

Abstract
B7-H1 and B7-DC are ligands for PD-1, a receptor implicated in negative regulation of T and B cell functions. These ligands, however, also costimulate T cell responses. It remains elusive whether or not costimulation is mediated through PD-1. By comparative molecular modeling and site-directed mutagenesis, we found that nonconserved residues between these ligands on the A′GFCC′C′′ face mediate interaction with PD-1. This indicates significant structural heterogeneity of the interactions between PD-1 and its ligands. Importantly, ligand mutants with abolished PD-1 binding capacity could still costimulate proliferation and cytokine production of T cells from normal and PD-1–deficient mice. Our results reveal unique binding characteristics of B7-H1 and B7-DC and provide direct evidence for an independent costimulatory receptor other than PD-1.
Keywords: PD-1 ligands, mutagenesis, costimulation, T cell activation
Introduction
Antigen-specific activation and proliferation of lymphocytes are regulated by both positive and negative signals from costimulatory molecules. The most extensively characterized T cell costimulatory pathway is B7-CD28, in which B7–1 (CD80) and B7–2 (CD86) can each engage two receptors, the stimulatory CD28 and the inhibitory CTLA-4 (CD152). In conjunction with signaling through the T cell receptor, CD28 ligation increases antigen-specific proliferation of T cells, enhances production of cyto-kines, stimulates differentiation and effector function, and promotes survival of T cells (1–3). In contrast, signaling through CTLA-4 is thought to deliver a negative signal that inhibits T cell proliferation, IL-2 production, and cell cycle progression (4, 5). Recently, new members of the B7 family have been identified, including B7-H1 (6, 7) and B7-DC (8, 9; both of which are ligands for PD-1), B7-H2 (10–12; a ligand for ICOS), and B7-H3 (13; an as of yet orphan ligand). Elucidation of functional characteristics of these ligands is currently the subject of intense studies.
The ligands of B7-CD28 family are expressed on the cell surface as homodimers with a membrane proximal IgC domain and a membrane distal IgV domain, while their receptors share a common extracellular IgV-like domain. Interactions of receptor–ligand pairs are mediated predominantly through residues in their IgV domains (14). In general, IgV domains are described as two-layered β-strands with “front” and “back” sheets (15). The front and back sheets of CTLA-4 IgV domain consist of strands A′GFCC′ and ABEDC′′, respectively (16, 17), whereas the front and back sheets of the B7 IgV domains are composed of strands AGFCC′C′′ and BED, respectively (18–20). Crystallographic analysis shows that CTLA-4/B7 binding interface is dominated by the interaction of the CDR3 analogous loop from CTLA-4, centered on the MYPPPY motif, with the surface on B7 formed predominately by the G, F, C, C′, and C′′ strands (18, 19). The structure of CD28 has not yet been determined, however, sequence homology analysis, mutagenesis studies, and computer modeling support the idea that the MYPPPY motif is also a major B7-binding site for CD28 (17, 21). Although the MYPPPY motif is not conserved in ICOS, our recent studies have indicated that a related FDPPPF sequence in the analogous position is also a major determinant for binding of ICOS to B7-H2 (22). In addition, crystal structures of CTLA-4/B7 complexes contain bivalent homodimers of CTLA-4 with B7-binding sites located distally to the CTLA-4 dimer interface, which suggests that the CTLA-4 homodimer can bind to noncovalent homodimers of B7–1 or B7–2 to form a lattice of CTLA-4/B7 interactions (18, 19). Formation of such a lattice is thought to trigger the formation of stable signaling complexes as part of the immunological synapse.
B7-H1 (also called PD-L1) and B7-DC (PD-L2) are new members of the B7 family and share 34% amino acid identity. Human and mouse orthologs of these ligands share 70% amino acid identity (7–9, 23). While transcripts of B7-H1 and B7-DC are found in various tissues (6, 9, 23), the expression profiles of their proteins are quite distinct. The expression of the B7-H1 protein, although virtually absent in normal tissues except macrophage-like cells, could be induced in a variety of tissues and cell types (6, 23, 24). In contrast, expression of B7-DC was only detected on dendritic cells and monocytes (8, 24). It has been shown that both B7-H1 and B7-DC bind to PD-1 (programmed cell death-1; references 7–9), a distant member of the CD28 family with an immunoreceptor tyrosine-based inhibitory motif (ITIM) in its cytoplasmic domain (25). PD-1 is expressed on a subset of thymocytes and up-regulated on T, B, and myeloid cells after activation (26). The phenotypes of PD-1−/− mice provide direct evidence for PD-1 being a negative regulator of immune responses in vivo. In the absence of PD-1, mice on the C57BL/6 background slowly develop a lupus-like glomerulonephritis and progressive arthritis (27). PD-1−/− mice on the BALB/c background rapidly develop a fatal autoimmune dilated cardiomyopathy (28). However, substantial evidence indicates that both B7-H1 and B7-DC can function to costimulate T cell responses. In the presence of suboptimal TCR signals, B7-H1 or B7-DC stimulate increased proliferation and production of cytokines in vitro (6, 8, 23). Infusion of B7-H1Ig increases CD4+ T cell responses and T helper cell–dependent humoral immunity (23). On the other hand, several in vitro studies indicate a negative regulatory role for B7-H1 and B7-DC in T cell responses (7, 9). These seemingly contradictory data could be best interpreted by expression of additional receptors on T cells other than PD-1. Indeed, we have recently shown that B7-H1 expressed on tumor cells can actively inhibit immune responses by promoting the apoptosis of effector CTL, and the apoptotic effect of B7-H1 is mediated largely by receptors other than PD-1 (29).
In this study, we pursued a structure-based approach to delineate binding sites of B7-H1 and B7-DC for PD-1. With the aid of three-dimensional models and site-specific mutagenesis, we have first identified residues that are important or essential for PD-1 binding and then mapped the receptor binding sites. Although the location of these binding sites on the AGFCC′C′′ face is similar to the one in CD80/CD86, residues forming the binding sites are not conserved in B7-H1/B7-DC, revealing unique receptor recognition structures. Mutants with impaired PD-1 binding characteristics were still capable of T cell costimulation, providing sound evidence for the presence of other costimulatory T cell receptors.
Materials and Methods
Mice and Cell Lines.
Female C57BL/6 (B6) mice were purchased from the National Cancer Institute (Frederick, MD). PD-1–deficient (PD-1−/−) mice were generated as described previously (30). Mice were used in accordance with NIH guidelines and under an animal study protocol approved by the Animal Care and Use Committee of the Mayo Clinic. A stably transfected Chinese hamster ovary (CHO)* cell clone secreting fusion proteins was maintained in CHO-S-SFM-II medium (GIBCO BRL) supplemented with 1% dialyzed FBS (HyClone). Lymphocytes and COS cells were grown in DMEM (GIBCO BRL) supplemented with 10% FBS, 25 mM HEPES, 2 mM l-glutamine, 1 mM sodium pyruvate, 1% MEM nonessential amino acids, 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate.
Ig Fusion Proteins.
The fusion proteins containing extracellular domain of mouse PD-1 linked to mouse IgG2a Fc portion (PD-1Ig; reference 29) were produced in stably transfected CHO cells and purified by protein G affinity column as described previously (22). Total RNA was isolated from mouse spleen cells and B7-DC cDNA was obtained by reverse-transcription PCR (8). B7-H1Ig and B7-DCIg were prepared by transiently transfecting COS cells with plasmid cDNA containing chimeric cDNA of the extracellular domain of mouse B7-H1 (23) or B7-DC in frame to the CH2-CH3 portion of human IgG1. The transfected COS cells were cultured in serum-free DMEM media and concentrated supernatants were used as sources for Ig fusion proteins in the initial binding assay (22). The Ig proteins were further purified with protein G column for BIAcore analysis and functional assays as described previously (22).
Molecular Modeling.
Molecular models of the Ig V-type domains of human B7-H1 (hB7-H1), mouse B7-H1 (mB7-H1), human B7-DC (hB7-DC), and mouse B7-DC (mB7-DC) were generated by homology (or comparative) modeling based on x-ray coordinates of human CD80 and CD86 as seen in the structures of the CD80/CTLA-4 and CD86/CTLA-4 complexes (18, 19). First, the V-domains of CD80 and CD86 were optimally superimposed and, based on this superposition, sequences of B7 family members were aligned. The superposition and initial alignments were performed using the sequence-structure alignment function of MOE (Molecular Operating Environment; Chemical Computing Group, Montreal, Quebec, Canada). The alignment was then manually adjusted to match Ig consensus positions (15) and map other conserved hydrophobic residues in the target sequences to core positions in the x-ray structures. Corresponding residues in the aligned sequences are thus predicted to have roughly equivalent spatial positions. This was done because taking structural information into account is a more reliable alignment criterion than sequence identity alone, if the primary identity is low, as in this case. In the aligned region, the average identity of the compared B7 sequences relative to the two structural templates, CD80 and CD86, is only ∼16%. The final version of the structure-oriented sequence alignment, which provided the basis for model building, is shown in Fig. 1 a. Following this alignment, core regions of the four models were assembled with MOE from the structural templates; insertions and deletions in loop regions were modeled by applying a segment matching procedure (31, 32). Side chain replacements were performed using preferred rotamer conformations seen in high-resolution protein databank structures (33, 34). In each case, 20 intermediate models were generated, average coordinates were calculated, and the resulting structures were energy minimized using a protein force field (35) until intramolecular contacts and stereochemistry of each model were reasonable. Graphical analysis of the models, including calculation of solvent-accessible surfaces (36), and residue mapping studies were performed with InsightII (Accelrys). Color figures were also generated with InsightII.
Figure 1.
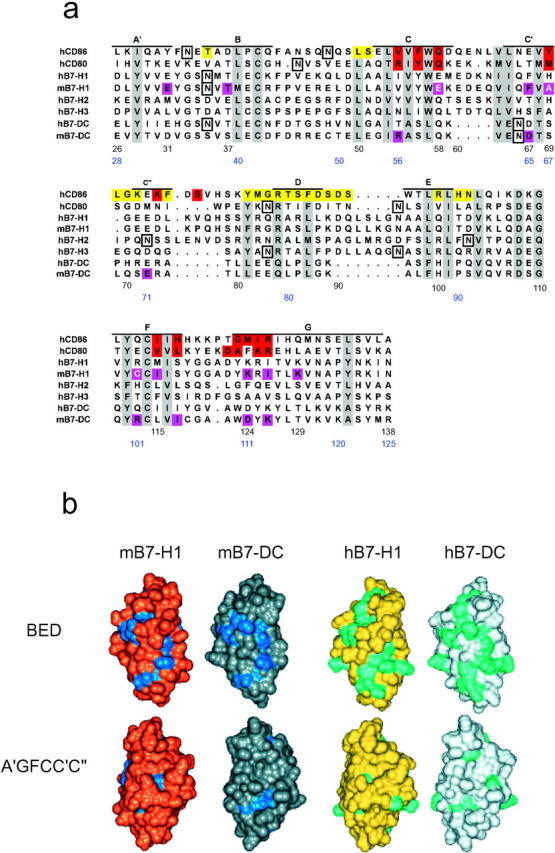
Molecular models and structural characteristics of B7-H1 and B7-DC. (a) Structure-oriented sequence alignment of B7 molecules. Sequences of the NH2-terminal IgV domains of human (h) and mouse (m) B7 family members were aligned based on a superposition of x-ray structures of human CD80 and CD86. The alignment provided the basis for modeling of human and mouse B7-H1 and B7-DC. Beta strands seen in the x-ray structures of CD80/86 are labeled (A′-G) and residue positions, which are most conserved across the B7 family (e.g., large hydrophobic, charged/polar, or cysteine residues), are shaded gray. Many of these residues map to immunoglobulin consensus and protein core positions and are thus important for structural integrity. Potential N-linked glycosylation sites in these proteins are boxed. CD86 residues shown on a yellow background are involved in the formation of the crystallographic homodimer interface, which is conserved in CD80, and residues highlighted in red participate in CTLA-4 binding in the structure of the complex with CD86. Residue positions in mB7-H1 and mB7-DC that are most important for PD-1 binding, based on our mutagenesis studies, are shown in magenta. Three residues in mB7-H1, the mutation of which increased the affinity of the B7-H1/PD-1 interactions, are shown in white on a magenta background. Residue numbers are provided for mB7-H1 (black) and B7-DC (blue). (b) Molecular models and conserved residues. Our V-domain models of mB7-H1 (gold), mB7-DC (silver), hB7-H1 (yellow), and hB7-DC (light gray) are shown with solvent-accessible surfaces and in two equivalent orientations. The top view focuses on the BED β-sheet domain surface and the bottom view on the opposite A′GFCC′C′′ surface (related by 180° rotation around the vertical axis). Residues conserved between mB7-H1 and mB7-DC are colored blue and residues conserved between hB7-H1 and hB7-DC are shown in cyan. As can be seen, conservation of residues on the protein surface (available for receptor binding) is more distinct on the BED β-sheet surface than the A′GFCC′C′′ face of the domains, in particular in the human forms.
Site-directed Mutagenesis.
All mutants of B7-H1Ig and B7-DCIg were constructed by two-step PCR where B7-H1Ig and B7-DCIg cDNA were used as templates, respectively (22). Overlapping oligonucleotide primers were synthesized encoding the desired mutations and two flanking 5′ and 3′ primers were designed to contain EcoRI and BglII restriction sites, respectively. Appropriate regions of cDNA were initially amplified using the corresponding overlapping and flanking primers. Then, using the flanking 5′ and 3′ primers, fragments whose sequences overlapped were fused together and amplified. PCR products were digested with EcoRI and BglII and ligated into EcoRI/BglII-digested pHIg vectors. To verify that the desired mutations were introduced, each mutant was sequenced using an ABI Prism 310 Genetic Analyzer. Plasmids were transfected into COS cells and serum-free supernatants were harvested and used as sources for in vitro binding assays or purified by protein G column for BIAcore analysis and functional assays.
ELISA.
Sandwich ELISA specific for B7-H1Ig and B7-DCIg was established as described previously (22). Briefly, microtiter plates were coated with 2 μg/ml goat anti–human IgG (Sigma-Aldrich) overnight at 4°C. Wells were blocked for 1 h with blocking buffer (10% FBS in PBS), and then washed with PBS containing 0.05% Tween 20 (PBS-Tween). COS cell culture supernatants were added and incubated for 2 h at room temperature. Known concentrations of purified B7-H1Ig were also added to separate wells on each plate for the generation of a standard curve. After extensive washing, horseradish peroxidase (HRP)-conjugated goat anti–human IgG (TAGO, Inc.) diluted at 1:2,000 was added and subsequently developed with TMB substrate before stopping the reaction by the addition of 0.5 M H2SO4. Absorbance was measured at wavelengths of 450 nm on a microtiter plate reader. Concentrations of mutant fusion proteins were determined by comparison with the linear range of a standard curve of B7-H1Ig. Data from triplicate wells were determined and the standard deviations from the mean were <10%. Experiments were repeated at least three times. The ability of the mutants and wild-type of B7-H1Ig and B7-DCIg to bind PD-1 was measured using a capture ELISA assay. Recombinant PD-1Ig fusion proteins were coated on microtiter plates at 5 μg/ml overnight at 4°C. The plates were blocked and washed, and then COS cell culture media was added, followed by incubation for 2 h at room temperature. After extensive washing, HRP-conjugated goat anti–human IgG was added, followed by TMB substrate, and absorbance was measured at 450 nm.
Flow Cytometry.
293 cells were transfected with a PD-1 GFP vector, which was constructed by fusing GFP in frame to C terminus of mouse PD-1 full-length cDNA. The cells were harvested at 24 h after transfection and incubated in flow cytometry buffer (PBS, 3% FBS, 0.02% NaN3) with an equal amount of fusion proteins in COS cell culture media on ice for 45 min. An unrelated fusion protein of human Ig was used as negative control. The cells were washed, further incubated with fluorescein isothiocyanate (PE)-conjugated goat anti–human IgG (BioSource International), and analyzed on a FACSCaliber™ (Becton Dickinson) with CELLQuest™ software (Becton Dickinson). GFP-positive cells were gated by FL1.
Surface Plasmon Resonance Analysis.
Affinity of purified wild-type and mutant B7-H1 and B7-DC proteins were analyzed on a BIAcore™ 3000 instrument (BIAcore) as described previously (22) with modifications. All reagents except fusion proteins were purchased in prefiltered and degassed form from BIAcore. All experiments were performed at 25°C using 0.1 M HEPES, 0.15 M NaCl (pH 7.4) as running buffer. Briefly, the PD-1Ig was first immobilized on a CM5 sensor chip (BIAcore AB) by amine coupling according to BIAcore protocol. A flow cell of the CM5 chip was derivatized through injection of a 1:1 EDC:NHS mixture for 7 min, followed by injection of 20 μg/ml of PD-1Ig at 10 μl/min diluted in 10 mM sodium acetate (pH 4.5). The PD-1Ig was immobilized at 2,000 RUs. This was followed by blocking the remaining activated carboxyl groups with 1 M ethanolamine (pH 8.5). A control flow cell was prepared in a similar fashion as above, substituting running buffer in place of PD-1Ig. The fusion proteins were diluted in running buffer in a concentration series starting from 3.75 μg/ml. The proteins were injected at a flow rate of 20 μl/min for 3 min, and then buffer was allowed to flow over the surface for 5 min for dissociation data. The flow cells were regenerated by a single 30 s pulse with10 mM NaOH. Data analysis was performed using BIAevaluation software package 3.1 (BIAcore).
T Cell Proliferation and Cytokine Assays.
T cells of wild-type B6 mice or PD-1−/− mice were purified by nylon wool columns (Robbins Scientific Co.) as described previously (10). The enriched T cells at 3 × 105 were cultured in flat-bottomed 96-well microplates that were precoated with anti-CD3 mAb (clone 145–2C11; BD Biosciences) in the presence of 5 μg/ml indicated Ig fusion proteins. Proliferation of T cells was determined by incorporation of 1 μCi/well 3H-TdR during the last 12 h of the 3-d culture. 3H-TdR incorporation was counted with a MicroBeta Trilux liquid scintillation counter (Wallac). To detect cytokine, supernatants were collected at the indicted time points of cultures, and the concentrations of IFN-γ were measured by sandwich ELISA following the manufacturer's instructions (BD Biosciences).
Results
Sequences, Structures, and Molecular Models.
The IgV-domains in CD80 and CD86 share only limited sequence identity of ∼20% but their three-dimensional structures are very similar, as revealed by independent crystallographic studies. As many core or Ig superfamily consensus residue positions seen in CD80/CD86 are also conserved or conservatively replaced in other B7 family members, including B7-H1 and B7-DC (Fig. 1 a), the significant structural similarity between CD80 and CD86 can be extended to other B7 proteins and exploited for model building and mapping of important residues. Accordingly, we have built molecular models of the mouse and human B7-H1 and B7-DC molecules, as shown in Fig. 1 b.
Sequence Analysis in Three Dimensions.
In the V-regions, B7-H1 and B7-DC share more sequence identity than average across the B7 family, ∼34%. As both B7-H1 and B7-DC bind PD-1, residue conservation could be significant for the formation of the receptor binding site. Therefore, we have initially used our models to compare the putative distribution of conserved residues that are exposed on the protein surface. Fig. 1 b shows a side-by-side comparison of these molecular models. Significant conservation of surface residues is indeed observed on the IgV BED faces of B7-H1 and B7-DC, more so in the human than the mouse proteins. By contrast, the opposite A′GFCC′C′′ face does not display significant residue conservation. This result is somewhat unexpected because the corresponding A′GFCC′C′′ face of both CD80 and CD86 contains the CD28/CTLA-4 binding site. These findings raised the questions whether or not conserved residues on the BED face of B7-H1 and B7-DC were important for receptor binding or, alternatively, whether the binding sites contained nonconserved residues.
Mutagenesis Analysis of Receptor Binding Sites.
With the aid of our molecular models, we scanned the IgV-domains of B7-H1 and B7-DC for conserved and nonconserved residues on both the BED and A′GFCC′C′′ faces and subjected them to site-specific mutagenesis. We mutated residues in the mouse molecules to enable subsequent functional studies of selected mutant proteins. All the mutants were expressed as human IgG1-tagged fusion proteins. The concentrations of Ig fusion proteins in serum-free COS cell culture media were determined by sandwich ELISA using two nonoverlapping anti–human IgG1 mAbs. The integrity of fusion proteins was confirmed by Western blotting analysis, in which each Ig fusion protein was probed with both anti–human Ig and anti-B7-H1 or anti-B7-DC mAbs. All analyzed fusion proteins appeared to be single band with the same size between wild-type and mutants (unpublished data). The binding characteristics of generated mutant proteins were evaluated for binding to PD-1 by ELISA and flow cytometry analysis. A total of 21 mB7-H1 and 17 mB7-DC mutants were prepared and tested. The results are summarized in Tables I and II . We only considered mB7-H1 and mB7-DC residues implicated in ligand–receptor interactions, if their mutation caused at least a 50% loss of binding by flow cytometry or at least one order of magnitude loss by ELISA. Mutation of about half of the selected residues significantly affected binding to mPD-1. For mB7-H1, these residues were F67, I115, K124, I126, and K129 and for mB7-DC, were R56, S67, E71, R101, I105, D111, and K113. All these important residues are located in the C′-, C′′- and F-, G-strands of A′GFCC′C′′ faces of the domains (Fig. 1 a). Interestingly, mutation of each of three residues in mB7-H1 (E58, A69, and C113) increased the binding to mPD-1 three- to fourfold by ELISA. Thus, these positions must at least be proximal or directly in interface to the receptor-ligand interface and have not only some tolerance for substitution but also potential optimization of binding interactions. Residues in mB7-H1 and mB7-DC that, when mutated, significantly reduce or abolish PD-1 binding are in equivalent regions of the A′GFCC′C′′ face of the domains (with only one exception in each case), although they are often distant in sequence (Fig. 2 a). By contrast, mutation of conserved residues on the BED face did not reduce binding. Thus, residue conservation on the BED face could be important for other effects such as ligand dimerization or recognition of other proteins.
Table I.
Summary of Amino Acid Substitutions and the Binding Characteristics of the Mouse B7-H1 Mutants
| Substitutionsb
|
PD-1 binding
|
||||
|---|---|---|---|---|---|
| Mutantsa | Sites | Nucleic acid(s) |
Amino acid |
FACSc | ELISA (%)d |
| B7-H1 | ++++ | 100 | |||
| L27A | A′ strand | TTG→GCC | Leu→Ala | ++++ | 100 |
| E31S | A′ strand | GAG→AGC | Glu→Ser | ++ | 50 |
| S34Y | B strand | AGC→TAC | Ser →Tyr | ++++ | 60 |
| T37Y | B strand | ACG→TAC | Thr→Tyr | ++ | 5 |
| D49S | B/C loop | GAC→AGC | Asp→Ser | ++++ | 30 |
| Y56S | C strand | TAC→AGC | Tyr→Ser | ++++ | 100 |
| E58S | C strand | GAA→AGC | Glu→Ser | +++++ | 300 |
| E62S | C/C′ loop | GAG→AGC | Glu→Ser | ++++ | 50 |
| F67A | C′ strand | TTT→GCC | Phe→Ala | +/− | 2 |
| A69F | C′ strand | GCA→TTC | Ala→Phe | +++++ | 300 |
| E72S | C′ strand | GAG→AGC | Glu→Ser | ++++ | 60 |
| K75S | C′′/D loop | AAG→AGC | Lys→Ser | ++++ | 100 |
| K89S | D strand | AAG→AGC | Lys→Ser | ++++ | 60 |
| A98F | E strand | GCC→TTC | Ala→Phe | ++++ | 40 |
| Q100S | E strand | CAG→AGC | Gln→Ser | ++++ | 100 |
| C113Y | F strand | TGC→TAC | Cys→Tyr | +++++ | 300 |
| I115A | F strand | ATA→GCC | Ile→Ala | +/− | 3 |
| S117Y | F strand | AGC→TAC | Ser→Tyr | ++++ | 100 |
| K124A | G strand | AAG→AGC | Lys→Ser | + | 3 |
| I126A | G strand | ATC→GCC | Ile→Ala | − | 1.4 |
| K129S | G strand | AAA→AGC | Lys→Ser | ++ | 35 |
Amino acids are numbered from the initiation methionine.
The nucleotides or amino acids in front of the “→'' symbol were replaced with the corresponding nucleotides or amino acids after the “→” symbol.
The ability of each mutant binding to PD-1 in the flow cytometry assay was scored as relative means of fluorescence compared to wild-type B7-H1Ig and categorized as follows: +++++, >120%; ++++, 80–120%; +++, 50–80%; ++, 20–50%; +, 5–20%; −, <5%.
Specific binding activities were determined for each of the indicated fusion proteins. The concentration of a mutant protein required giving an arbitrary A450 the same as that found for B7-H1Ig was determined from the linear region of binding curves. The value was calculated by the following formula: % wild-type B7-H1Ig binding = 100 × (1/A450mutants)/(1/A450B7-H1Ig) and was expressed as a percentage of specific binding activity of B7-H1Ig. Each value represents the average of three determinations from each binding curve and the data are representative of four experiments.
Table II.
Summary of Amino Acid Substitutions and the Binding Characteristics of the Mouse B7-DC Mutants
| Substitutionsb
|
PD-1 binding
|
||||
|---|---|---|---|---|---|
| Mutantsa | Sites | Nucleic acids(s) |
Amino acid |
FACSc | ELISA (%)d |
| B7-DC | ++++ | 100 | |||
| D33S | A′ strand | GAC→AGC | D→S | ++++ | 30 |
| S39Y | B strand | AGC→TAC | S→Y | ++++ | 60 |
| E41S | B strand | GAG→AGC | E→S | ++++ | 100 |
| R56S | C strand | AGA→TCT | R→S | +++/++ | 5 |
| S58Y | C strand | AGT→TAC | S→Y | ++++ | 170 |
| D65S | C′ strand | GAT→AGC | D→S | ++++ | 100 |
| S67Y | C′strand | TCT→TAC | S→Y | +++/++ | 3 |
| E71S | C′′ strand | GAA→AGC | E→S | +++/++ | 2 |
| R72S | C′′ strand | AGA→AGC | R→S | ++++ | 60 |
| K84S | D strand | AAG→AGC | K→S | +++/++++ | 13 |
| H88A | E strand | CAC→GCC | H→A | +++/++++ | 20 |
| R101S | F strand | CGT→AGC | R→S | +++ | 7 |
| L103A | F strand | CTG→GCC | L→A | +++ | 25 |
| I105A | F strand | ATC→GCC | I→A | ++ | 0.5 |
| D111S | G strand | GAC→AGC | D→S | ++ | 0.3 |
| K113S | G strand | AAG→AGC | K→S | −/+ | <0.1 |
| T116Y | G strand | ACG→TAC | T→Y | +++/++++ | 20 |
Amino acids are numbered from the initiation methionine.
The nucleotides or amino acids in front of the “→” symbol were replaced with the corresponding nucleotides or amino acids after the “→” symbol.
See Table I legend for the definition of binding activity to PD-1.
See Table I legend for the calculation of % specific binding activity to PD-1Ig. Each value represents the average of three determinations from each binding curve and the data are representative of four experiments.
Figures 2.

Mapping of B7-H1 and B7-DC mutants. (a) The models of mouse B7-H1 and B7-DC are shown with solvent-accessible surface and in the same orientation as in b. Residues in mB7-H1 and mB7-DC that, when mutated, displayed wild type-like or only partially (<50%) reduced binding by ELISA are colored in green. By contrast, residues whose mutations led to significantly (>50%) reduced or abolished binding by flow cytometry and/or ELISA are shown in magenta. With one exception in each case (top), these residues map to corresponding regions on the A′GFCC′C′′ faces (bottom) of the domains that are thus predicted to contain the PD-1 binding sites in these proteins. Residues in mB7-H1 whose mutation increased binding to PD-1 are shown in white. Seven of eight mB7-DC residues important for PD-1 binding are not conserved in mB7-H1, although they map to similar spatial positions. (b) Shown is a close-up view of the A′GFCC′C′′ faces, and important residues in mB7-H1 and mB7-DC are labeled (black and blue, respectively). (c) Models of mB7-H1 (gold), mB7-DC (silver), and the x-ray structures of human CD80 (hCD80; light gray) and human CD86 (hCD86; cyan) are shown in equivalent orientations, focusing on the A′GFCC′C′′ faces of their NH2-terminal Ig V-like domains. Residues in mB7-H1 and mB7-DC identified by mutagenesis as important for PD-1 binding are colored magenta. None of these residues are conserved. Residues in CD80 and CD86 that contact CTLA-4 in their respective crystallographic complexes are colored in pink (nonconserved) or red (identical or very similar). Whether or not mutation of each of these residues significantly affects CTLA-4 binding remains to be determined. However, the side-by-side comparison illustrates that the binding sites in these B7 family members map to corresponding regions, despite markedly different residue compositions (even if the same receptor is recognized).
The PD-1 binding sites in B7-H1 and B7-DC map to equivalent sites on the A′GFCC′C′′ face. Fig. 2 b shows a close-up of the predicted binding site regions. As can be seen, residues whose mutation negatively (or positively) affects PD-1 binding form a coherent surface in both ligands. The proximity of important residues and some residues not important for binding suggests that the observed effects are specific and not a consequence of global structural perturbation, which is further supported by our ability to produce higher avidity mutants of mB7-H1. Comparison of important residue positions confirms that the location of the putative binding sites in mB7-H1 and mB7-DC closely corresponds to the CD28/CTLA-4 binding sites in CD86 and CD80 (Fig. 2 c).
Surface plasmon resonance analysis of binding of wild-type and mutant proteins to PD-1 was largely consistent with the results from flow cytometry and ELISA (Tables I and II). B7-H1 mutant C113 has a similar or slightly higher maximal response (Rmax) value than the wild-type protein, while the mutant I126 shows minimal binding to mPD-1 (Fig. 3 a). Mutant B7-DC-K113 does not bind detectably to PD-1 (Fig. 3 b). These data demonstrate that the wild type B7-H1 and mutant C113 have greater steady-state affinity for PD-1 than mutant I126, similar to wild-type B7-DC compared with the mutant K113. B7-H1 mutant C113, wild-type B7-H1, and B7-DC all have similar on-rates and off-rates, while only the mutants I126 and K113 show slower and no detectable on- and off-rates, respectively.
Figure 3.

The surface Plasmon resonance analysis of B7-H1 and B7-DC binding to PD-1. The sensorgram overlay for binding of B7-H1Ig and its mutants (a) and for binding of B7-DCIg and its mutants (b) to immobilized PD-1Ig is shown. Flow cell 4 (Fc4) was immobilized with 2000 RU of PD-1Ig. Fc3 was prepared as a control using running buffer. The response unit is shown for all data, with Fc3 being subtracted from Fc4. Data was obtained by immobilizing a CM5 sensor chip with 2000 RU of PD-1Ig and by running a concentration series of analyte over the sensor surface starting at a concentration of 3.75 μg/ml up to 60 μg/ml.
Costimulatory Function of B7-H1 and B7-DC Mutants.
The costimulatory potential of wild-type B7-H1, B7-DC, and selected mutants were also tested. Similar to previously published data, wild-type B7-H1Ig and B7-DCIg increased proliferation and IFN-γ secretion in the presence of anti-CD3 mAb (Fig. 4 , a and b) and this effect could be largely abrogated by inclusion of specific mAb to mouse B7-H1 or B7-DC, respectively (unpublished data). In the absence of anti-CD3, B7-H1Ig and B7-DCIg up to 10 μg/ml in soluble or immobilized forms do not stimulate proliferation of T cells (unpublished data). We chose the mutants F67 and I126 of B7-H1 and K113 and D111 of B7-DC for further study because both F67 and I126 have minimal binding to PD-1 in both flow cytometry and ELISA (Table I). Similarly, K113 and D111 do not bind PD-1 (Table II). As shown in Fig. 4, a and b, these mutants were still able to costimulate proliferation and IFN-γ production of T cells in comparison with the wild-type of B7-H1 and B7-DC. In fact, mutants F67 and I126 of B7-H1 had even slightly increased costimulatory ability compared with wild-type B7-H1. Mutant D111 of B7-DC appeared to stimulate lower levels of IFN-g release than wild-type B7-DC in our assay. This difference, however, was not statistically significant (Fig. 4 b). Interestingly, mutant C113, which shows an approximately threefold better binding capacity in ELISA to PD-1 than binding of wild-type B7-H1 (Table I), also costimulated T cell proliferation and IFN-γ production. Thus, these results suggest that PD-1 is either not the only costimulatory receptor for B7-H1 and B7-DC or not a costimulatory receptor at all.
Figure 4.

T cell costimulatory effect of wild-type and mutants of B7-H1 and B7-DC. (a) Purified T cells from B6 mice were stimulated with immobilized wild-type or mutants of B7-H1 and B7-DC Ig fusion proteins at 5 μg/ml in the presence anti-CD3 mAb at indicated concentrations. Human Ig was used as negative control. The proliferation of T cells was determined by incorporation of 1 μCi/well of 3H-TdR during the last 12 h of the 3-d culture. Data depict one representative experiment of three. (b) For analysis of IFN-γ secretion, purified T cells were cultured in the presence of precoated anti-CD3 (1 μg/ml) and 5 μg/ml of immobilized Ig fusion proteins and control human Ig. Supernatants were collected after 48 and 72 h culture and assayed for IFN-γ using ELISA. Data depict one representative experiment of three.
Although B7-H1 and B7-DC might costimulate T cell growth through a yet unknown receptor, our findings could be interpreted as an integrated stimulatory effect of unidentified costimulatory receptor(s) and PD-1. Therefore, we tested costimulatory effects of our mutants in PD-1 deficient T cells. As shown in Fig. 5 , in general, PD-1−/− T cells had a much higher proliferative potential in response to anti-CD3 mAb than PD-1+/+ T cells (see Fig. 4). Interestingly, addition of wild-type B7-H1 and its mutants costimulated T cell proliferation equally well. Similar results were obtained using wild type B7-DC and its mutants. Thus, these observations strongly suggest that B7-H1 and B7-DC costimulates T cell growth through a non-PD-1 receptor.
Figure 5.

Costimulation of PD-1−/− T cells by B7-H1 and B7-DC mutants. Purified T cells from PD-1–deficient B6 mice were stimulated with immobilized wild-type or mutants of B7-H1 (a) and B7-DCIg (b) at 5 μg/ml in the presence anti-CD3 mAb at indicated concentrations. Human Ig (hIg) was used as negative control. The proliferation of T cells was determined by incorporation of 1 μCi/well of 3H-TdR during the last 12 h of the 3-d culture. Data depict one representative experiment of three.
Discussion
With the aid of comparative molecular model and site-directed mutagenesis, we have generated a first view of PD-1 binding sites in the NH2-terminal IgV-type domains of B7-H1 and B7-DC to PD-1. Our site-specific mutagenesis analysis has identified residues important for binding of B7-H1 and B7-DC to PD-1. On the basis of these studies, we have generated a preliminary outline of PD-1–binding sites in these two molecules by mapping mutated residues according to the binding characteristic of their mutants. Several lines of evidence indicate that it was indeed possible to select surface residues for mutagenesis from the models with a high degree of confidence. First, all mutants were expressed in sufficient quantities for characterization, ruling out the presence of grossly mis-folded mutant proteins. Second, the majority of mutants displayed wild-type–like or only slightly reduced binding. Third, and most importantly, costimulatory functions of the mutants with minimal binding to PD-1 remained intact, demonstrating that they were structurally sound. These observations essentially exclude the presence of significant structural perturbations, which would typically result from mutations of residues in the protein core regions.
By mapping of mutants, we could show that B7-H1 and B7-DC utilize nonconserved residues on their A′GFCC′C′′ face to bind PD-1. By contrast, notable amino acid conservation on the opposite BED faces was found to be irrelevant for PD-1 binding. Although the location of the PD-1 binding sites in B7-H1/B7-DC corresponds to the CD28/CTLA-4 binding sites in CD80/CD86, receptor binding residues are considerably more variable in the case of B7-H1 and B7-DC. Thus, PD-1 interactions with B7-H1 or B7-DC must vary significantly at the molecular level of detail. Considering the unusual degree of variability among residues important for binding to the same receptor, interactions between receptor and ligands should be more permissive to modulation in this case. This notion is consistent with the ability to generate mutant proteins with higher affinity than wild-type, as demonstrated here, at least for mB7-H1.
B7-H1 mutants F67, I126, and B7-DC mutants D111, K113, did not bind PD-1 but were still able to costimulate T cell proliferation and cytokine production with or without the expression of PD-1 on T cells. These data are consistent with our previous finding that costimulation of T cells by B7-H1Ig in the presence of anti-CD3 was not affected by the inclusion of soluble PD-1Ig (29). Therefore, binding to PD-1 and costimulatory functions of B7-H1 and B7-DC could be separated. These findings support that B7-H1 and B7-DC bind to additional activating receptor(s) other than PD-1. This scenario is similar to the B7-CD28 pathway where CD80 and CD86 bind two functionally distinct receptors, CD28 and CTLA-4. It remains to be determined, however, whether or not B7-H1 and B7-DC bind the same or different costimulatory receptor. Regardless, it is likely, based on our present study, that the B7-H1 and B7-DC binding sites for PD-1 and unknown costimulatory receptor(s) are distinct. Otherwise, mutants with substantially impaired PD-1 binding would be expected to show reduced costimulatory potential.
The studies including those from our laboratory have shown that both B7-H1 and B7-DC can provide a positive costimulatory signal for T cell proliferation and cytokine production (6, 8, 22). Several studies, however, suggested that engagement of B7-H1 or B7-DC with PD-1 results in inhibition of T cell proliferation, reducing cytokine production and cell cycle arrest (7, 9). We showed previously that costimulation by B7-H1Ig in the presence of immobilized anti-CD3 induced increased proliferation of human T cells in 48–72 h but a rapid decrease of T cell number was observed when the cultures were extended beyond 72 h (see supplemental data 1 in reference 29). Inhibition of T cell responses in such prolonged cultures is largely due to increased apoptosis. It is likely that PD-1 and additional costimulatory receptor may be expressed with different kinetics and/or on different subpopulations of T cells. If putative costimulatory receptor is expressed earlier than PD-1, engagement of this receptor with B7-H1 or B7-DC may be responsible for early T cell proliferation and cytokine production. Subsequently expressed receptors including PD-1 may mediate cytokine reduction and T cell apoptosis, which may lead to inhibited T cell responses. Our mutants thus may provide tools for further dissection of these experimental observations.
Our findings have important implications in the regulation of immune responses. For example, mutants with selective binding capacity for costimulatory receptor may be used to stimulate desired immune responses while avoiding potential inhibition through PD-1 signaling. On the other hand, mutants selectively binding to PD-1 but not costimulatory receptor could be useful for delivering negative signaling to down-regulate autoimmunity. Our observations provide a new possibility for the selective manipulation of ligand binding events and immune responses for therapy.
Acknowledgments
We thank Kathy Jensen for editing the manuscript.
This work was supported by National Institutes of Health grant CA97085, by the Mayo Foundation, and by Albany Molecular Research, Inc. (formerly New Chemical Entities, Inc.).
S. Wang and J. Bajorath contributed equally to this work.
Footnotes
Abbreviation used in this paper: CHO, Chinese hamster ovary.
References
- 1.Lenschow, D.J., T.L Walunas, and J.A. Bluestone. 1996. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14:233–258. [DOI] [PubMed] [Google Scholar]
- 2.Chambers, C.A., and J.P. Allison. 1997. Co-stimulation in T cell responses. Curr. Opin. Immunol. 9:396–404. [DOI] [PubMed] [Google Scholar]
- 3.Rathmell, J.C., and C.B. Thompson. 1999. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 17:781–828. [DOI] [PubMed] [Google Scholar]
- 4.Krummel, M.F., and J.P. Allison. 1996. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 183:2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walunas, T.L., C.Y. Bakker, and J.A. Bluestone. 1996. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 183:2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong, H., G. Zhu, K. Tamada, and L. Chen. 1999. B7-H1, a third member of the B7 family, co-stimulates T cell proliferation and interleukin-10 secretion. Nat. Med. 5:1365–1369. [DOI] [PubMed] [Google Scholar]
- 7.Freeman, G.J., A.J. Long, Y. Iwai, K. Bourque, T. Chernova, H. Nishimura, L.J. Fitz, N. Malenkovich, T. Okazaki, M.C. Byrne, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng, S.Y., M. Otsuji, K. Huang, J.E. Slansky, S.I. Pai, A. Shalabi, T. Shin, D.M. Pardoll, and H. Tsuchiya. 2001. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latchman, Y., C.R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova, Y. Iwai, A.J. Long, J.A. Brown, R. Nunes, et al. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2:261–269. [DOI] [PubMed] [Google Scholar]
- 10.Wang, S., G. Zhu, A.I. Chapoval, H. Dong, K. Tamada, J. Ni, and L. Chen. 2000. Costimulation of T cells by B7-H2, a B7-like molecule that binds ICOS. Blood. 96:2808–2813. [PubMed] [Google Scholar]
- 11.Swallow, M.M., J.J. Wallin, and W.C. Sha. 1999. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity. 11:423–432. [DOI] [PubMed] [Google Scholar]
- 12.Yoshinaga, S.K., J.S. Whoriskey, S.D. Khare, U. Sarmiento, J. Guo, T. Horan, G. Shih, M. Zhang, M.A. Coccia, T. Kohno, et al. 1999. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 402:827–832. [DOI] [PubMed] [Google Scholar]
- 13.Chapoval, A., J. Ni, J.S. Lau, R.A. Wilcox, D.B. Flies, D. Liu, H. Dong, G.L. Sica, G. Zhu, K. Tamada, and L. Chen. 2001. B7-H3: a costimulatory molecule for T cell activation and IFN-γ production. Nat. Immunol. 2:269–274. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz, J.C., X. Zhang, S.G. Nathenson, and S.C. Almo. 2002. Structural mechanisms of costimulation. Nat. Immunol. 3:427–434. [DOI] [PubMed] [Google Scholar]
- 15.Williams, A.F., and A.N. Barclay. 1998. The immunoglobulin superfamily-domains for cell surface recognition. Annu. Rev. Immunol. 6:381–405. [DOI] [PubMed] [Google Scholar]
- 16.Ostrov, D.A., W. Shi, J.C. Schwartz, S.C. Almo, and S.G. Nathenson. 2000. Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science. 290:816–819. [DOI] [PubMed] [Google Scholar]
- 17.Metzler, W.J., J. Bajorath, W. Fenderson, S.Y. Shaw, K.L. Constantine, J. Naemura, G. Leytze, R.J. Peach, T.B. Lavoie, L. Mueller, and P.S. Linsley. 1997. Solution structure of human CD152 and delineation of a CD80/CD86 binding site conserved in CD28. Nat. Struct. Biol. 4:527–531. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz, J.C., X. Zhang, A.A. Federov, S.G. Nathenson, and S.C. Almo. 2001. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature. 410:604–608. [DOI] [PubMed] [Google Scholar]
- 19.Stamper, C.C., Y. Zhang, J.F. Tobin, D.V. Erbe, S. Ikemizu, S.J. Davis, M.L. Stahl, J. Seehra, W.S. Somers, and L. Mosyak. 2001. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 410:608–611. [DOI] [PubMed] [Google Scholar]
- 20.Ikemizu, S., R.J. Gilbert, J.A. Fennelly, A.V. Collins, K. Harlos, E.Y. Jones, D.I. Stuart, and S.J. Davis. 2000. Structure and dimerization of a soluble form of B7-1. Immunity. 12:51–60. [DOI] [PubMed] [Google Scholar]
- 21.Bajorath, J., W.J. Metzler, and P.S. Linsley. 1997. Molecular modeling of CD28 and three-dimensional analysis of residue conservation in the CD28/CD152 family. J. Mol. Graph. Model. 15:135–139. [DOI] [PubMed] [Google Scholar]
- 22.Wang, S., G. Zhu, K. Tamada, L. Chen, and J. Bajorath. 2002. Ligand binding sites of inducible costimulator and high avidity mutants with improved function. J. Exp. Med. 195:1033–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura, H., H. Dong, G. Zhu, G.L. Sica, D.B. Flies, K. Tamada, and L. Chen. 2001. B7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood. 97:1809–1816. [DOI] [PubMed] [Google Scholar]
- 24.Ishida, M., Y. Iwai, Y. Tanaka, T. Okazaki, G.J. Freeman, N. Minato, and T. Honjo. 2002. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol. Lett. 84:57–62. [DOI] [PubMed] [Google Scholar]
- 25.Ishida, Y., Y. Agata, K. Shibahara, and T. Honjo. 1992. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11:3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agata, K., A. Kawasaki, H. Nishimura, Y. Ishida, T. Tsubata, H. Yagita, and T. Honjo. 1996. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8:765–772. [DOI] [PubMed] [Google Scholar]
- 27.Nishimura, H., M. Nose, H. Hiai, N. Minato, and T. Honjo. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 11:141–151. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura, H., T. Okazaki, Y. Tanaka, K. Nakatani, M. Hara, A. Matsumori, S. Sasayama, A. Mizoguchi, H. Hial, N. Minato, et al. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor deficient mice. Science. 291:319–322. [DOI] [PubMed] [Google Scholar]
- 29.Dong, H., S.E. Strome, D.R. Salomao, H. Tamura, F. Hirano, D.B. Flies, P.C. Roche, G. Zhu, K. Tamada, V.A. Lennon, et al. 2002. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 8:793–800. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura, H., N. Minato, T. Nakano, and T. Honjo. 1998. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 10:1563–1572. [DOI] [PubMed] [Google Scholar]
- 31.Levitt, M. 1992. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 226:507–533. [DOI] [PubMed] [Google Scholar]
- 32.Fechteler, T., U. Dengler, and D. Schomburg. 1995. Prediction of protein three-dimensional structures in insertion and deletion regions: a procedure for searching databases of representative protein fragments using geometric scoring criteria. J. Mol. Biol. 253:114–131. [DOI] [PubMed] [Google Scholar]
- 33.Ponder, J.W., and F.M. Richards. 1987. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structure classes. J. Mol. Biol. 193:775–791. [DOI] [PubMed] [Google Scholar]
- 34.Berman, H.M., J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, and P.E. Bourne. 2000. The protein data bank. Nucleic Acid Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engh, R.A., and R. Huber. 1991. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A47:392–400. [Google Scholar]
- 36.Connolly, M.L. 1983. Analytical molecular surface calculations. J. Appl. Crystallogr. 16:548–558. [Google Scholar]