

Abstract
Chronic airway rejection is characterized by prolonged inflammation, epithelial damage, and eventual luminal obliterative bronchiolitis (OB). In cardiac allografts, the inducible nitric oxide synthase (iNOS) promotes acute rejection but paradoxically reduces neointimal formation, the hallmark of chronic rejection. The specific roles of NOS isoforms in modulating lymphocyte traffic and airway rejection are not known. Using a double lumen mouse tracheal transplant model, tracheal grafts from B10.A (allo) or C57BL/6J (iso) mice were transplanted into cyclosporine-treated wild-type (WT) iNOS−/− or endothelial NOS (eNOS)−/− recipients. OB was observed in WT tracheal allografts at 3 weeks (53 ± 2% luminal occlusion vs. 17 ± 1% for isografts, P < 0.05) with sites of obstructive lesion formation coinciding with areas of CD3+ CD8+ T cell–rich lymphocytic bronchitis. In contrast, allografts in iNOS−/− recipients exhibited reductions in local expression of proinflammatory chemokines and cytokines, graft T cell recruitment and apoptosis, and luminal obliteration (29 ± 2%, P < 0.05 vs. WT allografts). Recipient eNOS deficiency, however, suppressed neither chemokine expression, lymphocyte infiltration, nor airway occlusion (54 ± 2%). These data demonstrate that iNOS exacerbates luminal obliteration of airway allografts in contrast with the known suppression by iNOS of cardiac allograft vasculopathy. Because iNOS−/− airways transplanted into WT allograft hosts are not protected from rejection, these data suggest that iNOS expressed by graft-infiltrating leukocytes exerts the dominant influence on airway rejection.
Keywords: lung transplantation, allograft, obliterative bronchitis, apoptosis, nitric oxide synthase
Introduction
Obliterative bronchiolitis (OB)* is an irreversible pathologic hallmark of chronic airway rejection, which has an immunologic basis and eventuates in fibrous obliteration of the terminal airways (1). Development of OB represents the primary cause of late morbidity and death after lung transplantation (2). Histologically, OB develops after an initial period marked by an influx of inflammatory cells within airway epithelium (3), recognized as a lymphocytic bronchitis/bronchiolitis (LB; 4). LB is thought to be a reversible airway inflammatory process linking acute rejection episodes to subsequent airway damage and fibrosis characteristic of OB, and is believed to be a harbinger of OB (5). Therefore, the appearance of LB serves as a window into ongoing and potentially reversible airway destruction. Despite histological recognition of LB and OB on the pathological spectrum of chronic airway rejection, little is known about potential molecular mechanisms linking the two processes.
One potential link, which served as the focus for this investigation, is endogenous formation of nitric oxide (NO) gas and the enzymes responsible for its synthesis. Because NO synthases (NOS) are expressed in abundance in the lungs (6–8), have known immunomodulatory functions, and pathological specimens taken from patients with OB exhibit increased expression of inducible NOS (iNOS; 9, 10), we hypothesized that NOS expression may play a critical role in the development of both LB and OB. Additional histological evidence for a pathological role for excessive NO in airway tissue damage and OB comes from the examination of pathological OB specimens, in which immunoreactivity for nitrotyrosine (11) is detected. This provides evidence of the fleeting presence of peroxynitrite (12), a cytotoxic reaction product of NO and superoxide that is capable of triggering apoptosis (13–15). Several circumstantial lines of evidence also point to a pathological role for excessive NO production in development of OB. NO promotes the recruitment of CD8+ T cells to inflammatory sites (16). In the airways, NO derived from iNOS appears to increase chemokine expression and thereby induce airway inflammation (17, 18). The CC chemokines (macrophage inflammatory protein-1α [MIP-1α] and regulated on activation normal T cell expressed and secreted [RANTES]) induce the directional migration of monocytes and T cells, which have been implicated as early mediators of lung allograft rejection (19). IL-1β, a major proinflammatory mediator and amplifier of the pulmonary immune response (20), induces apoptosis of lung fibroblasts via NO production (21). The tumor suppressor p53 accumulates in response to NO-induced apoptosis (22) and promotes apoptosis during cardiac allograft rejection (23). Theoretically, these chemotactic and proapoptotic actions of NO could account for several pathophysiologic aspects of the pulmonary allogeneic inflammatory response by regulating the recruitment of monocytes into the target sites (24, 25), inducing apoptosis of graft stromal or parenchymal cells, or activating apoptotic signaling pathways to delete activated lymphocytes to promote graft acceptance (26, 27).
In the general transplant literature, there are dichotomous reports as to the potential role of NO and NOS in rejection. iNOS appears to have a detrimental role in cardiac allograft rejection, as its expression is strongly associated with histologic evidence of rejection (28) and its blockade (with aminoguanidine) reduces the intensity of the cellular infiltrates and myocytolysis, pathological hallmarks of cardiac allograft rejection (29). However, rather than reducing chronic cardiac allograft rejection, absence of the iNOS gene exacerbated cardiac transplant arteriosclerosis in a murine model of cardiac transplantation (30). In a rat model of heterotopic tracheal transplantation, administration of aminoguanidine (to block NOS, particularly iNOS) resulted in accelerated lesion development although surprisingly, there was no affect on the number of graft-infiltrating CD4+ or CD8+ T cells (31). To reconcile these apparent discrepancies and to determine whether the endothelial NOS (eNOS) or inducible isoforms of NOS participate in LB or OB development, these studies used a new murine tracheal transplant model that permits airflow through the transplanted conduit airway.
From an immunological vantage point, apoptosis of effector T cell populations may serve an important immunomodulatory role. Apoptotic cells have been identified in rejecting allografts, including the heart (23, 32, 33), kidneys (34), and liver (35), and this process appears to be promoted by iNOS induction (23, 32). However, apoptosis of infiltrating inflammatory cells may not necessarily be a bad thing. Clearance of infiltrating T lymphocytes through apoptosis associated with iNOS expression may support a mechanism of allograft acceptance (36). These experiments were designed to determine the role of recipient iNOS or eNOS in the recruitment and apoptosis of infiltrating inflammatory cells and their impact on the subsequent development of LB and the establishment of OB.
Materials and Methods
Mice.
Inbred male B10.A (H2a), C57BL/6J (H2b), iNOS-deficient (C57BL/6-Nos2tm1Lau), or eNOS-deficient (C57BL/6J-Nos3tm1Unc) mice backcrossed over 10 generations on the C57BL/6J background were purchased from The Jackson Laboratory. All mice were from 7–10 wk old (25–30 g). Isogeneic tracheal transplants were performed using C57BL/6J mice as both recipients and donors. Allogeneic (B10.A) tracheal grafts were harvested and transplanted into WT (C57BL/6J), iNOS−/−, or eNOS−/− recipient mice. To ascertain the involvement of iNOS induction in the rejection process, WT or eNOS−/− recipient mice were given the selective iNOS inhibitor, N 6-(1-iminoethyl)-l-lysine dihydrochloride (L-NIL; Cayman Chemical), at a daily dose of 5 mg/kg intraperitoneally (37) for 3 wk after transplantation. Moreover, to specifically determine the role of epithelial versus leukocyte-derived iNOS, we also performed transplantation experiments using iNOS−/− donor (C57BL/6-Nos2tm1Lau) airway allografts transplanted into iNOS+/+ recipient (B10.A) mice. All experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee at Columbia University in accordance with American Association for Accreditation of Laboratory Animal Care guidelines.
Tracheal Transplantation and Immunosuppression.
After anesthetizing mice with 2 mg intraperitoneal ketamine (Fort Dodge Labs) and 0.2 mg xylazine (American Animal Labs), donor mice were exsanguinated and whole trachea was exposed with blunt dissection from surrounding tissue through a midline incision under sterile conditions. The trachea was then transected below the cricoid cartilage, distal to the carinal bifurcation. The tracheal graft was placed on a sterilized gauze moistened with saline until the time of transplantation (within 10 min of harvest).
Recipient mice were similarly anesthetized. After shaving a small area on the anterior neck and preparing with iodine alcohol solution, the whole trachea was exposed and two round buttons of tissue were removed (matched in size and aligned with the open ends of the donor tracheal segment; see Fig. 1 A). The distal recipient orifice was positioned in the sixth intercartilaginous space and the proximal orifice was placed immediately subjacent to the cricoid cartilage area. Distal and proximal tracheal anastomoses were performed end to side using continuous 10-0 polypropylene sutures. Surgical wounds were repaired with a two-layer closer technique (anterior cervical muscles and skin). This new model of tracheal transplantation permits airflow through the tracheal lumen as well as the elimination of inspissated mucous and is therefore more likely to be physiologic than the standard model (38, 39) in which a tracheal segment is transplanted into subcutaneous tissue in the recipient's back.
Figure 1.

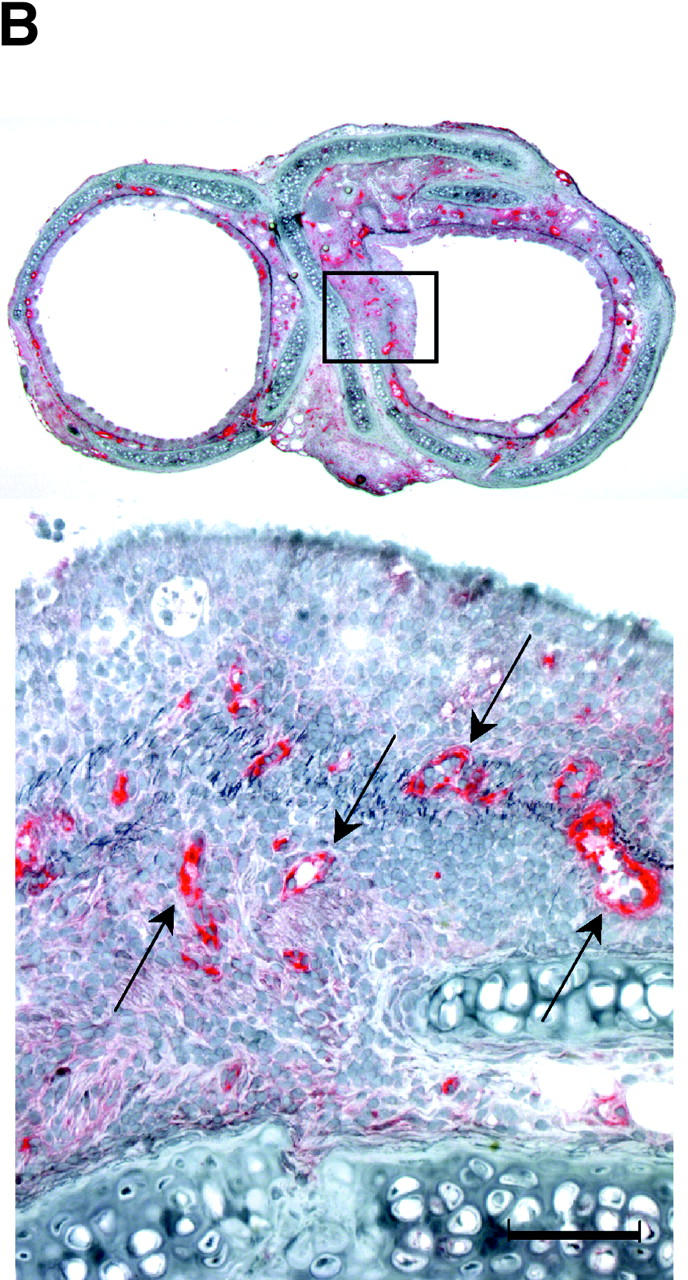


(A) New model of murine tracheal transplantation. The graft can fulfill its function as an airway by placing the proximal and distal graft anastomoses in the native trachea. This environment allows epithelium in the graft to be exposed to air and drain mucous through the anastomosis. Arrow, tracheal graft; arrowhead, native trachea. (B) Immunostaining for vWf in an allograft from a WT recipient. Arrows, anti–vWf-reactive capillaries. (C) Representative Verhoeff's-Van Gieson staining of transverse sections (low power magnification for a–d, high power magnification for e–h) through WT allografts (right side) with native trachea (left side) revealing the time course of the rejection process. After tracheal transplant (0 wk; a and e), mononuclear cells accumulated in the EL as well as SEL at 3 wk (b and f), with subsequent diminution by 6 wk (c and g). After 6 wk, epithelial cells were observed to dissociate from the basement membrane (BM). By 10 wk (d and h), epithelium appeared flattened with loss of ciliary structures and the subepithelial space was thickened with substantial collagen deposition. The time course of luminal occlusive lesions in the EL and SEL shown in D demonstrates that the intensity of the infiltrate corresponds with subsequent graft luminal (GL) occlusion. Comparing luminal patency with isografts, which exhibited near-complete patency, allografts were maximally occluded 3 wk after transplantation. n = 4 for each group; *, P < 0.05; bar, 50 μm.
All recipient animals received postoperative intramuscular immunosuppression (25 mg/kg/day cyclosporine; Bedford Laboratories) for 2 wk to model acute on chronic airway rejection, and 3 d of antibiotics (20 mg/kg/day cefazolin sodium; Apothecon) to suppress procedure-related infections. All recipient animals survived and were killed at a predefined interval of 3 wk after transplantation. This interval was chosen based on preliminary experiments suggesting that the most prominent cell infiltrates were likely to occur 3 wk after transplantation. Grafts were harvested and embedded together with the native tracheas in Tissue Freezing Medium™ (Triangle Biomedical Sciences) in a cryomold or snap frozen in liquid nitrogen and stored at −80°C until the time of histological or mRNA evaluation.
Cell Isolation.
Infiltrating inflammatory cells were isolated from transplanted airway graft tissue (40). In brief, tracheal grafts were removed from recipients and rinsed with RPMI 1640 medium (Life Technologies). Grafts were then minced using a scalpel in 3 ml RPMI 1640 medium containing 0.1% collagenase type 4 (Worthington Biochemical), 0.1% soybean trypsin inhibitor (Sigma-Aldrich), 50 U/ml DNase (Roche), 10% FBS, 10 μM Hepes (Life Technologies), and 1% penicillin-streptomycin (Life Technologies). The suspension was placed on an agitator at 37°C for 30 min and the digestion mixture was passed through a 70-μm nylon cell strainer (BD Biosciences) to remove undigested tissue and debris. Mononuclear cells were purified from the cell suspensions using density gradient centrifugation, using Lympholyte M (Accurate Chemicals). Recipient spleen cells were also prepared by isolation after centrifugation of cell suspensions and recovery of the pellet, followed by red cell lysis using ammonium chloride buffer.
FACS® Analysis.
Immunophenotyping of graft-infiltrating leukocytes was accomplished by FACS® analysis. Isolated mononuclear cells were stained for multicolor FACS® analysis using PE-labeled anti–mouse CD3 and FITC-labeled anti–mouse CD4, CD8 (BD Biosciences), and macrophage (F4/80; Caltag Laboratories). Cells were prepared in staining buffer that consisted of Ca2+- and Mg2+-free Dulbecco's PBS (Mediatech) with 0.05% sodium azide and 1% FBS. After a 5-min incubation at 4°C with purified anti-2.4G2 (BD Biosciences) to reduce nonspecific binding of FcII/III receptors, the cells were incubated for 30 min at 4°C in the dark with the indicated fluorescent antibodies (1:100). Next, cells were washed twice in staining buffer and populations of lymphocytes and macrophages were then identified using a FACSCalibur® system (BD Biosciences). Cell detector voltages were set using unstained lymphocytes isolated from the spleen or cultured murine macrophages (RAW 264.7; American Type Culture Collection). To determine the absolute numbers of the specific lymphocyte populations, percentages of cells established by FACS® analysis were multiplied by the total number of lymphocytes isolated from grafts determined by manual cell counts performed on a hemocytometer slide.
Histopathological Evaluation.
Frozen sections were cut 5-μm thick and placed on glass slides (Fisher Scientific), after which sections were air dried, fixed for 15 min in acetone at 4°C, and stored at −20°C until needed. Histochemical staining was performed for elastin (Accustain™; Sigma-Aldrich) to determine graft luminal occlusion. Immunostaining was performed for von Willebrand factor (vWf; rabbit anti–human vWf; Dako), smooth muscle cells (mouse anti–actin α-smooth muscle; Sigma-Aldrich), CD3, CD4, and CD8 (hamster anti–mouse CD3, rat anti–mouse CD4, and rat anti–mouse CD8; BD Biosciences), macrophage (rat anti–mouse F4/80; Caltag Laboratories), neutrophil (rat anti–mouse neutrophils; Cedarlane), iNOS and eNOS (polyclonal rabbit anti–mouse iNOS and polyclonal rabbit anti–mouse eNOS; Transduction Laboratories), and nitrotyrosine (polyclonal rabbit anti–human nitrotyrosine; Upstate Biotechnology). To ascertain cell proliferation in the transplanted grafts, in situ immunohistochemical detection of bromodeoxyuridine (BrdU; BD Biosciences) was performed. Recipient mice were injected intraperitoneally with 2.5 mg BrdU in PBS 24 and 2 h before sacrifice. After tissue harvest, slides were prepared from frozen sections in 0.01 M sodium citrate, pH 6.0, heated in a microwave to 90°C for 10 min, and cooled to room temperature followed by regular staining technique. For all immunohistochemistry done with a peroxidase readout, endogenous peroxidase was blocked with 0.3% H2O2. For all stains, nonspecific blocking was accomplished with normal serum derived from the host species in which the secondary antibody was prepared. Sections were then incubated with a suitable concentration of the primary antibody followed by the biotinylated secondary antibody. Immunodetection was performed using the Vectastain ABC Kit (Vector Laboratories) and developed in Vector substrate kits followed by the elastin stain, as well as a hematoxylin counterstain. As a negative control, each section was incubated with normal serum instead of the primary antisera.
Localization of intracellular chemokine expression (MIP-1α, RANTES, and IL-1β) in WT allografts was determined by confocal microscopic examination (LSM410; Carl Zeiss MicroImaging, Inc.) using multiparameter immunofluorescent staining techniques. After blocking nonspecific binding and Fc receptors with normal goat serum and the Fc receptor blocker anti-2.4G2 (BD Biosciences), CD8+ cells and macrophages were detected by FITC-conjugated antibodies (rat anti–mouse CD8 and F4/80; BD Biosciences and Caltag Laboratories, respectively). Sections were then fixed and permeabilized using a Cytofix/CytopermTM kit (BD Biosciences). Next, each section was incubated with rabbit anti–mouse antibody (MIP-1α, RANTES, and IL-1β; Cell Sciences) or rabbit IgG as a negative control at 4°C for 30 min in the dark. Intracellular chemokines were identified by incubating the sections with fluorescein-conjugated secondary antibody (Alexa Fluor® 594 goat anti–rabbit IgG; Molecular Probes) followed by a DAPI (4′,6-diamidino-2 phenylindole) counterstain.
To detect apoptosis in situ, the terminal deoxynucleotidyltransferase–mediated dUTP-digoxigenin nick-end labeling (TUNEL) method was performed according to the manufacturer's protocol (in situ Cell Death Detection Kit; Roche). TUNEL+ apoptotic cells were compared with the results obtained by the immunohistochemical detection of caspase-3 (polyclonal rabbit anti–human caspase-3, which detects the p17 subunit of caspase-3 but not the precursor form; R&D Systems) using adjacent serial sections for colocalization. A double staining technique was then performed to determine if the apoptotic cells were in fact T lymphocytes. After labeling immunohistochemically for T lymphocytes using a primary antibody to CD8 linked to alkaline phosphatase, apoptotic cells were detected by the TUNEL method (visualized with peroxidase substrate).
Morphometric measurements of cross-sectional areas and quantitative analysis for apoptotic cells in grafts was performed by blindly tracing both epithelial and subepithelial areas using a computer-assisted image analysis system (AxoCamHR; Carl Zeiss MicroImaging, Inc.) and counting positively staining apoptotic cells in five nonoverlapping high power (×400) fields from each of the five sections of each group and expressing both data as a percentage of whole luminal area (inside the area of the cartilage ring) and counting all cells identified by nuclear staining as the denominator.
Cytokine Assays.
To demonstrate the effect of recipient iNOS and eNOS deficiency and pharmacologic iNOS inhibition on serum levels of Th1 cytokines (TNF-α, IL-1β, IL-2, and total IL-12; Amersham Biosciences), a Th2 cytokine (IL-4; Amersham Biosciences) or two CC chemokines (MIP-1α and RANTES; R&D Systems), ELISAs were performed using commercial kits according to the manufacturer's instructions.
In Situ Hybridization.
To make RNA probes for in situ hybridization, RT-PCR was performed using Superscript™ II (Invitrogen) with total RNA as template. Total RNA was prepared from mouse tracheal tissue with TRIzol® Reagent (Invitrogen). Primers for mouse iNOS and eNOS were directed at 1055-1078/1791-1768 (these sequence data are available from GenBank/EMBL/DDBJ under accession no. NM 010927) and 1265-1287/2035-2015 (available from GenBank/EMBL/DDBJ under accession no. NM 008713) of the cDNA, respectively, producing 737- and 771-bp products. The forward and reverse primers sequence for iNOS were 5′-CCCCGCTATGGCCGCTTTGATGTG-3′, 5′-AGGACTGTGGCTCTGACCCGTGAA 3′. Those for eNOS were 5′-CACGCCGCCACAGCCTCCTTCAT-3′, 5′ CTCCCCGCCCAGCTCCTCCAG-3′. After reverse transcription, PCR was performed using the GeneAmp PCR System 9600 (PerkinElmer) with 35 cycles at 94°C annealling temperature. The PCR product mixture was run in a 1% agarose gel and was recovered using a Qiagen® II gel extraction kit (QIAGEN). This was then inserted into pCR®II Vector using the TA cloning method (Invitrogen). After the identity of the RNA expression plasmid was confirmed by enzyme cut and sequencing (ABI Prism 310 Genetic Analyzer; PE Corp.), the plasmid was linearized with SpeI and NotI enzymes to allow in vitro run-off synthesis of both sense- and antisense-oriented RNA probes. Both sense and antisense probes were labeled with a digoxigenin RNA labeling kit (Roche) and the labeled probes were then purified. In situ hybridization was performed as previously described (41). In brief, slides were prefixed in 4% paraformaldehyde and digested with 1 μg/ml proteinase K in Tris-EDTA, pH 8.0, followed by fixation again using 4% paraformaldehyde. Sections were acetylated with 0.1 mol/L triethanolamine, pH 8.0, with 0.25% (vol/vol) acetic anhydride, and then equilibrated for 60 min in hybridization buffer consisting of 4× SSC, 50% formamide, 5% dextran sulfate, 0.1 mg/ml yeast tRNA, and 0.05 mg/ml salmon sperm DNA. Hybridization was performed overnight at 45°C with either sense or antisense iNOS and eNOS probes (1:25 dilution in prehybridization buffer). Sections were subjected to stringent washes. After blocking with blocking buffer, sections were incubated with a 1:100 dilution of sheep anti–digoxigenin antibody (Digoxigenin detection kit; Roche) and color was allowed to develop for 1 h. Sections were covered with aqueous mounting medium until the time of inspection or photography.
Ribonuclease Protection Assay (RPA).
RPA was performed to evaluate the expression of proinflammatory cytokines and potential apoptotic mediators in the graft 3 wk after transplantation. Template RNA probes for RPA were designed based on the size of its probe so that multiple target mRNAs to be analyzed could be identified on a gel (Table I). The RNA probes were prepared as described above. Total RNA was isolated from each tracheal graft and 15 μg of each was used for the assay according to the manufacturer's protocol (RPA III™ kit; Ambion). In brief, in vitro transcription (MAXIscript™ kit; Ambion) was performed to generate antisense RNAs using T7 or SP6 RNA polymerase with [α- 32P]UTP (800 Ci/mmol; Amersham Biosciences). After gel purifying, ∼10 × 104 cpm each of full-length distinct probe transcripts were hybridized in excess at 42°C overnight to the target RNAs. Free probes and nonhybridized single stranded RNA were digested with RNase. Protected fragments were separated on a 5% denaturing acrylamide gel and quantified by autoradiography using BioMax™ film (Eastman Kodak Co.) at −80°C. Absorption values were obtained by densitometry scanning (National Institutes of Health (NIH) Image 1.62; NIH) of cDNAs. Expression of each band was normalized with its corresponding β actin band, and this ratio was then normalized with that of the isograft sample for each experiment.
Table I.
PCR Primer Sequences Were Synthesized to Detect the Indicated Mouse Cytokines and Apoptotic Mediators
| Template | Sequence Location | GenBank | Probe length (bp) |
|---|---|---|---|
| MIP-1α | 696-717 / 1047-1021 | X53372 | 352 |
| RANTES | 128-151 / 450-431 | M77747 | 323 |
| IFN-γ | 539-558 / 835-816 | K00083 | 297 |
| TNF-α | 1655-1678 / 1916-1893 | M38296 | 262 |
| β actin | provided by Ambion | — | 245 |
| iNOS | 1243-1270 / 1453-1426 | NM010927 | 211 |
| eNOS | 1319-1345 / 1504-1476 | NM008713 | 186 |
| nNOS | 3096-3119 / 3261-3240 | NM008712 | 166 |
| p53 | 1269-1292 / 1423-1400 | X01237 | 155 |
| IL-1β | 788-811 / 921-899 | M15131 | 134 |
Sequences used to make template RNA probes for RNase protection assays, probe lengths, GenBank accession numbers, and expected size of protected RNA fragments are indicated.
Statistics.
Data are expressed as mean ± SEM. All statistical comparisons were performed using a commercially available statistical package for the Macintosh personal computer (Stat View-J 5.0™; SAS Institute). One-way ANOVA and the Scheffe post-hoc test was used to compare conditions among the four groups. Differences were considered significant at the level of P < 0.05.
Results
Time Course of Graft Histopathology.
Pilot studies performed during the development of the airway transplant model failed to show histological evidence of tissue necrosis in microscopic sections evaluated at various time points. To determine angiogenesis in these airway allografts (transplanted without a vascular pedicle), capillarization was detected by immunostaining of endothelial cells with an antibody that recognizes vWf (Fig. 1 B). These data show many small vessels in the grafts, including in newly formed obstructing lesions. Histopathological evaluation of B10.A airway allografts harvested 4–6 h and 3, 6, and 10 wk after transplantation into C57BL/6J WT recipients (n = 4 each) demonstrated the sequential progression of airway allograft rejection. Mononuclear cell infiltration into the allografts was primarily observed in the subepithelial layer (SEL) as early as 2 wk (unpublished data), but the intensity of the infiltrate increased at both the epithelial layer (EL) and SEL during the period from 3–4 wk after transplantation. At these time points, both layers were thickened, leading to a partial concentric graft luminal occlusion (Fig. 1 C shows representative sections and pooled data are quantified in D). Most infiltrating cells in the SEL and EL regions of the allografts receded after 3–4 wk and at 6 wk, epithelial dissociation from the basement membrane was noted. After epithelial denudation, little if any inflammatory infiltrate was observed. By 10 wk after transplantation, epithelium appeared flattened, ciliary structures were no longer visible, and the subepithelial space was thickened with substantial collagen accumulation. As in human OB (5), immunohistochemical examination revealed little if any smooth muscle cells in the obstructive airway lesion, although as expected, the normal airway musculature stains quite clearly (unpublished data). The membranous and airway musculature areas of the cartilaginous “C” rings appeared relatively unaffected throughout the observation period. The result of assays for cell proliferation by in situ immunohistochemical detection of BrdU demonstrated positive staining in some (but not many) nuclei in the obstructive airway lesion (unpublished data). These results indicate that proliferative change is not a major culprit in the developing obstructive airway lesion. Based on these observations, to establish the role of NOS, grafts in subsequent experiments were harvested 3 wk after tracheal transplantation.
Quantification of Graft Mononuclear Cell Infiltration.
The prominent mononuclear cell infiltrate in airway allografts was comprised primarily of T cells, as determined by immunohistochemical analysis for the pan-T cell–marked CD3 (Fig. 2 , a–e). These airway inflammatory lesions with lymphocytic predominance are indistinguishable histologically from lymphocytic bronchitis observed in humans (5). Identification of subpopulations of graft-infiltrating CD4+ (Fig. 2, f–j) and CD8+ (Fig. 2, k–o) T cells, as well as F4/80+ macrophages (Fig. 2, p–t), was also performed. Absolute and relative numbers of allograft-infiltrating leukocytes, including CD3+ CD4+ T cells, CD3+ CD8+ T cells (Fig. 2 u), and F4/80+ macrophages (Fig. 2 v) were determined by FACS® analysis and manual cell counting. Concordant with the immunohistochemical data, analysis of cells extracted from graft tissue indicates that infiltrating lymphocytes in allografts from WT recipients (n = 7) are predominantly CD3+ CD8+ T cells (223 × 103 ± 9.9 × 103/graft for CD3+ CD8+, 151 × 103 ± 6.7 × 103/graft for CD3+ CD4+). In contrast, lymphocytic infiltration was markedly reduced with either recipient iNOS deletion (n = 5; 26-fold reduction for CD3+ CD8+ and 18-fold reduction for CD3+ CD4+) or pharmacologic inhibition of iNOS with L-NIL (n = 6; sevenfold reduction for CD3+ CD8+ and threefold reduction for CD3+ CD4+). Interestingly, pharmacological iNOS blockade showed a more potent suppression of CD3+ CD8+ T cells infiltration than that of CD3+ CD4+ T cells. iNOS−/− recipients exhibited similar degrees of suppression of both T cell subtypes. eNOS−/− recipients (n = 5), however, did not show any significant difference in graft CD4+ T cell, CD8+ T cell, or macrophage infiltration compared WT recipients. There was also a decrease in the number of macrophages from allografts from iNOS−/− recipients (eightfold reduction) and L-NIL–treated WT recipients (twofold reduction) compared with both allografts from WT and eNOS−/− recipients. These quantitative data suggest that iNOS expression, but not eNOS expression, increases absolute numbers of infiltrating T cell subpopulations and macrophages. Immunohistochemical staining revealed a relative paucity of neutrophils in all groups (unpublished data). Isografts demonstrated no obvious cellular infiltrates on histological examination, and correspondingly, no detectable infiltrates upon lymphocyte isolation.
Figure 2.
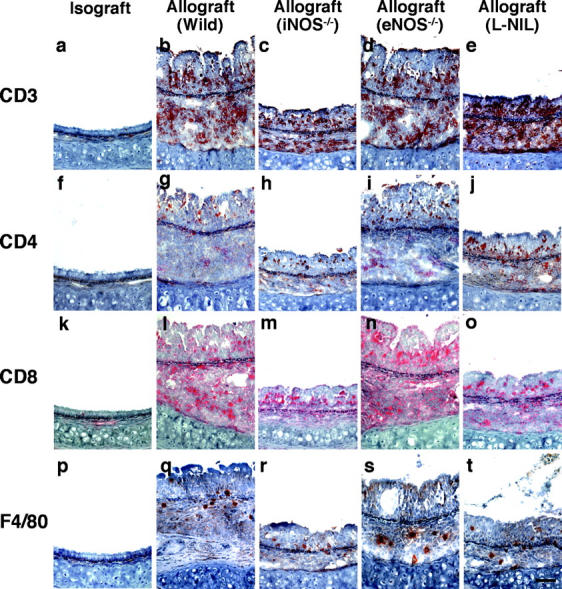


Immunohistochemical staining of the pan-T cell marker CD3 (a–e; brown), as well as CD4+ (f–j; brown), CD8+ (k–o; red) T cells, and F4/80+ (p–t; brown) macrophages in sections from grafts transplanted into isogeneic (a, f, k, and p), allogeneic WT (b, g, l, and q), iNOS−/− (c, h, m, and r), eNOS−/− (d, i, n, and s) recipients, and L-NIL–treated WT (e, j, o, and t) recipients 3 wk after transplantation. Quantitative analysis of mononuclear cell infiltrates by FACS®, shown as absolute numbers of recovered CD3+ CD4+ and CD3+ CD8+ T cells (u), and F4/80+ macrophages (v) in each graft from each group. Bar, 50 μm; *, P < 0.05 versus allografts from WT recipients; #, P < 0.05 versus allografts from iNOS−/− recipients; †, P < 0.05 versus allografts from eNOS−/− recipients.
Morphometric analysis of WT allografts revealed a concordance between the thickening of each of the airway layers leading to luminal occlusion and lymphocytic infiltrates (Fig. 3 a). Compared with these results in WT recipients, allografts transplanted into iNOS−/− recipients showed significantly less luminal narrowing with a concordant reduction in the numbers of infiltrating CD3+ CD4+ and CD3+ CD8+ T cells. On the other hand, eNOS−/− recipients showed neither any significant difference in luminal obstruction or influx of T lymphocytes compared with WT allograft recipients. Pharmacologic inhibition of recipient iNOS with L-NIL (5 mg/kg/day) 3 wk after transplantation to both WT (n = 10) and eNOS−/− (n = 5) recipients revealed significant reduction in the luminal obstruction of allografts compared with WT and eNOS−/− allograft recipients.
Figure 3.


(a) Analysis of tracheal graft sections 3 wk after transplantation. Representative sections (left) and morphometric analysis of sections (right) for each of the indicated conditions. Isografts were WT and allograft recipients were either WT, iNOS−/−, or eNOS−/−. The effect of pharmacologic recipient iNOS inhibition with L-NIL (5 mg/kg/day for 3 wk after transplantation) in WT and eNOS−/− recipients was also examined. Allografts placed in iNOS−/− or iNOS-inhibited recipients demonstrated significantly reduced luminal obliteration compared with WT and eNOS−/− allograft recipients. In contrast, recipient eNOS deficiency was associated with similar degrees of luminal occlusion as WT allograft recipients. *, P < 0.05 versus allografts from WT recipients; #, P < 0.05 versus allografts from iNOS−/− recipients; †, P < 0.05 versus allografts from eNOS−/− recipients. (b) The effect of epithelial versus leukocyte-derived iNOS on graft luminal occlusion. Donor tissue and recipient genotypes were either iNOS+/+ or iNOS−/−, as indicated. Effect of background strain is also indicated here. Genotypes that are underlined represent B10.A strain background. Genotypes that are not underlined represent C57BL/6J. Note that the leftmost three bars represent the same data as is shown in a, repeated here to facilitate comparison. *, P < 0.05.
In contrast to the original scenario, in which iNOS+/+ allografts transplanted into iNOS−/− recipients exhibited significantly reduced luminal occlusion (29 ± 2% vs. 53 ± 2% for iNOS+/+ allografts transplanted into iNOS+/+ recipients), graft luminal occlusion in iNOS−/− allografts transplanted into iNOS+/+ recipients (n = 5) 3 wk after transplantation was 49 ± 2%. These differences could not be accounted for by differences in background strain, because for iNOS+/+ trachea transplanted into iNOS+/+ recipients, luminal occlusion was similar when the reverse background strain combination (i.e., C57Bl/6J donors transplanted into B10.A recipients) was used (n = 5, luminal occlusion 49 ± 1%; Fig. 3 b). These data suggest that iNOS expressed by graft-infiltrating leukocytes, rather than by airway epithelium, exerts the dominant influence on airway rejection.
Quantification of Lymphocyte Apoptosis.
Apoptosis of T cells represents a major mechanism of immune modulation. Therefore, evidence for apoptosis of graft-infiltrating T cells was ascertained using the TUNEL method (Fig. 4 , a–d), and further corroborated immunohistochemically by colocalizing caspase-3 expression in the same cells in adjacent sections (Fig. 4, e–h). Caspases play an important role in the execution phase of apoptosis. Caspase-3 is one of the downstream effector caspases (42), activated by proapoptotic signals triggered by cytokines (43, 44) or through cell death receptors (45), which cleaves cytoskeletal and nuclear proteins to induce apoptosis. Both apoptotic cells and caspase-3 activation colocalize predominantly to the EL, moreso than the SEL. Most apoptotic cells were identified as CD8+ T lymphocytes with a double staining technique (Fig. 4, i–l). The frequency of apoptotic T lymphocyte TUNEL positivity in WT recipients of allografts was significantly higher in the EL than that seen in WT recipients of isografts (Fig. 4 m). Compared with WT allograft recipients, apoptotic cells were much less frequently observed in the EL of allografts placed in iNOS−/− recipients. eNOS−/− allograft recipients showed a very slight increase in apoptotic cells in the EL. These results indicate that apoptosis of graft-infiltrating T cells is increased in the EL of allografts, is markedly reduced by recipient iNOS deficiency, and slightly increased by recipient eNOS deficiency. Obliteration of the airway lumen appears to parallel quite closely the degree of lymphocytic infiltration as well as the apoptotic T cell activity detected in the lesions.
Figure 4.
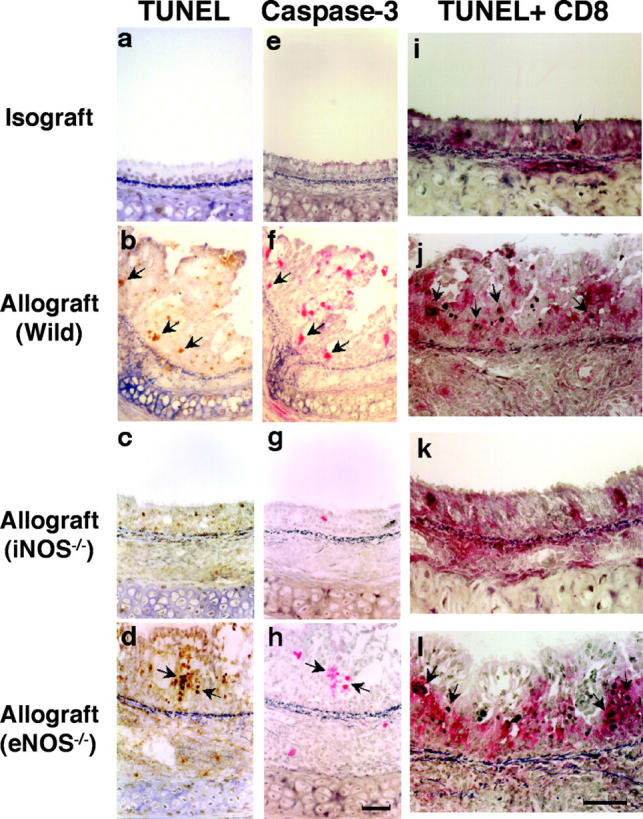

Apoptotic cells were detected using the in situ TUNEL labeling method (a–d; arrows) and further corroborated immunohistochemically by colocalizing caspase-3 expression in the same cells in adjacent sections (e–h; arrows). Double staining for apoptotic CD8+ T lymphocytes (i–l; arrows) is detected by black nuclear labeling for apoptosis and red cytoplasmic staining for CD8. Apoptosis of CD8+ T lymphocytes (arrows) was most abundant in the EL of allografts placed in WT and eNOS−/− recipients. The graph (m) shows quantitative T lymphocyte apoptotic activity in both the EL and SEL of the graft. Apoptosis of T lymphocytes was reduced by recipient iNOS deficiency. *, P < 0.05 versus allografts from WT recipients; #, P < 0.05 versus allografts from iNOS−/− recipients; bar, 50 μm.
Colocalization of iNOS mRNA/Protein and Nitrotyrosine Immunoreactivity.
In situ detection of iNOS mRNA as well as immunohistochemical detection of iNOS protein were performed to determine whether graft iNOS expression is predominantly a function of in situ induction within the allograft proper, or whether migrating leukocytes represent the predominant source. Although iNOS mRNA and protein were faintly detectable in isografts (Fig. 5 , a and e), they were prominently localized in the EL of allografts from both WT (Fig. 5, b and f) and eNOS−/− (Fig. 5, d and h) recipients. iNOS was strongly expressed in both the epithelial cells and infiltrating leukocytes, whereas eNOS immunoreactivity was limited to the surface epithelial cells of both isografts and allografts (unpublished data). There were no hybridization signals detected in adjacent serial sections hybridized with both sense iNOS and eNOS RNA probes (unpublished data). In iNOS−/− recipients, iNOS immunopositivity was limited to the surface epithelium of the iNOS+/+ allografts (Fig. 5 g). Interestingly, iNOS mRNA and protein expression were enhanced in grafts from eNOS−/− recipients, whereas there was no up-regulation of both eNOS mRNA and protein expression in grafts from iNOS−/− recipients (unpublished data). These data indicate that there is compensatory up-regulation of iNOS in allografts placed in eNOS null recipients.
Figure 5.
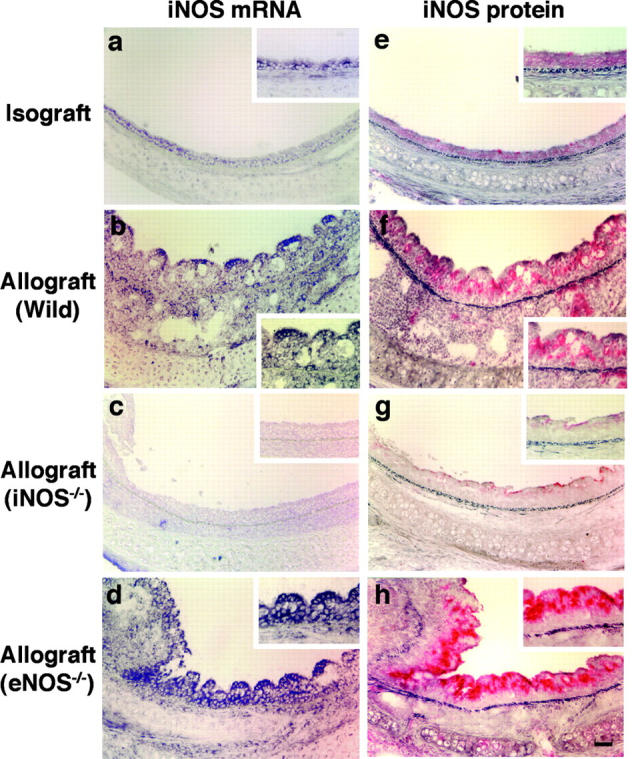
Analysis of iNOS mRNA expression by in situ hybridization and iNOS immunoreactivity in airway grafts. Both iNOS mRNA and protein production are modestly detected in the EL of isografts (a and e), but strongly detected in epithelial cells and inflammatory cells from allografts placed in WT recipients (b and f). Allografts from iNOS−/− recipients exhibited virtually no iNOS mRNA (c) or protein expression, except faint detection observed on the surface of the EL (g). However, elevated iNOS mRNA (d) and strong iNOS immunoreactivity (h) was identified in allografts from eNOS−/− recipients. Bar, 50 μm.
In a milieu in which oxygen radicals and NO are produced in abundance in very close proximity, such as in allograft rejection, peroxynitrite forms rapidly as the adduct of superoxide and NO. Peroxynitrite is a highly reactive compound that promotes the nitration of tyrosine residues in tissue proteins, which can then be detected immunohistochemically using an antibody that specifically recognizes nitrotyrosine (46). Immunohistochemical studies demonstrated abundant expression of nitrotyrosine in tracheal allografts, specifically in epithelial cells and graft-infiltrating lymphocytes from WT and eNOS−/− recipients, but not for iNOS−/− recipients (unpublished data). These data indicate that iNOS, rather than eNOS, is chiefly responsible for the production of NO that leads to the formation of peroxynitrite in tracheal allografts. Taken together with the data showing iNOS gene null or iNOS-inhibited allografts to be protected from luminal obliteration (Fig. 3 a), these data raise the specter of a causal link between peroxynitrite formation and the endoluminal obliterative process (47).
mRNA Expression of Chemokines, Proapoptotic Mediators, and NOS Isoforms.
To determine whether chemoattractant and apoptotic mediators were up-regulated in concordance with the influx and deletion of graft infiltrating lymphocytes, multiprobe RPAs were performed on tissue samples obtained 3 wk after transplantation (Fig. 6 a). Compared with isografts, normalized messages of the CC chemokines, MIP-1α (Fig. 6 b) and RANTES (Fig. 6 c), were significantly (P < 0.05) up-regulated in the allografts from WT recipients (33-fold increase for MIP-1α and 14-fold increase for RANTES) and eNOS−/− recipients (32-fold and 11-fold increase, respectively). Allografts from iNOS−/− recipients, however, showed significantly less expression of CC chemokines compared with the WT allografts. These findings suggest that iNOS, but not eNOS, plays a significant role in the CC chemokine up-regulation, which results in recruitment of CD8+ T cells. On these blots, IFN-γ mRNA was not detected and TNF-α was only faintly expressed (Fig. 6 d). Expression of mRNAs for the tumor suppressor p53 (Fig. 6 e) and IL-1β (Fig. 6 f) paralleled the results seen in histologic sections stained for apoptotic immunoreactivity. Compared with isografts, normalized message for p53 and IL-1β were up-regulated in the allografts from WT recipients (ninefold and fivefold increase, respectively; P < 0.05 each) and eNOS−/− recipients (11-fold and ninefold increase, respectively; P < 0.05 each). In contrast, allografts from iNOS−/− recipients showed significantly less expression of both mRNA species compared with WT allografts. These data, too, corresponded with the increased apoptotic immunoreactivity observed on histological sections of WT and eNOS−/− allografts. mRNA levels of p53 and IL-1β were diminished (to isograft levels) in allografts transplanted into iNOS null recipients, in concordance with the apoptosis data shown in Fig. 4.
Figure 6.
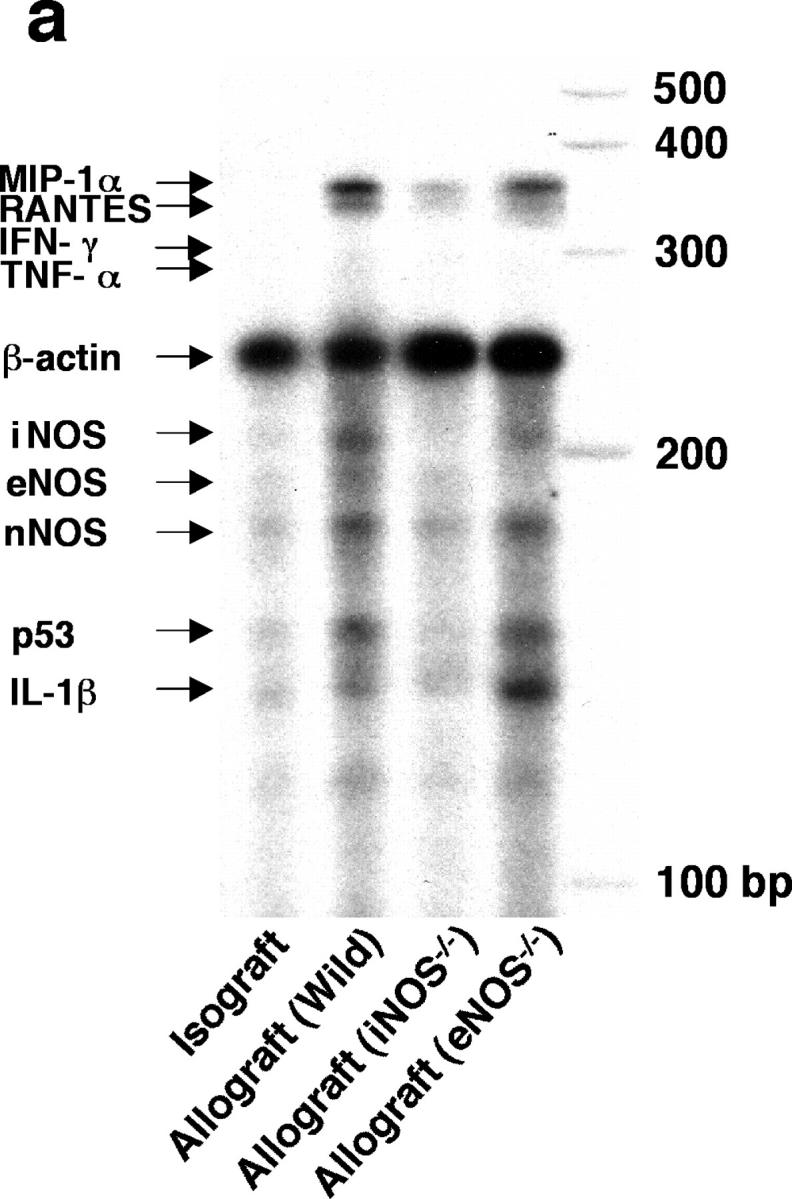

Simultaneous quantitation of multiple mRNA species encoding chemokines, proapoptotic mediators, and NOS isoforms in grafts from each group were examined using multiprobe RNase protection assays 3 wk after transplantation (n = 5 for each). 15 μg of total RNA was hybridized to each of the 10 antisense riboprobes (refer to Table I) (a) The intensity of each protected band was determined by densitometry, with values normalized first to the corresponding β actin band and then relative to the normalized value for each isograft experiment (b–i). *, P < 0.05 versus allografts from WT recipients; #, P < 0.05 versus allografts from iNOS−/− recipients; †, P < 0.05 versus allografts from eNOS−/− recipients.
Because the lack of one NOS isoform could theoretically, especially under stress conditions, lead to a compensatory induction of an alternate NOS isoform, mRNA levels for the three NOS isoforms were quantified by RNase protection assay in this tracheal transplant model. mRNA levels of iNOS were significantly increased in the allografts from WT and eNOS−/− recipients compared with isografts (15-fold and 12-fold increase, respectively; P < 0.05 each), whereas no significant increase was shown in the iNOS−/− recipients (Fig. 6 g; note that some baseline level of NOS may have been detected secondary to NOS expression in the allograft material rather than in infiltrating cells). eNOS mRNA expression in grafts was detected at the same low levels in all groups (Fig. 6 h). Expression of nNOS mRNA was increased in the allografts from WT and eNOS−/− recipients (sixfold increase for both; P < 0.05) compared with isografts (Fig. 6 i).
Cytokine Expression and Localization.
Recipient cytokine levels in serum samples (n = 10) from each group were measured 3 wk after transplantation (Fig. 7 , a–g). In contrast to the levels of cytokines that were observed in isogeneic recipients, the levels of chemokines, MIP-1α and RANTES, and IL-1β in WT and eNOS−/− allograft recipients were up-regulated after airway allograft transplantation. Recipient iNOS gene deficiency and pharmacologic inhibition, however, significantly suppressed MIP-1α production with trends toward reduced levels of other inflammatory chemokines/cytokines. Although some levels of some Th1 cytokines were not affected by either airway transplantation nor iNOS status (TNF-α, IL-12), there were strong trends shown toward up-regulation of IL-2. Unexpectedly, WT airway allograft transplantation caused an increase in serum levels of IL-4 (a Th2 cytokine) with decreased levels in iNOS gene null mice.
Figure 7.

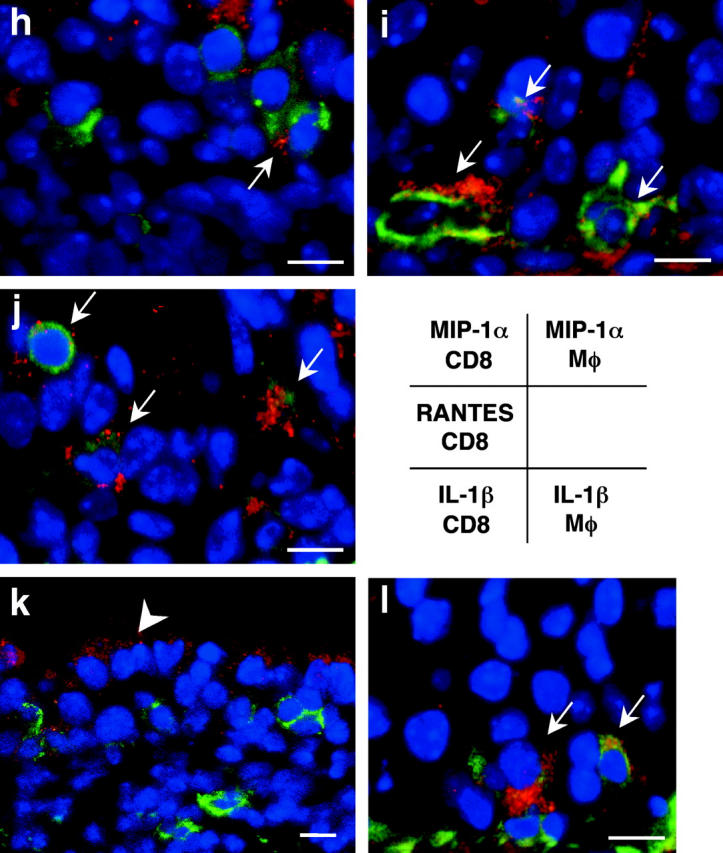
Serum cytokine levels measured by ELISA 3 wk after transplantation (n = 10 for each group; a–g.) Colocalization of intracellular chemokine expression (MIP-1α, RANTES, and IL-1β) and leukocyte immunophenotype was determined by confocal microscopic examination using immunofluorescent staining technique (h–l). Intracellular chemokines were identified by red emission after the application of an excitation wavelength of 594 nm. CD8+ T cells (h, j, and k) and F4/80+ macrophages (i and l) were labeled with FITC, which was detected as a green emission after the application of an excitation wavelength of 488 nm. Sites of colocalization (yellow) are indicated by arrows and epithelial cells that express IL-1β are indicated by the arrowhead. *, P < 0.05 versus allografts from WT recipients; #, P < 0.05 versus allografts from iNOS−/− recipients; †, P < 0.05 versus allografts from eNOS−/− recipients; bar, 5 μm.
Colocalization of intracellular chemokine (MIP-1α and RANTES) and proinflammatory cytokine (IL-1β) expression was determined by confocal microscopic examination of WT allograft tissue. Allograft-infiltrating cells that expressed MIP-1α were identified primarily as F4/80+ macrophages (Fig. 7 i) much more so than CD8+ T cells (Fig. 7 h). CD8+ T cells, however, expressed RANTES protein, evidenced by the colocalization of the fluorescent signals (Fig. 7 j). Although IL-1β expression was mainly localized in the epithelial lining (Fig. 7 k), it was also detected in F4/80+ macrophages (Fig. 7 l). CD8+ T cells do not appear to be sites of IL-1β synthesis (Fig. 7 k).
Discussion
To date, the underlying causes of OB remain elusive as they are likely multifactorial. Nevertheless, given the importance of OB as the prime limitation to long-term success with lung transplantation, understanding potential contributory mechanisms remains an important goal. Histologically, it appears that OB develops as a part of the immune response, as the result of repeated bouts of allograft airway inflammation that matures by tissue scarring, with resultant luminal obliteration. Because the bronchial/bronchiolar epithelium is one of the prime targets of lung rejection (48, 49), alloantigen-dependent airway epithelial damage occurring shortly after transplantation, during the lymphocytic bronchitis phase, can foreshadow the eventual loss of airway function.
NO is a well-recognized immunomodulatory gas that has special importance in the pathobiology of lung transplant rejection. It is exhaled in great quantities during lung transplant rejection, particularly during the lymphocytic bronchitis and early OB stages (50), and expression of the inducible enzyme responsible for its synthesis increases greatly during the active premonitory phases of OB, particularly in the bronchial epithelium (10). In contrast to the recognized up-regulation of iNOS during chronic lung transplant rejection, eNOS is down-regulated (9, 10).
These studies suggest that in striking contrast to cardiac allograft vasculopathy, where lack of the iNOS gene exacerbated cardiac transplant arteriosclerosis (30), in the airways, lack of iNOS is beneficial. iNOS can play a role as a potential mediator of cardiac chronic rejection by modulating mononuclear cell expansion and neointimal vascular smooth muscle migration or proliferation (28, 30). Endothelially derived NO appears to inhibit atherogenesis by suppressing expression of endothelial adhesion receptors (51), smooth muscle cell–derived chemokines (52), and activated leukocyte-derived superoxide radicals (53). Therefore, in a cardiac allograft model, it is not particularly surprising that iNOS suppresses vasculopathy. The data presented here, however, in transplanted airways, is diametrically opposite. The presence of the iNOS gene exacerbates LB and the luminal airway narrowing that is similar to OB. Our data show that recipient mice lacking the iNOS gene, or those treated with a specific iNOS inhibitor, exhibit significant protection against (a) lymphocytic bronchitis, (b) local induction of proinflammatory and proapoptotic mediators, (c) apoptosis of graft-infiltrating cells, and (d) late development of OB. In addition, leukocyte-derived iNOS rather than epithelium-derived iNOS appears to be most responsible for the development of the pathological airway lesion. These data indicate that chronic airway rejection and chronic vascular rejection differ fundamentally from each other, particularly with respect to the ultimate causes of luminal obstruction. There are several possible explanations for these differences. Koglin et al. (30) posit that iNOS prevents neointimal smooth muscle migration and hence, exerts a suppressive effect on vascular thickening. In contrast, our studies indicate that smooth muscle cells do not represent a significant component of the obstructive airway lesion. Rather, early on, there is a lymphocytic predominance and later, acellular collagenous material accumulates with ongoing epithelial damage. This is pathologically quite similar to the OB that develops in humans (5), in which fibrous scarring represents the dominant feature.
Chemotaxis for both CD4+ and CD8+ T cells is an important mechanism in allograft rejection. Local production of proiflammatory chemokine or cytokine derived from mononuclear cells have been suggested to trigger the rejection process observed in lung transplant recipients (54–57). Both Th1 and Th2 cytokines contribute to the development of allograft rejection and their transcript levels have been correlated with graft rejection severity (58), which is why helper T lymphocytes represent major targets of immunosuppressive therapy. In this study, we demonstrate that iNOS increases mRNA and protein for some, but not all, Th1 cytokines and also does so for a Th2 cytokine (IL-4). eNOS has essentially no effect on any measured cytokine in this model. Because of the striking recruitment of CD3+ CD8+ T cells to allografts, which was increased significantly by iNOS in parallel with increased induction of CC chemokines, it is reasonable to posit a causal relationship between iNOS expression, chemokine induction, and recruitment of alloeffector cytotoxic T lymphocytes.
Confocal microscopic studies showed that once recruited, graft-infiltrating lymphocytes and monocytes are likely to participate in the propagation and amplification of the alloimmune response to the transplanted airway. Cytotoxic T lymphocytes are known to participate in establishing the local cytokine environment by producing CC chemokines (59, 60), a result that is supported by the confocal microscopic studies that identify both CD8+ T cells and macrophages as sites of cytokine/chemokine production. Either recipient iNOS deficiency or pharmacologic inhibition of iNOS mitigated lymphocytic and macrophage infiltration significantly, and interestingly, changed the predominant T cell population from CD3+ CD8+ to CD3+ CD4+ T cells infiltrate, with a marked reduction in all T cells. These data indicate that iNOS, but not eNOS, plays a critical regulatory role in the recruitment of T lymphocytes, particularly CD3+ CD8+ T cells, during the process of airway rejection.
Some comment is necessary regarding the new model of airway transplantation presented in this study. Previous murine airway transplant models in mice (39) involved heterotopic airway implantation into subcutaneous tissue. However, the conditions of these grafts is airless, and in our own experience (unpublished data), we found that these heterotopic grafts became clogged intraluminally with secretions, which is less physiologic than the intraluminal air flow permitted by our model. This air flow–permissive model is likely to be more relevant in studies in which the role of a biologic gas, such as NO, is being examined. Biologically meaningful model-related differences may account for the divergence of our results with those reported in a rat tracheal transplant model. In studies in which rat tracheal segments were transplanted intraperitoneally (31), a relatively, but not completely, selective iNOS inhibitor (aminoguanidine) did not alter CD4+ nor CD8+ T cell infiltration, and although early luminal obliteration was accelerated, at 30 d, no differences in airway obliteration were seen between treated and untreated groups.
In summary, our results indicate that during the process of airway rejection, up-regulation of iNOS occurs in the allograft epithelium as well as in graft-infiltrating leukocytes. In this critical region, iNOS, especially that derived from leukocytes, promotes massive lymphocyte influx and their subsequent apoptosis, coinciding with the release of chemotactic as well as proapoptotic mediators, leaving a smoldering lesion in which fibrosis and eventual luminal obliteration take place. These data suggest that OB is fundamentally different than cardiac allograft vasculopathy, where iNOS induction appears to be protective, and suggest that iNOS inhibitors may actually have some role to play in OB prevention.
Acknowledgments
The authors thank Dr. Kim Olson for his expert assistance in sequencing the probes used for this study, and Drs. Hui Liao, Kenji Okada, and Yasushi Yoshikawa for expert technical advice.
This study was supported in part by the United States Public Health Service, grants R01 HL55397 and R01 HL 60900.
Footnotes
Abbreviations used in this paper: BrdU, bromodeoxyuridine; EL, epithelial layer; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; LB, lymphocytic bronchitis/bronchiolitis; L-NIL, N 6-(1-iminoethyl)-l-lysine dihydrochloride; MIP-1α, macrophage inflammatory protein 1α; NO, nitric oxide; NOS, NO synthase(s); OB, obliterative bronchiolitis; RANTES, regulated on activation normal T cell expressed and secreted; RPA, ribonuclease protection assay; SEL, subepithelial layer; TUNEL, terminal deoxynucleotidyltransferase–mediated dUTP-digoxigenin nick-end labeling; vWf, von Willebrand factor.
References
- 1.Berry, G.J., E.M. Brunt, D. Chamberlain, R.H. Hruban, R.K. Sibley, S. Stewart, and H.D. Tazelaar. 1990. A working formulation for the standardization of nomenclature in the diagnosis of heart and lung rejection: lung rejection study group. J. Heart Transplant. 9:593–601. [PubMed] [Google Scholar]
- 2.Trulock, E.P. 1997. Lung transplantation. Am. J. Respir. Crit. Care Med. 155:789–818. [DOI] [PubMed] [Google Scholar]
- 3.Hruban, R.H., W.E. Beschorner, W.A. Baumgartner, S.C. Achuff, T.A. Traill, B.R. Marsh, P.K. Gupta, G.M. Hutchins, and B.A. Reitz. 1988. Diagnosis of lung allograft rejection by bronchial intraepithelial Leu-7 positive T lymphocytes. J. Thorac. Cardiovasc. Surg. 96:939–946. [PubMed] [Google Scholar]
- 4.Yousem, S.A. 1993. Lymphocytic bronchitis/bronchiolitis in lung allograft recipients. Am. J. Surg. Pathol. 17:491–496. [DOI] [PubMed] [Google Scholar]
- 5.Yousem, S.A., G.J. Berry, P.T. Cagle, D. Chamberlain, A.N. Husain, R.H. Hruban, A. Marchevsky, N.P. Ohori, J. Ritter, S. Stewart, et al. 1996. Revision of the 1990 working formulation for the classification of pulmonary allograft rejection: lung rejection study group. J. Heart Lung Transplant. 15:1–15. [PubMed] [Google Scholar]
- 6.Grasemann, H., N. Knauer, R. Buscher, K. Hubner, J.M. Drazen, and F. Ratjen. 2000. Airway nitric oxide levels in cystic fibrosis patients are related to a polymorphism in the neuronal nitric oxide synthase gene. Am. J. Respir. Crit. Care Med. 162:2172–2176. [DOI] [PubMed] [Google Scholar]
- 7.Guo, F.H., H.R. De Raeve, T.W. Rice, D.J. Stuehr, F.B. Thunnissen, and S.C. Erzurum. 1995. Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proc. Natl. Acad. Sci. USA. 92:7809–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giaid, A., and D. Saleh. 1995. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 333:214–221. [DOI] [PubMed] [Google Scholar]
- 9.McDermott, C.D., S.M. Gavita, H. Shennib, and A. Giaid. 1997. Immunohistochemical localization of nitric oxide synthase and the oxidant peroxynitrite in lung transplant recipients with obliterative bronchiolitis. Transplantation. 64:270–274. [DOI] [PubMed] [Google Scholar]
- 10.Gabbay, E., E.H. Walters, B. Orsida, H. Whitford, C. Ward, T.C. Kotsimbos, G.I. Snell, and T.J. Williams. 2000. Post-lung transplant bronchiolitis obliterans syndrome (BOS) is characterized by increased exhaled nitric oxide levels and epithelial inducible nitric oxide synthase. Am. J. Respir. Crit. Care Med. 162:2182–2187. [DOI] [PubMed] [Google Scholar]
- 11.Mason, N.A., D.R. Springall, A. Pomerance, T.J. Evans, M.H. Yacoub, and J.M. Polak. 1998. Expression of inducible nitric oxide synthase and formation of peroxynitrite in posttransplant obliterative bronchiolitis. J. Heart Lung Transplant. 17:710–714. [PubMed] [Google Scholar]
- 12.Beckman, J.S., T.W. Beckman, J. Chen, P.A. Marshall, and B.A. Freeman. 1990. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA. 87:1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albina, J.E., S. Cui, R.B. Mateo, and J.S. Reichner. 1993. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J. Immunol. 150:5080–5085. [PubMed] [Google Scholar]
- 14.Pinsky, D.J., B. Cai, X. Yang, C. Rodriguez, R.R. Sciacca, and P.J. Cannon. 1995. The lethal effects of cytokine-induced nitric oxide on cardiac myocytes are blocked by nitric oxide synthase antagonism or transforming growth factor beta. J. Clin. Invest. 95:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinsky, D.J., W. Aji, M. Szabolcs, E.S. Athan, Y. Liu, Y.M. Yang, R.P. Kline, K.E. Olson, and P.J. Cannon. 1999. Nitric oxide triggers programmed cell death (apoptosis) of adult rat ventricular myocytes in culture. Am. J. Physiol. 277:H1189–H1199. [DOI] [PubMed] [Google Scholar]
- 16.Scheller, L.F., S.J. Green, and A.F. Azad. 1997. Inhibition of nitric oxide interrupts the accumulation of CD8+ T cells surrounding Plasmodium berghei-infected hepatocytes. Infect. Immun. 65:3882–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asano, K., C.B. Chee, B. Gaston, C.M. Lilly, C. Gerard, J.M. Drazen, and J.S. Stamler. 1994. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. USA. 91:10089–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trifilieff, A., Y. Fujitani, F. Mentz, B. Dugas, M. Fuentes, and C. Bertrand. 2000. Inducible nitric oxide synthase inhibitors suppress airway inflammation in mice through down-regulation of chemokine expression. J. Immunol. 165:1526–1533. [DOI] [PubMed] [Google Scholar]
- 19.Belperio, J.A., M.D. Burdick, M.P. Keane, Y.Y. Xue, J.P. Lynch, B.L. Daugherty, S.L. Kunkel, and R.M. Strieter. 2000. The role of the CC chemokine, RANTES, in acute lung allograft rejection. J. Immunol. 165:461–472. [DOI] [PubMed] [Google Scholar]
- 20.Kelley, J. 1990. Cytokines of the lung. Am. Rev. Respir. Dis. 141:765–788. [DOI] [PubMed] [Google Scholar]
- 21.Zhang, H.Y., M. Gharaee-Kermani, and S.H. Phan. 1997. Regulation of lung fibroblast α-smooth muscle actin expression, contractile phenotype, and apoptosis by IL-1β. J. Immunol. 158:1392–1399. [PubMed] [Google Scholar]
- 22.Messmer, U.K., and B. Brüne. 1996. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem. J. 319:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koglin, J., D.J. Granville, T. Glysing-Jensen, J.S. Mudgett, C.M. Carthy, B.M. McManus, and M.E. Russell. 1999. Attenuated acute cardiac rejection in NOS2−/− recipients correlates with reduced apoptosis. Circulation. 99:836–842. [DOI] [PubMed] [Google Scholar]
- 24.Russell, M.E., D.H. Adams, L.R. Wyner, Y. Yamashita, N.J. Halnon, and M.J. Karnovsky. 1993. Early and persistent induction of monocyte chemoattractant protein 1 in rat cardiac allografts. Proc. Natl. Acad. Sci. USA. 90:6086–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fairchild, R.L., A.M. VanBuskirk, T. Kondo, M.E. Wakely, and C.G. Orosz. 1997. Expression of chemokine genes during rejection and long-term acceptance of cardiac allografts. Transplantation. 63:1807–1812. [DOI] [PubMed] [Google Scholar]
- 26.Qian, S., L. Lu, F. Fu, Y. Li, W. Li, T.E. Starzl, J.J. Fung, and A.W. Thomson. 1997. Apoptosis within spontaneously accepted mouse liver allografts: evidence for deletion of cytotoxic T cells and implications for tolerance induction. J. Immunol. 158:4654–4661. [PMC free article] [PubMed] [Google Scholar]
- 27.Kabelitz, D. 1998. Apoptosis, graft rejection, and transplantation tolerance. Transplantation. 65:869–875. [DOI] [PubMed] [Google Scholar]
- 28.Russell, M.E., A.F. Wallace, L.R. Wyner, J.B. Newell, and M.J. Karnovsky. 1995. Upregulation and modulation of inducible nitric oxide synthase in rat cardiac allografts with chronic rejection and transplant arteriosclerosis. Circulation. 92:457–464. [DOI] [PubMed] [Google Scholar]
- 29.Worrall, N.K., W.D. Lazenby, T.P. Misko, T.S. Lin, C.P. Rodi, P.T. Manning, R.G. Tilton, J.R. Williamson, and T.B. Ferguson, Jr. 1995. Modulation of in vivo alloreactivity by inhibition of inducible nitric oxide synthase. J. Exp. Med. 181:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koglin, J., T. Glysing-Jensen, J.S. Mudgett, and M.E. Russell. 1998. Exacerbated transplant arteriosclerosis in inducible nitric oxide-deficient mice. Circulation. 97:2059–2065. [DOI] [PubMed] [Google Scholar]
- 31.Kallio, E.A., P.K. Koskinen, E. Aavik, K. Vaali, and K.B. Lemstöm. 1997. Role of nitric oxide in experimental obliterative bronchiolitis (chronic rejection) in the rat. J. Clin. Invest. 100:2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szabolcs, M., R.E. Michler, X. Yang, W. Aji, D. Roy, E. Athan, R.R. Sciacca, O.P. Minanov, and P.J. Cannon. 1996. Apoptosis of cardiac myocytes during cardiac allograft rejection. Relation to induction of nitric oxide synthase. Circulation. 94:1665–1673. [DOI] [PubMed] [Google Scholar]
- 33.Szabolcs, M.J., S. Ravalli, O. Minanov, R.R. Sciacca, R.E. Michler, and P.J. Cannon. 1998. Apoptosis and increased expression of inducible nitric oxide synthase in human allograft rejection. Transplantation. 65:804–812. [DOI] [PubMed] [Google Scholar]
- 34.August, C., K.W. Schmid, K.H. Dietl, and S. Heidenreich. 1999. Prognostic value of lymphocyte apoptosis in acute rejection of renal allografts. Transplantation. 67:581–585. [DOI] [PubMed] [Google Scholar]
- 35.Afford, S.C., S. Hubscher, A.J. Strain, D.H. Adams, and J.M. Neuberger. 1995. Apoptosis in the human liver during allograft rejection and end-stage liver disease. J. Pathol. 176:373–380. [DOI] [PubMed] [Google Scholar]
- 36.Russell, J.H. 1995. Activation-induced death of mature T cells in the regulation of immune responses. Curr. Opin. Immunol. 7:382–388. [DOI] [PubMed] [Google Scholar]
- 37.Ullrich, R., K.D. Bloch, F. Ichinose, W. Steudel, and W.M. Zapol. 1999. Hypoxic pulmonary blood flow redistribution and arterial oxygenation in endotoxin-challenged NOS2-deficient mice. J. Clin. Invest. 104:1421–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lane, B.P., G.S. Habicht, and G.S. Jasper. 1977. Lymphocyte-epithelium interaction during rejection of nonisogeneic rat tracheal grafts. Am. J. Pathol. 86:71–80. [PMC free article] [PubMed] [Google Scholar]
- 39.Hertz, M.I., J. Jessurun, M.B. King, S.K. Savik, and J.J. Murray. 1993. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am. J. Pathol. 142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 40.Jones, H.P., L. Tabor, X. Sun, M.D. Woolard, and J.W. Simecka. 2002. Depletion of CD8+ T cells exacerbates CD4+ Th cell-associated inflammatory lesions during murine mycoplasma respiratory disease. J. Immunol. 168:3493–3501. [DOI] [PubMed] [Google Scholar]
- 41.Okada, K., T. Fujita, K. Minamoto, H. Liao, Y. Naka, and D.J. Pinsky. 2000. Potentiation of endogenous fibrinolysis and rescue from lung ischemia/reperfusion injury in interleukin (IL)-10-reconstituted IL-10 null mice. J. Biol. Chem. 275:21468–21476. [DOI] [PubMed] [Google Scholar]
- 42.Ashkenazi, A., and V.M. Dixit. 1998. Death receptors: signaling and modulation. Science. 281:1305–1308. [DOI] [PubMed] [Google Scholar]
- 43.Tewari, M., D.R. Beidler, and V.M. Dixit. 1995. CrmA-inhibitable cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein during Fas- and tumor necrosis factor-induced apoptosis. J. Biol. Chem. 270:18738–18741. [DOI] [PubMed] [Google Scholar]
- 44.Saltzman, A., R. Munro, G. Searfoss, C. Franks, M. Jaye, and Y. Ivashchenko. 1998. Transforming growth factor-β-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp. Cell Res. 242:244–254. [DOI] [PubMed] [Google Scholar]
- 45.Schulze-Osthoff, K., D. Ferrari, M. Los, S. Wesselborg, and M.E. Peter. 1998. Apoptosis signaling by death receptors. Eur. J. Biochem. 254:439–459. [DOI] [PubMed] [Google Scholar]
- 46.Crow, J.P., and J.S. Beckman. 1995. Reactions between nitric oxide, superoxide, and peroxynitrite: footprints of peroxynitrite in vivo. Adv. Pharmacol. 34:17–43. [DOI] [PubMed] [Google Scholar]
- 47.De Andrade, J.A., J.P. Crow, L. Viera, C. Bruce Alexander, K. Randall Young, D.C. McGiffin, G.L. Zorn, S. Zhu, S. Matalon, and R.M. Jackson. 2000. Protein nitration, metabolites of reactive nitrogen species, and inflammation in lung allografts. Am. J. Respir. Crit. Care Med. 161:2035–2042. [DOI] [PubMed] [Google Scholar]
- 48.Ibrahim, L., M. Dominguez, and M. Yacoub. 1993. Primary human adult lung epithelial cells in vitro: response to interferon-gamma and cytomegalovirus. Immunology. 79:119–124. [PMC free article] [PubMed] [Google Scholar]
- 49.Ohori, N.P., A.T. Iacono, W.F. Grgurich, and S.A. Yousem. 1994. Significance of acute bronchitis/bronchiolitis in the lung transplant recipient. Am. J. Surg. Pathol. 18:1192–1204. [DOI] [PubMed] [Google Scholar]
- 50.Fisher, A.J., E. Gabbay, T. Small, S. Doig, J.H. Dark, and P.A. Corris. 1998. Cross sectional study of exhaled nitric oxide levels following lung transplantation. Thorax. 53:454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Caterina, R., P. Libby, H.B. Peng, V.J. Thannickal, T.B. Rajavashisth, M.A. Gimbrone, Jr., W.S. Shin, and J.K. Liao. 1995. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 96:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsao, P.S., B. Wang, R. Buitrago, J.Y. Shyy, and J.P. Cooke. 1997. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 96:934–940. [DOI] [PubMed] [Google Scholar]
- 53.Clancy, R.M., J. Leszczynska-Piziak, and S.B. Abramson. 1992. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J. Clin. Invest. 90:1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lega, M., J.H. Dauber, S.E. Urch, R. Banas, T.L. Whiteside, and B.P. Griffith. 1992. Tumor necrosis factor-alpha production by alveolar macrophages in heart-lung transplant recipients. Am. Rev. Respir. Dis. 145:1036–1041. [DOI] [PubMed] [Google Scholar]
- 55.Monti, G., A. Magnan, M. Fattal, B. Rain, M. Humbert, J.L. Mege, M. Noirclerc, P. Dartevelle, J. Cerrina, G. Simonneau, et al. 1996. Intrapulmonary production of RANTES during rejection and CMV pneumonitis after lung transplantation. Transplantation. 61:1757–1762. [DOI] [PubMed] [Google Scholar]
- 56.Zeevi, A., M. Pavlakis, K. Spichty, S. Chang, A. Iacono, J. Dauber, G. Burckart, K. McCurry, R. Keenan, W. Grgurich, et al. 2001. Prediction of rejection in lung transplantation. Transplant. Proc. 33:291–292. [DOI] [PubMed] [Google Scholar]
- 57.Agostini, C., F. Calabrese, F. Rea, M. Facco, A. Tosoni, M. Loy, G. Binotto, M. Valente, L. Trentin, and G. Semenzato. 2001. Cxcr3 and its ligand CXCL10 are expressed by inflammatory cells infiltrating lung allografts and mediate chemotaxis of T cells at sites of rejection. Am. J. Pathol. 158:1703–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cunningham, D.A., M.J. Dunn, M.H. Yacoub, and M.L. Rose. 1994. Local production of cytokines in the human cardiac allograft. A sequential study. Transplantation. 57:1333–1337. [DOI] [PubMed] [Google Scholar]
- 59.Conlon, K., A. Lloyd, U. Chattopadhyay, N. Lukacs, S. Kunkel, T. Schall, D. Taub, C. Morimoto, J. Osborne, J. Oppenheim, et al. 1995. CD8+ and CD45RA+ human peripheral blood lymphocytes are potent sources of macrophage inflammatory protein 1 α, interleukin-8 and RANTES. Eur. J. Immunol. 25:751–756. [DOI] [PubMed] [Google Scholar]
- 60.Biddison, W.E., D.D. Taub, W.W. Cruikshank, D.M. Center, E.W. Connor, and K. Honma. 1997. Chemokine and matrix metalloproteinase secretion by myelin proteolipid protein-specific CD8+ T cells: potential roles in inflammation. J. Immunol. 158:3046–3053. [PubMed] [Google Scholar]