

Abstract
Contact sensitivity (CS) is a classic example of in vivo T cell immunity in which skin sensitization with reactive hapten leads to immunized T cells, which are then recruited locally to mediate antigen-specific inflammation after subsequent skin challenge. We have previously shown that T cell recruitment in CS is triggered by local activation of complement, which generates C5a that triggers C5a receptors most likely on mast cells. Here, we show that B-1 cell–derived antihapten IgM antibodies generated within 1 day (d) of immunization combine with local challenge antigen to activate complement to recruit the T cells. These findings overturn three widely accepted immune response paradigms by showing that (a) specific IgM antibodies are required to initiate CS, which is a classical model of T cell immunity thought exclusively due to T cells, (b) CS priming induces production of specific IgM antibodies within 1 d, although primary antibody responses typically begin by day 4, and (c) B-1 cells produce the 1-d IgM response to CS priming, although these cells generally are thought to be nonresponsive to antigenic stimulation. Coupled with previous evidence, our findings indicate that the elicitation of CS is initiated by rapidly formed IgM antibodies. The IgM and challenge antigen likely form local complexes that activate complement, generating C5a, leading to local vascular activation to recruit the antigen-primed effector T cells that mediate the CS response.
Keywords: T cell recruitment, complement C5 and C5a, skin immunity, IgM response, T and B cell interactions
Introduction
Contact sensitivity (CS)* is a form of delayed-type hypersensitivity (DTH) that is a classic example of in vivo T cell immunity (1). In mice, CS responsiveness is induced by primary skin painting immunization on the body with a concentrated solution of a reactive hapten Ag. Subsequently, CS effector immune inflammatory responses are elicited by challenge, which consists of reexposure via painting a dilute solution of the same reactive hapten at a separate skin site, usually the ear. This challenge elicits local T cell–dependent inflammation that does not occur in similarly challenged nonimmune controls. Soon after the elicitation of the local secondary response by Ag challenge, very small numbers (2) of circulating sensitized Ag-specific T cells, which can be Th1 (1) or Tc1 (3), are recruited into the extravascular space at the skin challenge site.
A new round of local secondary T cell activation then occurs via T cell receptor binding to complexes of hapten conjugated to self- MHC on local APCs, stimulating production of effector cytokines like IFN-γ (4). The local inflammation caused by this T cell recruitment and extravascular activation on APC often peaks at 24 h and is a model for Th1-mediated inflammation in various diseases such as autoimmunity, allergies, and immune resistance responses to some microbes and tumors (1, 3).
The mechanisms involved in CS responses are currently assumed to operate independently of B cells, antibody, and complement (1). Under the prevailing paradigm, the initial sensitizing skin painting with hapten leads to activated epidermal Langerhans APCs with surface-bound complexes of the hapten self-peptides complexed with MHC. These Ag-charged APCs then traffic to draining LNs to sensitize recirculating specific T cells that can eventually mediate CS and some DTH effector T cell responses. The effector T cell responses are elicited after a secondary Ag challenge that as noted generates hapten–self-MHC complexes on skin APCs for activating the recruited CS effector T cells. Thus, according to current thinking, the local presence of the Ag-charged APCs is sufficient to activate the recruited sensitized T cells and trigger their production of cytokines, which are required to generate the full inflammatory phase of CS that peaks at 24 h (1).
However, our studies demonstrate that this paradigm is not correct. We have previously shown that the early recruitment of circulating T cells to the site of the extravascular APCs is an essential and regulated mechanism in CS. In addition, we have shown that this T cell recruitment after secondary Ag challenge requires early activation of complement (5–7) and generation of the C5a complement fragment, which can be detected locally within 1 h (8). Importantly, we showed that this locally generated C5a initiates a series of events that are required for recruitment of the T cells that mediate the CS effector response (5, 6, 8).
Discovery of this previously unappreciated and required local complement activation in CS led to this study. We now show that B cells and antibody, long excluded in concepts of CS and DTH, are in fact required to activate the complement that is needed for the early T cell recruitment needed to elicit the classical late phase of CS responses. Thus, we implicate B cells and antibodies in assisting T cell recruitment to mediate the CS effector T cell responses. In essence, we show that B cells produce specific antibodies that bind to the challenge Ag and form complement-activating Ag–antibody complexes required to initiate the elicitation of CS, a process we call CS initiation (5, 6, 8).
Our previous studies, which implicated C5/C5a in CS elicitation, provided an initial indication that B cells are required for this process (6). However, although we discussed this idea (9, 10), we did not present definitive evidence on this point. Thus, we demonstrate here for the first time that antibodies are clearly involved in initiating the elicitation of CS by showing that (a) specific IgM antibodies are required, (b) CS priming uniquely induces the production of these antibodies within only 1 d, and (c) B-1 cells produce the CS-initiating 1-d immune IgM antibody response.
In addition, we show that mice lacking B cells, including pan B cell–deficient JH−/− and CBA/N-xid mice, which lack B-1 and some other B cells, do not mount CS responses, measured either as macroscopic ear swelling or local 24 h IFN-γ elaboration in the ears. Nevertheless, we also show that priming the B cell–deficient mice generates CS effector T cells that can respond to secondary Ag challenge when transferred to an immunized intact host producing circulating IgM antibodies against the challenge Ag. Similarly, we show that the CS effector activity of these T cells is demonstrable in immunized B cell–deficient mice by injecting these hosts with monoclonal IgM antibodies specific for the challenge Ag, or by transferring B-1 cells from donors immunized with the challenge Ag only 1 d previously. Finally, we complete the demonstration that IgM antibodies are responsible for early CS initiation that begin the elicitation of CS, whereas T cells mediate the later occurring CS effector response. Thus, we separate the early CS initiation mechanism from the later CS effector response by showing that different Ag can be used to induce the initiating IgM antibodies and to prime the effector T cells mediating the classical late phase of CS response, as long as both Ag are used together for the secondary skin challenge.
Coupled with evidence from our previous studies that focused on C5/C5a, these new findings define the overall mechanism responsible for CS as follows: (a) B-1 cells respond within 1 d of CS priming (skin painting) by rapidly producing circulating IgM antibodies specific for the priming Ag, (b) these antibodies initiate the elicitation of CS responses to secondary challenge (ear painting) by binding to the challenge Ag and forming local Ag–antibody complexes, (c) the complexes activate complement to generate C5a, which initiates local Th1 cell recruitment, and (d) this enables the elicitation of the classical late (24-h) CS inflammatory response.
Materials and Methods
Mice and Reagents.
Specific pathogen-free male CBA/J mouse controls and 6–8-wk-old experimental male CBACa/HN-btk-xid/J (xid) mice deficient in B-1 cells, and female BALB/cJ (H-2d) (Jackson ImmunoResearch Laboratories) and control H-2d CB.17 mice (Taconic Farms) were rested at least 1 wk. B cell–deficient JH−/− mice (11) were bred and maintained in filtered microisolators in a bio-clean room and fed autoclaved food and water. Experiments were performed according to guidelines of the Animal Care and Use Committee at Yale University School of Medicine. Picryl chloride (PCl; Nacalai Tesque) was recrystallized twice and oxazolone (4-ethoxymethylene-2-phenyl-2-oxalolin-5-one) (OX) was obtained from Sigma-Aldrich. Both haptens were stored and protected from light.
Immunization and Elicitation of CS.
In brief, mice were immunized for CS by topical painting on the shaved chest and abdomen and the four feet with a total of 150 μl 5% PCl or 3% OX in 4:1 ethanol/acetone (5, 6, 8). 1 or 4 d later, secondary CS responses were elicited by topical ear application with 0.4–0.8% PCl or OX in 1:1 acetone/olive oil, and then observers often unaware of the groups measured ear thickness with a caliper (Ozaki) or micrometer (Mitutoyo) before and 2 and 24 h after challenge. The results from representative experiments using 4–6 mice per group are expressed as mean mm × 10−2 ± SE compared with age- and sex-matched syngeneic controls shaved and sham immunized by the application of vehicle alone, and then identically Ag challenged with measurement of the ears with the experimental groups.
Adoptive Cell Transfer of CS Reactivity.
Cell donors were actively sensitized with 5% PCl or 3% OX. On day 1 or 4, LNs and spleens were harvested, single cell suspensions prepared, and a mixture of 5–7 × 107 immune cells, or FACS® sorted cells in the numbers indicated, were adoptively transferred intravenously in 0.25 ml PBS into syngeneic recipients. The ear thickness of groups of 4–6 recipients was measured with a micrometer before the transfers. 24 h later, the recipients were challenged on each ear with 10 μl 0.4% PCl or OX in 1:1 olive oil/acetone. The subsequent increases in ear thickness were determined at 2 and 24 h after challenge and expressed as described above. Nonimmune mice similarly skin challenged and ear measured at 0, 2, and 24 h were controls. Their background ear swelling response was ∼1 unit × 10−2 mm at 2 h, and 2 units at 24 h. In some experiments, this control swelling was subtracted from experimental responses to yield net ear swelling.
Purification of Peritoneal B-1 Cells.
Peritoneal cells of 36 normal CBA/J were harvested by lavage with 4 ml cold 1% FBS-PBS, washed three times, and resuspended in RPMI 1640 containing 10% FBS (GIBCO BRL), 25 mM Hepes, 100 units/ml penicillin, and 100 μg/ml streptomycin. Macrophages were depleted by plastic adherence for 1 h at 37°C in 5% CO2. These normal nonimmune peritoneal B-1 cells were FACS® purified after staining with biotin-conjugated anti-B220 (CD45RA) mAb plus FITC-avidin and anti–CD5-PE mAb (BD Biosciences) using a FACStar™ (Becton Dickinson). Purities of B220+ CD5+ cells that were transferred intraperitoneally in 0.5 ml sterile PBS were ± 98%.
FACS® Sorting of Immune Lymphoid B-1 Cells.
Immune spleen and LN cells (LNCs) at 106–107/ml were stained with anti–CD5-CyChrome and anti–CD19-FITC at 0.025 μg per 106 cells and in some experiments also with anti-CD43 PE (BD Biosciences) for 30 min on ice, and then washed at 4°C with RPMI without FCS. Stained cells were sorted with a FACSVantage™ SE (Becton Dickinson) to obtain CD19+ CD5+ CD43+ cells versus CD19+ CD5+ CD43− cells, usually at a ratio of 10:1 CD43+/CD43− cells, or B-1 (CD5+ CD19+) cells versus B-2 (CD5− CD19+) cells (12, 13).
In Vitro T Cell Stimulation by Hapten-conjugated APCs.
As previously described (14), normal spleen cells treated with 100 μg/ml mitomycin C were incubated with 10 mM trinitrobenzene sulfonic acid to obtain TNP-APCs. 4 × 105 4-d TNP-Cl (PCl) immune LNCs were incubated in vitro in microwells at 37°C with varying numbers of TNP-APCs and supernatants were collected at 48 h for subsequent determination of IFN-γ content by ELISA.
In Vitro Chemotaxis Assay of CS Ear Extracts.
As previously described (6, 8), when mice were killed ear punch biopsies were frozen in liquid N2 and then thawed and extracted with a microhomogenizer on ice in a microfuge tube after centrifugation. To remove debris, separate ear extracts from individual mice were diluted two to four times in RPMI 1640 with RPMI gelatin and then placed in chemotaxis chambers (Neuroprobe) versus 2–5 × 106 target migrating macrophages of the J774A.1 monocyte/macrophage cell line (American Type Culture Collection), allowing cell migration through 5–8-μm pore PVP-free polycarbonate filters. Migrated cells attaching to the lower surface of the filter were fixed and DiffQuick (Fisher Scientific) stained and five filter spots were counted at ×400 or the filters were extracted with 4 M urea and absorbance of the extracts was measured at 650 nm. Migration was not due to chemokinesis because ear extracts added to the cells decreased migration. As previously described (6, 8), chemotactic activity in ears 2 h after challenge was due almost entirely to C5a and at 24 h was due to chemokines, including IP-10.
Quantitative Sandwich ELISA for IFN-γ.
As previously described (6, 8, 14), wells were coated overnight with capture anti–IFN-γ mAb. After blocking with 1% BSA, the ear extracts or control recombinant mouse IFN-γ (Genzyme) was added for 1 h at 25°C. After washing, 1 μg/ml of another biotinylated anti–IFN-γ mAb (BD Biosciences) and 1:3,000 horseradish peroxidase ELISA developing reagents were added.
Purification of Anti-TNP IgM.
Monoclonal anti-TNP IgM supernatant of 32.17 IgM-producing hybridoma (provided by F.T. Liu, University of California at Davis, Davis, CA) or pristane-induced ascites of the 13.4 anti-TNP IgM hybridoma (provided by J. Fleishman, Washington University, St. Louis, MO) were affinity purified on a TNP-BSA sepharose column followed by fast protein liquid chromatography gel filtration with resulting >98% IgM purity by SDS-PAGE, or purified by immune affinity chromatography with an anti–mouse μ chain column (Sigma-Aldrich). Control mouse myeloma IgM was obtained from Calbiochem-Novabiochem.
Statistics.
The paired two-tailed Student's t test with P < 0.05 was taken as the level of significance.
Results
B Cells Are Required for CS Responses.
Current views of the mechanisms responsible for CS exclude the participation of B cells. However, we found that CS responses fail in gene-targeted JH−/− mice (Fig. 1) , in which the disruption in IgH rearrangement specifically results in the absence of B cells (11). These mice are congenic to control H-2d CB.17 and BALB/c mice that have high CS responsiveness to PCl (TNP-Cl). In contrast, we previously showed that CS to PCl also fails in the absence of B cells in H-2b μMT mice (6), which also have a block in B cell development but are low CS responders to PCl. Thus, these studies with H-2d CB.17 background JH−/− mice (Fig. 1) confirm and extend prior findings by showing that CS responses fail in the absence of B cells from two different genetic lesions, rather than MHC-regulated responsiveness accounting for CS failure in B cell–deficient gene-targeted mice.
Figure 1.

(a and b) Impaired elicitation of CS and accompanying local generation of IFN-γ in B cell–deficient JH−/− and B-1 cell–deficient xid mice, despite adequate generation of Th1 effector T cells. (a) 24-h macroscopic ear swelling (left) and 24-h ear extract IFN-γ levels (right) were compared in pan-B cell–deficient JH−/− mice and normal CB.17 background controls. The pair of bars in each panel shows 4-d PCl contact-sensitized wild-type controls (left, open bars) compared with similarly PCl-sensitized JH−/− CB.17 mice (right, solid bars). Background responses to ear challenge in nonimmune controls of each strain were subtracted (n = 6 mice per group). (b) Ear swelling responses elicited at 2 h and later at 24 h in sensitized CBA/J (line 2) and B-1 cell–deficient CBA/N-xid male mice (line 4; left and middle) ear challenged with PCl 4 d later. The right shows the amount of IFN-γ in 24-h ear extracts (n = 6 mice per group). (c) Intact generation of Ag-specific IFN-γ–producing T cells in PCl-sensitized B cell–deficient xid mice. Bulk LNCs were prepared from PCl-sensitized CBA/J and xid mice. After harvest on day 4, cells containing equal numbers of T cells from each source were plated at 4 × 105 cells per well. The sensitized cells were stimulated in vitro with various numbers of indicated hapten-conjugated TNP-conjugated spleen cells as APCs, and 48 h later IFN-γ production in supernatants was measured by ELISA (n = 6 per group).
Thus, when CB.17 JH−/− mice are immunized and challenged with PCl they show virtually no CS ear swelling effector response at 24 h (Fig. 1 a, left, solid bar) and have greatly diminished local IFN-γ production as a measure of local CS effector T cell activation likely on APCs (Fig. 1 a, right, solid bar). In contrast, when CB.17 wild-type control mice are sensitized and challenged with PCl they show typical strong 24-h ear swelling (Fig. 1 a, left, open bar) and local IFN-γ production at 24 h (Fig. 1 a, right, open bar). Thus, the recruitment of CS effector inflammatory T cells and the subsequent activation for IFN-γ production is markedly impaired in B cell–deficient mice. Hence, B cells are required to enable CS responses.
As in JH−/− mice, CS responses fail in CBA/N-xid mice (Fig. 1 b) that have a partial B cell deficiency that includes all of the B-1 cells and a major subset of B-2 cells. Importantly, for these studies, the xid defect results in a virtually complete absence of B-1 cells in mature animals and consequently in very low serum IgM and minimal IgM responses (12), enabling us to test whether B-1 cell–derived IgM is involved in the elicitation of CS.
Using the standard CS protocol as outlined above, wild-type CBA/J mice elicit an early 2-h ear swelling component that we previously described as required for the subsequent elicitation of the classical 24-h component of CS (15), which is accompanied by local elaboration of IFN-γ assayed in ear extracts (6, 8). Thus, Fig. 1 b, line 2, shows the 2-h and subsequent 24-h CS ear swelling in wild-type CBA/J (left and middle), and in the right panel the accompanying 24-h ear extract IFN-γ responses are shown. In contrast, identical CS priming and challenge does not elicit CS in xid mice. Notably, both the elicited 2- and 24-h CS ear swelling responses are significantly lower in 4-d immune xid mice than in similarly immunized and challenged wild-type CBA/J mice (Fig. 1 b, left and middle, line 4 vs. 2). In addition, local production of IFN-γ at 24 h is decreased significantly in xid animals (Fig. 1 b, right, line 4 vs. 2). Thus, the elicitation of CS fails in partially B cell–deficient xid mice as in totally B cell–deficient JH−/− (Fig. 1 a) and μMT gene-targeted animals (6).
The xid Defect Prevents CS Initiation but Does Not Impair Priming of CS Effector T Cells.
Despite failure to elicit CS in xid mice, in vitro studies show that LNCs from PCl-sensitized xid mice respond to TNP-conjugated APCs (14) by producing strong IFN-γ responses comparable to those stimulated in LNCs from sensitized control wild-type CBA/J mice (Fig. 1 c). In addition, Ag-specific stimulation of T cell proliferation with LNCs from xid mice is comparable to control mice (unpublished data).
In vivo cell transfer studies similarly show that PCl priming induces normal CS effector activity in xid mice. These experiments take advantage of the substantially greater rapidity with which animals can be primed to produce the 2-h ear swelling response to secondary Ag challenge, which is B cell–dependent compared with the production of the full 2- plus 24-h CS response, which additionally depends on the subsequent generation of sensitized effector T cells. Thus, as previously shown, 1-d immune mice respond to secondary challenge with hapten ear painting by producing a strong 2-h ear swelling. However, the full 2- and 24-h CS response does not follow because CS effector T cells take 4 d to become fully sensitized after priming and are not yet functional at day 1 (Fig. 2 , line 1; reference 15). Because transferring only the isolated T cells from 4-d PCl immune donors into recipients primed 1 d previously with PCl can reconstitute the full CS response (16), the 1-d immune recipients enable testing the primed donors in the presence of generated functional CS effector T cells.
Figure 2.

Ag-specific CS reconstitution by transfer of 4-d PCl immune cells from xid donors into 1-d PCl immune CBA/J recipients. 4-d immune xid donors of cells for transfer were primed with contact sensitizers and 1-d PCl or OX immune wild-type CBA/J recipient mice were challenged with each Ag alone or with a mixture of PCl and OX. The xid PCl immune cells were transferred intravenously (see Materials and Methods; n = 4 mice per group). ***, P < 0.001 versus line 3. At 24 h, lines 3 and 4 were not significantly greater than line 1. P < 0.01, lines 4 and 5 versus line 3 at 2 h.
Results with this assay show that CS effector T cells are primed normally in the xid animals. Thus, when T cells from 4-d PCl immune xid mice are transferred to 1-d PCl immune wild-type CBA/J recipients, the xid T cells that show little response in the donors (Fig. 1 b, line 4) produce a strong 24-h CS effector T cell response in the 1-d immune recipients (Fig. 2, right, line 2). In fact, this response is similar to that mounted by similarly immunized wild-type CBA/J controls (Fig. 1 b, middle, line 2). Therefore, immunizing xid mice with PCl induces fully functional CS effector T cells whose activity is not expressed in the xid environment. However, T cell activity is readily demonstrable after transfer to 1-d immune wild-type recipients capable of mounting a B cell–dependent 2-h ear swelling response, which initiates the elicitation of a full 24-h CS response due to the immune xid CS effector T cells. In contrast, mice immunized 1 d previously with another Ag OX to control for nonspecific immunization effects, could not initiate for PCl immune xid CS effector T cells with PCl challenge (Fig. 2, line 3).
Thus, contrary to predictions based on current thinking, two distinct mechanisms cooperate to act in sequence to produce the classical 24-h CS response: one is required to initiate CS and is missing in xid mice, whereas the other, which induces the CS effector T cells, is fully functional but not expressed because the T cells that mediate CS cannot be recruited locally in the absence of the CS-initiating process.
Antigen Specificity Differences Confirm the Distinction between CS Initiation and CS Effector Mechanisms.
In typical CS studies, successful responses to skin challenge are elicited by using the same Ag for immunization and challenge, and the specificity of the response is demonstrated by failure to respond to challenge with a different hapten Ag. Thus, in the cell transfer studies described above, responses were successful when the same Ag was used to prime the 1-d immune CBA/J recipient and the 4-d immune CS effector T cell xid donor and when recipients were challenged after the cell transfer with this Ag (Fig. 2, line 2). In addition, responses failed appropriately when different Ag were used for priming or challenge (Fig. 2, lines 3 and 4). However, the studies presented below show that fully successful responses to challenge in transfer recipients can be obtained even when different Ag are used to immunize the effector T cell donor and the CS-initiating host, provided that the recipients are simultaneously challenged with a mixture of both immunizing antigens.
For these “dual-antigen” experiments, we transferred cells from 4-d PCl-immunized xid mice that have CS effector T cells responsive to PCl into 1-d OX-immunized CBA/J mice (a different Ag), which, because they are 1-d immune, did not yet have effector T cells capable of mounting a 24-h CS effector response to the OX Ag. Then, we challenged groups of such combined recipients with each Ag separately (Fig. 2, lines 3 and 4) and another group with both Ag together (Fig. 2, line 5). The results show that OX elicits only the 2-h response in the 1-d OX immune mice (Fig. 2, line 4) and 4-d PCl immune xid hardly elicit any response (Fig. 2, line 3). Thus, here both the 2- and the 24-h responses fail. However, in contrast, simultaneous challenge with both PCl and OX elicits the full 2- plus 24-h CS response (Fig. 2, line 5).
These findings demonstrate that successful elicitation of a 2-h CS initiation response, regardless of the Ag specificity of this response, is sufficient to support the 24-h response mediated by transferred CS effector T cells as long as the challenge also includes the Ag recognized by the late-acting effector T cells. The distinctive Ag specificities of the separate early and late components of CS responses show that separate mechanisms cooperate, acting in sequence to mediate the early initiation and the late effector phases of CS.
CS Responses Are Initiated by IgM Antibodies that React with the Challenge Ag.
The specificity and timing differences between the CS initiation and CS effector T cell mechanisms, coupled with the failure of CS initiation in B cell–deficient mice and our prior evidence indicating that local complement activation and subsequent Ag-specific C5a release are involved in CS initiation (5, 6, 8), suggest that Ag–antibody complexes might be central to CS initiation. This hypothesis is confirmed by studies in this section, which show that intravenous injection of monoclonal IgM antibodies reactive with the challenge Ag restores the initiation of CS effector T cell responses in immunized xid mice.
We primed xid mice with PCl to induce CS effector T cells that are ready to respond 4 d after priming but have absent CS initiation (Figs. 1 b and 2). On day 3, we injected purified IgM mAb from either of two anti-TNP IgM hybridomas (32.17 and 13.4), each reactive with the hapten self-products generated by PCl skin painting. On day 4, we then challenged the mice with PCl (TNP-Cl).
Injection of 100 μg of these mAbs enabled immunized xid mice to elicit strong 24-h CS ear swelling responses (Fig. 3 a, line 5 and b, line 4). In fact, injection of only 20 μg anti-TNP IgM was sufficient (Fig. 3 b, line 3). In contrast, no significant 24-h CS responses were detectable when a similarly purified mouse myeloma IgM that does not react with the challenge Ag was injected (Fig. 3 a, line 6), indicating that the initiation of the CS response requires antibody specific for the challenge Ag.
Figure 3.
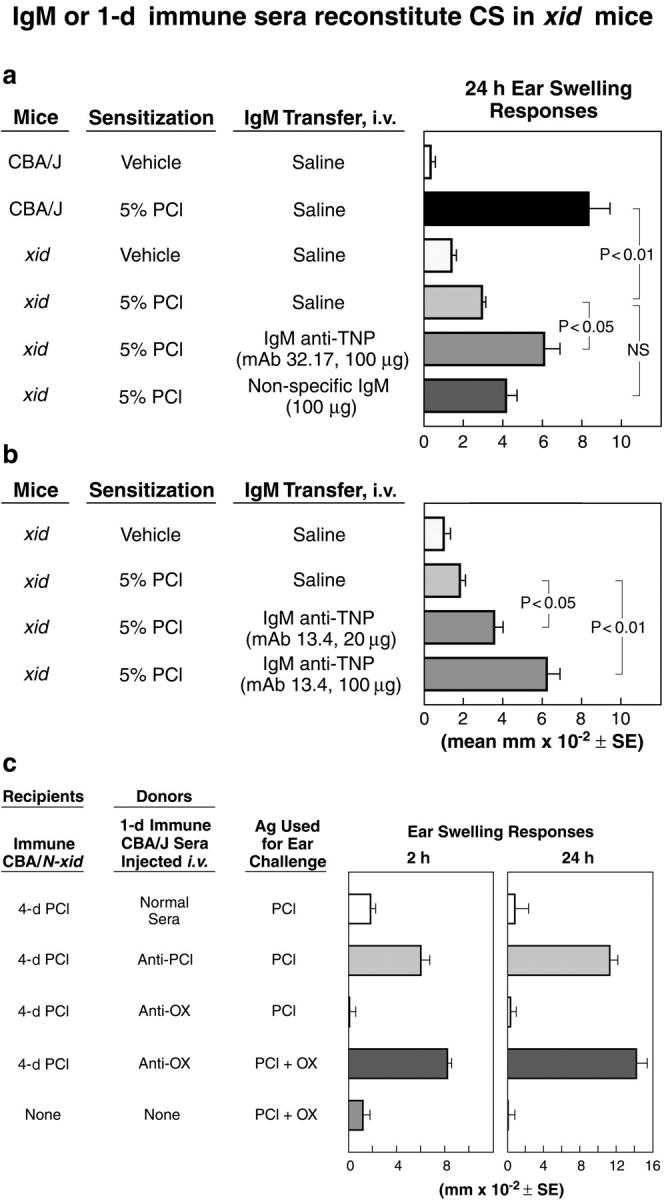
Antigen-specific monoclonal IgM or specific 1-d immune sera reconstitute CS in sensitized xid mice. (a and b) Ag-specific anti-TNP IgM reconstitutes 24-h CS ear swelling in immunized xid mice. Normal CBA/J wild-type and B-1 cell–deficient xid mice were contact sensitized on day 0 and ear challenged with PCl on day 4 to elicit CS (a, line 2 vs. 1 and line 4 vs. 5). Results are shown for two purified monoclonal anti-TNP IgM antibodies, 32.17 IgM (a) and 13.4 IgM (b) injected intravenously (n = 6 mice per group). 100 μg 32.17 IgM versus 100 μg nonspecific IgM were injected intravenously 1 d before Ag challenge on the ears with 0.4% TNP-Cl of 4-d PCl immune xid mice (a). Either 20 or 100 μg 13.4 IgM (lines 3 and 4) were injected intravenously 1 d before Ag challenge into xid recipients that were immunized 3 days previously (b). (c) Antigen-specific reconstitution of 24-h CS in immunized CBA/N-xid with 1-d immune sera. Transfer of 1-d PCl immune CBA/J sera (0.7 ml) mediated 2-h ear swelling and reconstituted 24-h CS in 4-d PCl immune xid mice (line 2), whereas normal sera was inactive (line 1). 1-d OX immune sera did not have this activity (line 3). However, the combination of active 4-d PCl CS of xid recipients and the transfer of 1-d OX immune sera resulted in 2-h ear swelling and reconstituted 24-h CS when the PCl immune xid mice were challenged with a mixture of PCl and OX (line 4) compared with double challenged controls (line 5; n = 4 mice per group).
Taken together, these findings demonstrate that CS effector T cells develop normally in xid mice and directly show that the CS failure in these mice is due to a B cell defect that interferes with their ability to produce IgM antibody. These findings displace the current paradigm that states T cells alone mediate CS by showing that IgM antibodies are necessary and sufficient to initiate CS effector responses for the recruitment of the primed CS effector T cells.
Rapid IgM Antibody Responses After Immunization Act to Initiate CS.
Because we showed that CS responses are initiated in 1-d primed animals, IgM antibodies to the priming Ag should therefore be detectable in the 1-d immune mice and should be capable of initiating CS. Accordingly, the injection of serum harvested from 1-d PCl immunized wild-type mice is highly effective in enabling the elicitation of both the 2- and 24-h CS response in PCl-challenged 4-d PCl immune xid mice (Fig. 3 c, line 2). Importantly, serum from nonimmune CBA/J mice is inactive (Fig. 3 c, line 1), whereas serum from 1-d immunized animals is highly effective (Fig. 3, line 2). In addition, passage of this latter immune serum through an anti-IgM column removes activity, suggesting that IgM antibodies in the 1-d immune serum are responsible for initiating CS. Thus, the 2-h activity produced by 1-d PCl immune sera in xid recipients is 9.9 ± 0.4 mm × 10−2 ± SE compared with ear swelling activity transferred by normal sera of 1.8 ± 0.4. Importantly, passage through an anti-IgM column reduced the 1-d immune sera activity to 2.6 mm × 10−2 ± 0.7 (P < 0.05) and transfer of the base eluate from this column restored the response to 10.3 ± 1.7. Therefore, we conclude that the production of CS-initiating IgM antibodies is stimulated by skin painting immunization and surprisingly begins within 1 d of immunization.
Ag specificity studies confirm and extend this conclusion. Serum from 1-d OX immune animals is inactive in PCl-immunized xid mice that are challenged with PCl (Fig. 3 c, line 3). However, injection of this anti-OX 1-d immune serum into similar 4-d PCl-sensitized xid mice and subsequent challenge with a mixture of PCl and OX results in 2-h ear swelling that is CS-initiating, thus allowing the elicitation of a full 24-h CS effector T cell response (Fig. 3 c, line 4). These findings confirm within the system the key role that early Ag-specific antibodies play in initiating CS. In addition, they reveal the mechanism underlying the specificity distinction between early CS initiation and late CS effector responses by showing that CS initiation is mediated by antibody, whereas CS effector responses are mediated by sensitized T cells.
1-d Immune B-1 Cells Produce CS-initiating Activity.
Although B-1 cells have been shown to produce IgM antibodies in response to a variety of immunization protocols, current thinking often relegates them either to a nonresponsive role as producers of natural or unstimulated constitutive antibodies, or to a minimal role as producers of germ line–encoded antibody responses to a very limited set of Ag. However, as we show in this section, B-1 cells respond to CS immunization by rapidly producing CS-initiating activity likely due to IgM antibodies.
We transferred 106 FACS®-sorted peritoneal B-1 cells (98% pure CD5+ B220+) from nonimmune CBA mice intraperitoneally into unimmunized CBA/N-xid mice, primed the recipients by skin painting with PCl the next day, and then skin challenged them with PCl 4 d later. CS responsiveness measured at 2 and 24 h is restored in usually unreactive xid, although incompletely (Fig. 4 a, group E vs. C). In addition, there is fully restored 24-h chemotactic activity assayed in CS ear extracts (Fig. 4 b, right, group E vs. D and C), which we previously showed depends on chemokines like IP-10 that are induced locally by IFN-γ (8). Similarly, 2-h chemotactic activity, which we previously showed to be dependent on local generation of C5 (8) and thus is an indicator of CS initiation, is also restored strongly (Fig. 4 b, left, group E vs. B). Thus, by all of these macroscopic and biochemical measures, the transfer of FACS®-purified B-1 cells isolated from unimmunized wild-type mice into xid before immunization, largely restores CS responsiveness after priming and challenge.
Figure 4.

Normal B-1 cell reconstitution before immunization restores 2- and 24-h CS in CBA/N-xid and restores elaborated 2- and 24-h chemotactic activity. (a) FACS®-sorted peritoneal B-1 cells (98% CD5+ B220+) obtained from intact wild-type CBA/J mice were transferred intraperitoneally to xid the day before PCl sensitization and 2- and 24-h CS responses were elicited on day 4 (n = 6 mice per group). ***, P < 0.001 group B versus A, and group E versus F at 2 and 24 h; *, P < 0.01 versus group E versus F. (b) Data show chemotactic activity for migrated J774A.1 macrophages in the extracts of ears at 2 and 24 h in the indicated mouse strains that also were shown in a, and were immunized and challenged as indicated (n = 6 mice per group). ***, P < 0.001 group B versus A, and group E versus F; **, P < 0.02 versus group B versus A, and group E versus F at 24 h.
The ability to initiate CS in response to secondary Ag challenge is also restored by transfers of 1-d immune lymphoid cells (LN plus spleen) before challenge of primed B cell–deficient recipient mice. First, the transfer of lymphoid cells from 1-d PCl or OX immune CBA/J donors to unimmunized xid mice enables 2-h CS responses to the cognate priming Ag PCl or OX, but without 24-h responses due to T cells that are not generated until day 4 (Fig. 5 a, left, groups A and B). Second, the transfer of these 1-d immune lymphoid cells into 4-d immune xid mice mediates a 2-h CS initiation response that enables elicitation of the 24-h CS effector response to the homologous TNP Ag (Fig. 5 a, right, group C vs. E and group G vs. H). However, transfers of wild-type lymphoid cells that are 1-d immune to another Ag (OX) into xid mice that are immune to a different Ag (PCl) and challenge with the cognate Ag PCl, fails to restore 24-h CS (Fig. 5 a, right, groups D and E). Thus, transferring B cell–containing lymphoid populations from 1-d immune donors is equivalent to injecting IgM antibodies from these donors in that both provide strong Ag-specific support for CS initiation.
Figure 5.

1–d immune B-1 cells reconstitute CS in 4-d immune xid or JH−/−mice. (a) 1-d PCl or OX immune cells transfer 2 but not 24 h reactivity (groups A and B). Reconstitution of 2- and 24-h CS in 4-d immune CBA/N-xid mice by transfer of these 1-d wild-type immune cells. Per Ag specificity, the 1-d PCl immune CBA/J wild-type cells were transferred to the indicated 4-d OX immune xid recipient mice and challenged as indicated either with OX (group F) or 1-d OX immune cells were transferred to 4-d PCl immune xid recipients challenged with PCl (group D; n = 4 mice per group). P < 0.001, group C versus A, D, and E at 24 h. P < 0.001, group G versus F and H at 2 and 24 h. (b) Reconstitution of CS by transfer of 1-d PCl immune B-1 cells to 4-d PCl immune B cell–deficient recipients. 1-d immune CBA/J mixed cells (group C) or FACS®-sorted B-1 cells (groups E–H) transferred to 4-d immune xid mice mediates CS. For dose response, 3 × 104 − 3 × 106 B-1 cells (CD19+CD5+ CD43+ or CD19+CD5+ CD43−) that were FACS®-sorted 1-d immune spleen and LNCs were transferred to 4-d immune xid mice. The recipients were tested for CS elicitation, showing reconstitution of CS by B-1 cells at all doses tested to 3 × 104 cells (n = 4 mice per group). ***, P < 0.001; **, P < 0.01; *, P < 0.05 versus group B. (c) Transfer of 1-d immune FACS®-sorted CD43+ and CD43− BALB/c B-1 cells (CD19+ CD5+) to 4-d PCl immune JH−/− recipient mice reconstitutes CS (n = 4 mice per group). P < 0.001, groups D–F versus C at 24 h.
FACS® sorting studies show that the responding B cells within the lymphoid populations from 1-d immunized donors are small numbers of B-1 cells. To isolate these cells, we used the CD19 and CD5 markers, which are present on B-1 (B-1a) cells (12) and the CD43 marker, which has been shown to be present on most but not all CD19+ CD5+ B-1 cells (13). Consistent with the known distribution of these markers, ∼90% of the cells in the small fraction of CD19+ CD5+ B-1 cells (∼1–2% of all cells) detected in the LNs and spleen were CD43+. Both of the CD43+ (300 × 103) and the CD43− (160 × 103) B-1 subpopulations enabled 2-h CS-initiating responses to Ag challenge when transferred to xid mice that were immunized 4 d previously with the Ag used for challenge (Fig. 5 b, left, groups E and F). In addition, both of these B-1 subpopulations mediate the initiation of elicitation of strong 24-h CS responses (Fig. 5 b, right, groups E and F).
Dose response transfer of the 1-d immune CD19+ CD5+ CD43+ lymphoid cells showed that 100 × 103, and as few as 30 × 103 FACS®-sorted CD43+ B-1 cells, transferred to 4-d PCl immune xid mice the ability to elicit strong 24-h responses (Fig. 5 b, right, group H). In contrast, the transferred 2-h reactivity decreased progressively with the number of transferred B-1 cells until 2-h reactivity was insignificant at 30 × 103 (Fig. 5 b, left, groups F–H). In contrast, the mediation of 24-h CS was maintained, demonstrating that optimal macroscopic 24-h CS effector T cell responses do not require optimal macroscopic CS initiation responses, which are likely based on microvascular activation. Thus, macroscopic suboptimal 2-h responses due to B-1 cell IgM antibody can support effective 24-h T cell responses.
We tested whether these results were special to xid mice, as we considered that residual B cells in xid mice might contribute to our findings. Thus, we used pan B cell–deficient JH−/− mice in similar experiments. The transfer of mixed lymphoid cells, or 100 × 103 FACS®-purified B-1 cells (CD19+CD5+subdivided into CD43+or CD43−), into immunized JH− / −recipients again showed that B-1 cells in the LN and spleen from 1-d immune BALB/c H-2d donors are sufficient to restore both the 2- and 24-h CS responses in these fully B cell–deficient animals (Fig. 5 c, groups D–F). Thus, B-1 cells in 1-d immunized wild-type mice are necessary and sufficient for mediating Ag-specific initiation for CS effector T cell responses in either xid or JH−/− mice. Because monoclonal IgM antibody or antibody produced by 1-d immunized animals is also necessary and sufficient to enable CS initiation, we conclude that transferred B-1 cells produce these IgM antibodies in the B cell–deficient recipients and thereby enable the CS initiation process.
Interestingly, transferred B-1 cells fully reconstituted the 24-h CS response in JH− / −mice (Fig. 5 c, groups D–F), whereas in xid mice they only partially restore 24-h macroscopic CS responses (Fig. 4 a). This might be due to other defects specific to xid mice that could contribute to elicitation of CS responses, such as in mast cells (17) and macrophages (18). However, the difference may also be due to difficulties in effectively transferring B-1 cells into an environment that already contains B cells because the long-term establishment of B-1 cells in unirradiated xid mice is difficult to achieve (unpublished data).
Later in the Immune Response, B-2 Cells Provide Support for CS Initiation.
Although FACS®-purified B-1 cells harvested from 1-d immune donors readily enable CS initiation in immunized B cell–deficient mice, B-2 cells harvested at this early time are unable to do so. However, several days later, at a time when B-2 cells typically begin differentiating into cells producing antibodies to the immunizing Ag, B-2 cells also become capable of restoring CS initiation in B cell–deficient recipients.
In these studies, we transferred FACS®-purified B-1 and B-2 cells obtained from BALB/c mice at various days after PCl immunization into 4-d PCl-immune JH−/− mice, which are congenic to BALB/c and can therefore accept BALB/c cell transfers (Fig. 6) . As in the xid transfers, B-1 cells (CD19+ CD5+) from 1-d immune donors readily reconstitute 24-h CS (Fig. 6, right, group C) to thus reconstitute both the 2- or 24-h CS response (Fig. 6, group C).
Figure 6.

Comparing the ability of wild-type immune B-1 versus B-2 cells harvested various days after PCl sensitization to reconstitute CS in JH−/− mice. 1-, 4-, or 8-d PCl immune FACS®-sorted B-1 cells (CD19+ CD5+) or 1-, 4-, or 8-d immune FACS®-sorted B-2 cells (CD19+ CD5−) all at 150 × 103 cells per recipient from PCl-immunized BALB/c donors were transferred to indicated groups of 4-d PCl immune JH−/− recipients, which were challenged with PCl Ag and elicited 2- and 24-h measured CS ear swelling responses (n = 4 mice per group).
In contrast, 4 d after immunization, both the B-1 and B-2 cells from the immunized donors could initiate CS (Fig. 6, groups E and F), but by 8 d after immunization the activity had shifted entirely into the B-2 population (Fig. 6, groups G and H). Thus, B-2 cells, like B-1 cells, are able to mediate the initiation of CS. However, they require a longer time after immunization for this activity to develop and as a population retain this activity for a longer time once it develops.
Because we concluded that CS initiation by B-1 cells is due to the production of IgM antibodies to the challenge Ag, these findings indicate that B-1 cells develop the ability to produce the necessary antibodies within 1 d of immunization and lose that ability ∼1 wk later. In contrast, B-2 cells only develop the ability to produce the necessary antibodies 4 d after immunization, but they seem to retain that ability longer than B-1 cells. Thus, with respect to the classical murine CS response, which is elicited by Ag priming on day 0 and challenging on day 4, most or perhaps all of the antibody that participates in CS initiation needed for elicitation of the CS effector T cell response is produced by B-1 cells.
Discussion
Synopsis.
These studies make four related observations that together demonstrate for the first time that B cells, via the antibodies they produce, are responsible for initiating the elicitation of a classical T cell–mediated in vivo response. CS as an example of DTH has universally been considered to be a purely T cell response that does not require the participation of B cells. However, here we show that (a) both the 2-h CS initiation phase and the subsequent 24-h effector T cell phase of the elicited CS response fails in B cell–deficient mice primed via standard contact sensitization with the Ag then used for secondary stimulation via skin challenge, (b) both responses are restored in these mice by the introduction of monoclonal IgM antibodies specific for the challenge Ag, and (c) the 2- and 24-h CS responses are similarly restored by the introduction of either B-1 cells or serum from wild-type mice primed just 1 d previously with the challenge Ag. Thus, we conclude that IgM antibodies initiate the postulated cascade of events that culminates in the successful elicitation of CS effector T cell responses and that B-1 cells responding rapidly to the primary immunization produce the IgM antibodies that initiate the elicitation of CS.
Relation to Prior Findings in CS.
These novel findings, summarized in Fig. 7 , were foreshadowed by our previous studies findings. Those studies showed in a model system that IgE or IgG1 antibodies reactive with the challenge Ag were required to initiate 24-h CS responses mediated by isolated CS effector T cells (19). Because both of these isotypes are known to bind to Fc receptors on mast cells, we interpret findings in this system as indicating that local Ag challenge stimulated mast cells via the bound antibodies to release vasoactive mediators that initiated the recruitment of the CS effector T cells (19).
Figure 7.

B-1 cell CS initiation for the recruitment of CS effector T cells. (1) Primary sensitization for the future elicitation of CS responses by painting reactive chemical hapten Ag (e.g., PCl) on the skin of the body. (2) Local secondary Ag challenge by painting the ears with a dilute solution of the sensitizing hapten Ag PCl to elicit CS responses. On day 1 after sensitization, Ag challenge elicits 2-h ear swelling without 24-h swelling (reference 16), due to the development of immune B-1 cells producing IgM antibody in the absence of sensitized T cells. (3) Local complement is activated (references 5 and 6) by immune complexes of B-1 cell IgM plus Ag to generate C5a (references 7 and 8). (4) Mast cells (reference 20) and platelets (reference 21) are activated by C5a by stimulating C5a receptors (reference 8) to release serotonin (references 1 and 19) and TNF-α (references 8 and 22) that activate local endothelial cells (reference 22) to generate the 2-h CS-initiating response. Released TNF-α induces endothelial expression of ICAM-1 and VCAM-1 (reference 22) to facilitate local recruitment of the CS effector T cells (references 15 and 16) to produce inflamatory cytokines like IFN-γ (reference 6) that induce chemokines like IP-10 (reference 8). (5) This allows the recruitment of the CS effector T cells developed by day 4 at the site of challenge to mediate the classical 24-h ear swelling response. In addition, C5/C5a-independent CS-initiating pathways, e.g., challenge Ag that binds to IgE antibody bound to Fcɛ receptors on mast cells (reference 19), can provide similar CS-initiating function at later times after immunization. (6) CS initiation leads to the recruitment of circulating CS effector T cells that were sensitized at least 4 d previously to the same Ag presented by the secondary challenge. Note that the Ag on the APCs recognized by the T cells need not be the same as that recognized by the IgM antibodies that initiate the elicitation of the response. Also, immune T cells transferred intravenously before challenge in recipients with a source of CS-initiating IgM antibody can substitute for the native endogenous CS effector T cell population.
More recently, in studies that led to the work presented here, we showed that secondary Ag challenge in 4-d contact-sensitized mice results in the Ag-specific local, early (1–2 h) generation of complement-derived C5a (8). Because C5a binds to C5a receptors (8) and most likely acts indirectly in CS to trigger both mast cells (20) and platelets (21), we suggested that C5a therefore leads to local endothelial activation (22), which results in the recruitment of the CS effector T cells responsible for eliciting the full 24-h CS response. These earlier findings imply the participation of complement-binding Ag–antibody complexes in CS initiation because such complexes are the most likely mechanism for generating C5a. In contrast to the IgM-facilitated and C5a-mediated CS initiation described here, Arthus hypersensitivity reactions, although also dependent on complement, are instead mediated by very large amounts of high affinity hyperimmune IgG antibody. Alternatively, IgG can also act in DTH via Langerhans cells to aid the induction of immunization (23) rather than the elicitation of responses. Finally, IgG1 can act via mast cell Fc receptors to elicit late phase cutaneous basophil DTH (24, 25).
These studies test and confirm the hypothesis that IgM–Ag complexes generate C5a that initiates CS for T cells by showing directly that IgM antibodies specific for the challenge Ag are required to elicit CS. Neither the challenge Ag alone nor the antibody alone is sufficient for this purpose. Both must be present. Thus, taking our earlier findings into account, we propose that the binding of IgM antibodies to the challenge Ag at the challenge site results in the formation of local, complement-activating Ag–antibody complexes that generate C5a to trigger the rest of the CS initiation pathway. For these studies we focused on the transfer of IgM just before the elicitation of CS, rather than before immunization to rule out any possible role of transferred IgM in antigen presentation for sensitization. However, the transfer of IgM-producing B-1 cells (Fig. 4), or even the transfer of purified hybridoma anti-TNP IgM before sensitization (unpublished data), also reconstituted CS in immunized xid mice, presumably via IgM availability to complex with challenge Ag and generate local C5a.
Others have suggested that local Ag challenge used for the elicitation of CS can lead to early activation of keratinocytes, which then produce factors that are responsible for initiating T cell recruitment (26). Our data argue against this formulation because keratinocyte development and function is not likely to be impaired in B cell–deficient mice and because the B-1 cell–derived IgM antibody that we show reconstitutes CS has no known activating effects on keratinocytes.
One wonders how the key role that IgM antibodies play in initiating CS could have been missed in over 50 yr of study. Part of the explanation may lie in the general acceptance of the “solely due to T cells” paradigm, which impeded investigations that might have tested whether B cells or antibodies also could participate. We also tended to go along with this view, but only began to examine B cell and antibody participation in the elicitation of CS responses when we were led to this idea by the surprising finding that complement C5 (6, 7) and Ag-specific local generation of C5a (8) were involved in CS initiation. In addition, we were unwilling to accept this new idea that CS is antibody dependent until we had thoroughly explored potential limitations in our data.
Proof that B-1 Cell IgM Plays a Crucial Role in CS Initiation.
For example, the demonstration here that the injection of monoclonal IgM antibody reactive with the challenge Ag restores CS initiation in B cell–deficient mice should in principle be sufficient to solidly implicate antibodies in this process. However, establishing that antibodies play a role in CS initiation under normal conditions requires the demonstration that primary immunization by skin painting can stimulate antibody production rapidly enough to be effective, because animals are typically challenged only 4 d after priming (1, 15). This very early production of IgM seemed unlikely because IgM antibody responses to cell-bound Ag such as those on sheep erythrocytes are seldom detectable until at least 4 d after immunization and IgM responses to soluble proteins arise even later at 5–7 d after priming (27, 28). Nevertheless, as we have shown, direct evaluation of antibody responses to skin painting with a contact sensitizing hapten demonstrates that this mode of immunization induces IgM antibody production so rapidly that antibody levels sufficient to initiate CS are already present in the circulation 1 d after priming. Thus, we conclude that skin painting immunization clearly induces B cells to produce enough specific IgM antibody for CS initiation 4 d after priming, when the effector T cells are generated.
To identify the B cell subset responsible for this very early production of antibody, we evaluated FACS®-sorted B-1 and B-2 cell populations from wild-type immunized donors for their ability to restore CS initiation in 4-d immune B-1 (xid) and pan B cell–deficient (JH−/−) recipients, which we show have generated sensitized CS effector T cells but lack ability to produce antibodies needed to locally recruit these cells. We found that both B-1 and B-2 cells produce sufficient amounts of antibody to enable CS initiation when transferred to these B cell–deficient recipients. Significantly, however, only the B-1 cells respond rapidly enough to be active when taken from 1-d immunized donors. In fact, as few as 30,000 FACS®-purified B-1 cells from these 1-d immune donors is sufficient to restore CS initiation.
Relation to Prior Findings in B-1 Cells.
Together, these findings introduce a new role for immunized B-1 cells as rapid producers of IgM antibodies that initiate CS and potentially other T cell–dependent inflammatory responses. This novel function for B-1 cells is surprising because B-1 cells are usually known as producers of natural IgM antibodies and are commonly thought either to be unable to respond to immunization or to respond only to T-independent Ag. However, a closer examination of the literature shows that B-1 cells have been shown to produce antibodies to the DNP/TNP hapten that is used here to induce CS when the hapten is presented on an appropriate carrier (28). In perhaps the best example of the potential breadth of B-1 antibody responses, B-1 cells produce antibodies to phosphorylcholine presented either in its natural T-independent form as an Ag coat of pneumoccocal polysaccharide or in a T-dependent form as a hapten linked to certain carrier proteins such as keyhole limpet hemocyanin (28–32). The rapidity of the B-1 response we noted also has precedents with stimulation via nonspecific LPS (33–35) and pneumococcal polysaccharide (36, 37).
Our identification of functional antibody-producing cells in the spleen and LN rapidly after contact sensitization, which likely reflects the migration of activated peritoneal B-1 cells to these peripheral lymphoid sites, has parallels in more recent work. Studies in Ig transgenic mice demonstrate that some B-1 cells can migrate from the peritoneal cavity to the lamina propria of the gastrointestinal tract where they produce IgA antibodies (38, 39). Thus, there are precedents to our demonstration that B-1 cells can rapidly produce IgM antibodies and can migrate to peripheral lymphoid sites for antibody production.
It is not known how hapten-specific peritoneal B-1 cells are rapidly stimulated to produce CS-initiating IgM antibody. However, we know that hapten–self-protein complexes emanate from the skin site of contact sensitization for distribution throughout the body via the circulation (40). Thus, it is likely that Ag is distributed from the skin to the peritoneal cavity where B-1 cells are found preferentially (12). In fact, our results show that B-1 cells in the peritoneal cavity are activated for CS initiation within hours after immunization (unpublished data). We postulate that IgM anti-TNP Ig receptors of higher affinity on these B-1 cells are cross-linked by multivalent Ag dispersed from the skin site of sensitization. This may stimulate Ag-specific IgM production by activated B-1 cells migrating to the lymphoid tissues, as shown for IgA-producing B-1 cells in the GI tract (38, 39). In addition, we find that IL-4 is needed as a costimulus for IgM production by the B-1 cells and appears to be released rapidly after sensitization by NKT cells (41, 42).
Thus, these findings introduce a new view of B-1 cell responsiveness and of the previously unsuspected importance of these cells to in vivo T cell–mediated immune effector responses. As we have shown, the rapid antibody response mounted by B-1 cells in contact-sensitized mice constitutes the first and required step in what was previously considered to be an exclusively T cell immune response. Without the antibody produced by B-1 cells to initiate this required sequence, the traditional CS response fails despite the development of CS effector T cells, which, although present, cannot be recruited locally. Thus, our findings are a surprising inversion of the classical immunological paradigm in which T cells help B cells in antibody responses, which still stands. However, our findings insert antibody production by B-1 and sometimes by other B cells as a necessary first step in the elicitation of a classical in vivo effector T cell CS response, i.e., B cells can help T cells.
Biologic Relevance of B-1 Cell IgM Mediation of CS Initiation.
From a clinical perspective, our findings may offer a new pathway for triggering delayed and contact hypersensitivity and related DTH reactions and therefore potentially provide new routes for therapeutic intervention. B cells are in fact already known to participate in a variety of T cell–mediated disease models of mice including autoimmunity in collagen arthritis (43), nonobese diabetes (44), lupus-like lesions of lpr/lpr mice (45), encephalomyelitis (46), and also in T cell protection against infections (47). However, to date, the participation of B cells in these disease responses has largely been interpreted as due to their afferent APC function needed to induce the sensitized T cells (48, 49). In contrast, our findings show that B-1 cell IgM can mediate the required early steps in the efferent phase of CS with no role in the induction of the CS effector T cells. This suggests that antibodies may participate in the elicitation of the efferent T cell responses in at least some of the clinical conditions listed above, particularly in disease models like collagen arthritis and encephalomyelitis, where B cells (46), antibodies (50, 51), complement (52, 53), and mast cells (54–57) have all been implicated in mediation of the efferent T cell responses.
Importantly, we have shown that the Ag recognized by the antibodies capable of forming early phase complement-activating complexes and leading to the recruitment of T cells to the CS site, need not be of the same Ag specificity as the Ag recognized by the CS effector T cells that cause late phase inflammation and possibly damage at the site. T cells sensitized to a particular Ag can in fact be recruited by complement-activating antibodies like IgM that recognize and form complexes with any Ag that is locally concentrated or by antibodies of other isotypes like IgE that directly activate mast cells (19). If the Ag that the T cells recognize is also present at this site along with the Ag recognized by the antibody, then diverse, available sensitized T cells can be recruited and selected subsets can be activated by their Ag expressed as peptides on local APCs. This dual Ag effect can thus produce a classical CS response regardless of whether the T cells recognize the same Ag as the initiating antibodies. Therefore, antibodies to a variety of locally present Ag (bacterial, viral, or allergens) could in theory initiate CS, DTH, or other T cell responses in which the effector T cells are activated by responding locally to another Ag that is expressed on the local APCs. This dual Ag CS initiation mechanism, therefore, could provide the basis for responses that contribute to the well-known synergy between infections and allergy (58) or autoimmunity (59).
As a practical matter, the dissociation of the specificity of the response trigger from the specificity of the response effector mechanism that acts in an obligate sequence, may make the etiology of disease processes initially more difficult to discern. However, once this Ag duality of separate and sequential antibody-mediated initiating and T cell–dependent late effector phases is recognized, it may open the way to unraveling cause and effect in complex immunological disease situations and therefore offer new targets and routes for therapeutic intervention.
Acknowledgments
The authors thank Drs. Mark Shlomchik, Noriko M. Tsuji, and Alfred Bothwell for their valuable advice, and Charles Janeway and Jordan Pober for helpful discussions while reviewing the manuscript. We are also most grateful to Ometa Herman and Gina Jager for expert technical help, Marilyn Avallone for her superb secretarial skills, and Tom Taylor for his skillful FACS® cell sorting. Finally, we especially thank John Mantovani for excellent assistance in the referencing and preparation of this manuscript.
This work is supported by grants DK-34989 and AR-41942 to P.W. Askenase and grant AI-34762 to L.A. Herzenberg from the National Institutes of Health, a Rockefeller Culpepper grant and a grant from the American Academy of Allergy, Asthma and Immunology to P.W. Askenase, and a grant from the Polish Committee of Scientific Research.
L.A. Herzenberg and P.W. Askenase contributed equally to this work.
Footnotes
Abbreviations used in this paper: CS, contact sensitivity; DTH, delayed-type hypersensitivity; LNC, LN cell; OX, oxazolone (4-ethoxymethylene-2-phenyl-2-oxazolone-5-one); PCl, picryl chloride; TNP, trinitrophenol.
References
- 1.Askenase, P.W. 1998. Effector and regulatory molecules and mechanisms in delayed hypersensitivity. In Allergy: Principles and Practice 5th Edition. N.F. Adkinson, Jr., editor. Mosby Co., St. Louis. 323–341.
- 2.Marchal, G., M. Seman, G. Milon, P. Truffa-Bachi, and V. Zilberfarb. 1982. Local adoptive transfer of skin delayed-type hypersensitivity initiated by a single T lymphocyte. J. Immunol. 129:954–958. [PubMed] [Google Scholar]
- 3.Kalish, RS, and P.W. Askenase. 1999. Molecular mechanisms of CD8+ T cell-mediated delayed hypersensitivity: implications for allergies, asthma, and autoimmunity. J. Allergy Clin. Immunol. 103:192–199. [DOI] [PubMed] [Google Scholar]
- 4.Fong, T.A., and T.R. Mosmann. 1989. The role of IFN-gamma in delayed-type hypersensitivity mediated by Th1 clones. J. Immunol. 143:2887–2893. [PubMed] [Google Scholar]
- 5.Tsuji, R.F., M. Kikuchi, and P.W. Askenase. 1996. Possible involvement of C5/C5a in the efferent and elicitation phases of contact sensitivity. J. Immunol. 156:4444–4450. [PubMed] [Google Scholar]
- 6.Tsuji, R.F., G.P. Geba, Y. Wang, K. Kawamoto, L.A. Matis, and P.W. Askenase. 1997. Required early complement activation in contact sensitivity with generation of local C5-dependent chemotactic activity, and late T cell interferon gamma: a possible initiating role of B cells. J. Exp. Med. 186:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuji, R.F., M. Uramoto, H. Koshino, N.M. Tsuji, J. Magae, K. Nagai, and M. Yamasaki. 1992. Preferential suppression of delayed-type hypersensitivity by L-156,602, a C5a receptor antagonist. Biosci. Biotechnol. Biochem. 56:1686–1689. [DOI] [PubMed] [Google Scholar]
- 8.Tsuji, R.F., I. Kawikova, R. Ramabhadran, M. Akahira-Azuma, D. Taub, T.E. Hugli, C. Gerard, and P.W. Askenase. 2000. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J. Immunol. 165:1588–1598. [DOI] [PubMed] [Google Scholar]
- 9.Askenase, P.W., I. Kawikova, V. Paliwal, M. Akahira-Azuma, C. Gerard, T. Hugli, and R. Tsuji. 1999. A new paradigm of T cell allergy: requirement for the B-1 cell subset. Int. Arch. Allergy Appl. Immunol. 118:145–149. [DOI] [PubMed] [Google Scholar]
- 10.Askenase, P.W., and R.F. Tsuji. 2000. B-1 B cell IgM antibody initiates T cell elicitation of contact sensitivity. Curr. Top. Microbiol. Immunol. 252:171–179. [DOI] [PubMed] [Google Scholar]
- 11.Chen, J., M. Trounstine, F.W. Alt, F. Young, C. Kurahara, J.F. Loring, and D. Huszar. 1993. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int. Immunol. 5:647–656. [DOI] [PubMed] [Google Scholar]
- 12.Hardy, R.R., and K. Hayakawa. 1994. CD5 B cells, a fetal B cell lineage. Adv. Immunol. 55:297–339. [DOI] [PubMed] [Google Scholar]
- 13.Wells, S.M., A.B. Kantor, and A.M. Stall. 1994. CD43 (S7) expression identified peripheral B cell subsets. J. Immunol. 153:5503–5515. [PubMed] [Google Scholar]
- 14.Ushio, H., R.F. Tsuji, M. Szczepanik, K. Kawamoto, H. Matsuda, and P.W. Askenase. 1998. IL-12 reverses established antigen-specific tolerance of contact sensitivity by affecting costimulatory molecules B7-1 (CD80) and B7-2 (CD86). J. Immunol. 160:2080–2088. [PubMed] [Google Scholar]
- 15.Van Loveren, H., and P.W. Askenase. 1984. Delayed-type hypersensitivity is mediated by a sequence of two different T cell activities. J. Immunol. 133:2397–2401. [PubMed] [Google Scholar]
- 16.Ptak, W., W.R. Herzog, and P.W. Askenase. 1991. Delayed-type hypersensitivity initiation by early-acting cells that are antigen mismatched or MHC incompatible with late-acting, delayed-type hypersensitivity effector T cells. J. Immunol. 146:469–475. [PubMed] [Google Scholar]
- 17.Hata, D., Y. Kawakami, N. Inagaki, C.S. Lantz, T. Kitamura, W.N. Khan, M. Maeda-Yamamoto, T. Miura, W. Han, S.E. Hartman, et al. 1998. Involvement of Bruton's tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187:1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukhopadhyay, S., P.K. Sahoo, A. George, V. Bal, S. Rath, and B. Ravindran. 1999. Delayed clearance of filarial infection and enhanced Th1 immunity due to modulation of macrophage APC functions in xid mice. J. Immunol. 163:875–883. [PubMed] [Google Scholar]
- 19.Ptak, W., G.P. Geba, and P.W. Askenase. 1991. Initiation of delayed-type hypersensitivity by low doses of monoclonal IgE antibody. Mediation by serotonin and inhibition by histamine. J. Immunol. 146:3929–3936. [PubMed] [Google Scholar]
- 20.Askenase, P.W., S. Bursztajn, M.D. Gershon, and R.K. Gershon. 1980. T cell–dependent mast cell degranulation and release of serotonin in murine delayed-type hypersensitivity. J. Exp. Med. 152:1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geba, G.P., W. Ptak, G.A. Anderson, R.E. Ratzlaff, J. Levin, and P.W. Askenase. 1996. Delayed-type hypersensitivity in mast cell deficient mice: dependence on platelets for expression of contact sensitivity. J. Immunol. 157:557–565. [PubMed] [Google Scholar]
- 22.McHale, J.F., O.A. Harari, D. Marshall, and D.O. Haskard. 1999. Vascular endothelial cell expression of ICAM-1 and VCAM-1 at the onset of eliciting contact hypersensitivity in mice: evidence for a dominant role of TNF-α. J. Immunol. 162:1648–1655. [PubMed] [Google Scholar]
- 23.Bhutto, A.M., M. Honda, K. Maeda, S. Anan, and H. Yoshida. 1992. Contact hypersensitivity reaction to ovalbumin in newborn guinea pigs from maternally sensitized animals. J. Dermatol. Sci. 4:156–165. [DOI] [PubMed] [Google Scholar]
- 24.Haynes, J.D., R.W. Rosenstein, and P.W. Askenase. 1978. A newly described activity of guinea pig IgG1 antibodies: transfer of cutaneous basophil reactions. J. Immunol. 120:886–894. [PubMed] [Google Scholar]
- 25.Moore, K.G., and A.M. Dannenberg, Jr. 1993. Immediate and delayed (late-phase) dermal contact sensitivity reactions in guinea pigs. Passive transfer by IgG1 antibodies, initiation by mast cell degranulation, and suppression by soybean proteinase inhibition. Int. Arch. Allergy Immunol. 101:72–81. [DOI] [PubMed] [Google Scholar]
- 26.Grabbe, S., and T. Schwartz. 1998. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol. Today. 19:37–44. [DOI] [PubMed] [Google Scholar]
- 27.Hayakawa, K., R.R. Hardy, L.A. Herzenberg, A.D. Steinberg, and L.A. Herzenberg. 1984. Ly-1 B: a functionally distinct B-cell subpopulation. In Progress of Immunology V. Academic Press, Tokyo. 661–668.
- 28.Silverman, G.J., S.P. Cary, D.C. Dwyer, L. Luo, R. Wagenknecht, and V.E. Curtiss. 2000. A B cell superantigen-induced persistent “hole” in the B-1 repertoire. J. Exp. Med. 192:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw, P.X., S. Horkko, M.K. Chang, L.K. Curtiss, W. Palinski, G.J. Silverman, and J.L. Witztum. 2000. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Invest. 105:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masmoudi, H., T. Mota-Santos, F. Huetz, A. Coutinho, and P.A. Cazenave. 1990. All T15 Id-positive antibodies (but not the majority of VHT15+ antibodies) are produced by peritoneal CD5+ B lymphocytes. Int. Immunol. 2:515–520. [DOI] [PubMed] [Google Scholar]
- 31.Briles, D.E., C. Forman, S. Hudak, and J.L. Claflin. 1982. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J. Exp. Med. 156:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallick, S., J.L. Claflin, and D.E. Briles. 1983. Resistance to Streptococcus pneumoniae is induced by a phosphocholine-protein conjugate. J. Immunol. 130:2871–2875. [PubMed] [Google Scholar]
- 33.Cunningham, A.J. 1974. Large numbers of cells in normal mice produce antibody components of isologous erythrocytes. Nature. 252:749–751. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham, A.J., and E.J. Steele. 1981. Ontogeny of the autoimmune reaction in normal mice to antigens in erythrocytes and gut. Clin. Exp. Immunol. 44:38–48. [PMC free article] [PubMed] [Google Scholar]
- 35.Hayakawa, K., R.R. Hardy, M. Honda, L.A. Herzenberg, and A.D. Steinberg. 1984. Ly-1 B cells: functionally distinct lymphocytes that secrete IgM autoantibodies. Proc. Natl. Acad. Sci. USA. 81:2494–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerman, R., and D. Segre. 1970. Anti-pneumococcal polysaccharide type 3 hemolytic plaques in mice: cellular response in immunity and immunologic paralysis. J. Immunol. 104:1262–1266. [PubMed] [Google Scholar]
- 37.Baker, P.H., and P.W. Stashak. 1969. Quantitative and qualitative studies on the primary antibody response to pneumococcal polysaccharides at the cellular level. J. Immunol. 103:1342–1348. [PubMed] [Google Scholar]
- 38.Fagarasan, S., K. Kinoshita, M. Muramatsu, K. Ikuta, and T. Honjo. 2001. In situ class switching and differentiation of IgA-producing cells in the gut lamina propria. Nature. 413:639–643. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe, N., K. Ikuta, S. Fagarasan, S. Yazumi, T. Chiba, and T. Honjo. 2000. Migration and differentiation of autoreactive B-1 cells induced by activated γ/δ T cells in antierythrocyte immunoglobulin transgenic mice. J. Exp. Med. 192:1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pior, J., T. Vogl, C. Sorg, and E. Macher. 1999. Free hapten molecules are dispersed by way of the bloodstream during contact sensitization to fluorescein isothiocyanate. J. Invest. Dermatol. 113:888–893. [DOI] [PubMed] [Google Scholar]
- 41.Campos, R., M. Ahahira-Azuma, M. Szczepanik, and P.W. Askenase. 2001. IL-4−/− and Stat-6−/− mice have defective contact sensitivity (CS): the cause is absent IL-4 induced activation of CS-initiating B-1 cells. Immunology. 104:7 (Abstr. [Google Scholar]
- 42.Askenase, P.W., M. Azuma-Akahira, M. Szczepanik, M. Kronenberg, and R. Campos. 2001. Liver NK T cells are activated early in contact sensitivity (CS) to release IL-4 that activates B-1 cells that initiate elicited responses. Immunology. 104:14 (Abstr. [Google Scholar]
- 43.Svensson, L., J. Jirholt, R. Holmdahl, and L. Jansson. 1998. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin. Exp. Immunol. 111:521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akashi, T., S. Nagafuchi, K. Anzai, S. Kondo, D. Kitamura, S. Wakana, J. Ono, M. Kikuchi, Y. Niho, and T. Watanabe. 1997. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int. Immunol. 9:1159–1164. [DOI] [PubMed] [Google Scholar]
- 45.Chan, O.T., M.P. Madaio, and M.J. Shlomchik. 1999. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immunol. 163:3592–3596. [PubMed] [Google Scholar]
- 46.Lyons, J.A., M. San, M.P. Happ, and A.H. Cross. 1999. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 29:3432–3439. [DOI] [PubMed] [Google Scholar]
- 47.Vordermeier, H.M., N. Venkataprasad, D.P. Harris, and J. Ivanyi. 1996. Increase of tuberculous infection in the organs of B cell-deficient mice. Clin. Exp. Immunol. 106:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang, X., and R.C. Brunham. 1998. Gene knockout B cell-deficient mice demonstrate that B cells play an important role in the initiation of T cell responses to Chlamydia trachomatis (mouse pneumonitis) lung infection. J. Immunol. 161:1439–1446. [PubMed] [Google Scholar]
- 49.Falcone, M., J. Lee, G. Patstone, B. Yeung, and N. Sarvetnick. 1998. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response GAD65 antigen in nonobese diabetic mice. J. Immunol. 161:1163–1168. [PubMed] [Google Scholar]
- 50.Linington, C., B. Engelhardt, G. Kapocs, and H. Lassman. 1992. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J. Neuroimmunol. 40:219–224. [DOI] [PubMed] [Google Scholar]
- 51.Litzenburger, T., R. Fassler, J. Bauer, H. Lassmann, C. Linington, H. Wekerle, and A. Iglesias. 1998. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J. Exp. Med. 188:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, Y., S.A. Rollins, J.A. Madri, and L.A. Matis. 1995. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc. Natl. Acad. Sci. USA. 92:8955–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, Y., J. Kristan, L. Hao, C.S. Lenkoski, Y. Shen, and L.A. Matis. 2000. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J. Immunol. 164:4340–4347. [DOI] [PubMed] [Google Scholar]
- 54.Dietsch, G.N., and D.J. Hinrichs. 1989. The role of mast cells in the elicitation of experimental allergic encephalomyelitis. J. Immunol. 142:1476–1481. [PubMed] [Google Scholar]
- 55.Bebo, B.F., Jr., T. Yong, E.L. Orr, and D.S. Linthicum. 1996. Hypothesis: a possible role for mast cells and their inflammatory mediators in the pathogenesis of autoimmune encephalomyelitis. J. Neurosci. Res. 45:340–348. [DOI] [PubMed] [Google Scholar]
- 56.Kakizoe, E., S.H. Li, Y. Kobayashi, Y. Nishikori, S. Dekio, and H. Okunishi. 1999. Increases in mast cells and chymase in fibroproliferative paws of collagen-induced arthritic mice. Inflamm. Res. 48:318–324. [DOI] [PubMed] [Google Scholar]
- 57.Secor, V.H., W.E. Secor, C.A. Gutekunst, and M.A. Brown. 2000. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 191:813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gern, J.E., R.F. Lemanske, and W.W. Busse. 1999. Early life origins of asthma. J. Clin. Invest. 104:837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Regner, M., and P.H. Lambert. 2001. Autoimmunity through infection or immunization? Nat. Immunol. 2:185–188. [DOI] [PubMed] [Google Scholar]