD Biosciences) as appropriate. Data was acquired using a FACSCalibur™ (Becton Dickinson) and analyzed with FlowJo™ software (Treestar, Inc.). Forward and side scatter gates included only nucleated viable cells, dead cells were excluded based on light scatter, and propidium iodide (PI) or 7-AAD uptake as possible.
To measure the activity of a pro-apoptotic pathway, cells were permeabilized and stained with specific affinity-purified rabbit IgG anti-activated caspase 3 (Cell Signaling Technology) and detected with PE goat anti–rabbit IgG (Southern Biotechnology Associates, Inc.). Alternatively, DNA fragmentation was determined by the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (Roche) and changes in mitochondrial transmembrane potential (ΔΨm) were detected by staining with JC-1 (Intergen Company; reference 9).
Histology.
Spleens were placed in OCT compound embedding media (Fisher Scientific) and frozen in a 2-methylbutane/dry ice bath. 7-μM sections were cut and mounted on Superfrost Plus Slides (Fisher Scientific). Slides were fixed in 3.7% formaldehyde in PBS, pH 7.4, for 20 min at room temperature, blocked in PBS, 5% FCS for 1 h at room temperature, and then stained with either PE-labeled anti-B220 or anti-CD3 (diluted in blocking buffer), washed, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice, washed, and then stained with the TUNEL assay kit (Roche). After mounting with Prolong (Molecular Probes), digitized images were captured at ×10 or ×40.
Adoptive Transfer Studies.
Isolated splenocytes were suspended at 5 × 107/ml in RPMI, labeled with 1 μM carboxyfluorescein diacetate succinimidylester (CFSE; Molecular Probes), and incubated at 37°C for 10 min. The cells were washed three times in ice cold RPMI with 5% FCS and resuspended in PBS. A total of 3 × 107 splenocytes were transferred intravenously through the tail vein. After 24 h, mice received either SpA or control protein as described above, or 500 μg of the immunostimulatory CpG phosphorothioate oligonucleotide (5′ TGA CTG TGA ACG TTC GAG ATG A 3′), which was used as a positive control for proliferation (i.e., mitogen; reference 10). Control studies confirmed that CFSE treatment did not compromise the ability of B cells to respond to SpA or mitogens (unpublished data).
In Vitro Stimulation Studies.
In certain studies, freshly isolated splenocytes were placed in culture of 5% CO2 in RPMI with 5% FCS, supplemented with l-glutamine, 10 mM Hepes, amino acid solution, 1 mM sodium pyruvate, 60 μM β-mercaptoethanol, and penicillin/streptomycin at 2 × 106 cell/ml at 37°C. In some studies, B cells were first enriched by magnetic negative selection (StemCell Technologies Inc.) to attain >92% B220+ cells. In other studies, freshly isolated cells were first labeled with 5 μM CFSE, washed twice in IMDM, and placed in culture for 72–96 h as described above, with or without various stimulants: 10 μg/ml Escherichia coli LPS (Sigma-Aldrich), 10 ng/ml IL-4 (BD Biosciences), or 5 ng/ml soluble mCD40L-mCD8α fusion ligand (provided by P. Lane, University of Birmingham, Birmingham, UK; reference 11) cross-linked with 5 μg/ml rat anti–murine CD8 (53-6.72; BD Biosciences). To assess the role of the caspase pathway, splenocytes were cultured with 25 μM of the pan-caspase inhibitor, Z-Val-Ala-Asp(OMe)-fluoromethyl ketone (Z-VAD) or control compound and buffer alone according to the manufacturer's directions (Enzyme Systems Products).
Statistical Analysis.
Comparisons between different groups used the two tailed Student's t test with P values of <0.05 taken as significant.
Results
SpA Induces Loss of Genomic S107 Rearrangements.
In earlier reports, we have demonstrated that in vivo treatment of mice with SpA leads to a rapid loss of detectable SpA-reactive B cells in both the peripheral and central compartments, with the lowest levels occurring ∼4–6 d after exposure (5). We also found that in vivo exposure results in a concurrent selective depletion of splenic S107-μ transcripts whereas μ transcripts from other VH families were unaffected (6). The persistent selective loss of S107 transcripts after SpA exposure was interpreted as a reflection of the special immunobiology of B-1 cells and spontaneous IgM-secreting cells (6, 7). These findings, however, also suggest that splenic transcript levels may not be the best marker of the magnitude of supraclonal B cell deletion.
To address concerns that the tolerizing properties of SpA may derive in part from cellular regulatory effects on Ab gene transcription, we assessed the outcome of in vivo SpA exposure on the representation of genomic S107 rearrangements in the spleen, which should avoid a bias for detection of cells with disproportionately higher content of Ab gene transcripts. The results from these PCR-based assays were also normalized for content of J558 (clan I) genomic rearrangements. As shown in Fig. 1 , SpA treatment of adult C57BL/6 mice resulted in a greater than a sixfold reduction of the amplimers of genomic S107 rearrangements. Furthermore, because this assay also detects the nonproductive VH genes that represent ∼15% of adult splenic S107 rearrangements (Feeney, A., personal communication), these findings likely underestimate the level of VH-selective B cell loss in the periphery induced by SpA with sequential treatments. These results provide independent confirmation that in vivo exposure can result in the selective cellular deletion of a large supraclonal set of S107-expressing lymphocytes, which is targeted based on VH region expression.
Figure 1.

In vivo SpA exposure results in a selective loss of genomic S107 rearrangements. C57BL/6 mice received control or SpA intraperitoneal treatment. Each VH family–specific blot included amplifications of the serial amounts of genomic DNA. As illustrated, the amount of genomic template is indicated above the resultant blot, with relative values normalized by densitometric quantitation using the J558 rearrangements shown below. +, a positive control of a S107 gene–containing plasmid; −, a negative control of a J558 gene–containing plasmid. The data are representative of two independent studies of two mice in each group that used JH1- or JH2-specific antisense primers.
SpA Modulates the Surface Phenotype of B Cells Targeted for Deletion.
To begin to elucidate the process by which SpA induces the deletion of B cells, we evaluated the response of T15i Ig “knockin” mice, in which almost all B cells express an S107 transgene that conveys the high affinity VH framework-associated SpA-binding motif (2). In these studies, 1 mg SpA in buffered saline was instilled into the peritoneum and mice were killed at intervals to survey for subsequent induced changes. 2 h after treatment, we first detected down-regulation of surface Ig on splenic T15i transgene–expressing (B220+) B cells (unpublished data). Beginning at 4 h after treatment, sequential increases in the representation of T15i VH-expressing B cells were demonstrated in the spleen. In untreated T15i+/+ mice, the total number of mononuclear splenocytes was 62 × 106 ± 4.4 (mean ± SEM) with 45 ± 2.2% B cells whereas at 4 h after treatment both the total number of splenocytes and the representation of B cells increased to 82 × 106 ± 8.5 and 50 ± 0.7%, respectively. A further increase was observed at 12 h after treatment with 96 × 106 ± 4.8 splenocytes and 58 ± 0.7% B cells whereas at 16 h we found 92 × 106 ± 4.8 splenocytes and 51 ± 5.0% B cells. As these early changes began so rapidly, and cell cycle analysis failed to detect significant induced proliferation after these brief periods of exposure (unpublished data), we therefore believe this in part reflects altered trafficking into the spleen.
In vivo SpA exposure also induced a concurrent comodulation of other B cell surface molecules. As illustrated in Fig. 2 A, 16 h after in vivo instillation, the B cells from the SpA-treated T15i+/+ mice displayed reduced levels of sIgM and sIgD and also the BCR coreceptor, CD19. Levels of CD21 were also reduced (probably as it acts as a receptor for complementized SpA; 12). In contrast, levels of B220 (a CD45 isoform) were unaffected (Fig. 2 A), which is consistent with reports that B220 is not comodulated after in vitro anti-IgM treatment (13).
Figure 2.
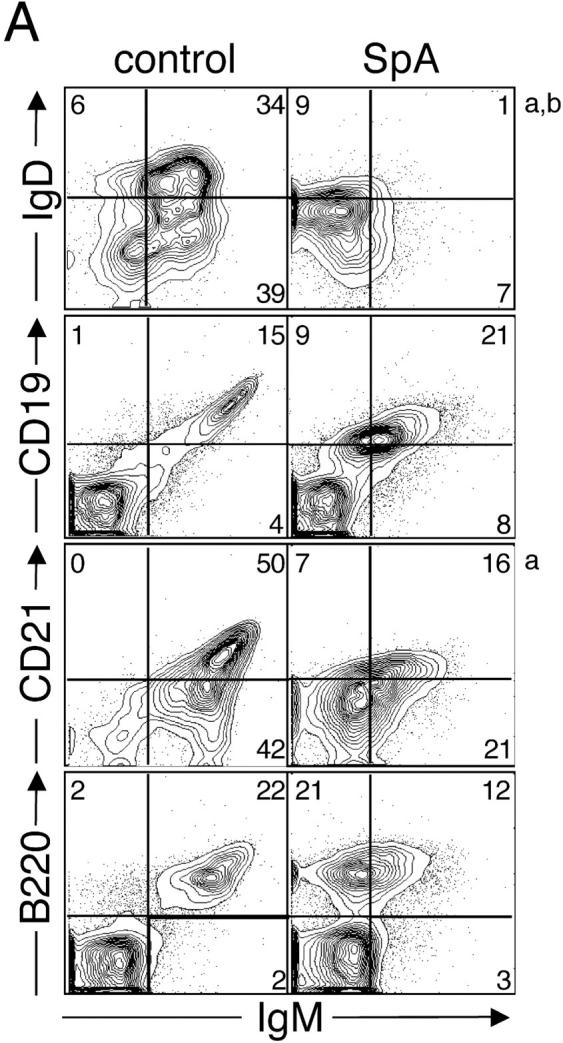

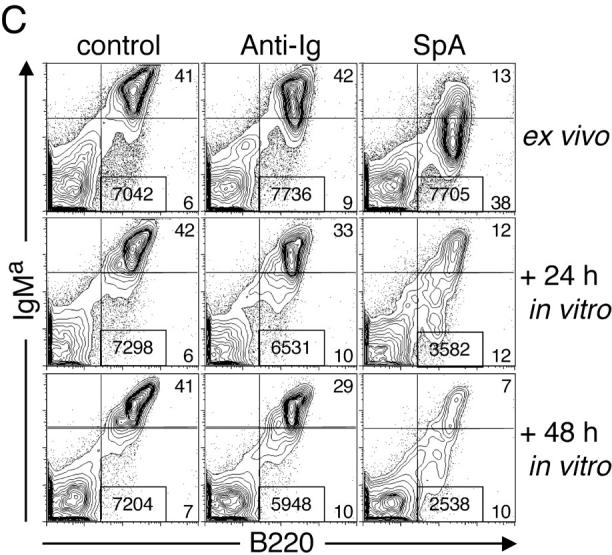
Phenotypic changes of the initiation phase and cell death induced in splenic T15i+/+ B cells. (A) SpA induces down-regulation of membrane-associated BCR, CD19, and CD21 on splenic T15i+/+ B cells at 16 h after treatment. Based on staining for CD19 or B220, SpA treatment was shown to result in an increased representation of B lymphocytes in the spleen at this time point. Panels depicted were first gated based on B220 expression (a). Studies depicted were from a different experiment than in other panels (b). (B) In vivo SpA exposure induces an activation phenotype on T15i+/+ B cells. Shaded regions depict results after treatment with control protein and those from SpA-treated mice are indicated by the bold line. Values are indicated for mean intensity of fluorescence, with significant difference indicated by *. For these studies, nonviable cells were gated out based on scatter and PI uptake. Results are also depicted after in vitro culture of splenocytes for different periods, as indicated. (C) SpA treatment induces accelerated loss of splenic T15i+/+ B cells after in vitro culture. Panels depict the representation of splenic B cells in samples obtained at time of harvest (ex vivo, 16 h) from mice that received 0.5 mg OVA or SpA, or 2 mg of anti-Ig, and after sequential periods in culture in the absence of additional stimulants. In each panel, the total number of B cells (boxed numbers) was determined for a total of 15,000 viable (i.e., PI−) mononuclear cells. The data are representative of three experiments.
Splenic B cells from treated T15i+/+ mice were also evaluated for other induced phenotypic changes. As illustrated in Fig. 2 B, when harvested 16 h after in vivo exposure, B cells from SpA-treated mice displayed increased levels of CD69 and CD86 (top), which have previously been described as early markers of specific B cell activation. After sequential periods in culture without additional stimulants, surface expression of these molecules peaked and then waned whereas levels of CD80 and CD95 were shown to increase at later time points (Fig. 2 B) with up-regulation of CD40, CD54, and MHC II (Fig. 3 C; unpublished data). In general, B cell phenotypic changes induced by in vivo SpA exposure reiterate the patterns first described to occur after in vitro stimulation with experimental BCR ligand analogs (14) or antigenic exposure.
Figure 3.

VH-susceptible splenic B cells in adult T15i+/− mice undergo induced phenotypic changes after in vivo SpA exposure. (A) After treatments as described in Fig. 2, at 16 h after in vivo exposure sIg levels on IgMa-bearing T15i+/− B cells are greatly depressed after SpA treatment but not control treatment. (B) sIg levels on IgMb-bearing splenic B cells from these mice are unaffected. (C) Levels of surface molecules associated with activation of splenic mononuclear cells gated on B220 are depicted for Ig transgene–linked IgMa-bearing B cells. (D) Levels of these surface molecules are demonstrated for splenic B cells bearing IgMb, which identifies the endogenous polyclonal B cells in the same samples. Here, results are shown for CD86, CD54, and MHC II for splenic B cells at time of harvest whereas the results for CD80 and CD40 are depicted after an additional 24 h in culture. Results after treatment with control protein are shaded and those from SpA-treated mice are indicated by the bold line. Mean intensity of fluorescence values are indicated. For these studies, nonviable cells were gated out based on scatter and PI uptake. The data are representative of three experiments.
Time course analyses of cellular loss of VH-targeted B lymphocytes from these T15i+/+ mice demonstrated that after only 16 h of in vivo SpA exposure more than half of these B cells were fated for cell death, which progressed during the subsequent 48 h of in vitro culture as determined by PI staining (Fig. 2 C). By contrast, after in vivo treatment with anti-Ig, far fewer (only ∼10%) of B cells were fated to die. Hence, despite their early removal from the SpA- or anti-Ig–treated host, these B cells were still committed to a fate of activation-associated accelerated cell death (Fig. 2 C). These observations are consistent with findings that the overwhelming majority of VH-susceptible B cells are depleted after 4–6 d of repeated in vivo SpA doses (5 and unpublished data). Cellular death induced by SpA was most pronounced in B cells with the most down-regulated levels of sIg (unpublished data). Equivalent findings were also documented in studies of splenic B cells that were enriched by negative selection before in vitro culture (unpublished data). Significantly, akin to findings from an in vivo model system that used anti-IgD as a surrogate Ag (14), we could not detect evidence of induced B cell loss in the spleen in surveys at or before 16 h after in vivo SpA administration.
To confirm that these induced phenotypic changes occurred only on SpA-binding B cells, equivalent treatment studies were performed in hemizygotic T15i mice (i.e., T15i+/−) that have two distinct mature B cell populations. In these mice, only ∼30% of splenic B cells express the T15 VH region linked to the IgMa allotype. In the remainder of T15i+/− B cells, this transgene has been inactivated by secondary recombination, leading to diverse endogenous VH rearrangements distinguished by the IgMb allotype (15).
When the SpA-treated T15i hemizygotic mice were evaluated after in vivo challenge, only the IgMa-bearing B cells that express the S107 transgene–encoded VH region had a marked down-regulation of surface Ig (Fig. 3 A). In contrast, in these same samples no global changes were detected for the levels of membrane-associated IgMb that express predominantly nonsusceptible VH regions (Fig. 3 B). Flow cytometric surveys demonstrated the IgMa-bearing B cells from SpA-treated T15i+/− mice displayed the same sequential induction of markers of lymphocyte activation. At time of harvest 16 h after in vivo exposure, there were increased levels of CD86, CD54 (ICAM-1), and MHC II on IgMa-bearing B cells from SpA-treated mice (Fig. 3 C) compared with the IgMb-bearing B cells from the same mice (Fig. 3 D). After an additional 24 h in culture, levels of CD80 and CD40 were significantly elevated on IgMa-bearing B cells from the SpA-treated T15i+/− mice (Fig. 3 C). By comparison, although SpA treatment did not induce generalized changes on the splenic IgMb-bearing B cells, a small proportion (i.e., <10%) of the IgMb-bearing B cells from SpA-treated mice were found to have up-regulated their surface MHC II (Fig. 3 D). Furthermore, after an additional 24 h in culture we also detected an increase in CD80 expression on this small subset of IgMb-bearing B cells from these mice (Fig. 3 D). In explanation, this minor subpopulation of IgMb-bearing B cells is at the same frequency that we have previously documented for the expression of SpA-binding clan III VH regions among the polyclonal B cells in C57BL/6 mice (5, 6).
Taken together, these studies indicate that in vivo exposure to SpA specifically targets certain B cells based on VH usage. This results in the induction of a phenotype of down-regulated BCR and associated cellular activation with later progression to the triggering of an intracellular death pathway.
In Vivo Clonal Fate after SpA Treatment.
To assess directly the in vivo fate of susceptible B cells after SpA treatment and determine whether exposure induces B cell proliferation, T15i+/+ splenocytes were labeled with CFSE and adoptively transferred into congenic C57BL/6 recipients (16). As illustrated in Fig. 4 , at 48 h after treatment, transferred T15i+/+ B cells recovered from the spleens of SpA-treated recipient mice were found to have undergone up to two to three rounds of proliferation whereas there was no evidence of proliferation in splenocytes from control-treated mice. Significantly, despite this evidence of proliferation, in this experiment there was still a mean 36% decrease of B cells in the SpA-treated T15i+/+ mice compared with the control-treated mice (P < 0.015). By contrast, there was a mean 173% increase of T15i+/+ B cells in the spleens of the mitogen-treated mice (Table I) . In the LNs of these SpA-treated mice there was a >58% mean reduction of T15i+/+ B cells compared with control-treated mice (Fig. 4; unpublished data). Analysis of a similar group at 96 h after SpA treatment demonstrated an even greater loss (i.e., mean 54% deletion) in the spleen compared with control-treated mice (P < 0.01; Table I). Intracellular staining studies for IgMa provided independent confirmation of the same relative levels of deletion (unpublished data). These studies document that although some proliferation was induced by SpA, it was insufficient to replace the B cells targeted for in vivo deletion.
Figure 4.
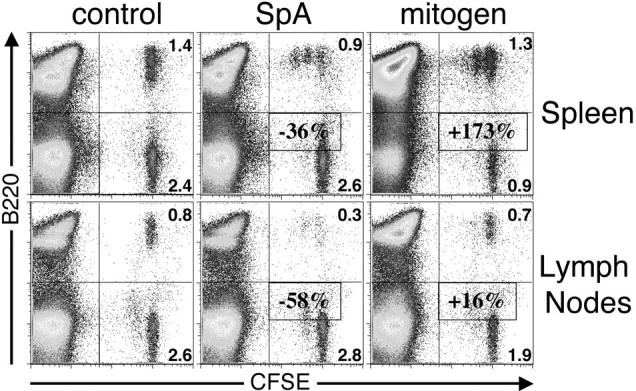
In vivo clonal fate after SpA treatment. T15i+/+ splenocytes were labeled with CFSE and adoptively transferred into congenic C57BL/6 recipients. Here, CFSE identifies mononuclear cells derived from the T15i+/+ donor and dilution of staining intensity identifies cells that have undergone cell division. B cells are identified as B220+. The in vivo outcome to treatments with OVA (control), SpA, and immunostimulatory DNA (mitogen) are depicted in cells recovered from the spleens and LNs. Mean values for B cell deletion (−) or proliferation (+) in treatment groups compared with control mice are indicated and determined as described in Table I. The data are representative of three experiments.
Table I.
Outcome of In Vivo Treatment after Adoptive Transfer
| T15i+/+→C57BL/648 ha
|
T15i+/+→C57BL/696 h
|
T15i+/+→Fasl
gld48 h
|
||||
|---|---|---|---|---|---|---|
| Treatment | B:Tb | Percent Δc | B:T | Percent Δ | B:T | Percent Δ |
| Total cells | ||||||
| control | 0.61 ± 0.05 | — | 0.71 ± 0.08 | — | 1.2 ± 0.1d | — |
| SpA | 0.39 ± 0.04 | ↓36 | 0.32 ± 0.04 | ↓54 | 0.6 ± 0.07 | ↓45 |
| mitogen | 1.7 ± 0.005 | ↑173 | 1.7 ± 0.4 | ↑138 | 2.5e | ↑108 |
| Progenitor cells | ||||||
| control | 0.6 ± 0.05 | — | NAf | — | 1.3 ± 0.2 | — |
| SpA | 0.28 ± 0.04 | ↓54 | NA | — | 0.5 ± 0.09 | ↓57 |
| mitogen | 1.4 ± 0.08 | ↑133 | NA | — | 2.5 | ↑92 |
The number of hours of in vivo exposure to SpA, OVA (control), or CpG (mitogen).
The B:T ratio of isolated T15i+/+ splenocytes was unchanged from that found in control-treated mice. In the CFSE-labeled cells, the relative loss of transferred B cells was determined after various durations in vivo using the assumption that the T cell population remains unchanged in all treatment groups. There was no evidence of T cell proliferation.
Indicates the percentage change, either increase (↑) or decrease (↓), compared to the control-treated group whose value is 100%.
Adoptive transfer of labeled T15i+/+ splenocytes resulted in reconstitution of distinct and reproducible B:T ratios in different untreated/control-treated immunodeficient strains (unpublished data).
Each group contains n = 3 with this exception in which n = 1. Otherwise, mean values ± SEM are shown for each group. To estimate the number of original transferred cells (i.e., progenitor cells) that survived treatment, calculations were based on the evaluation of sequential rounds of proliferation, as previously described (reference 66), using the equation: Progenitor cells = Round 0 + (Round 1/2) + (Round 2/4) + (Round 3/8).
NA, not applicable as proliferation/daughter cells were not discernible.
The effect of SpA treatment was also evaluated at other sites in the peripheral immune system, which demonstrated that after 6 d the absolute numbers of B cells were similarly reduced in the spleen, peripheral LNs, mesenteric LNs, and Peyer's patches (i.e., mean 68–88% loss; Fig. 5) . We also found a comparable reduction in B220high B cells in the bone marrow (5 and unpublished data) whereas there were no significant differences in the absolute numbers of T cells at any of these sites (unpublished data). The representation of B cells was also reduced in the blood, liver, and lung (i.e., mean 78–85% loss; Fig. 5), which further suggested that SpA-induced B cell depletion was unlikely to represent redistribution or sequestration.
Figure 5.

The representation of B cells at various lymphoid and nonlymphoid sites after SpA treatment. Adult T15I+/+ mice were treated on days 0 and 4, harvested on day 6, and evaluated by flow cytometry. The percentage of viable B and T cells in the mononuclear compartment were derived by B220 and CD3 staining, and B/T ratios were calculated to estimate the percentage change (an indication of B cell deletion). a, the mean absolute number ± SD of B cells at each site where n = 3; b, the P values derived from the absolute number of B cells for spleen, peripheral LNs, mesenteric LNs (MLN), and Peyer's patches (PP); c, P values derived from the mononuclear percentages for blood, lung, and liver samples.
Mechanisms of Apoptotic Death Induced by SpA.
To characterize the intracellular mechanism(s) responsible for SpA-induced B cell deletion, we looked for induction of pathways of programmed cell death and first evaluated the levels of cellular DNA fragmentation in targeted B cells using the TUNEL assay, which detects DNA 3′-hydroxyl ends resulting from caspase-activated DNA cleavage (17, 18). We found that in T15i+/+ spleens, B220+ TUNEL+ apoptotic bodies were deposited in B zones of primary follicles 48 h after SpA treatment (Fig. 6, A–D) and these B220+ apoptotic bodies were also detected in the B zones in the peripheral LNs, mesenteric LNs, and Peyer's patches (unpublished data). These apoptotic bodies appeared to be excluded from the T zones (Fig. 6 B and unpublished data). The highest frequencies were detected at 48 h after SpA treatment with decreased representation at 72 h, and these apoptotic bodies were barely detectable at 96 h (unpublished data).
Figure 6.




Apoptotic pathways induced in VH-susceptible T15i+/+ B cells during the degradation phase that follows in vivo treatment with SpA. At time of harvest (48 h ex vivo), there is evidence of increased DNA fragmentation detected by the TUNEL assay (green) in B cells from T15i+/+ mice that received SpA (B and D) compared with control (A and C). A and B were stained with B220 and C and D were stained with CD3 (red). (E) After in vivo treatment of Fas ligand–deficient (gld) or congenic (C57BL/6) mice that received CFSE-labeled T15i+/+ splenocytes, at 48 h the intracellular levels of activated caspase 3 are greatly enriched in susceptible B cells (i.e., B220+) from SpA-treated mice (bold line) compared with those receiving control treatment (shaded). By contrast, differences are not seen in non-B cells (i.e., B220−). Panels depict data gated on CFSE+ events (refer to methods in Table I). (F) Results are depicted for cytometric assays of ΔΨm. Splenic B cells are identified by gating on B220+ cells or T cells by CD3+ staining. Herein, the dissipation of ΔΨm is identified based on increased shift in FL-1 and lower levels of staining with the specific fluorescent dye detected in FL-2. Results of splenic lymphocytes are depicted for mice that received SpA treatments at 0, 4, 8, 16, 48, and 72 h before they were killed. (G) The caspase pathway contributes to SpA-induced apoptotic B cell death. After 16 h of in vivo exposure, splenocytes from control or SpA-treated T15i+/+ mice were placed in culture with or without a general caspase inhibitor, Z-VAD. After in vitro culture, although the VH-susceptible B cells from SpA-treated mice undergo accelerated apoptotic death after an additional 24 h in culture, compared with those from control (OVA) mice, the addition of the caspase inhibitor enhances the survival of SpA-exposed T15i-expressing splenic B cells. The data are representative of three or more experiments.
Studies of T15+/+ splenocytes harvested 16 h after in vivo SpA exposure demonstrated the same viability and absence of TUNEL staining as splenocytes from control-treated mice. However, after 24 h in culture, these B cells from SpA-treated mice displayed a rapid progression of DNA fragmentation, which was significantly greater than in B cells from control-treated mice or in the non-B cells in the same culture (unpublished data). These in vitro changes progressed with kinetics comparable to those documented for SpA-induced in situ deposition of B cell apoptotic bodies in peripheral lymphoid tissues (Fig. 6, A–D, and unpublished data).
Next, we looked for involvement of the caspase pathway, which are cysteine proteases that play a central role in apoptotic death systems. Akin to the TUNEL studies, we found that immediately after 16 h of in vivo exposure, levels of activated caspase 3 were not altered in B220+ splenocytes cells from SpA-treated T15i+/+ mice (unpublished data). However, after the placement in culture, we found that intracellular activated caspase 3 levels progressively increased in the SpA-treated B cells in parallel with the enhanced induction of DNA fragmentation. Moreover, throughout the 72-h culture period, these levels increased at a much greater rate in the B cells from SpA-treated T15i+/+ mice, which also were found to rapidly progress to cytolysis (unpublished data).
To verify that these findings accurately reflect the pathway of in vivo death in VH-targeted B cells, the same studies were performed after adoptive transfer of CFSE-labeled T15+/+ splenocytes into C57BL/6 recipients. As shown in Fig. 6 E, at 48 h after treatment, compared with B cells in control-treated mice and non-B cells in SpA-treated mice, the levels of intracellular activated caspase 3 were significantly increased only in the T15i+/+ B cells of SpA-treated mice.
To further dissect the temporal relationship between B cell triggering and the induction of the death pathways, we also surveyed for induced changes in mitochondrial membrane potential that reflect the early phase of the “intrinsic” apoptosis pathway. As shown in Fig. 6 F, compared with splenic B cells from an untreated T15i+/+ mouse, at 4 and 8 h after SpA treatment, differences in ΔΨm were already detectable in B cells and much greater proportions of affected B cells were detectable at 16–48 h (Fig. 6 F and unpublished data). By 72 h the percentage of affected B cells greatly decreased, but this likely in part represented the in vivo clearance of apoptotic B cells. We also found that T15i+/+ splenic B cells, harvested 4–24 h after in vivo SpA exposure and when subsequently placed in culture, demonstrated these same progressive increases in ΔΨm compared with lymphocytes from control-treated mice (unpublished data).
To investigate the relationship between these different induced apoptotic pathways, we directly tested the contribution of caspase-dependent mechanisms to the sequence of events that leads to cellular deletion by repeating the 16-h ex vivo/in vitro survival studies with the inclusion of Z-VAD, a potent pan-caspase inhibitor (Fig. 6 G). As expected, Z-VAD had no effect on the cell death of primary splenocytes that naturally occurs in vitro. However, when examined after 24 h in culture, Z-VAD enhanced the survival of the SpA-targeted T15i+/+ B cells to a level indistinguishable from that of B cells from control-treated mice, blocking the accelerated DNA fragmentation that reflects the action of caspase-activated DNase (unpublished data). The B cells from SpA-treated mice, however, still displayed the same induced loss of ΔΨm (unpublished data). Notably, when evaluated after 48 h and later times in culture, compared with the rapid apoptotic death that occurred in the absence of the inhibitor, there was still an accelerated (albeit slower) loss of viability for the B cells from SpA-treated mice cultured with the caspase inhibitor (unpublished data). Although this is consistent with evidence that Z-VAD does not prevent all death pathways leading to cytolysis (19, 20), these findings indicate that there is not an absolute requirement for a caspase pathway for induction by SpA of mitochondrial changes that lead to accelerated cell death.
Factors that Rescue B Cells Targeted by SpA.
To better understand the mechanism(s) responsible for the loss of the SpA-targeted T15i+/+ B cells, we evaluated how their fate in culture would be affected by supplemental stimulants. As illustrated in Fig. 7 A, when harvested after 16 h of in vivo exposure to SpA, we confirmed that there was an accelerated loss of viable T15i VH-expressing B cells in the absence of an additional factor in culture. After 72 h in culture, this represented an 82% relative depletion of these B cells (i.e., from 46% of mononuclear cells at ex vivo harvest to 8% of residual splenic mononuclear cells after culture). In contrast, B cells from the control-treated mouse displayed only a 46% decrease (i.e., from 37 to 20% of mononuclear cells). In cultures containing LPS, a type I thymus-independent mitogen, a pronounced B cell pro-survival effect was demonstrated for B cells previously exposed to SpA. Significantly, pro-survival effects of SpA were also demonstrated in cultures that contained either soluble CD40L or IL-4. In addition to significantly enhancing the survival of B cells from SpA-treated T15i+/+ mice, culturing with these latter stimulants also normalized sIg levels on responding B cells (Fig. 7 A).
Figure 7.
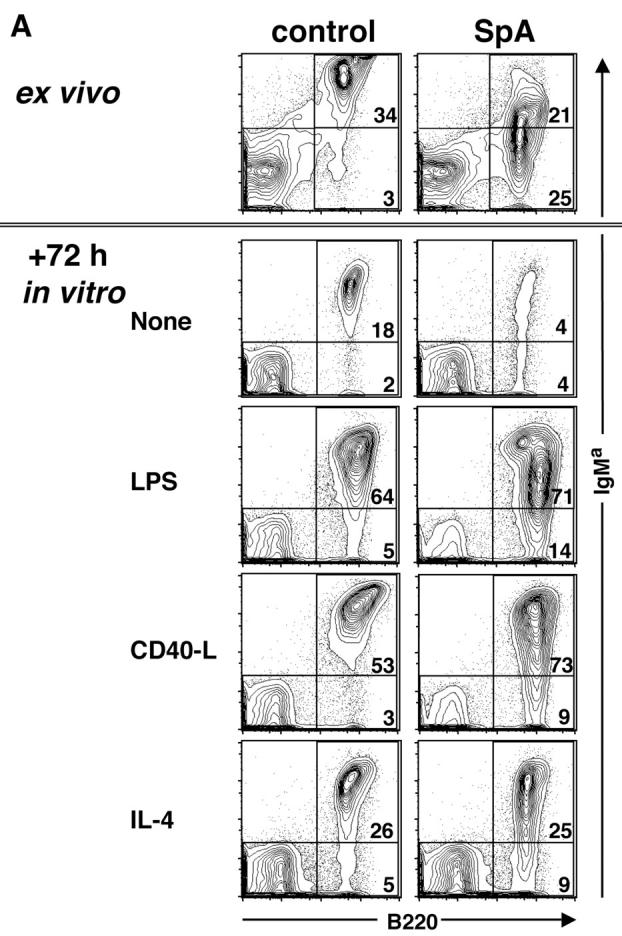

Supplemental factors can rescue B cells from death and induce enhanced proliferation. Splenocytes from T15i+/+ mice were analyzed 16 h after treatment (ex vivo) and culture with or without additional stimuli. (A) After 72 h in culture, the B cells affected by SpA (i.e., IgMa bearing) display increased representation after in vitro culture with CD40L or IL-4. (B) Results are depicted following the same experimental design, except that the ex vivo splenocytes were labeled with CFSE and then placed in culture for 72 h. As shown, after gating on IgMa-bearing B cells, cells that have not undergone a single round of proliferation display the highest intensity of fluorescence (i.e., CFSE high) whereas after each round of proliferation the fluorescence is decreased. The bar graphs depict the percentage of cells that have undergone proliferation. Solid bars depict SpA treated and open bars represent B cells from control treated. After culture with CD40L or IL-4, there were no significant differences in the number of total mononuclear cells recovered from the OVA- or SpA-exposed cell cultures. The data are representative of three experiments.
To determine if the increased representation of B cells in these cultures was in part due to enhanced survival or whether it reflected enhanced proliferation, we repeated these studies with splenocytes labeled with CFSE before placement in culture (16). As illustrated in Fig. 7 B, after only 16 h of in vivo exposure, when labeled splenocytes were placed in culture for 72 h without the addition of stimulants, neither B cells from control- nor SpA-treated mice displayed evidence of proliferation. In contrast, proliferation was induced by the addition of any of these stimulants. Significantly, between each of these paired cultures, more B cells from the SpA-treated mice underwent proliferation (and fewer also remained without a single round of proliferation). Of special interest, although CD40L was generally much more effective than IL-4 at driving B cell proliferation, with the inclusion of either of these stimulants, the B cells from SpA-treated mice also appeared to have more rapid initiation and progression of proliferation (Fig. 7 B and unpublished data). Although in earlier studies we showed that SpA-induced deletion is unimpaired in T cell–deficient mice (6), the current findings document that targeted B cells can be rescued by high local levels of the B cell pro-survival factors, IL-4 and CD40L, which are naturally provided by helper T cells. It also shows that although a large proportion of B cells display mitochondrial membrane permeabilization at 16 h (Fig. 6 F), the in vitro addition of a second signal results in greatly enhanced survival and proliferation, suggesting that at this early time point mitochondrial membrane changes may not inevitably lead to B cell death.
Bcl-2 Overexpression Blocks SpA-induced B Cell Death.
In other systems the pro-survival influence of Bcl-2 has been shown to oppose apoptotic signals affecting the mitochondrial pathway (21). To determine whether overexpression of Bcl-2 can affect the cellular fate of B cells triggered by SpA, we challenged TgN (Bcl-2)22 transgenic mice (22) and compared their responses to those of congenic BALB/c mice (Fig. 8) . Although in these mice only a limited percentage of their polyclonal peripheral B cells express VH genes conveying susceptibility to SpA (∼5%), the fate of these targeted B cells can be readily monitored (3, 5, 6). Reiterating earlier findings, when examined after 16 h of SpA exposure there were significant decreases in the proportion of splenic B cells with detectable Fab-mediated SpA-binding activity in mice from both strains (Fig. 8), which reflected SpA-induced depression of sIg levels that was limited to affected B cells (unpublished data). After an additional 96 h in culture, compared with splenic B cells from the control-treated BALB/c mice that included 5.0 ± 0.7% (mean ± SD) SpA-reactive B cells, B cells from SpA-treated BALB/c mice exhibited a significant deletion (62%) with only 1.9 ± 1% of residual SpA-reactive B cells (P < 0.004). Although showing the same trend, compared with the representation in control-treated Bcl-2 transgenic mice (5.6 ± 1.3%), a smaller percentage of splenic SpA-reactive Bcl-2 transgenic mice B cells (28%) were deleted after SpA treatment (4.1 ± 0.8%; P < 0.06). Comparisons also demonstrated that SpA exposure resulted in a significantly greater deletion of SpA-reactive B cells in the BALB/c than in Bcl-2 transgenic mice (P < 0.0095). Hence, the current data indicate that overexpression of Bcl-2 impairs the induction of apoptotic cell death in B cells exposed in vivo to SpA. These findings most likely reflect the interference by Bcl-2 overexpression on the threshold for the induction of death pathways that first affects ΔΨm.
Figure 8.

Effect of Bcl-2 overexpression on SpA-induced B cell deletion. The outcome of in vivo treatments with SpA or a control protein are depicted for Bcl-2 transgenic (TgN(Bcl-2)22) and BALB/c mice. When evaluated ex vivo, 16 h after treatment, SpA exposure greatly decreases the representation of detectable SpA-reactive B cells in either strain of mice. After subsequent in vitro culture, SpA exposure results in the deletion of most of the SpA-reactive BALB/c B cells whereas comparable deletion does not occur in the Bcl-2 transgenic B cells. Analyses were performed on groups of OVA-treated BALB/c mice (n = 5), SpA-treated BALB/c mice (n = 6), OVA-treated Bcl-2 transgenic mice (n = 5), and SpA-treated Bcl-2 transgenic mice (n = 4).
Apoptotic B Cell Death Does Not Require the Fas Death Receptor.
Although the data described above implicate the mitochondria-associated “intrinsic pathway” in the B cell death induced by SpA, there is also extensive evidence that “death receptors” and most notably Fas, can be involved in the regulation of clonal expansion after certain types of B cell activation (for review see 23). However, when CFSE-labeled T15i+/+ splenocytes were adoptively transferred into Fasl gld mice (in which a mutation has inactivated the Fas ligand; 24), we found that there was no impairment in the efficiency of SpA-induced death in Fas-deficient mice (Table I). Moreover, in surveys of apoptotic pathways, comparable levels of activated caspase 3 were induced after SpA treatment of Fas ligand–deficient mice compared with control mice (Fig. 6 E), and there were also no significant differences detected by the TUNEL assay (unpublished data). To also assess the functional outcome of exposure, BALB/c FasLlpr/lpr mice (in which a mutation has inactivated the Fas receptor; 25) and control groups of six mice received 1 mg SpA or control protein and then were assessed 1 mo later. The results from ELISPOT studies of SpA-treated Fas-deficient mice reiterated values determined from previous studies in BALB/c and C57BL/6 mice (5, 6), demonstrating that in vivo SpA exposure induced similar long-term depletion of splenic SpA-reactive IgM-secreting cells in both Fas-sufficient and Fas-deficient mice (unpublished data). Taken together, the data indicate that the Fas ligand/receptor pathway is not required for the induction of apoptotic supraclonal B cell death by SpA.
Discussion
In these studies we have investigated the mechanisms by which protein A of S. aureus acts as a natural B cell toxin to induce the programmed cell death of susceptible VH-targeted B cells. We found that apoptotic B cell death induced by SpA has three distinct phases (Fig. 9 ; reference 26). During the initiation phase, which begins shortly after infusion, there is a rapid down-regulation of surface Ig. Within a few hours this is followed by an up-regulation of surface phenotypic markers of activation on the B cells destined for deletion. These up-regulated B cell surface molecules included CD40, CD95, and MHC II, which have been implicated in interactions with T cells. There is also evidence of induced rapid dissipation of ΔΨm in the targeted B cells.
Figure 9.

Mechanisms of death induced by a B cell superantigen. Encounter of susceptible B cells with pentameric natural SpA is mediated by a nonimmune interaction with BCR that include clan III VH regions. (1) During the initiation phase, this encounter results in down-regulation of BCR and the coreceptors that include CD19 and CD21, and the induction of activation markers (not depicted). (2) A decision phase, lasting many hours follows subsequently, during which B cells might be rescued by second signal. (3) The degradation phase is presumably mediated by the apoptosome, with release of cytochrome C, and eventually leading to the apoptotic caspase-dependent death pathways.
The outcome of the decision phase is seen after 36–48 h of in vivo exposure when the rapid dissipation of ΔΨm in the targeted B cells may eventually lead to the activation of DNases and the “point of no return” (27). However, our in vitro studies suggest that during the decision phase this clonal fate can be aborted by addition of the T cell factors, IL-4 or CD40L (refer to Fig. 7).
The subsequent degradation phase is associated with a full induction of catabolic hydrolases, leading to the activation of caspase 3 and the caspase-associated DNase responsible for TUNEL staining. Cumulatively, from the time of initial exposure, SpA-induced VH-targeted cellular deletion progressed to completion over 4–6 d (5, 6).
Our studies detected SpA-induced mitochondrial changes after only 4 h whereas induction of activated caspase 3 was not detected until >16 h after treatment. Confirming that this BCR-mediated cell death pathway involves separate but sequential phases of induced death, we found that culture of in vivo–exposed B cells in the presence of the pan-caspase inhibitor, Z-VAD, prevented the induction of both the caspase pathway and TUNEL+ DNA fragmentation whereas the in vitro progression of ΔΨm was unaffected. These findings, akin to those from recent in vitro B cell stimulation studies (28), document the caspase independence of the mitochondrial effector phase of this induced death process. Also consistent with this model, we found that the in vivo overexpression of Bcl-2 hindered the apoptosis-specific degradation phase associated with the SpA-induced deletion. These findings therefore document yet another system for which certain Bcl-2 members, which are inserted into the outer mitochondrial membrane, appear to maintain mitochondrial integrity and prevent apoptosis by suppressing mitochondrial membrane permeabilization (22). The overexpression of Bcl-2 may also interfere with this BCR-mediated death by decreasing the rise in calcium after activation. Hence, these findings confirm the early and central role of mitochondria (and perhaps the ER) in this BCR-mediated death process induced by SpA.
The apoptotic pathways demonstrated for SpA-induced B cell deletion reflect features previously reported for activation-induced cell death (AICD) that in other settings has been posited to be the cause of lymphocyte death initiated by engagement of the Ag receptor. Studies of Ig transgenic mice have shown that B lymphocytes, especially centrocytes in a germinal center response, die in response to stimulation by soluble Ag with insufficient BCR cross-linking properties (29–32). This process is believed to reflect physiologic mechanisms for maintaining peripheral tolerance, as it enables newly generated self-reactive cells to be deleted after encounter with their specific auto-Ag (30, 33, 34). Akin to our findings, in vitro studies using human Burkitt's lymphoma cell lines, believed to be representative of germinal center B cells, are also sensitive to apoptosis induced by soluble anti-IgM Abs (35–38) due to the activation of caspase 3 (39, 40). Indeed, anti-IgM–induced apoptosis in lymphoma cell lines is reported to also be initiated at the mitochondrial level by a caspase-independent induced loss of ΔΨm and cytochrome c release, that only later leads to the execution of apoptosis (39). Overexpression of Bcl-2 has been shown to also render Burkitt's cell lines and germinal center B cells resistant to BCR-mediated apoptosis in vitro (39, 41–44) and in vivo (45). Studies of cell lines have suggested that arrest of cell cycle progression contributes to B cell AICD (46, 38). Overall, our findings are consistent with evidence from in vitro B cell systems in that there is a distinct temporal sequence of events after certain pro-apoptotic death stimuli, in which the mitochondrial pathway is affected before the induction of pro-apoptotic caspases and DNases.
Adoptive transfer studies demonstrated that 48 h of in vivo exposure to SpA induced a limited level of in vivo proliferation (Fig. 4) that we did not detect in the splenocytes cultured after only 16 h of in vivo exposure (Fig. 7 B). We postulate that in the former the proliferation was detectable either because of the more prolonged period of in vivo exposure to SpA, or due to the in vivo influence of cytokine and/or cognate help from other bystander mononuclear cells. Despite this induced proliferation, at 48 h after in vivo treatment there was still a mean 36% loss of T15i+/+ splenic B cells in SpA-treated mice. We also enumerated the remaining sets of B cells from each sequential round of proliferation to estimate the number of transferred B cells from which they derived (i.e., progenitors; Table I). This indicated that SpA treatment deleted 54% of the transferred T15i+/+ progenitors (P < 0.0017). By comparison, analogous studies have shown that the T cell superantigen, staphylococcal enterotoxin B, induced a much more vigorous period of in vivo proliferation, which peaked at an ∼66% increase in the representation of susceptible T cells at 72 h after treatment, and apoptotic T cell deletion only followed after a discrete number of cell divisions (47). Hence, although in vivo exposure to either type of superantigen can induce proliferation of the lymphocytes targeted for death, the proliferative response to this staphylococcal B cell superantigen is far more limited than has been reported for a staphylococcal T cell superantigen.
In studies of B cell lines, the BCR-mediated apoptotic pathway has been shown to be independent of the CD95 and TNF-R death receptor systems (38, 39) that have been implicated in the AICD pathways of peripheral CD4+ and CD8+ T lymphocytes (48; for review see 23). In our investigations, although other death domain signaling pathways were not examined, we did confirmed that SpA-induced deletion was unimpaired in Fas-deficient mice (Fig. 6 E). We also found that SpA induced the same death pathways as were induced by in vivo treatment with anti-Ig in the same systems, although the latter was far less efficient (Fig. 2 C and unpublished data). Therefore, our studies confirm that the B cell death pathway induced by SpA is the same as that induced by BCR triggering, which is distinct from that reported to be initiated by Fas triggering (49).
Extending our earlier evidence that deletion is unimpaired in TCRβδ-deficient mice (6), our demonstration of the rescue of SpA-targeted B cells by IL-4 (or CD40L) is also in agreement with comparable recent studies in hen egg lysozyme double transgenic mice (50). From a classical perspective, this clearly indicates that the process of SpA-induced B cell deletion embodies features predicted for the “two signal” model (51) in which an “imbalance” of these signals leads to lymphocyte death. To provide a biologic context for this finding, in an earlier point we demonstrated that naive mice do not mount a T cell response to SpA but immunization with small doses of SpA in adjuvant can induce specific T cell sensitization with a frequency of responder T cells comparable to that associated with other conventional protein Ags (5). Even a robust SpA-specific T cell response is orders of magnitude lower than the very high frequency of B cells targeted for death by SpA. Based on the cumulative findings, we wonder whether a SpA-specific conventional T cell response would be sufficient to affect the fate of a substantial number of the B cells targeted by SpA. However, the outcome of SpA exposure may also be different during staphylococcal infection when T cell superantigens might be coexpressed.
The nature of the VH-targeted BCR-binding interaction of SpA conveys special features that are well suited for the induction of cell death. The immunobiologic properties of SpA are aided by its oligovalent structure, as this secreted membrane protein contains five homologous Ig-binding domains in tandem, and although the native pentameric Ig-binding protein (or even a tetramer) displays deletional properties, we have previously shown that a monomeric form does not (6). In solution, the 42-kD native SpA molecule takes on an extended structure and in every SpA molecule there are at least two accessible Fab-binding sites capable of micromolar binding interactions. In vitro studies have suggested that due to apparent affinity thresholds and differences in effective valency, certain anti-IgM reagents might be more effective at inducing BCR-mediated apoptosis (52). In vivo, the activities of infused Ab-based anti-Ig agents can be greatly impaired by interactions with circulating Ig (Finkelman, F., personal communication). By comparison, the actual in vivo BCR ligand that forms after SpA infusion might be quite complex, as intravenous administration of SpA rapidly results in the formation of high molecular weight complexes with IgG, which can persist for at least 24 h in the circulation (53), and our recent in vitro studies suggest that the interactions of SpA with VH-targeted B cells may in fact be enhanced by the presence of soluble Ig (unpublished data). We speculate that the nature of these in vivo–formed complexes may facilitate signal spreading (for review see 54) that contributes to the in vivo deletional properties of SpA. The different properties of the effective in vitro SpA complex or the inclusion of additional stimulants may explain the very different outcome previously reported for in vitro stimulation with SpA preparations, which are reputed to instead convey pro-survival (i.e., anti-apoptotic) effects for VH-susceptible human peripheral B cells (55, 56). This may reiterate lessons learned from studies of the properties of potential anti-lymphoma Ab agents, as the outcome of in vitro studies of BCR-targeted agents, like SpA, may not be an accurate predictor of in vivo biologic activity (57).
Although they are believed in general to be less vulnerable than B cell precursors to negative selection, mature B cells have also been shown to undergo deletion after in vitro (38, 40, 58, 59, 60) or in vivo (14) treatment with experimental anti-Ig reagents. Therefore, we attempted to determine whether VH-targeted B cells within distinct peripheral subpopulations might be differentially affected by in vivo SpA exposure. Instead, we found that SpA rapidly induced changes in essential phenotypic markers that interfered with our first attempts to make meaningful quantitative measurements of the effects on defined B cell subsets (such as conventional follicular B cells or the CD21hi marginal zone B cells; Fig. 2). In any case, the genomic rearrangement surveys documented a >85% loss of susceptible B cells (Fig. 1) and transcript and cellular analyses have shown comparable or even greater levels of induced deletion (5–7). The cumulative evidence, therefore, indicates that most immature and peripheral mature B cells, including follicular B cells (5), B-1 cells (6, 7), and marginal zone B cells (unpublished data) that bear high affinity VH clan III BCR, are susceptible to SpA-induced deletion.
Although the central features are concordant with those revealed in earlier studies of BCR-mediated negative selection of B lymphocytes, here we have characterized the outcome of in vivo exposure to a natural toxin that induces death in B lymphocytes via targeting of a conserved surface on the VH region. In general, apoptosis is accepted to be important for controlling the size of lymphocyte populations and maintaining homeostasis within the immune system, as it occurs during lymphopoiesis and during antigenic challenges, contributing to the maintenance of self-tolerance and the specificity of immune responses (29, 61). These studies demonstrate that SpA acts by co-opting these physiologic pathways of BCR-mediated apoptotic pathways, and we have recently shown that this can result in the induction of immunologic tolerance (5, 6). These mechanisms are therefore not unique to SpA, but are likely also common to other BCR-targeting toleragens, from those reported in the earliest studies of parenteral administration with anti-Ig reagents (14, 62) to anti-idiotypic Abs (63) and other experimental antigenic systems (64).
In conclusion, these studies extend evidence that SpA has properties with many parallels with those of bacterial and viral proteins classified as superantigens for T cells. In addition, these data also provide an example in which SpA can be used to elucidate the special cellular and molecular pathways of B lymphocytes, and to better understand their roles in the regulation of host immunity. Furthermore, this prototypic B cell superantigen may also have the same affect in the human immune system, for which the invariant VH3-encoded SpA binding site is represented on up to half of all mature B lymphocytes (for reviewed see 1), as recent studies indicate that clan III VH-expressing primate B cells are also susceptible to SpA (65). Therefore, now armed with an understanding of these underlying principles, we postulate that SpA, or analogs, may also prove useful in therapeutic interventions designed to target the B cells that mediate certain diseases.
Acknowledgments
We thank Drs. Thomas Rothstein, Tim Behrens, Robert Ashman, Ann Feeney, Fred Finkelman, and Robert Rickert for suggestions and critical review of the manuscript, and Tatiana Povali for technical assistance with the histology. We also thank the reviewers.
This work was supported by grants AI40305, AR47360, and AI46637 from the National Institutes of Health, and a grant from the Alliance for Lupus Research. C.S. Goodyear is supported by a fellowship from The Cancer Research Institute.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; BCR, B cell receptor; CFSE, carboxyfluorescein diacetate succinimidylester; ΔΨm, mitochondrial transmembrane potential; PI, propidium iodide; SpA, protein A of Staphylococcus aureus; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; Z-VAD, Z-Val-Ala-Asp(OMe)-fluoromethyl ketone.
References
- 1.Silverman, G.J., and C.S. Goodyear. 2002. A model B-cell superantigen and the immunobiology of B lymphocytes. Clin. Immunol. 102:117–134. [DOI] [PubMed] [Google Scholar]
- 2.Graille, M., E.A. Stura, A.L. Corper, B.J. Sutton, M.J. Taussig, J.B. Charbonnier, and G.J. Silverman. 2000. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA. 97:5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cary, S.P., J. Lee, R. Wagenknecht, and G.J. Silverman. 2000. Characterization of superantigen-induced clonal deletion with a novel clan III-restricted avian monoclonal antibody: exploiting evolutionary distance to create antibodies specific for a conserved VH region surface. J. Immunol. 164:4730–4741. [DOI] [PubMed] [Google Scholar]
- 4.Cary, S., M. Krishnan, T.N. Marion, and G.J. Silverman. 1999. The murine clan V(H) III related 7183, J606 and S107 and DNA4 families commonly encode for binding to a bacterial B cell superantigen. Mol. Immunol. 36:769–776. [DOI] [PubMed] [Google Scholar]
- 5.Silverman, G.J., J.V. Nayak, K. Warnatz, S. Cary, H. Tighe, and V.E. Curtiss. 1998. The dual phases of the response to neonatal exposure to a VH family-restricted staphylococcal B-cell superantigen. J. Immunol. 161:5720–5732. [PubMed] [Google Scholar]
- 6.Silverman, G.J., S.P. Cary, D.C. Dwyer, L. Luo, R. Wagenknecht, and V.E. Curtiss. 2000. A B cell superantigen-induced persistent “Hole” in the B-1 repertoire. J. Exp. Med. 192:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman, G.J. 2001. Adoptive transfer of a superantigen-induced “hole” in the repertoire of natural IgM-secreting cells. Cell. Immunol. 209:76–80. [DOI] [PubMed] [Google Scholar]
- 8.Feeney, A.J. 1992. Predominance of VH-D-JH junctions occurring at sites of short sequence homology results in limited junctional diversity in neonatal antibodies. J. Immunol. 149:222–229. [PubMed] [Google Scholar]
- 9.Bernardi, P., L. Scorrano, R. Colonna, V. Petronilli, and F. Di Lisa. 1999. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264:687–701. [DOI] [PubMed] [Google Scholar]
- 10.Martin-Orozco, E., H. Kobayashi, J. Van Uden, M.D. Nguyen, R.S. Kornbluth, and E. Raz. 1999. Enhancement of antigen-presenting cell surface molecules involved in cognate interactions by immunostimulatory DNA sequences. Int. Immunol. 11:1111–1118. [DOI] [PubMed] [Google Scholar]
- 11.Lane, P., T. Brocker, S. Hubele, E. Padovan, A. Lanzavecchia, and F. McConnell. 1993. Soluble CD40 ligand can replace the normal T cell–derived CD40 ligand signal to B cells in T cell–dependent activation. J. Exp. Med. 177:1209–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozlowski, L.M., A.M. Soulika, G.J. Silverman, J.D. Lambris, and A.I. Levinson. 1996. Complement activation by a B cell superantigen. J. Immunol. 157:1200–1206. [PubMed] [Google Scholar]
- 13.Phee, H., W. Rodgers, and K.M. Coggeshall. 2001. Visualization of negative signaling in B cells by quantitative confocal microscopy. Mol. Cell. Biol. 21:8615–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkelman, F.D., J.M. Holmes, O.I. Dukhanina, and S.C. Morris. 1995. Cross-linking of membrane immunoglobulin D, in the absence of T cell help, kills mature B cells in vivo. J. Exp. Med. 181:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taki, S., M. Meiering, and K. Rajewsky. 1993. Targeted insertion of a variable region gene into the immunoglobulin heavy chain locus. Science. 262:1268–1271. [DOI] [PubMed] [Google Scholar]
- 16.Lyon, A., and C. Parish. 1994. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171:131–137. [DOI] [PubMed] [Google Scholar]
- 17.Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 391:43–50. [DOI] [PubMed] [Google Scholar]
- 18.Liu, X., H. Zou, C. Slaughter, and X. Wang. 1997. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 89:175–184. [DOI] [PubMed] [Google Scholar]
- 19.Xiang, J., D.T. Chao, and S.J. Korsmeyer. 1996. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 93:14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy, N.J., M.K. Whyte, C.S. Gilbert, and G.I. Evan. 1997. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 136:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu, S., A. Konishi, T. Kodama, and Y. Tsujimoto. 2000. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA. 97:3100–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Budd, R.C. 2001. Activation-induced cell death. Curr. Opin. Immunol. 13:356–362. [DOI] [PubMed] [Google Scholar]
- 24.Davidson, W.F., K.L. Holmes, J.B. Roths, and H.C. Morse, III. 1985. Immunologic abnormalities of mice bearing the gld mutation suggest a common pathway for murine nonmalignant lymphoproliferative disorders with autoimmunity. Proc. Natl. Acad. Sci. USA. 82:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe-Fukunaga, R., C.I. Brannan, N.G. Copeland, N.A. Jenkins, and S. Nagata. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 356:314–317. [DOI] [PubMed] [Google Scholar]
- 26.Kroemer, G., and J.C. Reed. 2000. Mitochondrial control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- 27.Ferri, K.F., and G. Kroemer. 2000. Control of apoptotic DNA degradation. Nat. Cell Biol. 2:E63–E64. [DOI] [PubMed] [Google Scholar]
- 28.Berard, M., P. Mondiere, M. Casamayor-Palleja, A. Hennino, C. Bella, and T. Defrance. 1999. Mitochondria connects the antigen receptor to effector caspases during B cell receptor-induced apoptosis in normal human B cells. J. Immunol. 163:4655–4662. [PubMed] [Google Scholar]
- 29.Goodnow, C.C., J.G. Cyster, S.B. Hartley, S.E. Bell, M.P. Cooke, J.I. Healy, S. Akkaraju, J.C. Rathmell, S.L. Pogue, and K.P. Shokat. 1995. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 59:279–368. [DOI] [PubMed] [Google Scholar]
- 30.Shokat, K.M., and C.C. Goodnow. 1995. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature. 375:334–338. [DOI] [PubMed] [Google Scholar]
- 31.Pulendran, B., G. Kannourakis, S. Nouri, K.G. Smith, and G.J. Nossal. 1995. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature. 375:331–334. [DOI] [PubMed] [Google Scholar]
- 32.Han, S., B. Zheng, J. Dal Porto, and G. Kelsoe. 1995. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J. Exp. Med. 182:1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami, M., T. Tsubata, M. Okamoto, A. Shimizu, S. Kumagai, H. Imura, and T. Honjo. 1992. Antigen-induced apoptotic death of Ly-1 B cells responsible for autoimmune disease in transgenic mice. Nature. 357:77–80. [DOI] [PubMed] [Google Scholar]
- 34.Homburg, C.H., M. de Haas, A.E. von dem Borne, A.J. Verhoeven, C.P. Reutelingsperger, and D. Roos. 1995. Human neutrophils lose their surface Fc gamma RIII and acquire Annexin V binding sites during apoptosis in vitro. Blood. 85:532–540. [PubMed] [Google Scholar]
- 35.Peter, M.E., J. Dhein, A. Ehret, S. Hellbardt, H. Walczak, G. Moldenhauer, and P.H. Krammer. 1995. APO-1 (CD95)-dependent and -independent antigen receptor-induced apoptosis in human T and B cell lines. Int. Immunol. 7:1873–1877. [DOI] [PubMed] [Google Scholar]
- 36.An, S., and K.A. Knox. 1996. Ligation of CD40 rescues Ramos-Burkitt lymphoma B cells from calcium ionophore- and antigen receptor-triggered apoptosis by inhibiting activation of the cysteine protease CPP32/Yama and cleavage of its substrate PARP. FEBS Lett. 386:115–122. [DOI] [PubMed] [Google Scholar]
- 37.Falk, M.H., B.C. Trauth, K.M. Debatin, C. Klas, C.D. Gregory, A.B. Rickinson, A. Calender, G.M. Lenoir, J.W. Ellwart, P.H. Krammer, et al. 1992. Expression of the APO-1 antigen in Burkitt lymphoma cell lines correlates with a shift towards a lymphoblastoid phenotype. Blood. 79:3300–3306. [PubMed] [Google Scholar]
- 38.Lens, S.M., B.F. den Drijver, A.J. Potgens, K. Tesselaar, M.H. van Oers, and R.A. van Lier. 1998. Dissection of pathways leading to antigen receptor-induced and Fas/CD95-induced apoptosis in human B cells. J. Immunol. 160:6083–6092. [PubMed] [Google Scholar]
- 39.Bouchon, A., P.H. Krammer, and H. Walczak. 2000. Critical role for mitochondria in B cell receptor-mediated apoptosis. Eur. J. Immunol. 30:69–77. [DOI] [PubMed] [Google Scholar]
- 40.Rickers, A., E. Brockstedt, M.Y. Mapara, A. Otto, B. Dorken, and K. Bommert. 1998. Inhibition of CPP32 blocks surface IgM-mediated apoptosis and D4-GDI cleavage in human BL60 Burkitt lymphoma cells. Eur. J. Immunol. 28:296–304. [DOI] [PubMed] [Google Scholar]
- 41.Liu, Y.J., D.Y. Mason, G.D. Johnson, S. Abbot, C.D. Gregory, D.L. Hardie, J. Gordon, and I.C. MacLennan. 1991. Germinal center cells express bcl-2 protein after activation by signals which prevent their entry into apoptosis. Eur. J. Immunol. 21:1905–1910. [DOI] [PubMed] [Google Scholar]
- 42.Pittner, B.T., and E.C. Snow. 1998. Strength of signal through BCR determines the fate of cycling B cells by regulating the expression of the Bcl-2 family of survival proteins. Cell. Immunol. 186:55–62. [DOI] [PubMed] [Google Scholar]
- 43.Knox, K.A., M. Finney, A.E. Milner, C.D. Gregory, M.J. Wakelam, R.H. Michell, and J. Gordon. 1992. Second-messenger pathways involved in the regulation of survival in germinal-centre B cells and in Burkitt lymphoma lines. Int. J. Cancer. 52:959–966. [DOI] [PubMed] [Google Scholar]
- 44.Ning, Z.Q., J.D. Norton, J. Li, and J.J. Murphy. 1997. Distinct mechanisms for rescue from apoptosis in Ramos human B cells by signalling through CD40 and interleukin-4 receptor: role for inhibition of an early response gene, Berg36. Biochem. Soc. Trans. 25:306S. [DOI] [PubMed] [Google Scholar]
- 45.Lang, J., B. Arnold, G. Hammerling, A.W. Harris, S. Korsmeyer, D. Russell, A. Strasser, and D. Nemazee. 1997. Enforced Bcl-2 expression inhibits antigen-mediated clonal elimination of peripheral B cells in an antigen dose-dependent manner and promotes receptor editing in autoreactive, immature B cells. J. Exp. Med. 186:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andjelic, S., and H.C. Liou. 1998. Antigen receptor-induced B lymphocyte apoptosis mediated via a protease of the caspase family. Eur. J. Immunol. 28:570–581. [DOI] [PubMed] [Google Scholar]
- 47.Renno, T., A. Attinger, S. Locatelli, T. Bakker, S. Vacheron, and H.R. MacDonald. 1999. Cutting edge: apoptosis of superantigen-activated T cells occurs preferentially after a discrete number of cell divisions in vivo. J. Immunol. 162:6312–6315. [PubMed] [Google Scholar]
- 48.Dhein, J., H. Walczak, C. Baumler, K.M. Debatin, and P.H. Krammer. 1995. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature. 373:438–441. [DOI] [PubMed] [Google Scholar]
- 49.Rothstein, T.L., J.K. Wang, D.J. Panka, L.C. Foote, Z. Wang, B. Stanger, H. Cui, S.T. Ju, and A. Marshak-Rothstein. 1995. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 374:163–165. [DOI] [PubMed] [Google Scholar]
- 50.Morris, S.C., N.L. Dragula, and F.D. Finkelman. 2002. IL-4 promotes Stat6-dependent survival of autoreactive B cells in vivo without inducing autoantibody production. J. Immunol. 169:1696–1704. [DOI] [PubMed] [Google Scholar]
- 51.Bretscher, P., and M. Cohn. 1970. A theory of self-nonself discrimination: paralysis and induction involve the recognition of one or two determinants on antigen, respectively. Science. 163:1042–1049. [DOI] [PubMed] [Google Scholar]
- 52.Mongini, P.K., Q. Liu, M.A. Vilensky, P.F. Highet, and J.K. Inman. 1998. Evidence for an upper affinity threshold for anti-IgM-induced apoptosis in a human B-cell lymphoma. Blood. 92:3756–3771. [PubMed] [Google Scholar]
- 53.Matsuuchi, L., and M.R. Gold. 2001. New views of BCR structure and organization. Curr. Opin. Immunol. 13:270–277. [DOI] [PubMed] [Google Scholar]
- 54.Das, C., and J.J. Langone. 1987. Correlation between antitumor activity of protein A and in vivo formation of defined high molecular weight complexes with immunoglobulin G in BALB/c mice. Cancer Res. 47:2002–2007. [PubMed] [Google Scholar]
- 55.Kristiansen, S.V., V. Pascual, and P.E. Lipsky. 1994. Staphylococcal protein A induces biased production of Ig by VH3-expressing B lymphocytes. J. Immunol. 153:2974–2984. [PubMed] [Google Scholar]
- 56.Kozlowski, L.M., S.R. Kunning, Y. Zheng, L.M. Wheatley, and A.I. Levinson. 1995. Staphylococcus aureus Cowan I-induced human immunoglobin responses: preferential IgM rheumatoid factor production and VH3 mRNA expression by protein A-binding B cells. J. Clin. Immunol. 15:145–151. [DOI] [PubMed] [Google Scholar]
- 57.Tutt, A.L., R.R. French, T.M. Illidge, J. Honeychurch, H.M. McBride, C.A. Penfold, D.T. Fearon, R.M. Parkhouse, G.G. Klaus, and M.J. Glennie. 1998. Monoclonal antibody therapy of B cell lymphoma: signaling activity on tumor cells appears more important than recruitment of effectors. J. Immunol. 161:3176–3185. [PubMed] [Google Scholar]
- 58.Parry, S.L., J. Hasbold, M. Holman, and G.G. Klaus. 1994. Hypercross-linking surface IgM or IgD receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and anti-CD40. J. Immunol. 152:2821–2829. [PubMed] [Google Scholar]
- 59.Kozono, Y., B.L. Kotzin, and V.M. Holers. 1996. Resting B cells from New Zealand Black mice demonstrate a defect in apoptosis induction following surface IgM ligation. J. Immunol. 156:4498–4503. [PubMed] [Google Scholar]
- 60.Parry, S.L., M.J. Holman, J. Hasbold, and G.G. Klaus. 1994. Plastic-immobilized anti-mu or anti-delta antibodies induce apoptosis in mature murine B lymphocytes. Eur. J. Immunol. 24:974–979. [DOI] [PubMed] [Google Scholar]
- 61.Green, D.R., and D.W. Scott. 1994. Activation-induced apoptosis in lymphocytes. Curr. Opin. Immunol. 6:476–487. [DOI] [PubMed] [Google Scholar]
- 62.Lawton, A.R., and M.D. Cooper. 1974. Modification of B lymphocyte differentiation by anti-immunoglobulins. Contemp. Top. Immunobiol. 3:193–225. [DOI] [PubMed] [Google Scholar]
- 63.Kohler, H., D.R. Kaplan, and D.S. Strayer. 1974. Clonal depletion in neonatal tolerance. Science. 186:643–644. [DOI] [PubMed] [Google Scholar]
- 64.Dintzis, H.M., R.Z. Dintzis, and B. Vogelstein. 1976. Molecular determinants of immunogenicity: the immunon model of immune response. Proc. Natl. Acad. Sci. USA. 73:3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silverman, G.J., and C.S. Goodyear. 2001. In vivo VH targeted B-cell apoptotic deletion in mice and non-human primates. FASEB J. 15:A694. [Google Scholar]
- 66.Parish, C.R., and H.S. Warren. 2001. Use of the intracellular fluorescent dye CFSE to monitor lymphocyte migration and proliferation. In Current Protocols in Immunology. J.E. Coligan, editor. John Wiley & Sons, Inc., New York. 4.9.1–4.9.8. [DOI] [PubMed]