

Abstract
Stromal-derived factor (SDF)-1 and its G protein–coupled receptor, CXCR4, regulate stem/progenitor cell migration and retention in the marrow and are required for hematopoiesis. We show here an interaction between CXCR4 and the Src-related kinase, Lyn, in normal progenitors. We demonstrate that CXCR4-dependent stimulation of Lyn is associated with the activation of phosphatidylinositol 3-kinase (PI3-kinase). This chemokine signaling, which involves a Src-related kinase and PI3-kinase, appears to be a target for BCR/ABL, a fusion oncoprotein expressed only in leukemia cells. We show that the binding of phosphorylated BCR/ABL to Lyn results in the constitutive activation of Lyn and PI3-kinase, along with a total loss of responsiveness of these kinases to SDF-1 stimulation. Inhibition of BCR/ABL tyrosine kinase with STI571 restores Lyn responsiveness to SDF-1 signaling. Thus, BCR/ABL perturbs Lyn function through a tyrosine kinase-dependent mechanism. Accordingly, the blockade of Lyn tyrosine kinase inhibits both BCR/ABL-dependent and CXCR4-dependent cell movements. Our results demonstrate, for the first time, that Lyn-mediated pathological crosstalk exists between BCR/ABL and the CXCR4 pathway in leukemia cells, which disrupts chemokine signaling and chemotaxis, and increases the ability of immature cells to escape from the marrow. These results define a Src tyrosine kinases-dependent mechanism whereby BCR/ABL (and potentially other oncoproteins) dysregulates G protein–coupled receptor signaling and function of mammalian precursors.
Keywords: BCR/ABL, Src, chemokine receptors, leukemia, chemotaxis
Introduction
The BCR/ABL oncogenic tyrosine kinase is responsible for initiating and maintaining the leukemic phenotype of chronic myelogenous leukemia (CML)* cells. This oncoprotein is responsible for the phosphorylation, activation, and dysregulation of intracellular signaling proteins that regulate proliferation and survival of progenitor cells in the marrow (1–4). The ability of immature leukemia cells to leave the marrow and accumulate in high numbers in blood and spleen suggests that the signaling events that regulate migration and retention of progenitor cells in marrow are also effected by this oncoprotein. Hematopoietic precursors need to be held in close contact to bone marrow stromal cells for the extensive interactions necessary for proper growth and maturation. Several targets of BCR/ABL, associated with integrin regulation, such as focal adhesion proteins paxillin, talin, tensin, and FAK have already been identified (4, 5). It has been also shown that CML cells fail to respond to at least two different chemokines: macrophage inflammatory protein (MIP)-1α and stromal-derived factor (SDF)-1 (6–8). Chemokines are known to play an important role in regulating movement and retention of progenitor cells within the bone marrow microenvironment. Intracellular signaling pathways that mediate these effects are poorly understood.
SDF-1 is a chemokine which acts through CXCR4, the only known receptor for SDF-1 (9, 10). G protein–coupled CXCR4 receptor, expressed on hematopoietic cells, has been reported to mediate chemotaxis of CD34+ stem cells, and to play a critical role in the homing of these cells in the bone marrow microenvironment (11, 12). Mice lacking SDF-1 protein or the CXCR4 receptor show lethal abnormalities in bone marrow myelopoiesis and B cell lymphopoiesis (13, 14). Recently, SDF-1 was reported to function as both a chemoattractant and as a modulator of cellular growth/survival (15–20). The phosphatidylinositol 3-kinase (PI3-kinase) and RAS-to-MAPK pathways are essential routes through which membrane-bound G protein–coupled receptors convey either chemotactic, survival, or proliferative/differentiative signals to the nucleus. Consequently, G protein–coupled CXCR4 receptor was reported to activate the PI3-kinase and mitogenic MAPK cascade (15, 18, 21). The Src family kinases have been known to play a key role in connecting G protein–coupled receptors to RAS-activating proteins and MAPK (22–26). Interestingly, a Src tyrosine kinase was recently shown to be a novel direct effector of G proteins (27), and the direct binding of activated c-Src to G protein–coupled receptors was required for MAPK activation (28). Despite the fact that the chemokine receptor, CXCR4, is known to regulate the PI3-kinase and RAS-to-MAPK cascade, the means by which this receptor couples to these signaling cascades and modulates movement, survival, and proliferation remains unclear.
It was recently reported that BCR/ABL completely blocks CXCR4-stimulated calcium flux and phosphorylation of p70 S6 kinase in hematopoietic transformed cells (8). In this study, we show that BCR/ABL strongly activates a new CXCR4-dependent signaling component through the Src family tyrosine kinase, Lyn. These opposing results indicate that the BCR/ABL oncoprotein alters the chemotactic response to SDF-1 through a complex mechanism involving both inhibitory and stimulatory effects on different signaling molecules. Crosstalk between BCR/ABL and G protein–coupled CXCR4 signaling through Lyn may allow this oncoprotein to couple simultaneously to PI3-kinase and the RAS-to-MAPK cascade and to take over this chemokine pathway. Our results define a molecular mechanism for the disruption of chemotaxis by oncoproteins in hematopoietic progenitor cells, and potentially, in other cellular systems. They also provide further insight into the striking ability of Src family kinases to transform mammalian cells.
Materials and Methods
Primary Cells and Cell Lines.
Bone marrow samples were obtained from normal individuals and CML patients after informed consent was obtained using guidelines approved by the Institutional Review Board of the University of Pennsylvania. CD34+ marrow cells were obtained from healthy individuals using immunoaffinity chromatography with magnetic beads (MACS® cell isolation kits; Miltenyi Biotec). Preparations were >95% viable CD34-expressing undifferentiated cells. Primary leukemic blasts were isolated by apheresis and Percoll density gradient centrifugation (29) from patients with BCR/ABL-positive CML. Human myeloid HL-60, Mo7e, and K562 cells were obtained from the American Type Culture Collection. BCR/ABL tyrosine kinase inhibitor STI 571 (Gleevec, imatinib mesylate) is currently widely distributed by Novartis Pharmaceuticals Corporation. A 10 mM stock solution of STI 571 in distilled water was prepared from which fresh working solutions were prepared in RPMI 1640 medium before the experiments. Cells were incubated in fresh medium with or without 1 μM STI 571 for 20 h.
Animals.
Lyn-deficient and wild-type mice were maintained in a pathogen-free environment at the National Cancer Institute-Frederick. Animal care was provided in accordance with the procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice between the ages of 8 and 12 wk were used for the study. Bone marrow was harvested from mouse femurs, and mononuclear cells were isolated using Ficoll-Paque TM Plus (Amersham Pharmacia Biotech AB) (36). Bone marrow–derived Lyn-deficient and wild-type mononuclear cells were maintained in culture and evaluated with regard to Lyn protein expression as described previously (36). It was confirmed that cells derived from Lyn-deficient mice did not express Lyn protein at all. In contrast, cells derived from wild-type mice expressed high levels of Lyn protein (unpublished data).
Retroviral Production and HL-60 Infection Protocol.
The retroviral vectors MigR1 and Mig210 expressing the enhanced green fluorescent protein (eGFP) and BCR/ABL proteins, respectively, were gifts of W. Pear from the University of Pennsylvania, Philadelphia, PA. 293T cells were transfected with MigR1 or Mig-210 using SuperFect Reagent (QIAGEN). The supernatant was collected 48 h after transfection. The virus was titred against 3T3-NIH cells. Subsequently, HL-60 cells were transduced by spinoculation and the infected cells were analyzed for the expression of GFP by flow cytometry as described previously (30). GFP/BCR/ABL positive HL60 cells were sorted with purity of 97%.
Immunoprecipitation and Reimmunoprecipitation Experiments.
Cells were stimulated for various times with 100–300 ng/ml SDF-1β or SDF-1α (UBI). After stimulation, cells were lysed with buffer (0.8–2% NP-40, 10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 30 mM sodium pyrophosphate, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 0.25 U/ml aprotinin, 10 μg/ml leupeptin, and 1 μM pepstatin). Lysates (10 min on ice) containing identical amounts of protein per sample were cleared by centrifugation, and then incubated with the indicated antibody: anti-Lyn; anti-p85 PI3-kinase; and anti-ABL SH2 domain (Upstate Biotechnology, Inc.) for 4 h at 4°C. Preblocked pansorbin beads were added for 30 min, and washed twice with lysis buffer and once with 10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1 mM Na3VO4. In vitro kinase assays were performed directly on beads as we described previously (23). For secondary immunoprecipitation, Lyn immunoprecipitates were washed twice to remove free ATP and the beads were then boiled in 1 volume of 20 mM Tris-HCl, pH 8.0, 0.5% SDS, and 1 mM dithiothreitol to disrupt protein–protein interaction, as previously described in detail (31). After centrifugation, the diluted supernatant (after reducing the SDS concentration to 0.1%) was reimmunoprecipitated with anti-ABL antibodies. The secondary immunoprecipitates containing BCR/ABL proteins were then washed in lysis buffer, boiled in sample buffer, and loaded on 8% SDS-PAGE.
Assay for Lyn and BCR/ABL Kinase Activity.
A modification of our previous protocol for Lyn kinase autophosphorylation was employed (23). To measure kinase activity of Lyn or BCR/ABL, precipitates were washed as indicated and then incubated in 25 mM Hepes, pH 7.1, 10 mM MnCl2, 1 μM ATP, and 10 μCi γ-32[P] ATP (6,000 μCi/mmol; DuPont NEN). In some experiments, 5 μg of acid-denaturated enolase was added to the kinase reaction mixture as an exogenous substrate. Kinase assays were performed at room temperature. The reaction was stopped by the addition of 2× Laemmli sample buffer and boiling. Samples were analyzed by 8% SDS-PAGE, followed by autoradiography on Kodak X AR-5 film at –70°C, and quantitated using densitometry.
Assay for PI3-Kinase Activity.
PI3-kinase activity was assayed directly on beads of anti–PI3-kinase immunoprecipitates with antibodies directed against the 85-kD regulatory subunit (Upstate Biotechnology, Inc.). The reaction was performed for 10 min at room temperature in a buffer containing 40 mM Hepes, pH 7.2, 6 mM MgCl2, and 1 mM EDTA, 20 μg of PI (Avanti Polar Lipids), 10 μM ATP, and 10 μCi γ-32[P] ATP (6,000 Ci/mmol; DuPont NEN). 0.2 mM adenosine was added to the reaction mixture to inhibit residual PI4-kinase activity. After incubation, the reaction was stopped with methanol plus 2.4 N HCl, 1/1 (vol/vol), lipids were extracted, analyzed by thin-layer chromatography, and then quantified as we described earlier (32).
Chemotaxis Assay.
SDF-1–induced chemotaxis of BCR/ABL-negative HL-60 cells (MigR1) and their BCR/ABL-transformed counterpart (Mig210) was determined in six independent experiments with Costar Transwell cell culture chambers according to the manufacturer's recommendations (Corning Inc.). HL-60 cells (both MigR1 and Mig210) were preincubated for 17 h in serum-free medium (RPMI 1640) in 37°C. Subsequently, cells were incubated for 30 min in the presence or the absence of 10 μM PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolol[3,4-d]pyramidine) (Calbiochem) before stimulation with SDF-1 (Upstate Biotechnology, Inc.). This concentration of PP2 was found to be effective in inhibiting Lyn activity in our preliminary dose–response experiments. Cells were tested for their ability to migrate in response to medium with SDF-1 (100–300 ng/ml) or medium alone for 1–3 h at 37°C. Migrating cells monolayers were fixed and stained using standard cytological techniques (Wright-Giemsa), and subsequently cells were counted. Growth factor–dependent cell line Mo7e, grown in DMEM with 500 ng/ml GM-CSF, was used as an additional control. These cells were preincubated for 2.5 h in GM-CSF–deficient media before treatment with PP2, stimulation with SDF-1 and chemotaxis assay. Bone marrow–derived CD34-positive cells were preincubated for 2.5 h in serum-free Iscove modified Dulbecco medium (IMDM; JRH Biosciences) before treatment for 30 min with 10 mM PP2 or its inactive analogue PP3 (4-amino-7-phenylpyrazol[3,4-d]pyramidyne) (Calbiochem), stimulation with 200 ng/ml SDF-1 or 100 ng/ml PMA (Sigma-Aldrich) and chemotaxis assay. PP3 and PMA were used as controls for PP2 and SDF-1, respectively. Bone marrow–derived Lyn-deficient and wild-type mononuclear cells were isolated, maintained in culture, and evaluated with regard to Lyn protein expression as described previously (36). These cells were preincubated for 2.5 h in serum-free IMDM (JRH Biosciences) before stimulation with 200 ng/ml SDF-1 or 100 ng/ml phorbol PMA (Sigma-Aldrich) and chemotaxis assay. In all chemotactic experiments, inserts were loaded with 100 μl of medium containing 50,000–100,000 cells (input population). The transmigrated cells which were visible in six randomly selected fields were counted (in different experiments, the number of control unstimulated transmigrating cells was 299–700 in six fields, which is represented as 100% on our figures).
Results
SDF-1 Regulates the Activity of the Lyn Protein Tyrosine Kinase in Hematopoietic Cells.
The chemokine receptor CXCR4 and its ligand, SDF-1, not only regulate movement (11, 12, 15), but are also able to modulate hematopoietic cell proliferation and survival (16–20). To evaluate the possible role of nonreceptor Src family tyrosine kinases in the movement, proliferation, and survival process, we screened SDF-1–stimulated CD34+ progenitor cells, and HL-60 cells which have been shown to express CXCR4 on their surface (33), for the activities of known Src-like kinases. We observed that the activity of the Lyn tyrosine kinase was significantly increased in SDF-1–stimulated myeloid cells, as recently reported in Jurkat T cells (34). Fig. 1 shows a representative time course for Lyn activation by SDF-1 in HL-60 cells (A) and CD34+ primary cells (B). Both Lyn autophosphorylation and its ability to stimulate phosphorylation of the exogenous substrate protein enolase increased within minutes of SDF-1 stimulation with maximum activity being detected after 5 min. Lyn activity declined slowly after the 5–10 min peak and returned to baseline levels after 60 min. Two major isoforms of Lyn, 53 kD and 56 kD, are formed due to alternative splicing (35). Autophosphorylation of both isoforms was detected in HL-60 cells and the more abundant 53-kD form was detectable in CD 34+ primary bone marrow cells. There was no change in the actual levels of Lyn proteins during the SDF-1 treatment of HL-60 or CD34+ cells, as determined by Western blot analysis (unpublished data). The stem cell factor/kit ligand (SCF/KL), a cytokine critical for normal hematopoiesis, is known to activate Lyn in hematopoietic progenitor cells (36). Therefore, stimulation with SCF/KL was used as a positive control in the kinase assays. Fig. 1 A shows a representative time course for Lyn activation induced by SCF/KL in HL-60 cells. Results obtained were similar to those obtained with SDF-1 stimulation. These results indicate that SDF-1 and SCF/KL share the Lyn signaling pathway in hematopoietic cells. Interestingly, SCF/KL has been recently reported to enhance chemotaxis induced by SDF-1. Also, cooperativity in downstream signaling pathways, including MAPK, has been demonstrated with SDF-1 in combination with SCF/KL (37).
Figure 1.


SDF-1 stimulates Lyn kinase activity in hematopoietic cells. (A) Lyn autophosphorylation is increased after treatment of HL-60 cells with SDF-1; SDF-1 induces an increase in Lyn phosphorylation of substrate enolase. A similar time course was obtained using SCF as a positive control. (B) Lyn and enolase phosphorylation are stimulated after treatment of CD34+ marrow progenitors with SDF-1. Cells in all experiments were stimulated for the indicated times with either SDF-1 or SCF, lysed, and immunoprecipitated with anti-Lyn antibodies. Immune complex in vitro kinase assays were performed as described in Materials and Methods. The changes in Lyn autophosphorylation are indicated (by fold increase) based on the densitometry. The results shown are representative of two experiments.
SDF-1 Regulates the Activity of the PI3-Kinase in Hematopoietic Cells.
The Lyn activation time course described above correlates with some of the earliest signaling events known to occur after SDF-1 stimulation. In particular, it corresponds to the previously observed time course for PI3-kinase activation (21). We detected an SDF-1 induced increase of PI3-kinase activity in p85 immunoprecipitates as reported previously (15, 21). Fig. 2, A and B shows a representative time course for PI3-kinase activation by the CXCR4 receptor in HL-60 and CD34+ precursor cells. PI3-kinase activity increased in p85 immunoprecipitates within minutes, with maximum activity occurring between 5 and 10 min. Given that concurrent activation of PI3-kinase and Lyn was detected, we asked whether these kinases could physically associate in SDF-1–stimulated cells. Lyn has been shown to couple (directly or indirectly) to p85 subunit-dependent PI3-kinase in B lymphocytes activated through the B cell antigen receptor (38), in human neutrophils activated through the N-formyl peptide chemoattractant G protein–coupled receptor (23), in human monocytes stimulated with bacterial lipopolysaccharide via the CD14 receptor (39), and in human platelets activated through the thrombin receptor (40). A relatively small proportion of cellular PI3-kinase, that which is tyrosine phosphorylated, binds to the SH2 domain of activated Src-related kinases (31). Accordingly, only slight coprecipitation of p53/56 Lyn and p85-PI 3-kinase was demonstrated by simple IP and subsequent immunoblotting in our experiments (Fig. 2 C-b). Thus, a more sensitive technique was chosen wherein Lyn immunoprecipitates were directly subjected to an in vitro PI3-kinase assay. We detected PI3-kinase activity in Lyn immunoprecipitates from SDF-1–stimulated cells (Fig. 2 C-a). In contrast, we observed no PI-kinase activity in control immunoprecipitates with Lck antibody, ruling out nonspecific binding as a possible source of PI3-kinase activity. The amount of PI3-kinase activity specifically associated with Lyn was ∼10% of the total activity measured in p85 subunit antibody precipitates. The time course of Lyn-associated PI3-kinase activity corresponded to that of Lyn tyrosine kinase activity (Fig. 1, A and B) as well as for total cellular PI3-kinase activation (Fig. 2, A and B). These data indicate that a link exists between Lyn and PI3-kinase activation in SDF-1–stimulated hematopoietic cells.
Figure 2.




SDF-1 stimulates PI3-kinase activity in hematopoietic cells. (A) Cellular p85 subunit-dependent PI3-kinase activity is increased after SDF-1 treatment of HL-60 cells. (B) Cellular p85 subunit-dependent PI3-kinase activity is stimulated after SDF-1 treatment of CD34+ bone marrow progenitors. (C) Lyn-associated PI3-kinase activity is increased in SDF-1–stimulated HL-60 cells. Cells were stimulated for the indicated times with SDF-1, lysed, and immunoprecipitated with anti-PI3-kinase p85 antibodies (A, B, and C-a 1), anti-Lyn antibodies (2C-a, 2-5, and 2C-b), or control anti-Lck antibodies. PI3-kinase activity was measured in immunoprecipitates as described in Materials and Methods. The indicated changes in PIP are based on densitometry values. Aliquots of total cell lysates were subjected to either Lyn immunoprecipitation and immunoblotting with anti-p85PI3K Ab or immunoblotting with anti-Lyn ab Similar amounts of Lyn protein were present in stimulated and control cells (C-b). The results shown are representative of two experiments.
Defect in the SDF-1–induced Lyn/PI3-Kinase Pathway in CML.
Previous studies have shown that BCR/ABL forms complexes with Src-related kinases in IL-3–dependent myeloid murine cell lines (41). To determine whether these signaling complexes can perturb G protein–coupled CXCR4 chemokine receptor-regulated pathways, we performed several experiments in human hematopoietic cells, which are described below.
As Lyn and PI3-kinase are regulated by SDF-1 in normal hematopoietic cells, we evaluated the effect of SDF-1 on Lyn and PI3-kinase activity in BCR/ABL-positive blasts of CML patients. For direct comparison, we used material from the same patients for both Lyn and PI3-kinase experiments. Portions from the same cell cultures were treated with SDF-1 and used for either Lyn or PI3-kinase analysis. Both Lyn and PI3-kinase were highly active in blasts from all CML patients (4 out of 4 patients). Basal activity of Lyn and PI3-kinase increased by several fold in leukemic blasts as compared with progenitor CD34+ cells derived from the bone marrow of healthy individuals. Moreover, the activity of the kinases did not increase in SDF-1– treated blasts (Fig. 3, A and B) as compared with normal cells (Fig. 1 B and Fig. 2 B). We conclude that both Lyn and PI3-kinase are constitutively active, and thus insensitive to stimulation through CXCR4 in transformed, as opposed to normal progenitor cells.
Figure 3.


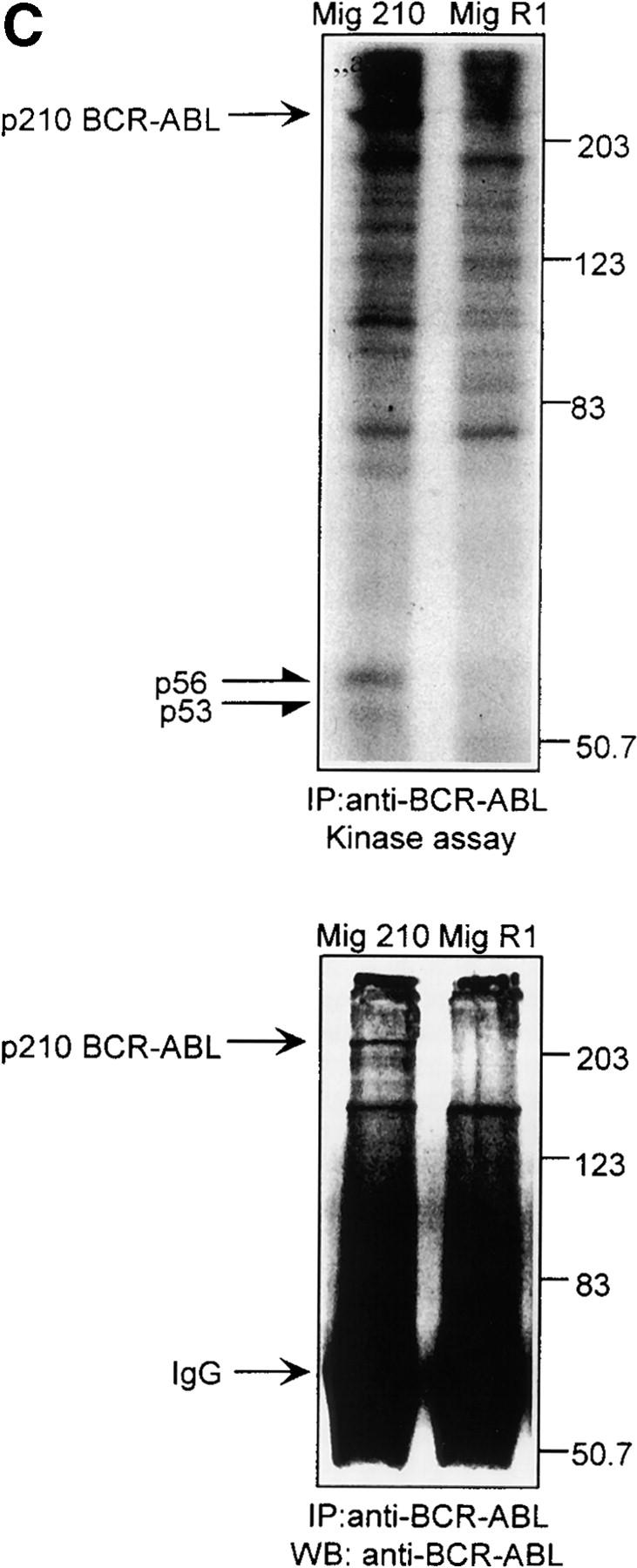



Defect of SDF-1–induced Lyn/PI3-kinase pathway in chronic myeloid leukemia. (A and B) Representative time course of Lyn and p85-dependent PI3-kinase phosphorylation in SDF-1–stimulated BCR/ABL-positive leukemic blasts (CML). Lyn tyrosine kinase activity (A) and PI3-kinase activity (B) are not increased after SDF-1 stimulation. This is in contrast to time courses observed in normal CD34+ cells and BCR/ABL-negative HL-60 cells, which are presented in Fig. 1, 2, and 3, E and F. Results are representative of four experiments. (C) Expression of exogenous BCR/ABL in HL-60 cells. Infected cells were analyzed for protein levels of exogenous BCR/ABL by Western blot and for BCR/ABL tyrosine kinase activity by immune complex in vitro kinase assays, as described in Materials and Methods. P56/53-kD tyrosine phosphorylated protein is detectable in BCR/ABL precipitates from BCR/ABL-infected Mig210 cells, but not from cells infected with control vector (MigR1). (D) Expression of endogenous Lyn in BCR/ABL-infected HL-60 cells. P210-kD tyrosine phosphorylated protein is coimmunoprecipitated with anti-Lyn antibody in Mig210, but not MigR1 cells. This protein comigrates with endogenous BCR/ABL in K562 cells (positive control). The phosphorylated 210-kD protein was identified as BCR/ABL by secondary p210 BCR/ABL immunoprecipitations on primary Lyn precipitates. The secondary IPs were performed after the disruption of protein complexes by disruption buffer (see Material and Methods). The position of BCR/ABL is indicated by the arrow. The relatively weak signal from secondary IPs is explained by the loss of precipitated proteins during disruption and reprecipitation. Results are representative of two experiments (D-a). (E and F) Comparison of SDF-1–stimulated Lyn and PI3-kinase activities and protein levels in BCR/ABL-negative HL-60 cells and BCR/ABL-positive HL-60 cells. Lyn autophosphorylation is inducible by SDF-1 in MigR1, but not in Mig210 cells (E). P85 subunit-dependent PI3-kinase activity is stimulated by SDF-1 in control cells, but not in Mig210 cells (F).
The use of primary cells can be problematic due to their limited viability in vitro, limited sample size and patient variation. Hence, we also used a myeloid human cell line to relate our findings to CML phenotype. HL-60 cells normally do not contain the Philadelphia chromosome and are thus BCR/ABL-negative. Therefore, these cells were infected with a retroviral vector coexpressing p210 BCR/ABL and GFP as described previously (30). After infection, green positive cells were sorted by FACS® yielding a >95% pure GFP-positive cell population. Immunoblotting and in vitro kinase assays were performed to confirm the expression and enzymatic activity of the kinase. Infected HL-60 cells possessed high levels of protein expression and tyrosine kinase activity of the expected 210-kD BCR/ABL protein (Fig. 3 C).
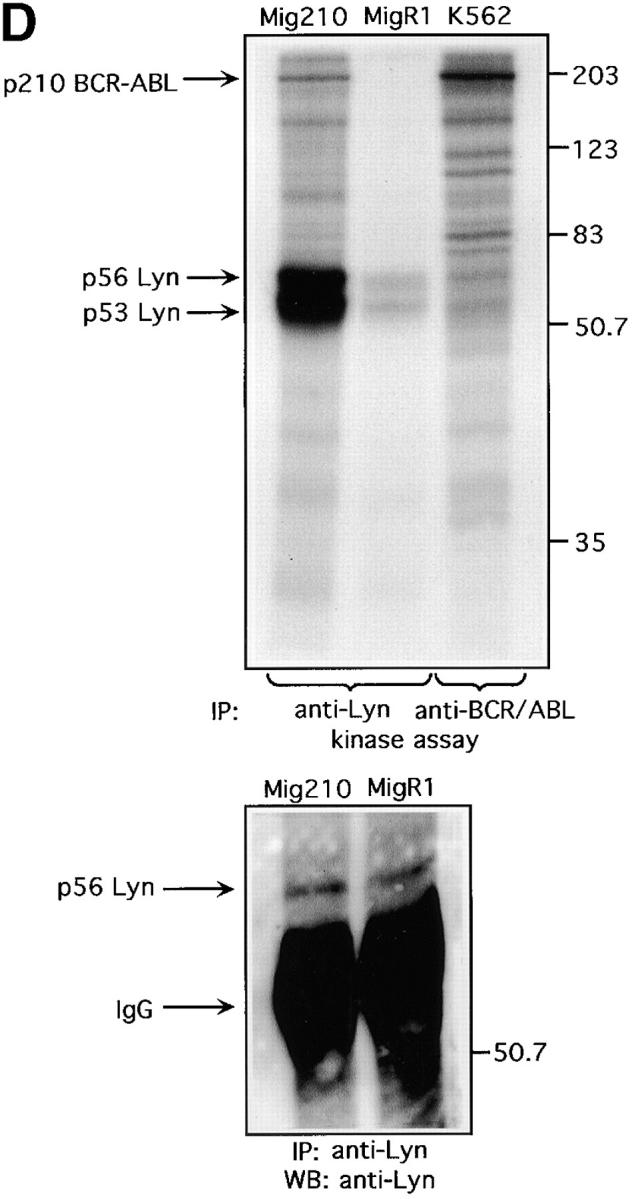
We evaluated BCR-ABL protein expression and its tyrosine kinase activity in infected HL-60 cells by Western blot and an immune complex in vitro kinase assay. We observed the presence of a p56/53-kD tyrosine phosphorylated protein in BCR-ABL precipitates (Fig. 3 C). This presence was detectable only in Mig210 BCR/ABL-positive cells, but not in control-vector infected MigR1 cells. The possible association of BCR/ABL with Lyn was further probed by looking for the presence of BCR/ABL in Lyn immunoprecipitates. Fig. 3 D shows that we could precipitate phosphorylated 210-kD protein with the anti-Lyn antibody and that this association took place only in BCR/ABL-infected HL-60 cells (Mig210). The phosphorylated 210-kD protein comigrated with BCR/ABL in the positive control (BCR/ABL-positive K562 cell line). Levels of tyrosine phosphorylation of Lyn and other associated proteins were elevated in the BCR/ABL-positive cells as compared with control cells.
Since the p210 and p56/53 proteins were detected in Lyn and BCR/ABL precipitates, respectively, it strongly suggested that constitutively phosphorylated BCR/ABL couples to Src-related kinase Lyn in transformed human cells. The identity of the 210-kD protein as BCR/ABL was confirmed by reimmunoprecipitating the Lyn precipitates with anti-ABL antibody. For this purpose, Lyn immunoprecipitates from Mig210 and MigR1 cells were subjected to in vitro kinase assays, treated with disruption buffer to dissociate protein complexes, and subsequently reprecipitated with anti-ABL antibodies as described previously (31, 41). Fig. 3 D-a shows that we could specifically reprecipitate phosphorylated 210-kD protein with anti ABL-antibodies and that this precipitation occurred in BCR/ABL-infected, Mig210, but not in control, MigR1 cells. The reimmunoprecipitated protein comigrated with p210 BCR/ABL, isolated through anti-ABL immunoprecipitates of control K562 cells. These results directly show that the p210 protein in Lyn immune complexes from human hematopoietic cells is BCR/ABL.
Lyn and PI3-kinase were shown to be excessively active in CML blasts (Fig. 3, A and B). Therefore, it was of interest to measure whether activities of these kinases were upregulated in BCR/ABL-infected HL-60 cells. Infection of cells did not alter Lyn and PI3-kinase protein levels, as determined by Western blot analysis (Fig. 3, E and F). However, basal enzymatic activities of Lyn and PI3-kinase were increased by an average of fivefold and threefold, respectively, in BCR/ABL-infected cells as compared with control cells. (Fig. 3, E and F). Consequently, SDF-1 had no significant effect on Lyn and PI3-kinases activities in BCR/ABL-positive cells. This was in contrast to control cells, which showed clear time-dependent changes for Lyn and PI3-kinase activities after SDF-1 stimulation. These findings suggest that BCR/ABL tyrosine kinase activity perturbs the SDF-1–regulated Lyn transduction pathway in hematopoietic transformed cells. To make certain that the observed alteration in Lyn and PI3-kinase activities are caused by BCR/ABL tyrosine kinase-dependent mechanism, we measured Lyn and PI3-kinases activation in STI 571-pretreated cells. STI 571 is a protein-tyrosine kinase inhibitor that strongly inhibits the BCR/ABL tyrosine kinase, and is well selective (42). As shown in Fig. 4, A and B , pretreatment of Mig210 cells with STI 571 partially restores Lyn and PI3-kinase responsiveness to SDF-1. Thus, CXCR4 receptor cannot regulate Lyn and PI3-kinase in BCR/ABL-transformed cells apparently due to perturbations caused by tyrosine kinase activity of this oncoprotein.
Figure 4.


Effect of STI-571 on SDF-1–regulated Lyn/PI3-kinase pathway in BCR/ABL-positive cells. (A) Comparison of SDF-1–stimulated Lyn activity and protein levels in BCR/ABL-positive HL-60 cells exposed or not exposed to 1.0 μM STI-571. (B) Comparison of SDF-1–stimulated PI3-kinase activity and protein levels in BCR/ABL-positive HL-60 cells exposed or not exposed to 1.0 μM STI-571. The changes in Lyn or PI3-kinase phosphorylation are indicated (by fold increase) based on the densitometry.
Src Family Tyrosine Kinase Pathway Is Required for both SDF-1–dependent and BCR/ABL-dependent Hematopoietic Cell Movements.
It was previously established that Lyn is required for normal stem cell factor-induced chemotaxis of primary hematopoietic cells in mice (36). To address the question of the involvement of Lyn in CXCR4-regulated chemotaxis in BCR/ABL-positive and BCR/ABL-negative control cells, we examined the effect of pyrazolopirimidine PP2 on SDF-1–induced chemotaxis. PP2 was designed and synthesized for inhibition of Src family kinases and appeared to be selective for these kinases (43). We confirmed that PP2 effectively inhibited tyrosine phosphorylation of Lyn at concentrations from 0.1 to 10 μM. In the presence of 0.1 and 1 μM PP2, Lyn phosphorylation was blunted by 74 and 90%, respectively, with 100% inhibition seen at 10 μM (unpublished data). Indeed, if the decrease in Lyn-stimulated phosphorylation of enolase is considered, the level of inhibition is even greater (100% inhibition seen at 1 μM PP2) (unpublished data). This is similar to the effective in vivo doses of PP2 reported in the literature for Src family kinases (43).
As shown in Fig. 5 A the initial number of BCR/ABL-negative MigR1 cells that migrated to medium alone was arbitrarily set to 100%. The number of MigR1 cells that migrated to medium plus SDF-1 was approximately fivefold higher. Similar results were obtained using another Lyn-expressing (unpublished data), BCR/ABL-negative hematopoietic cell line, M07e (Fig. 5 B). In contrast, BCR/ABL-positive Mig210 cells had a strong chemotactic response to both medium alone or medium plus SDF-1. The presence of SDF-1 in the medium had no significant effect on Mig210 cell migration. This result indicates that BCR/ABL oncoprotein blocks CXCR4-regulated chemotaxis, but increases spontaneous cell motility. Chemotaxis, both CXCR4-dependent (in MigR1 or M07e cells) and BCR/ABL-dependent (in Mig210 cells), was effectively blocked by PP2 pretreatment. It indicates that a Src family kinase is required for chemotaxis and is shared between CXCR4 signaling and BCR/ABL signaling in hematopoietic cells. These results are highly consistent with participation of Lyn in BCR/ABL and CXCR4 signal transduction as demonstrated in Figs. 1–4.
Figure 5.




Effect of PP2 on CXCR4-dependent and BCR/ABL-dependent cell movements. Inhibition of CXCR4-dependent cell movements in Lyn-deficient primary cells. (A) The inhibition of SDF-1–regulated chemotaxis by PP2 was determined as described under Materials and Methods and Results in BCR/ABL-negative HL-60 cells (MigR1) and BCR/ABL-positive HL-60 cells (Mig210). The initial number of BCR/ABL-negative MigR1 cells that migrated to medium alone was set to 100%. The results shown (mean ± SD) are averages of six experiments. (B) The inhibition of SDF-1–regulated chemotaxis by PP2 was determined in hematopoietic precursor cells Mo7e as described under Materials and Methods and Results. The results shown are representative of six experiments. (C) The suppression of CXCR4-dependent chemotaxis by PP2, but not by its inactive analogue PP3, in CD34+ human primary myeloid cells. PMA-induced migration is normal in PP2-pretreated CD34+ cells. Treatment and migration were performed as described in Materials and Methods. The results shown represent the average ± range of four separate determinations with different normal cell donors. (D) The reduction of SDF-1-induced migration in Lyn-deficient mononuclear bone marrow cells from knock-out mice. PMA-induced migration appears to be normal in these cells. Data represents the mean and standard deviation of eight samples with eight different animals. Treatment with factors and migration assays were performed as described in Materials and Methods.
We demonstrated using both BCR/ABL gene transfer and the BCR/ABL inhibitor STI-571, that BCR/ABL severely perturbs CXCR4-regulated Lyn signaling pathway and cellular migration (Figs. 3 E, 4 A, and 5 A). The specificity and relevance of Lyn and other Src-family kinases to hematopoietic cell migration is a complex issue which is further addressed here. The specificity of the Src-family kinase-requiring SDF-1 pathway is indicated by the data we obtained with primary human stem/progenitor cells. We showed that PP2, a specific inhibitor of Src family kinases, but not PP3, an inactive analoque of PP2 (43), inhibits the effects of SDF-1 in CD 34+ cells (Fig. 5 C). To further demonstrate that the blockade of migration in PP2-treated CD34+ cells was specific for SDF-1 signaling and Src kinases, we examined migration in response to PMA. PMA is a direct activator of the protein kinase C signaling pathway that works separately from the Src-family kinases. Fig. 5 C shows that, in contrast to SDF-1-induced migration, PMA-induced migration was not inhibited at all in PP2-treated CD34+ human cells. By using PP3 and PMA, we showed that PP2 did not exert any nonspecific effects on chemotactic signaling or cytotoxic effects that could account for some of the blockade of SDF-1–regulated migration. Thus, the inhibitory effect of PP2 on cellular migration was due to the specific blockade of Src-family kinases.
Since PP2 acts on a broad range of Src-family kinases, it is possible that other Src kinases are also involved in the interactions shown for Lyn. To make certain that Lyn is important for SDF-1-regulated migration, we measured the effect of SDF-1 on Lyn-deficient mononuclear cells derived from marrow of knock-out mice (36). Although SDF-1 increased migration of wild-type mice-derived cells by an average of sixfold, these increases were blunted in Lyn-deficient mice-derived cells by 77.7% (Fig. 5 D). Thus, cellular migration was inhibited by nearly 80% in Lyn-deficient cells, indicating that the Lyn pathway is quantitatively predominant for SDF-1–regulated hematopoietic cell migration. PMA-induced migration was not inhibited in Lyn-deficient cells ruling out cytotoxic effects that could account for the inhibition of migration. Lyn deficiency did not completely prevent SDF-1–induced migration. It is possible that other Src-related kinases are able to partially compensate for the loss of Lyn in knock-out mice. Multiple Src-family kinases are able to compensate for the loss of one related kinase (58, 59). In conclusion, the data we generated using both pharmacological and genetic approaches showed that Lyn-requiring pathway is important for hematopoietic cell migration during stimulation by SDF-1. Since Lyn is stimulated by BCR/ABL oncoprotein in leukemia cells, these cells confuse signals from BCR/ABL as signals from a chemokine receptor. This results in inappropriate, receptor-independent cell migration.
Discussion
In this work, we show a direct functional and physical association between the oncogenic BCR/ABL tyrosine kinase, Src-related tyrosine kinase, Lyn, and the CXCR4 G protein-coupled receptor-induced chemokine pathway in hematopoietic cells. The Lyn tyrosine kinase is rapidly and transiently activated in SDF-1–stimulated normal hematopoietic cells as well as in the BCR/ABL-negative HL-60 cell line. However, BCR/ABL-positive leukemic blasts of CML patients and BCR/ABL-infected HL-60 cells demonstrate constitutive Lyn activity. Lyn, and the Lyn-associated PI3-kinase, are not SDF-1 responsive in these cells. The regulatory effect of SDF-1 on Lyn and PI3-kinase is restored in STI-571–treated cells indicating that BCR/ABL interacts with G protein-coupled CXCR4 signaling in a tyrosine kinase-dependent manner. As a further indication of the involvement of BCR/ABL in CXCR4 signaling, we also observed a physical association between BCR/ABL and Lyn. Thus, we demonstrate for the first time that BCR/ABL perturbs, via Lyn, a CXCR4-regulated pathway. While the binding of BCR/ABL to Src family kinases has been reported previously (41), to the best of our knowledge it has not been reported that this interaction alters the G protein–coupled chemokine receptor signaling pathways. On first examination our results seem to be in contrast to the data of Salgia et al. (8), who showed that BCR/ABL strongly blocks, but does not activate, CXCR4 regulated calcium flux and p70 S6 kinase phosphorylation in transformed cells. However, since Lyn and PI3-kinase are directly associated with and strongly activated by BCR/ABL, it is possible that Lyn and PI3-kinase are simply sequestered by this oncoprotein. Without sufficient amounts of these signaling proteins for use by CXCR4, SDF-1 signaling would be shut down. Since we also show that Lyn and PI3-kinase are continuously activated by BCR/ABL, from inside of the cell, it is possible that the paradoxical cellular responses we describe in BCR/ABL-transformed cells, e.g., reduced chemotactic response to SDF-1 accompanied by increased spontaneous motility (Fig. 5), could result. Since Lyn is a target for both BCR/ABL oncoprotein and CXCR4 receptor, hematopoietic cells may “mistake” signals from BCR/ABL as signals from the CXCR4 chemokine receptor. This could lead to inappropriate cell migration in CML and explain this long-observed phenomenon. Our findings, along with those of Salgia et al. (8), are of particular interest since they provide a plausible explanation for some of the homing and retention defects characteristic of leukemia, (i.e., the ability of CML cells to leave the marrow, circulate in high numbers in the blood, and infiltrate other tissues).
Effectors involved in SDF-1–induced PI3-kinase activation and RAS-to-MAPK activation are currently unknown. The data presented here imply a mechanism that explains the ability of the G protein–coupled CXCR4 receptor and BCR/ABL oncoprotein to stimulate many intracellular signaling cascades, which were described previously in normal and malignant hematopoietic cells (4, 15, 21, 23). Different G protein–coupled signaling pathways synergize with other receptor pathways to enhance or inhibit signals elicited by these various receptors. Numerous reports describe Src as a go-between that relays messages from G protein–coupled receptors to the growth factor receptor tyrosine kinase signaling complex (22, 23, 26, 44). It has been suggested that the Src-related tyrosine kinase Lyn is a signaling intermediate between G protein-coupled chemoattractant receptors and PI3-kinase as well as the RAS-to-MAPK pathway in human neutrophils and eosinophils (23, 32, 45). Analogous roles for Src family members were proposed in other cell types (24–26, 28, 39–40). Since we have found that Lyn and PI3-kinase are stimulated by SDF-1 and are physically associated, it is logical to suggest a similar role for Lyn in signal propagation from the G protein–coupled CXCR4 chemokine receptor to PI3-kinase and RAS-to-MAPK activation in normal hematopoietic cells. Since we also have demonstrated that Lyn is a direct signaling target for BCR/ABL in transformed cells, it is reasonable to propose an analogous role for Lyn in signal transmission from BCR/ABL to PI3-kinase and RAS/MAPK. At the present time, it would be difficult to accept the notion that activation of PI3-kinase by BCR/ABL occurs exclusively through Lyn. There are a several of other pathways proposed in the past through which this oncoprotein could potentially activate PI3-kinase (46, 47). In any event, according to our present model, both the CXCR4 G protein-coupled receptor and BCR/ABL oncoprotein may gain access to intracellular cascades through Src-related tyrosine kinase Lyn in progenitor hematopoietic cells (Fig. 6) . This signaling paradigm would be consistent with our present data and with the previous observation of Hallek et al. (46), who discovered that the signaling complex of receptor tyrosine kinase p145c-kit can be activated by the p210 BCR/ABL oncoprotein in hematopoietic cells. Lyn associates with the juxtamembrane region of the c-kit receptor (48). This receptor is critical in the movement, proliferation, and survival of hematopoietic progenitor cells (36). Nonetheless, we do not dispute the possibility that other than Src kinases-dependent crosstalk pathways might also exist. This is the legitimate object of additional studies.
Figure 6.

Proposed model for interaction between oncoproteins and G protein-coupled signal transduction pathways in transformed mammalian cells. BCR/ABL oncoprotein interferes with the Src-related kinase Lyn signaling, which is regulated by G protein–coupled chemokine receptors in hematopoietic cells. A previously defined direct interaction of G protein subunits with Src family kinases (references 24 and 27) and linkage of G protein–coupled receptor via Src kinases to the receptor tyrosine kinase complex and its downstream signaling cascades (references 22–28 and 44) is indicated, as described in the text. The signaling paradigm we have depicted provides a mechanism to explain the ability of BCR/ABL, and potentially other oncoproteins, to couple simultaneously to multiple signaling transducers (e.g., PI3-kinase, Shc adaptor protein, receptor tyrosine kinase, and the RAS-to-MAPK cascade). This paradigm may also provide further insight into the remarkable ability of Src family kinases to transform various cell types.
In addition to our present results, alteration of Lyn/PI3-kinases' activities may be relevant to the pathogenesis of human leukemia for the following reasons: (i) we described several years ago a key role for Lyn kinase in propagation signals from G protein–coupled chemoattractant receptors to p85 subunit-dependent PI3-kinase, Shc/RAS-to-MAPK pathway, and to the interior of blood cells (23, 32). Then, we proposed that G protein–coupled chemoattractant receptors crosstalk to tyrosine phosphorylation signaling through Lyn. It was shown recently, in Lyn-deficient mice, that Lyn is also required for normal stem cell factor–regulated chemotaxis and proliferation in progenitor hematopoietic cells (36). Moreover, regulation of Lyn phosphorylation is an important antiapoptotic event in blood cells (49). These findings suggest that Lyn is a signaling element that brings chemotaxis, proliferation, and survival to a point of convergence in developing hematopoietic cells. Src-family tyrosine kinases are a major group of cellular signal transducers that are involved in proliferation/survival and oncogenity in various tissues. In fact, c-Src was the first identified protein tyrosine kinase and the first oncogene identified (50, 51). Tyrosine kinases of the Src family play a critical role in crosstalk between G protein–coupled receptors, integrins, and tyrosine kinase growth factor signaling. Src family kinases pleiotropically link these receptors to the mitogenic MAPK cascade, which may provide an oncogenicity mechanism (23, 26). (ii) PI3-kinase, which is often physically and/or functionally associated with Src-family tyrosine kinases, is an important regulator of progenitor mammalian cells movement, differentiation, proliferation, and survival (52, 53), and is also able to transform cells (54). A new oncogene derived from the p85 regulatory subunit of PI3-kinase was recently identified (55). This oncogene is responsible for the malignant transformation of lymphoid cells in mice. In addition, it was shown that the gene encoding the catalytic subunit of PI3-kinase is also able to transform cells (56). It was previously demonstrated that PI3-kinase is a downstream effector of ABL oncogenic variants (57). The p85 subunit-dependent PI3-kinase plays a major role in the BCR/ABL-induced proliferation and survival of CML cells (47). Thus, a constitutive activation of PI3-kinase that appears simultaneously with excessive Lyn activity in BCR/ABL-positive hematopoietic cells could potentially play important role in the pathogenesis of CML, or at the least in determining the malignant phenotype.
In conclusion, we describe the CXCR4-unregulatable Lyn and PI3-kinase activities in BCR/ABL-positive cells, as opposed to the CXCR4-regulatable Lyn and PI3-kinase activities in BCR/ABL-negative control cells. Our results demonstrate that the SDF-1 chemokine-regulated Lyn pathway is a target for the BCR/ABL oncoprotein in transformed hematopoietic cells. Thus, disruption of G protein–coupled chemokine receptor signaling by the oncogenic tyrosine kinases, via Src-family kinase-dependent mechanism, may be involved in the pathogenesis of human myeloid leukemia. It is tempting to suggest that the BCR-ABL/Lyn pathway is coupled to transcriptional regulation in hematopoietic cells. Future studies directed at the mechanisms by which G protein–coupled chemokine receptors contribute to cell transformation should prove informative, as will investigation of downstream transcriptional elements linked to the BCR-ABL/Lyn pathway in hematopoietic cells. An important goal is to determine whether analogous Src family kinases-mediated mechanisms operate in different types of precursor cells and G protein–coupled receptors. Identification of signaling molecules involved in the movement, growth, and survival of normal progenitor and malignant cells should lead to therapeutic targets of great efficiency, as has been demonstrated in the case of STI571 and the inhibition of BCR/ABL kinase in CML (42). Interestingly, a dominant role for Lyn kinase in K562 cells selected for resistance to STI571 was reported recently (60). Our present results extend this new observation. They both strongly suggest that combination of STI571 with inhibitors of Lyn or/and PI3K may be considered as therapeutic alternative that could circumvent resistance to STI571, particularly in patients with advanced disease. Experiments to test this possibility are in progress in our laboratories.
Acknowledgments
We are grateful to Dr. Warren S. Pear for providing us with MigR1 and Mig210 constructs. We thank Drs. Alan D. Schreiber and Stephen G. Emerson for their valuable comments. We also thank Dr. Cezary Swider for sharing with us his unpublished flow cytometry results, Dolores Winterstein and Anna Kalota for technical assistance. Finally, we thank Dr. Anna Kowalska for her advice in chemotaxis assay.
This work was supported in part by Program Project 1PO1DK52558-03 (to S.G. Emerson and A.M. Gewirtz). Dr. Gewirtz is a Distinguished Clinical Scientist of the Doris Duke Charitable Foundation.
S. Chinta and E. Urbanowska contributed equally to this paper.
Footnotes
Abbreviations used in this paper: CML, chronic myelogenous leukemia; PI3-kinase, phosphatidylinositol 3-kinase; SDF, stromal-derived factor.
References
- 1.Groffen, J., J.R. Stephenson, N. Heisterkamp, A. de Klein, C.R. Bartram, and G. Grosveld. 1984. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 36:93–99. [DOI] [PubMed] [Google Scholar]
- 2.Daley, G.Q., and N.Y. Ben. 1991. Implicating the bcr/abl gene in the pathogenesis of Philadelphia chromosome-positive human leukemia. Adv. Cancer Res. 57:151–184. [DOI] [PubMed] [Google Scholar]
- 3.Tauchi, T., H.S. Boswell, D. Leibowitz, and H.E. Broxmeyer. 1994. Coupling between p210bcr-abl and Shc and Grb2 adaptor proteins in hematopoietic cells permits growth factor receptor-independent link to Ras-activation pathway. J. Exp. Med. 179:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabarowski, J.H.S., and O.N. Witte. 2000. Consequences of BCR-ABL expression within the hematopoietic stem cell in chronic myeloid leukemia. Stem Cells. 18:399–408. [DOI] [PubMed] [Google Scholar]
- 5.Salgia, R., J.-L. Li, S.H. Lo, B. Brunckhorst, G.S. Kansas, S. Sobhany, Y. Sun, E. Pisick, M. Hallek, T. Ernst, et al. 1995. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by p210Bcr/ABL. J. Biol. Chem. 270:5039–5047. [DOI] [PubMed] [Google Scholar]
- 6.Eaves, C., J. Cashman, S. Walpe, and A. Eaves. 1993. Unresponsiveness of primitive chronic myeloid leukemia cells to macrophage inflammatory protein 1 α, an inhibitor of primitive normal hematopoietic cells. Proc. Natl. Acad. Sci. USA. 90:12015–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wark, G., C. Heyworth, E. Spooncer, L. Czaplewski, J. Francis, T. Dexter, and A. Whetton. 1998. Abl protein kinase abrogates the response of multipotent hematopoietic cells to the growth inhibitor macrophage inflammatory protein-1 α. Oncogene. 16:1319–1324. [DOI] [PubMed] [Google Scholar]
- 8.Salgia, R., E. Quackenbush, J. Lin, N. Souchkova, M. Sattler, D.S. Ewaniuk, K.M. Klucher, G.Q. Daley, S.K. Kraeft, R. Sackstein, et al. 1999. The BCR/ABL oncogene alters the chemotactic response to stromal-derived factor-1. Blood. 94:4233–4246. [PubMed] [Google Scholar]
- 9.Oberline, E., A. Amara, F. Bachelerie, C. Bessiac, J.L. Vereliezier, O. Schwartz, J.M. Heard, I. Clark-Lewis, D.F. Legler, and B. Moser. 1996. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 382:833–835. [DOI] [PubMed] [Google Scholar]
- 10.Bleul, C.C., M. Farzan, H. Choe, C. Parolin, I. Clark-Lewis, J. Sodroski, and T.A. Springer. 1996. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 382:829–833. [DOI] [PubMed] [Google Scholar]
- 11.Aiuti, A., I.J. Webb, C. Bleul, T. Springer, and J.C. Gutierrez-Ramos. 1997. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 185:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim, C.H., and H.E. Broxmeyer. 1998. In vitro behavour of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow microenvironment. Blood. 91:100–110. [PubMed] [Google Scholar]
- 13.Nagasawa, T., S. Hirota, K. Tashibana, N. Takakura, S. Nishikawa, Y. Kitamura, N. Yoshida, H. Kikutani, and T. Kishimoto. 1996. Defects of B-cell lymphopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 382:635–638. [DOI] [PubMed] [Google Scholar]
- 14.Zou, Y.R., A.H. Kottmann, M. Kuroda, I. Taniuchi, and D.R. Littman. 1998. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 393:595–599. [DOI] [PubMed] [Google Scholar]
- 15.Ganju, R.K., S.A. Brubaker, J. Meyer, P. Dutt, Y. Yang, S. Qin, W. Newman, and J. Groopman. 1998. The chemokine, stromal cell-derived factor-1, binds to the transmembrane G protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 273:23169–23173. [DOI] [PubMed] [Google Scholar]
- 16.Hodohara, K., N. Fuji, N. Yamamoto, and K. Kaushansky. 2000. Stromal cell-derived factor-1 (SDF-1) acts together with trombopoetin to enhance the development of megakaryocytic progenitor cells (CFU-MK). Blood. 95:769–775. [PubMed] [Google Scholar]
- 17.Lataillade, J.J., D. Clay, C. Dupuy, S. Rigal, C. Jasmin, P. Bourin, and M.C. Le Bousee-Kerdiles. 2000. Chemokine SDF-1 enhances circulating CD34+ cell proliferation in synergy with cytokines: possible role in progenitor survival. Blood. 95:756–768. [PubMed] [Google Scholar]
- 18.Yonezawa, A., T. Hori, H. Sakaida, and T. Uchiyama. 2000. SDF-1 has costimulatory effects on human T cells: possible involvement of MAPK (ERK2) activation. Microbiol. Immunol. 44:135–141. [DOI] [PubMed] [Google Scholar]
- 19.Burger, J.A., N. Tsukada, M. Burger, N.J. Zvaifler, M. Dell'Aquila, and T.J. Kipps. 2000. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 96:2655–2663. [PubMed] [Google Scholar]
- 20.Lataillade, J.J., D. Clay, P. Baurin, F. Herodin, C. Dupuy, C. Jasmin, and M.C. Le Bousse-Kerdiles. 2002. Stromal cell-derived factor 1 regulates primitive hematopoiesis by suppressing apoptosis and by promoting G0/G1 transition in CD34+ cells: evidence for an autocrine/paracrine mechanism. Blood. 99:1117–1129. [DOI] [PubMed] [Google Scholar]
- 21.Wang, J.-F., I.-W. Park, and J.E. Groopman. 2000. Stromal cell-derived factor-1 stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: role of phosphoinositide-3 kinase and protein kinase C. Blood. 95:2505–2513. [PubMed] [Google Scholar]
- 22.Daub, H., C. Wallash, A. Lankenau, A. Herrlich, and A. Ullrich. 1997. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 16:7032–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ptasznik, A., A. Traynor-Kaplan, and G.M. Bokoch. 1995. G-protein coupled chemoattractant receptors regulate Lyn tyrosine kinase-Shc adapter protein signaling complexes. J. Biol. Chem. 270:19969–19973. [DOI] [PubMed] [Google Scholar]
- 24.Luttrell, L.M., B.E. Hawes, T. van Biesen, D.K. Luttrell, T.J. Lansing, and R.J. Lefkowitz. 1996. Role of c-Src tyrosine kinase in G protein-coupled receptor and G β/γ subunit-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 271:19443–19450. [DOI] [PubMed] [Google Scholar]
- 25.Daulhac, L., A. Kowalski-Chauvel, L. Pradayrol, N. Vaysse, and C. Seva. 1999. Src-family tyrosine kinases in activation of ERK-1 and p85/p110-phosphatidylinositol 3-kinase by G/CCKB receptors. J. Biol. Chem. 274:20657–20663. [DOI] [PubMed] [Google Scholar]
- 26.Della Rocca, G.J., S. Maudsley, Y. Daaka, R.J. Lefkowitz, and L.M. Luttrell. 1999. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. J. Biol. Chem. 274:13978–13984. [DOI] [PubMed] [Google Scholar]
- 27.Ma, Y.C., J. Huang, S. Ali, W. Lowry, and X.Y. Huang. 2000. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 102:635–646. [DOI] [PubMed] [Google Scholar]
- 28.Cao, W., L.M. Luttrell, A.V. Medvedev, K.L. Pierce, K.W. Daniel, T.M. Dixon, R.J. Lefkowitz, and S. Collins. 2000. Direct binding of activated c-Src to the adrenergic receptor is required for MAP kinase activation. J. Biol. Chem. 275:38131–38134. [DOI] [PubMed] [Google Scholar]
- 29.Dobos, G.J., J. Norgauer, M. Eberle, P.J. Schollmeyer, and A.J. Traynor-Kaplan. 1992. C5a reduces formyl peptide-induced actin polymerization and phosphatidylinositol 3,4,5-trisphosphate formation, but not phosphatidylinositol 4,5-bisphophate hydrolysis and superoxide production in human neutrophils. Immunol. 149:609–614. [PubMed] [Google Scholar]
- 30.Pear, W.S., J.P. Miller, L. Xu, J.C. Pui, B. Soffer, R.C. Quackenbush, A.A. Pendergast, R. Brousan, J.C. Aster, M.L. Scott, et al. 1998. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving p210 bcr/abl-transduced bone marrow. Blood. 92:3780–3792. [PubMed] [Google Scholar]
- 31.Pleiman, C.M., M.R. Clark, L.K. Timson, S. Winitz, K.M. Coggeshall, G.L. Johnson, A.S. Shaw, and J.C. Cambier. 1993. Mapping of sites on the Src family protein tyrosine kinases p55blk, p59fyn, and p56Lyn which interact with the effector molecules phospholipase Cγ2, microtubule-associated protein kinase, GTPase-activating protein and phosphatidylinositol 3 kinase. Mol. Cell. Biol. 9:5877–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ptasznik, A., E.R. Prossnitz, D. Yoshikawa, A. Smrca, A. Traynor-Kaplan, and G.M. Bokoch. 1996. A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J. Biol. Chem. 271:25204–25207. [DOI] [PubMed] [Google Scholar]
- 33.Gupta, S.K., K. Pillarisetti, and P.G. Lysko. 1999. Modulation of CXCR4 expression and SDF-1 functional activity during differentiation of human monocytes and macrophages. J. Leuk. Biol. 66:135–143. [DOI] [PubMed] [Google Scholar]
- 34.Chernock, R.D., R.P. Cherla, and R.K. Ganju. 2001. SHP2 and cbl participate in chemokine receptor CXCR4-mediated signaling pathways. Blood. 97:608–615. [DOI] [PubMed] [Google Scholar]
- 35.Yamanashi, Y., S. Mori, M. Yoshida, T. Kishimoto, K. Onowe, T. Yomamoto, and K. Toyoshima. 1989. Selective expression of a protein tyrosine kinase, p56Lyn, in hematopoietic cells and association with production of human T-cell lymphotropic virus type I. Proc. Natl. Acad. Sci. 86:6538–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Laughlin-Brunner, B., N. Radosevic, M. Taylor, L. Shivakrupa, C. DeBerry, D.D. Metcalfe, M. Zhou, C. Lowell, and D. Linnekin. 2001. Lyn is required for normal stem cell factor-induced proliferation and chemotaxis of primary hematopoietic cells. Blood. 98:343–350. [DOI] [PubMed] [Google Scholar]
- 37.Dutt, P., J.-F. Wang, and J.E. Groopman. 1998. Stromal cell-derived factor-1 and stem cell factor/kit ligand share signaling pathways in hemopoietic progenitors: a potential mechanism for cooperative induction of chemotaxis. J. Immunol. 161:3652–3658. [PubMed] [Google Scholar]
- 38.Pleiman, C.M., W.M. Hertz, and J.C. Cambier. 1994. Activation of phophatidylinositol 3-kinase by Src family kinase SH3 binding to the 85 subunit. Science. 263:1609–1612. [DOI] [PubMed] [Google Scholar]
- 39.Herrera-Velit, P. and N.E. Reiner. 1996. Bacterial lipopolysaccharide induces the association and coordinate activation of p53/56 Lyn and phosphatidylinositol 3-kinase in human monocytes. J. Immunol. 156:1157–1165. [PubMed] [Google Scholar]
- 40.Pigazzi, A., S. Heydrick, F. Folli, S. Benoit, A. Michelson, and J. Loscalzo. 1999. Nitric oxide inhibits thrombin receptor-activating peptide-induced phosphoinositide 3-kinase activity in human platelets. J. Biol. Chem. 274:14368–14375. [DOI] [PubMed] [Google Scholar]
- 41.Dauhauser-Riedl, S., M. Warmuth, B.J. Druker, B. Emmerich, and M. Hallek. 1996. Activation of Src kinases p53/56 lyn and p59 hck by p210 Bcr/Abl in myeloid cells. Cancer Res. 56:3589–3596. [PubMed] [Google Scholar]
- 42.Michael, W., N. Deiminger, J.M. Goldman, N. Lydon, and J.V. Melo. 1997. The tyrosine kinase inhibitor CGP57148 selectively inhibits the growth of Bcr/Abl-positive cells. Blood. 90:3691–3698. [PubMed] [Google Scholar]
- 43.Hanke, J.H., J.P. Gardner, R.L. Dow, P.S. Changelian, W.H. Brissette, E.J. Weringer, B.A. Pollok, and P.A. Connelley. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 271:695–701. [DOI] [PubMed] [Google Scholar]
- 44.Daub, H., F.U. Weiss, C. Wallasch, and A. Ullrich. 1996. Role of transactivation of the EGF receptor in signalling by G protein-coupled receptors. Nature. 379:557–560. [DOI] [PubMed] [Google Scholar]
- 45.Lynch, O.T., M.A. Giembycz, I. Daniels, P.J. Barnes, and M.A. Lindsay. 2000. Pleiotropic role of Lyn kinase in leukotriene B4-induced eosinophil activation. Blood. 95:3541–3547. [PubMed] [Google Scholar]
- 46.Hallek, M., S. Danhauser-Riedl, R. Herbst, M. Warmuth, A. Winkler, H.J. Kolb, B. Druker, J.D. Griffin, B. Emmerich, and A. Ullrich. 1996. Interaction of the receptor tyrosine kinase p145c-kit with the p210bcr/abl kinase in myeloid cells. Br. J. Haematology. 94:5–16. [DOI] [PubMed] [Google Scholar]
- 47.Skorski, T., P. Kanakaraj, M. Nieborowska-Skorska, M.Z. Ratajczak, S.-C. Wen, G. Zon, A.M. Gewirtz, B. Perussia, and B. Calabretta. 1995. Phosphatidylinositol 3-kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 86:726–736. [PubMed] [Google Scholar]
- 48.Linnekin, D., C.S. DeBerry, and S. Mou. 1997. Lyn associates with the juxtamembrane region of c-kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 272:27450–27455. [DOI] [PubMed] [Google Scholar]
- 49.Daigle, I., S. Yousefi, M. Colonna, D.R. Green, and H.U. Simon. 2002. Death receptors binds SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat. Med. 8:61–67. [DOI] [PubMed] [Google Scholar]
- 50.Bishop, J.M. 1983. Cellular oncogenes and retroviruses. Annu. Rev. Biochem. 52:301–354. [DOI] [PubMed] [Google Scholar]
- 51.Hunter, T., and J.A. Cooper. 1985. Protein tyrosine kinases. Annu. Rev. Biochem. 54:897–930. [DOI] [PubMed] [Google Scholar]
- 52.Sotsios, Y., and S.G. Ward. 2000. Phosphoinositide 3-kinase: a key biochemical signal for cell migration in response to chemokines. Immunol. Rev. 177:217–235. [DOI] [PubMed] [Google Scholar]
- 53.Ptasznik, A., G.M. Beattie, M.I. Mally, V. Cirulli, A. Lopez, and A. Hayek. 1997. Phosphatidylinositol 3-kinase is a negative regulator of cellular differentiation. J. Cell Biol. 137:1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cantley, L.C., K.R. Auger, C. Carpenter, B. Duckworth, A. Graziani, R. Kapeller, and S. Soltoff. 1991. Oncogene and signal transduction. Cell. 64:281–302. [DOI] [PubMed] [Google Scholar]
- 55.Jimenez, C., D.R. Jones, P. Rodriques-Viciana, A. Garcia, E. Leonardo, S. Wennstrom, C. von Kobbe, J.L. Toran, L. R-Barlado, V. Calso, S.G. Copin, et al. 1998. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphatidyinositol 3-kinase. EMBO J. 17:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang, H.W., M. Aoki, D. Fruman, K.R. Auger, A. Bellacosa, P.N. Tsichlis, L.C. Cantley, T.M. Roberts, and P.K. Vogt. 1997. Transformation of cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 276:1648–1650. [DOI] [PubMed] [Google Scholar]
- 57.Varticovski, L., G.Q. Daley, P. Jackson, D. Baltimore, and L.C. Cantley. 1999. Activation of phosphatidylinositol 3-kinase in cells expressing abl oncogene variants. Mol. Cell. Biol. 11:1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas, S.M., and J.S. Brugge. 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13:513–609. [DOI] [PubMed] [Google Scholar]
- 59.Stein, P.L., H. Vogel, and P. Soriano. 1994. Combined deficiencies of Src, Fyn and Yes tyrosine kinases in mutant mice. Genes and Developments. 8:1999–2007. [DOI] [PubMed] [Google Scholar]
- 60.Donato, N.J., J.Y. Wu, G.E. Gallick, R.B. Arlinghaus, and M. Talpaz. 2001. A dominant role for Lyn kinase in K562 cells selected for resistance to STI571. Blood. 98:839a. [DOI] [PubMed]