

Abstract
Lymph node (LN) function depends on T and B cell compartmentalization, antigen presenting cells, and high endothelial venules (HEVs) expressing mucosal addressin cell adhesion molecule (MAdCAM-1) and peripheral node addressin (PNAd), ligands for naive cell entrance into LNs. Luminal PNAd expression requires a HEV-restricted sulfotransferase (HEC-6ST). To investigate LTαβ's activities in lymphoid organogenesis, mice simultaneously expressing LTα and LTβ under rat insulin promoter II (RIP) control were compared with RIPLTα mice in a model of lymphoid neogenesis and with LTβ−/− mice. RIPLTαβ pancreata exhibited massive intra-islet mononuclear infiltrates that differed from the more sparse peri-islet cell accumulations in RIPLTα pancreata: separation into T and B cell areas was more distinct with prominent FDC networks, expression of lymphoid chemokines (CCL21, CCL19, and CXCL13) was more intense, and L-selectin+ cells were more frequent. In contrast to the predominant abluminal PNAd pattern of HEV in LTβ−/− MLN and RIPLTα pancreatic infiltrates, PNAd was expressed at the luminal and abluminal aspects of HEV in wild-type LN and in RIPLTαβ pancreata, coincident with HEC-6ST. These data highlight distinct roles of LTα and LTαβ in lymphoid organogenesis supporting the notion that HEC-6ST–dependent luminal PNAd is under regulation by LTαβ.
Keywords: lymphoid organogenesis, chemokines, sulfotransferase, inflammation, high endothelial venules
Introduction
Members of the TNF/lymphotoxin (LT)* ligand-receptor families and lymphoid chemokines play critical roles in LN development. The design of LNs serves adaptive immunity by facilitating interactions between antigen-presenting cells and responsive lymphocytes. Thus, LN function depends on the presence of naive, L-selectin+ lymphocytes, distinct T and B cell compartments, antigen presenting cells, stromal cells, and specialized blood vessels called high endothelial venules (HEVs). The cells of HEV express peripheral node addressin (PNAd) and mucosal addressin cell adhesion molecule (MAdCAM-1), adhesion molecules that serve as ligands for L-selectin and α4β7, respectively, expressed by blood-borne T and B cells required for their extravasation into LNs (1).
During early development, HEVs of all LNs express MAdCAM-1, but soon after birth peripheral LN-HEVs (PLN-HEVs) switch to PNAd, whereas mesenteric LN-HEVs (MLN-HEVs) continue to express MAdCAM-1 in addition to PNAd (2). PNAd, an L-selectin ligand, detected by the prototypic antibody MECA 79 (3, 4), is a sulfo sialyl-Lewisx determinant common to a defined set of glycoproteins (GlyCAM-1, CD34, podocalyxin, or MAdCAM-1; reference 5). A HEV-specific sulfotransferase, abbreviated here as HEC-6ST, that mediates the sulfation at the C6-position of GlcNAc residues of these glycoproteins was described by two groups (6, 7). This enzyme is necessary for luminal expression of PNAd, as LNs of HEC-6ST–deficient mice lack apical, luminal PNAd expression (8). However, PLN- and MLN-HEV of HEC-6ST–deficient mice retain basal, abluminal PNAd expression, a pattern similar to that seen in Peyer's patches (PP). HEC-6ST–deficient mice also exhibit a dramatic reduction in LN cell number consistent with the view that luminal PNAd expression has an important role in LN cell recruitment (8). The requirement of HEC-6ST for luminal PNAd expression and its importance in cell recruitment to LNs indicates a crucial role for this enzyme in LN development. Upstream factors that regulate HEC-6ST expression remain to be elucidated. We have previously suggested that the LTαβ complex may be involved (9).
Several lines of evidence demonstrate that LTα and LTαβ play crucial, nonredundant roles in lymphoid organ development. Mice deficient in LTα, a homotrimeric soluble factor that binds to the two TNF receptors, TNFRI and TNFRII (10), lack all LNs and PP, and have highly disorganized spleens (11, 12). Furthermore, transgenic expression of LTα under the control of the rat insulin gene promoter element (RIPLTα) partially restores lymphoid organs and function to LTα−/− mice (13). LTβ, a membrane-bound protein, on its own is nonfunctional, but can form heterotrimeric complexes with LTα (10, 14). While LTα2β1 can bind TNFRI and TNFRII, its signaling capacity through these receptors has not yet been elucidated. LTα1β2 signals through another member of the TNF receptor family, LTβR (15, 16). LTβ−/− mice lack all PLNs and PP, but retain MLNs and cervical LNs, and their splenic defects are less pronounced than those of LTα−/− mice (17, 18). In addition, treatment of pregnant mice with LTβR-Ig fusion protein results in progeny that lack most PLNs, but retain MLNs (19).
Chemokines characteristic of LNs include CCL19 (EBV-induced molecule 1 ligand chemokine [ELC]) and CCL21 (secondary lymphoid chemokine [SLC]) that are constitutively expressed by T cell zone stromal cells (20). CCL21 mRNA is also expressed by HEVs in PP and LNs (21, 22). In addition, CXCL13 (B lymphocyte chemoattractant [BLC]) is constitutively expressed by stromal cells in B cell follicles (23). As the spleens of LTα−/− and LTβ−/− mice exhibit a dramatic reduction in lymphoid chemokines (24), LT-regulated expression of these chemokines is one likely key mechanism in lymphoid organ development.
Transgenic mice that express TNF/LT ligands or either of the above mentioned lymphoid chemokines under RIP control develop cellular accumulations with characteristics of lymphoid organs at the sites of transgene expression (25–31). This ectopic lymphoid tissue, organized through a process called lymphoid neogenesis, contains elements of both chronic inflammation and lymphoid organs and has been observed in several autoimmune diseases (32). Interestingly, in some cases, these cellular accumulations are vascularized with endothelia showing morphologic characteristics of HEV expressing PNAd and MAdCAM-1. For example, RIPLTα mice show HEV-like vessels within pancreatic infiltrates that express MAdCAM-1 and low levels of PNAd (27, 33). These cellular accumulations are reminiscent of a developing LN, and are predominantly mediated by LTα3 through TNFRI (34). In these RIPLTα mice LTβ, most likely in the form of LTαβ expressed by infiltrating lymphocytes, is implicated in PNAd expression since the infiltrates of RIPLTα.LTβ−/− mice lack PNAd reactivity and exhibit a reduction in L-selectin+ cells, although MAdCAM-1 expression remains prominent (33). Furthermore, LTα3 treatment can induce MAdCAM-1 in an endothelial cell line in vitro (35). These data are consistent with the previously articulated hypothesis that LTα alone suffices for MAdCAM-1 expression and MLN organogenesis but LTαβ is necessary for PLN development and their population by L-selectin+ cells through regulation of PNAd (9). The mechanisms by which LTαβ regulates PNAd expression have not been elucidated. Here we investigated the contributions of LTα and LTαβ to the process of lymphoid neogenesis with particular attention to the HEV phenotype in mice simultaneously transgenic for LTα and LTβ, each under RIP control and in LTβ−/− MLN. Coexpression of LTα and LTβ in the pancreas resulted in invasive, intra-islet cellular accumulations that differed both qualitatively and quantitatively from those seen in RIPLTα transgenics: separation into T–B cell areas was more distinct, expression of lymphoid chemokines was more intense, and L-selectin+ cells made up a much higher proportion of the mononuclear infiltrate. Most notably, PNAd expression in HEV was prominent in luminal locations. With a recently developed antibody, we showed that expression of HEC-6ST depended on coexpression of LTα and LTβ, and correlated with luminal expression of PNAd in HEV.
Materials and Methods
Mice.
To generate the RIPLTβ transgene construct, the rat insulin promoter II was recloned in Bluescript SK+ (RIP-BSK) from a RIP-TNFα construct provided by Dr. Allison Greene (Section of Immunobiology, Yale University School of Medicine, New Haven, CT). A genomic mouse LTβ DNA fragment was generated by anchored PCR using a genomic clone PNN03/PL001 provided by Dr. Jeffery Browning (Biogen, Cambridge, MA) as template, introducing an NheI cloning site and Kozak consensus sequence upstream of the initiation codon and a HindIII site downstream of the polyadenylation sequence. This fragment was inserted in RIP-BSK downstream of RIP, and recloned. The RIPLTβ fragment was excised from a preparative gel, purified according to standard protocol to apparent homogeneity and confirmed by resequencing. RIPLTβ DNA was injected into SJLBL/6 F2 oocytes by the Yale Animal Resource Center Transgenics Mouse Facility as described previously (26). Positive founders were identified by PCR and Southern blot analysis of tail DNA using a 32P-labeled mouse LTβ probe prepared by random-primer labeling (Amersham Biosciences).
Mice transgenic for RIPLTα have been described previously (26, 27). These mice were bred to RIPLTβ transgenic mice. Progeny that were heterozygous for both transgenes were used for further study. C57BL/6 mice were purchased from the National Cancer Institute of the National Institutes of Health. LTβ-deficient mice have been described previously (18). All mice were studied under a protocol approved by the Yale University Institutional Animal Care and Use Committee.
Preparation of HEC-6ST Antibody.
Polyclonal rabbit anti-sera specific for mouse sulfotransferase was generated by immunization with three different peptides derived from mouse HEC-6ST. Peptides were specifically chosen for sequences lacking similarity to previously described murine sulfotransferases (36). The peptides were: (a) CHMSVHRHLSQREESRR-COOH; (b) KIICKSQVDIVKAIQTLPE-COOH; (c) RGKGMGQHAFHTNC-COOH. Peptides were synthesized at the W.M. Keck Foundation facility at Yale University and included a cysteine residue for conjugation to keyhole limpet hemocyanin (KLH; Calbiochem). Peptide-KLH conjugation was performed using the chemical cross-linker SPDP (Pierce Chemical Co.) according to manufacturers' instructions. Pooled peptide–KLH conjugates (2 mg/ml) were mixed with TiterMax Gold (CytRx Corporation) in a 1:1 ratio and injected subcutaneously into New Zealand white rabbits. After the third boost, the serum was found to specifically stain HEV in sections of PLNs, and the binding was inhibited by incubation with “peptide c” (unpublished data). The antiserum was then purified by affinity chromatography using “peptide c” as sorbent, and tested in indirect immunofluorescence.
Histologic Analysis.
Standard hematoxylin and eosin staining procedures were used on formalin fixed, paraffin embedded tissue for histological analysis. Slide preparation and staining was performed by the Dermatopathology Laboratory at the Yale University School of Medicine.
For immunohistochemistry, pancreas and kidney tissue were dissected and immediately frozen in OCT compound (Tissue-Tek) on dry ice. Sections of 7 μm were cut onto poly-l-lysine–coated glass slides (Sigma Diagnostics), fixed in 100% cold acetone for 10 min and stored at −70°C. For staining, slides were air-dried at room temperature and blocked in 0.5% TNB (NEN Life Science Products) in TRIS-HCl for 45 min, followed by incubation in 1% mouse serum in PBS containing 0.1M Tris/0.1% Tween 20 (blocking solution) for 30 min. Sections were stained in a humidified tray with 2 μg/ml of anti-B220, anti-CD4, anti-CD8, anti-CD35 (CR1), anti-PNAd (MECA 79), and anti–MAdCAM-1 (MECA 367) primary antibodies diluted in blocking solution. Biotinylated species-specific secondary antibodies were diluted 1:250 in blocking solution and incubated on sections for 30 min. All antibodies were obtained from BD Biosciences. Slides were treated with streptavidin-conjugated alkaline phosphatase VectaStain reagent (Vector Laboratories), according to the manufacturer's protocol (Vector Laboratories). Enzyme reactivity was detected using Vector Red (Vector Laboratories) containing 100 mM levamisole to inhibit endogenous alkaline phosphatase activity. In all experiments, species-specific isotype-matched irrelevant antibody was used as a control. Sections were counterstained, as needed, with 0.05% methyl green (Sigma-Aldrich) and mounted in Crystal/Mount (Biomeda).
For immunofluorescence, PLN and pancreas tissue were isolated and slides prepared as above. Sections were blocked with 5% mouse serum/3% BSA in PBS, pH 7.4. Purified rabbit anti-HEC-6ST antibody was diluted 1:1,000 in blocking solution, and MECA 79 antibody (BD Biosciences) was used at 2 μg/ml. Anti-HEC-6ST and MECA 79 primary antibodies were incubated on slides for 1 h at ambient temperature. For HEC-6ST detection, sections were incubated with biotinylated goat anti–rabbit IgG (Jackson ImmunoResearch Laboratories) followed by incubation with Cy2-conjugated streptavidin (Jackson ImmunoResearch Laboratories). MECA 79 was detected by incubation with Cy3-conjugated goat anti-Rat IgM antibody (Jackson ImmunoResearch Laboratories). Sections were counterstained with 20% Harris' hematoxylin (Sigma Diagnostics) and mounted with Fluorosave (Calbiochem). Slides were analyzed by fluorescence microscopy using a Carl Zeiss Microimaging, Inc. Axioskop microscope.
In Situ Hybridization.
The technique previously described by Hjelmstrom et al. (37) was used. Briefly, pancreas and kidney were isolated and fixed in 4% paraformaldehyde/0.14 M Sorenson's phosphate buffer overnight followed by 30% sucrose/4% paraformaldehyde in 0.14 M Sorenson's phosphate buffer. Tissue was frozen in OCT compound (Tissue-Tek) on dry ice and sections of 7 μm were cut onto poly-l-lysine coated slides. Sections were washed in PBS, prehybridized for 1–2 h, and hybridized overnight at 54°C with sense or antisense digoxigenin (DIG)-labeled riboprobes in hybridization solution. Sections were subjected to high stringency washes followed by overnight incubation with alkaline phosphatase–conjugated sheep anti-DIG antibody (Roche) and developed with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP; Boehringer). Signal development was stopped by washing slides in 10 mM Tris/1 mM EDTA, pH 8.0. Sections were counterstained with 0.05% methyl green and mounted in Crystal Mount (Biomeda). Riboprobes used for in situ hybridization included: LTα, LTβ, CCL21, CCL19, and CXCL13 sense and antisense probes (37, 38). CCL19 cDNA was a generous gift from Dr. Jason Cyster, University of California, San Francisco, San Francisco, CA. All DIG-labeled probes were prepared as described previously (37). In all experiments, sense probes were used as negative controls.
Preparation of Lymphocytes.
PLN and pancreas tissue were harvested from mice after intracardial perfusion with HBSS (GIBCO BRL). PLN was homogenized in HBSS and passed over a 40 μm cell strainer (Becton Dickinson). Pancreatic infiltrates were prepared by digestion of pancreas in RPMI 1640 (GIBCO BRL) supplemented with penicillin/streptomycin, 0.01% DNase I (Roche), and 0.15% Collagenase P (Roche). Tissue was incubated for 15 min in a 37°C shaking waterbath. Collagenase digestion was stopped by washing cells with excess cold HBSS. Digested tissue was passed over a 40 μm cell strainer to isolate pancreatic-infiltrating cells. Isolated cells, from all tissues, were resuspended in HBSS and quantitated using a hemacytometer.
Flow Cytometric Analysis.
FACS® staining was performed by conventional staining procedures using either FITC- or PE-conjugated anti-CD4, anti-CD8, anti-B220, anti-CD11c, anti-CD11b, and anti-CD62L antibodies (all from BD Biosciences). Data were collected on a Becton Dickinson FACScan™ flow cytometer and analyzed with FlowJo software.
Results
Generation of RIPLTβ Transgenic Mice.
The RIPLTβ transgenic construct was designed with the rat insulin promoter II (RIP) driving expression of murine LTβ. Southern blot analysis and PCR identified 14 transgene positive mice of 62 mice (unpublished data). Germline transmission was confirmed in F1 crosses of RIPLTβ transgenic mice to C57BL/6 mice. By in situ hybridization with a DIG-labeled LTβ anti-sense riboprobe, LTβ transgene transcription was apparent in the islets of Langerhans, presumably in the insulin-producing β cells (Fig. 1 A). Low LTβ mRNA was also apparent in the kidney in proximal convoluted tubules (Fig. 1 B) in a pattern similar to the LTα expression associated with the promoter in the RIPLTα kidney (27). Two lines of transgenic mice were selected for further study: line 9311 carries approximately two copies and line 9327 carries 20 copies of the transgene. These lines were crossed to RIPLTα mice to generate RIPLTαβ mice. Comparable results were obtained with the progeny of the two lines.
Figure 1.
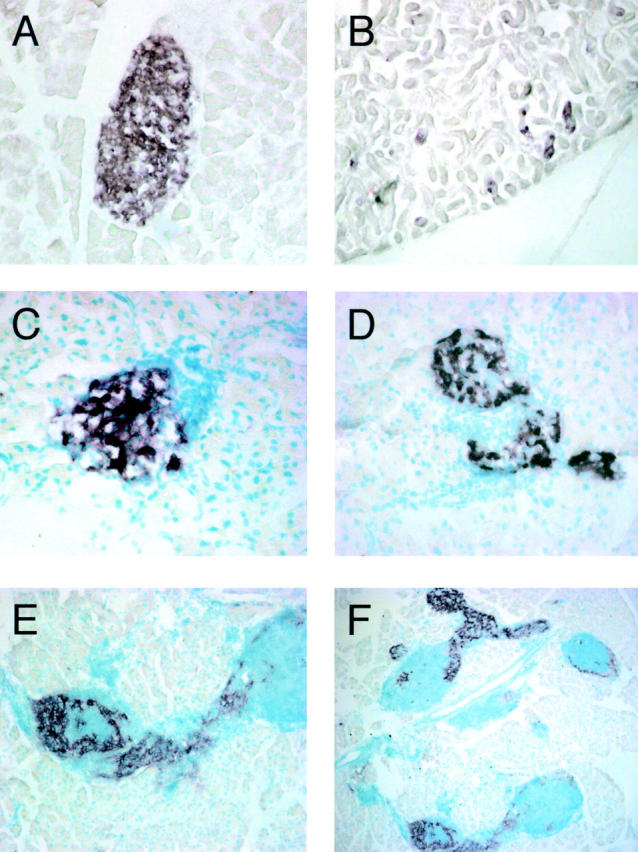
LT expression in RIPLTβ and RIPLTαβ mice. In situ hybridization for RIPLTβ transgene transcription with a DIG-labeled antisense LTβ riboprobe on RIPLTβ pancreas (A) and kidney (B). In situ hybridization with DIG-labeled LTα (C and D) and LTβ (E and F) antisense riboprobes on RIPLTα (C) and RIPLTαβ (D–F) pancreas. E is a higher magnification of F showing the extensive cellular accumulation in RIPLTαβ pancreatic tissue. Objective 40× (A–E); objective 5× (F).
Extensive Intra-islet Invasion by Cellular Infiltrates in RIP-LTαβ Pancreata.
RIPLTα, RIPLTβ, and RIPLTαβ pancreata were analyzed by in situ hybridization with LTα and LTβ antisense riboprobes, and tissue sections were counterstained with methyl green to evaluate leukocytic infiltration. When examined at 5–7 wk of age, high levels of LTα mRNA accumulation were found in RIPLTα pancreata (Fig. 1 C). In addition, a very low signal for LTβ was seen within the cellular infiltrate associated with the islets, most likely resulting from transcriptional activity of infiltrating T and B cells (unpublished data). As noted above, RIPLTβ pancreata revealed LTβ mRNA, but no LTα mRNA. In RIPLTαβ mice, both LTα and LTβ were transcribed selectively in the islets (Fig. 1, D–F). A peri-islet leukocyte accumulation in the periductal areas of the pancreas just outside the islet developed in ∼40% of the islets in RIPLTα mice, confirming previous reports (Fig. 2 B; reference 34). Consistent with the fact that LTβ in the absence of LTα does not form a functional ligand, no alterations were seen in any tissue investigated in RIPLTβ mice (Fig. 2 C). However, RIPLTαβ mice, when examined as early as 5 wk of age, exhibited extensive mononuclear cell infiltrates in the pancreas at the sites of transgene expression (Fig. 2 D). Fig. 1 F shows multiple LT β positive islets encompassed and invaded by cellular infiltrates. Unlike the peri-islet infiltrate observed in RIPLTα pancreata (Fig. 2 B), the cellular accumulations in RIPLTαβ pancreata massively invaded the islet distorting normal architecture (Figs. 1 F and 2 D). Moreover, >90% of the islets in RIPLTαβ pancreata showed at least some pathology. The extent of individual islet involvement within the same pancreas ranged from almost total obliteration to a relatively intact islet as shown in Fig. 1 F. In every RIPLTαβ pancreas evaluated at least some β cells remained, as detected by transgene transcription (Fig. 1, D–F) and insulin staining (unpublished data). Consistent with the much higher level of cellular accumulation, ∼10 times more mononuclear cells (4.5 × 106 cells at 5 wk of age) were recovered from individual RIPLTαβ pancreata than from RIPLTα pancreata.
Figure 2.

Histologic comparison of C57BL/6 and RIPLT pancreata. Hematoxylin and eosin stain of pancreas of C57BL/6 (A), RIPLTα (B), RIPLTβ (C), and RIPLTαβ (D) mice. RIPLTβ pancreas is indistinguishable from that of nontransgenic mice. RIPLTα exhibit a peri-islet cellular accumulation in contrast to the extensive invasive intra-islet cellular accumulation observed in RIPLTαβ mice. Objective 20×.
The Cellular Phenotypes and Compartmentalization of RIP-LTαβ Infiltrates Are Similar to a Lymph Node.
To compare the cellular composition of pancreatic infiltrates of RIPLTαβ mice with those of RIPLTα mice, we performed immunohistochemistry and FACS® analyses of infiltrating cells recovered from RIPLTαβ and RIPLTα pancreata. The cellular composition of RIPLTαβ infiltrates was highly similar to that of PLNs; all cell populations of a typical PLN were represented, although there was a higher proportion of CD11c+ cells and a lower proportion of B220+ cells (Table I) . Immunohistochemistry of serial tissue sections reacted with anti-B220, -CD4, and -CD8 and -CR1 antibodies (Fig. 3) revealed a distinct delineation between B and T cell compartments and the presence of follicular dendritic cells in B cell areas of RIPLTαβ infiltrates, indicating a cellular compartmentalization characteristic of secondary lymphoid tissue. One hallmark of PLNs is a high proportion of L-selectin+ cells. L-selectin is expressed on white blood cells, including naive T and B lymphocytes, and is essential for cell recruitment into the PLN. The relative number of L-selectin+ cells in RIPLTα pancreata ranged from 28% in young mice to 55% in 1-yr-old mice (n = 6) similar to previous reports of L-selectin expression in RIPLTα kidney (33). In contrast, RIPLTαβ pancreatic infiltrates contained 79% L-selectin+ cells as early as 7 wk of age, much more comparable to the 85% L-selectin+ cells in PLN of C57BL/6 mice (Table I) supporting the hypothesis that LTαβ contributes to the recruitment of L-selectin+ cells.
Table I.
Cellular Composition of RIPLTαβ Infiltrates
| Source of cells
|
||
|---|---|---|
| C57BL/6 PLN | RIPLTαβ pancreas | |
| Cell type | ||
| CD4+ | 45.5 | 44.9 |
| CD8+ | 20.0 | 15.3 |
| B220+ | 36.1 | 17.1 |
| CD11b+ | 8.9 | 5.3 |
| CD11c+ | 4.2 | 9.6 |
| L-selectin+ | 85.3 | 79.2 |
Leukocytes were isolated from C57BL/6 PLN and RIPLTαβ pancreata. Cells were stained with FITC- or PE-conjugated antibodies specific for B220, CD4, CD8, CD11c, CD11b, and L-selectin as described in the Materials and Methods. The percentage of positive cells was determined by flow cytometry.
Figure 3.

Cellular compartmentalization of RIPLTαβ infiltrates. RIPLTαβ pancreatic tissue were analyzed by immunohistochemistry to detect B cells, T cells, and FDCs within RIPLTαβ infiltrates. Serial sections (A–C) were stained with anti-B220 (A), anti-CD4 (B), and anti-CD8 (C). Serial sections (D and E) were stained with anti-B220 (D) and anti-CR1 (E). All were visualized with Vector Red substrate. Objective 40× (A–C); objective 20× (D and E). I, Islet tissue detected by anti-insulin staining (not shown).
Transcription of Lymphoid Chemokines in RIPLTαβ Pancreatic Infiltrates.
To gain insight into the mechanisms driving the cell compartmentalization in RIPLTαβ pancreatic infiltrates, we investigated chemokine expression by in situ hybridization analysis. Previous studies have shown CCL21 and CXCL13 lymphoid chemokine mRNA in RIPLTα kidney infiltrates (37). The more striking T and B cell compartmentalization in RIPLTαβ pancreatic infiltrates suggested a further investigation of CCL21, CCL19, and CXCL13. All three lymphoid chemokine transcripts were substantially more intense and extensive in RIPLTαβ than in the previously reported RIPLTα pancreata (Fig. 4) . This is particularly striking in the case of CCL21 and CCL19. CCL21 mRNA was detected in HEV-like structures within RIPLTαβ infiltrates (Fig. 4 B, inset). CCL19 transcripts were localized in the infiltrate in a reticular pattern similar to that observed in C57BL/6 PLN (Fig. 4, C and D). It was notable that CCL19 was also expressed within the remaining islet tissue, likely by islet infiltrating dendritic cells. High expression of CCL21 and CCL19 correlated with the high number of T cells and dendritic cells observed in RIPLTαβ infiltrates. CXCL13 mRNA was detected at relatively low levels within the infiltrates (Fig. 4 F), consistent with the lower proportion of B cells (Table I). Even so, CXCL13 mRNA localized to regions distinct from those in which CCL19 and CCL21 were found, consistent with the organization of RIPLTαβ infiltrates into separate B and T cell compartments. No mRNA of CCL21, CCL19, and CXCL13 was detected by in situ hybridization studies of C57BL/6 and RIPLTβ pancreas (unpublished data).
Figure 4.
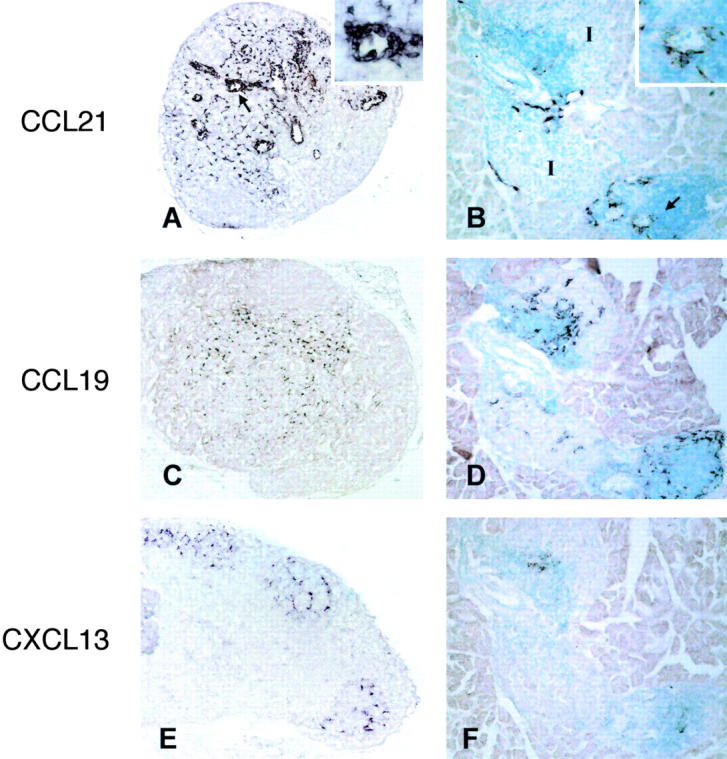
In situ hybridization analysis of lymphoid chemokine transcription in LN and RIPLTαβ pancreas. C57BL/6 PLN (A, C, and E) and serial sections of RIPLTαβ pancreas (B, D, and F) were probed with DIG-labeled antisense CCL21 (A and B), CCL19 (C and D), and CXCL13 (E and F) riboprobes. Positive signal is seen as dark purple staining. Arrows in A and B denote high magnification inset. I, Islet.
LTαβ Regulates Luminal PNAd Expression.
Previous studies of the RIPLTα mouse had revealed MAdCAM-1 and abluminal PNAd expression on HEV-like vessels at the sites of transgene expression (27, 33; Fig. 5, C and D) . In the RIPLTαβ pancreas immunohistochemistry with MECA 367 antibody also revealed high levels of MAdCAM-1 within and around the infiltrates of the islet (unpublished data). Furthermore, consistent with the high number of L-selectin+ cells (Table I), there was intense luminal MECA 79 staining in RIPLTαβ pancreata with a few vessels displaying only abluminal MECA 79 (Fig. 5, E and F) as determined by immunohistochemistry. This is in contrast to the predominately abluminal MECA 79 pattern in the RIPLTα pancreas (Fig. 5, C and D; reference 33). This pericellular (i.e., luminal and abluminal) MECA 79 staining pattern in RIPLTαβ infiltrates was clearly different from that of RIPLTα pancreata and highly reminiscent of a mature LN (Fig. 5, A and B). In addition, RIPLTαβ pancreatic infiltrates exhibited an increase in the number of MECA 79+ vessels when compared with RIPLTα infiltrates. To further investigate a role for LTβ in luminal PNAd expression, immunohistochemistry with MECA 79 was performed on LTβ−/− MLNs. In contrast to the pericellular MECA 79 staining pattern on almost all wild-type LN–HEV (Fig. 5, A and B), abluminal MECA 79 staining was seen on many LTβ−/− MLN–HEV and there was a corresponding 20% reduction in L-selectin+ cells in the MLN (Fig. 5, G and H, and unpublished data).
Figure 5.

LTαβ contributes to luminal PNAd expression on HEV. PNAd expression was detected by immunohistochemistry analysis with MECA 79 antibody in C57BL/6 PLN (A and B), RIPLTα pancreas (C and D), RIPLTαβ pancreas (E and F), and LTβ−/− MLN (G and H). LTβ−/− MLN exhibited a reduction in luminal MECA 79 expression. RIPLTαβ pancreata exhibited an increase in the number of MECA 79+ vessels and in luminal PNAd expression compared with the predominately abluminal pattern observed in RIPLTα pancreatic infiltrates. Objective 20× (A, C, E, and G); objective 40× (B, D, F, and H).
LTαβ Regulates Luminal PNAd through Induction of HEC-6ST.
Given the luminal PNAd expression and high content of L-selectin+ cells observed in RIPLTαβ pancreatic infiltrates and the reduction in luminal PNAd staining in LTβ−/− MLNs, we investigated the potential mechanisms by which LTαβ could influence expression of luminal PNAd. We focused on expression of HEC-6ST, a key enzyme of the PNAd-generating biosynthetic pathway that previous studies have shown to be selectively transcribed in LN HEVs (6, 7) and to regulate luminal PNAd expression (8). Using a recently developed antibody to HEC-6ST, we evaluated its expression in HEVs of C57BL/6 PLNs, LTβ−/− MLNs, and in the infiltrates of RIPLTα and RIPLTαβ pancreata. Double staining of C57BL/6 PLN-HEVs with anti-HEC-6ST and MECA 79 revealed concomitant luminal PNAd and HEC-6ST expression (Fig. 6) . The inset in Fig. 6 shows that MECA 79 staining was confined to the cell surface, whereas HEC-6ST was expressed intracellularly, consistent with its expected location in the Golgi apparatus (39). HEC-6ST expression was reduced in LTβ−/− MLN HEVs, particularly when abluminally stained vessels were evaluated (Fig. 6). However, we noted that luminal MECA 79 staining could be found on some HEV of LTβ−/− (Fig. 5, G and H). Interestingly, HEC-6ST staining was also positive in those latter vessels (unpublished data). HEC-6ST expression was apparent in MECA 79+ vessels of RIPLTαβ infiltrates but not detected in RIPLTα pancreatic infiltrates (Fig. 7) , consistent with the increase in luminal PNAd expression in the former (Figs. 5 and 7). Nevertheless, even in RIPLTαβ infiltrates, there were still some abluminally stained vessels that were negative for HEC-6ST.
Figure 6.

LTαβ–induced luminal PNAd correlates with HEC-6ST expression. Fixed tissue sections of C57BL/6 PLN (top panel) and LTβ−/− MLN (bottom panel) were analyzed by two-color immunofluorescence staining with MECA 79 (red) and anti-HEC-6ST (green) antibodies. C57BL/6 PLN exhibited pericellular PNAd expression with concomitant HEC-6ST expression in HEV. Top panel inset of C57BL/6 PLN-HEV demonstrates intracellular HEC-6ST expression and cell surface PNAd expression. Two-color analysis of LTβ−/− MLN, with particular attention to vessels that show only abluminal MECA 79+ staining, reveals the absence of HEC-6ST expression. Objective 40×.
Figure 7.

RIPLTαβ-HEV show increased luminal PNAd and HEC-6ST expression. RIPLTα (top panel) and RIPLTαβ (bottom panel) pancreatic tissue sections were analyzed by two-color immunofluorescence analysis with MECA 79 (red) and anti-HEC-6ST (green) antibodies. The H&E stained image was merged with MECA 79 staining to orient the histologic location of the vessels; objective 20×. RIPLTα infiltrates exhibited predominately abluminal MECA 79+ staining vessels and no detectable HEC-6ST expression similar to LTβ−/− MLN-HEV (Fig 6, bottom panel). In contrast, analysis of RIPLTαβ pancreata revealed both luminal and abluminal MECA 79+ vessels with coincident HEC-6ST expression on those luminal, pericellular MECA 79+ HEV. Bottom panel inset of RIPLTαβ HEV demonstrates intracellular HEC-6ST expression and cell surface PNAd expression–a pattern more reminiscent of C57BL/6 PLN (Fig 6, top panel) than RIPLTα infiltrates. Objective 40×.
Discussion
The cell infiltrates recruited by ectopic LTα and LTαβ expression and their organized tissue architecture share many characteristics with LN structure and chronic inflammatory foci in autoimmune diseases. The present study elucidates a key mechanism in lymphoid neogenesis (i.e., generation of the vascular signals directing recruitment of the appropriate cells to the site of transgene expression) and thus by extrapolation is key to understanding LN development and inflammatory disease processes. A mechanism for LTα in lymphoid neogenesis, particularly in the induction of MAdCAM-1 expression on endothelial cells, had been established in RIPLTα transgenic mice (26, 27, 33, 34) and by in vitro endothelial cell treatment with this cytokine (35). Although LTβ had been implicated in the generation of the MECA 79 epitope, the mechanism underlying PNAd regulation remained unknown (33). The crucial role of LTβ, in the form of the LTαβ complex, which is revealed here by its coincident expression with HEC-6ST, points to a mechanism of HEV differentiation in LNs and in chronic inflammation.
The RIPLTαβ mouse presents a unique opportunity to evaluate the complementary roles of LTα and LTαβ in lymphoid neogenesis. The quantitative and qualitative differences in the lymphoid accumulations, chemokine expression, and adhesion molecules between RIPLTα and RIPLTαβ mice provide insight into the selective requirements for LTα and LTβ in MLNs and PLNs. The differences in the extent and location of PNAd expression in RIPLTαβ mice when compared with RIPLTα mice correlated with the expression of HEC-6ST, implicating LTαβ in the induction of PNAd through its regulation of this important enzyme. These studies emphasize that LTα is absolutely critical by itself in its induction of MAdCAM-1, and in its contribution to the LTαβ complex, as LTβ alone had no discernible effect on cellular accumulation or expression of chemokines or adhesion molecules. However, LTαβ also contributes a further element to cell recruitment, as demonstrated by the 10-fold higher cell numbers in RIPLTαβ pancreatic infiltrates compared with RIPLTα infiltrates.
Previous studies of Mebius et al. (2), demonstrating the change in PLN HEV vascular addressins from predominant MAdCAM-1 to PNAd, and the present data support a LT driven kinetic developmental model of endothelial addressin expression on PLN-HEV. First, MAdCAM-1, which is induced by LTα3 (33, 35), is expressed on HEV. Second, abluminal PNAd is expressed. Third, luminal PNAd is induced dependent on LTαβ and HEC-6ST expression. Thus, HEV of RIPLTα infiltrates resemble the second stage of this model, whereas RIPLTαβ infiltrates represent a later stage in HEV vascular addressin expression. The role of the individual cytokines in regulation of other molecules that contribute to PNAd expression, such as core proteins CD34 and GlyCAM-1, modifying enzymes FucTIV and FucTVII and additional sulfotransferases, needs further study.
The identity of the cell that responds to LTαβ is not addressed in the present studies, nor is it revealed whether HEV respond directly or indirectly. The simplest scenario is that an endothelial cell expresses the LTβR and responds to the membrane-associated cytokine; HEC-6ST is induced and modifies the oligosaccharide side chain of the core protein in the Golgi apparatus, giving rise to the MECA 79 epitope. Alternatively, LTαβ may activate a stromal cell or another intermediate LTβR expressing cell that induces HEC-6ST by means of additional factors.
Recently, it has been shown that LTβR signaling induces gene expression via two NF-κB pathways (40, 41). Signaling through the TNF receptors activates the classical pathway resulting in nuclear translocation of p50:p65 heterodimers and expression of proinflammatory genes (VCAM-1, MIP-1β, MIP-2). In addition to activation of the canonical NF-κB pathway, the LTβR can also engage the alternative pathway, which regulates NF-κB–inducing kinase (NIK) activity and subsequent p100 processing resulting in nuclear translocation of RelB:p52 heterodimers (40, 42). Both NIK and multiple alternative pathway target genes such as CCL21, CCL19, and CXCL13 play crucial roles in secondary lymphoid organogenesis (43, 44). It is proposed that transcriptional activation of lymphoid chemokine genes and HEC-6ST observed in RIPLTαβ transgenic infiltrates could occur through the alternative NF-κB pathway.
The ratio of LTα to LTβ, presumably determining the ratios of trimeric forms LTα3, LTα1β2, LTα2β1 and usage of different signaling pathways, remains unknown at different stages of development and in RIPLTαβ mice. A plausible developmental hypothesis is that there is a progression from LTα3 to the LTαβ complex. It is likely that there is a mixture of LTα3, LTα2β1, and LTα1β2 in the RIPLTαβ pancreas, explaining the continued high expression of MAdCAM-1, along with PNAd and HEC-6ST. Such a pattern may prevail in the MLN which coexpresses MAdCAM-1 and PNAd on the HEV. The ratio may be skewed toward LTα1β2 in the adult PLN with expression of PNAd and HEC-6ST, and little or no MAdCAM-1. In PP, there may be a different ratio with a predominance of LTα3 and LTα2β1 and a corresponding preponderance of MAdCAM-1 with abluminal PNAd.
Ectopic lymphoid accumulations have been termed tertiary lymphoid organs to distinguish them from secondary lymphoid organs (LN, PP, and spleen; reference 45). Development of these ectopic lymphoid accumulations has been noted in the RIPLTα mouse (26, 27), the nonobese diabetic (NOD) mouse (46, 47), and in human disease such as rheumatoid arthritis (48–50) and thyroiditis (51). While the initiating event in tertiary lymphoid organ development in these human pathologies has not been elucidated, the fact that similar lymphoid structures are induced by transgenic expression of TNF/LT family members (26, 27) or lymphoid chemokines (25–31) is intriguing. These studies have shown that multiple mechanisms can induce lymphoid accumulations. The present and other studies have shown that LTα3 and the LTαβ complex induce chemokines. Conversely, RIP driven expression of CXCL13 induces expression of the LTαβ complex (29). Furthermore, the expression of PNAd and MAdCAM-1 in RIPCCL21 infiltrates is reduced by treatment with an LTβR-Ig (31). Collectively, these studies reemphasize the parallels between chronic inflammation and lymphoid organ development and suggest that lymphoid neogenesis mimics, at least in part, the developmental program of secondary lymphoid organs. The function of tertiary lymphoid organs was suggested but not proven in our previous studies of RIPLTα mice. The RIPLTαβ mice, with their high number of L-selectin+ cells, CD11c+ cells, extensive FDC network and prominent luminal PNAd expression, provide a superior model to address this phenomenon.
Tertiary lymphoid organs could be either beneficial in setting up a local site for antigen presentation or detrimental, leading to tissue injury and promoting epitope spreading in autoimmune disease. In several aspects, the RIPLTα mouse has the appearance of the early stages of Type I diabetes with a peri-islet leukocytic accumulation that does not progress to an invasive insulitis or diabetes unless an additional signal is provided in the form of B7.1 coexpression (52). The RIPLTαβ mouse appears to represent a later stage of disease since the infiltrate invades the islet with clear evidence of islet distortion. Nevertheless, some islet function remains, indicating these animals have not progressed to insulin-dependent disease. The precise signal that drives the cells into the RIPLTαβ islets remains unknown, but could be the intra-islet expression of CCL19. The provision of putative antigen presenting cells (CD11c+, CR1+) and of naive, L-selectin+ cells, attracted by the luminal expression of PNAd and CCL21 and CXCL13 may set the stage for responsiveness to autoantigen leading to overt disease.
Acknowledgments
We thank Myriam Hill for invaluable technical assistance, Jeffrey Browning (Biogen) for the LTα genomic clone, Allison Greene (Yale University) for the RIPTNFα construct, Jason Cyster (UCSF) for the CCL19 construct, Priti Shenoy and Naveen Bangia (Yale University) for the design and initial implementation of the immunization strategy for generating the HEC-6ST antibody, and Mark Singer, Steve Rosen, Annette Bistrup, and Durwin Tsay (UCSF) for collaboration in the purification and analysis of the HEC-6ST antibody.
Supported by National Institutes of Health (NIH) RO1 CA 16885 (N.H. Ruddle), NIH RO1 DK57731 (N.H. Ruddle), and NIH F31 GM 20919 (D.L. Drayton).
Footnotes
Abbreviations used in this paper: HEC-6ST, high endothelial cell sulfotransferase; HEV, high endothelial venule; LT, lymphotoxin; MAdCAM-1, mucosal addressin cell adhesion molecule; MLN, mesenteric lymph node; PLN, peripheral lymph node; PNAd, peripheral node addressin; PP, Peyer's patch; RIP, rat insulin promoter.
References
- 1.Girard, J.P., and T.A. Springer. 1995. High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol. Today. 16:449–457. [DOI] [PubMed] [Google Scholar]
- 2.Mebius, R.E., P.R. Streeter, S. Michie, E.C. Butcher, and I.L. Weissman. 1996. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3− cells to colonize lymph nodes. Proc. Natl. Acad. Sci. USA. 93:11019–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Streeter, P.R., B.T. Rouse, and E.C. Butcher. 1988. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J. Cell Biol. 107:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hemmerich, S., E.C. Butcher, and S.D. Rosen. 1994. Sulfation-dependent recognition of HEV-ligands by L-selectin and MECA-79, an adhesion-blocking mAb. J. Exp. Med. 180:2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen, S.D. 1999. Endothelial ligands for L-selectin: from lymphocyte recirculation to allograft rejection. Am. J. Pathol. 155:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bistrup, A., S. Bhakta, J.K. Lee, Y.Y. Belov, M.D. Gunn, F.R. Zuo, C.C. Huang, R. Kannagi, S.D. Rosen, and S. Hemmerich. 1999. Sulfotransferases of two specificities function in the reconstitution of high endothelial cell ligands for L-selectin. J. Cell Biol. 145:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiraoka, N., P. Bronislawa, J. Nakayama, S. Tsuboi, M. Suzuki, J.C. Yeh, D. Izawa, T. Tanaka, M. Miyasaka, J.B. Lowe, and M. Fukuda. 1999. A novel, high endothelial venule-specific sulfotransferase expresses 6-sulfo sialyl Lewis(x), an L-selectin ligand displayed by CD34. Immunity. 11:79–89. [DOI] [PubMed] [Google Scholar]
- 8.Hemmerich, S., A. Bistrup, M.S. Singer, A. van Zante, J.K. Lee, D. Tsay, M. Peters, J.L. Carminati, T.J. Brennan, K. Carver-Moore, et al. 2001. Sulfation of L-selectin ligands by an HEV-restricted sulfotransferase regulates lymphocyte homing to lymph nodes. Immunity. 15:237–247. [DOI] [PubMed] [Google Scholar]
- 9.Drayton, D.L., K. Chan, W. Lesslauer, J. Lee, X.Y. Ying, and N.H. Ruddle. 2002. Lymphocyte traffic in lymphoid organ neogenesis: differential roles of LTalpha and LTalphabeta. Adv. Exp. Med. Biol. 512:43–48. [PubMed] [Google Scholar]
- 10.Ware, C.F., T.L. Vanarsdale, P.D. Crowe, and J.L. Browning. 1995. The ligands and receptors of the lymphotoxin system. Pathways for Cytolysis. G.M. Griffiths and J. Tschopp, eds. Springer-Verlag, Basel, Switzerland. 175–218. [DOI] [PubMed]
- 11.Banks, T.A., B.T. Rouse, M.K. Kerley, P.J. Blair, V.L. Godfrey, N.A. Kuklin, D.M. Bouley, J. Thomas, S. Kanangat, and M.L. Mucenski. 1995. Lymphotoxin-alpha-deficient mice: effects on secondary lymphoid organ development and humoral immune responsiveness. J. Immunol. 155:1685–1693. [PubMed] [Google Scholar]
- 12.De Togni, P., J. Goellner, N.H. Ruddle, P.R. Streeter, A. Fick, S. Mariathasan, S.C. Smith, R. Carlson, L.P. Shornick, J. Strauss-Schoenberger, et al. 1994. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 264:703–707. [DOI] [PubMed] [Google Scholar]
- 13.Sacca, R., S. Turley, L. Soong, I. Mellman, and N.H. Ruddle. 1997. Transgenic expression of lymphotoxin restores lymph nodes to lymphotoxin-alpha-deficient mice. J. Immunol. 159:4252–4260. [PubMed] [Google Scholar]
- 14.Browning, J.L., A. Ngam-ek, P. Lawton, J. DeMarinis, R. Tizard, E.P. Chow, C. Hession, B. O'Brine-Greco, S.F. Foley, and C.F. Ware. 1993. Lymphotoxin beta, a novel member of the TNF family that forms a heteromeric complex with lymphotoxin on the cell surface. Cell. 72:847–856. [DOI] [PubMed] [Google Scholar]
- 15.Crowe, P.D., T.L. VanArsdale, B.N. Walter, C.F. Ware, C. Hession, B. Ehrenfels, J.L. Browning, W.S. Din, R.G. Goodwin, and C.A. Smith. 1994. A lymphotoxin-beta-specific receptor. Science. 264:707–710. [DOI] [PubMed] [Google Scholar]
- 16.Force, W.R., B.N. Walter, C. Hession, R. Tizard, C.A. Kozak, J.L. Browning, and C.F. Ware. 1995. Mouse lymphotoxin-beta receptor. Molecular genetics, ligand binding, and expression. J. Immunol. 155:5280–5288. [PubMed] [Google Scholar]
- 17.Alimzhanov, M.B., D.V. Kuprash, M.H. Kosco-Vilbois, A. Luz, R.L. Turetskaya, A. Tarakhovsky, K. Rajewsky, S.A. Nedospasov, and K. Pfeffer. 1997. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc. Natl. Acad. Sci. USA. 94:9302–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koni, P.A., R. Sacca, P. Lawton, J.L. Browning, N.H. Ruddle, and R.A. Flavell. 1997. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta in lymphotoxi-beta deficient mice. Immunity. 6:491–500. [DOI] [PubMed] [Google Scholar]
- 19.Rennert, P.D., D. James, F. Mackay, J.L. Browning, and P.S. Hochman. 1998. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 9:71–79. [DOI] [PubMed] [Google Scholar]
- 20.Luther, S.A., H.L. Tang, P.L. Hyman, A.G. Farr, and J.G. Cyster. 2000. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA. 97:12694–12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunn, M.D., K. Tangemann, C. Tam, J.G. Cyster, S.D. Rosen, and L.T. Williams. 1998. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 95:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warnock, R.A., J.J. Campbell, M.E. Dorf, A. Matsuzawa, L.M. McEvoy, and E.C. Butcher. 2000. The role of chemokines in the microenvironmental control of T versus B cell arrest in Peyer's patch high endothelial venules. J. Exp. Med. 191:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legler, D.F., M. Loetsher, R.S. Roos, I. Clark-Lewis, M. Baggiolini, and B. Moser. 1998. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 187:655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ngo, V.N., H. Korner, M.D. Gunn, K.N. Schmidt, D.S. Riminton, M.D. Cooper, J.L. Browning, J.D. Sedgwick, and J.G. Cyster. 1999. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 189:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picarella, D.E., A. Kratz, C.B. Li, N.H. Ruddle, and R.A. Flavell. 1993. Transgenic tumor necrosis factor (TNF)-alpha production in pancreatic islets leads to insulitis, not diabetes. Distinct patterns of inflammation in TNF-alpha and TNF-beta transgenic mice. J. Immunol. 150:4136–4150. [PubMed] [Google Scholar]
- 26.Picarella, D.E., A. Kratz, C.B. Li, N.H. Ruddle, and R.A. Flavell. 1992. Insulitis in transgenic mice expressing tumor necrosis factor beta (lymphotoxin) in the pancreas. Proc. Natl. Acad. Sci. USA. 89:10036–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kratz, A., A. Campos-Neto, M.S. Hanson, and N.H. Ruddle. 1996. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 183:1461–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan, L., C.R. Reilly, Y. Luo, M.E. Dorf, and D. Lo. 2000. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J. Immunol. 164:3955–3959. [DOI] [PubMed] [Google Scholar]
- 29.Luther, S.A., T. Lopez, W. Bai, D. Hanahan, and J.G. Cyster. 2000. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 12:471–481. [DOI] [PubMed] [Google Scholar]
- 30.Chen, S.C., G. Vassileva, D. Kinsley, S. Holzmann, D. Manfra, M.T. Wiekowski, N. Romani, and S.A. Lira. 2002. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J. Immunol. 168:1001–1008. [DOI] [PubMed] [Google Scholar]
- 31.Luther, S.A., A. Bidgol, D.C. Hargreaves, A. Schmidt, Y. Xu, J. Paniyadi, M. Matloubian, and J.G. Cyster. 2002. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J. Immunol. 169:424–433. [DOI] [PubMed] [Google Scholar]
- 32.Weyand, C.M., P.J. Kurtin, and J.J. Goronzy. 2001. Ectopic lymphoid organogenesis: a fast track for autoimmunity. Am. J. Pathol. 159:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuff, C.A., R. Sacca, and N.H. Ruddle. 1999. Differential induction of adhesion molecule and chemokine expression by LTalpha3 and LTalphabeta in inflammation elucidates potential mechanisms of mesenteric and peripheral lymph node development. J. Immunol. 162:5965–5972. [PubMed] [Google Scholar]
- 34.Sacca, R., C.A. Cuff, W. Lesslauer, and N.H. Ruddle. 1998. Differential activities of secreted lymphotoxin-alpha3 and membrane lymphotoxin-alpha1beta2 in lymphotoxin-induced inflammation: critical role of TNF receptor 1 signaling. J. Immunol. 160:485–491. [PubMed] [Google Scholar]
- 35.Cuff, C.A., J. Schwartz, C.M. Bergman, K.S. Russell, J.R. Bender, and N.H. Ruddle. 1998. Lymphotoxin alpha3 induces chemokines and adhesion molecules: insight into the role of LT alpha in inflammation and lymphoid organ development. J. Immunol. 161:6853–6860. [PubMed] [Google Scholar]
- 36.Hemmerich, S., J.K. Lee, S. Bhakta, A. Bistrup, N.R. Ruddle, and S.D. Rosen. 2001. Chromosomal localization and genomic organization for the galactose/N-acetylgalactosamine/N-acetylglucosamine 6-O-sulfotransferase gene family. Glycobiology. 11:75–87. [DOI] [PubMed] [Google Scholar]
- 37.Hjelmstrom, P., J. Fjell, T. Nakagawa, R. Sacca, C.A. Cuff, and N.H. Ruddle. 2000. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am. J. Pathol. 156:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Millet, I., and N.H. Ruddle. 1994. Differential regulation of lymphotoxin (LT), lymphotoxin-beta, and TNF-alpha in murine T cell clones activated through the TCR. J. Immunol. 152:4336–4346. [PubMed] [Google Scholar]
- 39.Hemmerich, S. 2001. Carbohydrate sulfotransferases: novel therapeutic targets for inflammation, viral infection and cancer. Drug Discov. Today. 6:27–35. [DOI] [PubMed] [Google Scholar]
- 40.Dejardin, E., N.M. Droin, M. Delhase, E. Haas, Y. Cao, C. Makris, Z.W. Li, M. Karin, C.F. Ware, and D.R. Green. 2002. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 17:525–535. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-kappaB puzzle. Cell. 109:S81–96. [DOI] [PubMed] [Google Scholar]
- 42.Shinkura, R., K. Kitada, F. Matsuda, K. Tashiro, K. Ikuta, M. Suzuki, K. Kogishi, T. Serikawa, and T. Honjo. 1999. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 22:74–77. [DOI] [PubMed] [Google Scholar]
- 43.Fagarasan, S., R. Shinkura, T. Kamata, F. Nogaki, K. Ikuta, K. Tashiro, and T. Honjo. 2000. Alymphoplasia (aly)-type nuclear factor kappaB-inducing kinase (NIK) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med. 191:1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ansel, K.M., and J.G. Cyster. 2001. Chemokines in lymphopoiesis and lymphoid organ development. Curr. Opin. Immunol. 13:172–179. [DOI] [PubMed] [Google Scholar]
- 45.Ruddle, N.H. 1999. Lymphoid neo-organogenesis: lymphotoxin's role in inflammation and development. Immunol. Res. 19:119–125. [DOI] [PubMed] [Google Scholar]
- 46.Hanninen, A., C. Taylor, P.R. Streeter, L.S. Stark, J.M. Sarte, J.A. Shizuru, O. Simell, and S.A. Michie. 1993. Vascular addressins are induced on islets vessels during insulitis in nonobese diabetic mice and are involved in lymphoid cell binding to islet endothelium. J. Clin. Invest. 92:2509–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faveeuw, C., M.C. Gagnerault, and F. Lepault. 1994. Expression of homing and adhesion molecules in infiltrated islets of Langerhans and salivary glands of nonobese diabetic mice. J. Immunol. 152:5969–5978. [PubMed] [Google Scholar]
- 48.Schroder, A.E., A. Greiner, C. Seyfert, and C. Berek. 1996. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 93:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young, C.L., T.C.I. Adamson, J.H. Vaughan, and R.I. Fox. 1984. Immunohistologic characterization of synovial membrane lymphocytes in rheumatoid arthritis. Arthritis Rheum. 27:32–39. [DOI] [PubMed] [Google Scholar]
- 50.Takemura, S., A. Braun, C. Crowson, P.J. Kurtin, R.H. Cofield, W.M. O'Fallon, J.J. Goronzy, and C.M. Weyand. 2001. Lymphoid neogenesis in rheumatoid synovitis. J. Immunol. 167:1072–1080. [DOI] [PubMed] [Google Scholar]
- 51.Mooij, P., H.J. de Wit, and H.A. Drexhage. 1993. An excess of dietary iodine accelerates the development of a thyroid-associated lymphoid tissue in autoimmune prone BB rats. Clin. Immunol. Immunopathol. 69:189–198. [DOI] [PubMed] [Google Scholar]
- 52.Guerder, S., D.E. Picarella, P.S. Linsley, and R.A. Flavell. 1994. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc. Natl. Acad. Sci. USA. 91:5138–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]