

Abstract
In spontaneous inflammatory arthritis of K/BxN T cell receptor transgenic mice, the effector phase of the disease is provoked by binding of immunoglobulins (Igs) to joint surfaces. Inflammatory cytokines are known to be involved in human inflammatory arthritis, in particular rheumatoid arthritis, although, overall, the pathogenetic mechanisms of the human affliction remain unclear. To explore the analogy between the K/BxN model and human patients, we assessed the role and relative importance of inflammatory cytokines in K/BxN joint inflammation by transferring arthritogenic serum into a panel of genetically deficient recipients. Interleukin (IL)-1 proved absolutely necessary. Tumor necrosis factor (TNF)–α was also required, although seemingly less critically than IL-1, because a proportion of TNF-α–deficient mice developed robust disease. There was no evidence for an important role for IL-6. Bone destruction and reconstruction were also examined. We found that all mice with strong inflammation exhibited the bone erosion and reconstruction phenomena typical of K/BxN arthritis, with no evidence of any particular requirement for TNFα for bone destruction. The variability in the requirement for TNF-α, reminiscent of that observed in treated rheumatoid arthritis patients, did not appear genetically programmed but related instead to subtle environmental changes.
Keywords: transgenic, cytokine, knockout, inflammatory, TNF
Introduction
Inflammatory arthritides, in particular rheumatoid arthritis, have been the focus of intense investigation, but their etiology and pathogenesis remain controversial. There is no consensus on what initiates rheumatoid arthritis (RA)*; i.e., whether it is primarily an autoimmune response, an inflammatory response to some persisting microbial invasion, or a combination of the two. There is also dispute over the leukocyte populations that are involved in the initiation of joint inflammation. The paradigm currently dominating the field portrays antigen-specific T cells in the joint as inciting the inflammatory cascade by triggering macrophages and synoviocytes (1, 2), but this scenario has been questioned for a lack of direct experimental demonstration of certain of its key points, and because of some discordant observations, such as the paucity of T cell–derived cyto-kines in inflamed joints (3). In contrast, a role for inflammatory cytokines like TNF-α and IL-1 is well established (4), most demonstratively by the impressive effect of therapeutic protocols that block TNF–TNF-R interactions (1). There has also been debate on the relative importance of the IL-1 and TNF-α pathways (4). It has also been noted that, even in the best of trial outcomes, arthritis is not fully reversed and roughly one third of RA patients are refractory to TNF–TNFR-blocking drugs.
The K/BxN TCR transgenic mouse is a recently developed model of inflammatory arthritis (5–9). All K/BxN animals spontaneously show an autoimmune disease with most (although not all) of the clinical, histological, and immunological features of RA in humans. The disorder is critically dependent on both T and B cells. Although the pathologic manifestations are joint-specific, the process is initiated, and then perpetuated, by dual T/B cell autoreactivity to a ubiquitously expressed antigen, glucose-6-phosphate isomerase (GPI). Transfer of anti-GPI Igs from arthritic K/BxN mice into healthy animals provokes arthritis within days, even when the recipients are devoid of lymphocytes. GPI–anti-GPI immune complexes (Ics) are the link between the systemic T and B lymphocyte autoreactivity and the ensuing joint-specific inflammation and destruction; the joint specificity is perhaps a reflection of the presence of GPI on the articular cavity surface (10). Initiation of the inflammatory effector phase requires both the complement network and Fc receptors (11). The relevance of the K/BxN model to human RA is supported by a recent report that serum from almost two thirds of RA patients contained anti-GPI Abs, absent from serum of normal individuals or of patients with Lyme arthritis or Sjogren's syndrome (12), although more recent data show less obvious a correlation (unpublished data). The observation of GPI and GPI–anti-GPI complexes on cartilage surfaces of human joints is also of interest (10).
Our early studies on K/BxN mice revealed augmented local synthesis of inflammatory cytokines, such as IL-6 and TNF-α, in arthritic joints (5). However, the functional relevance of this observation was not tested, other than a report that failed to demonstrate a required role for TNF-α (13). The role of inflammatory cytokines is an important element to consider in attempting to relate the mechanistically defined mouse model to human RA patients. For example, does Ig-induced arthritis correspond to the TNFα-dependent form of the human disease or rather to the variants resistant to TNFα/TNFR blockade?
Here, we apply the K/BxN serum transfer system to a panel of mice deficient in one or more inflammatory cytokines or their receptors. A critical role for IL-1 is established, along with a strong, but not absolute, requirement for TNFα. Interestingly, we find that the requirement for TNFα varies markedly from individual to individual, as it does in humans.
Materials and Methods
Mice.
The knockout mice used for serum transfer were obtained from the Jackson Laboratory, brought to our animal facility at the Harvard Medical School animals facility at 4–5 wk of age, and used 1–3 wk later (in rare exceptions, the mice were bred in our colony). These mice include the following: IL-6−/− (14) on a B6 background; IL-1r1−/− on both B6 (15) and (B6 × 129)F2 (16) backgrounds; TNFα−/− (17) on a (B6 × 129)F2 background; Lta−/− on the (B6 × 129)F2 background (18); TNFR1−/− (19) and TNFR2−/− (20) on a B6 background; and TNFR1/2−/− (21) on a (B6 × 129)F2 background.
RNA Analysis.
RNA was prepared from ankle tissue by a modification of the LiCl/urea technique (22), designed to avoid contamination of the joint RNA with bone marrow–derived material by leaving the bone intact. After dissection of ankles (sectioned at the long bones of the lower leg and in the metatarsal area), the tissue was freed of skin and superficial tendons. The joint was immersed in 1 ml RNA solubilization solution (6 M urea, 2% SDS). Articular cavities were opened with a scalpel and were exposed to the medium to release the cellular contents. After 10-min incubation, the fragment was removed, and an equal volume of concentrated LiCl solution (6 M LiCl, 6 M urea, and 10 mM sodium acetate, pH 5) was added to precipitate the RNA. cDNA was synthesized from these RNAs by MuLV reverse transcriptase (GIBCO BRL).
Cyclophilin was used as an endogenous control using a probe concentration of 200 and 400 nM for each primer in each reaction. The probe and primers sequences used are as follows: probe, 5′ VIC CTTGGGCCGCGTCTCCTT TAMRA 3′; forward primer, 5′ CAGACGCCACTGTCGCTTT 3′; and reverse primer, 5′ TGTCTTTGGAACTTTGTCTGCAA 3′. For the quantification of TNFα and IL6, the TaqMan predeveloped assay reagents were used (PE, Applied Biosytems). For IL1 β, the probe and primer concentrations per reaction were the same as those used for cyclophilin. The probe and primers sequences used are as follows: probe, 5′ FAM TGCAGCTGGAGAGTGTGGATCCCA TAMRA 3′; forward primer, 5′ TGAAAGACGGCACACCCA 3′; and reverse primer, 5′ AAACCGCTTTTCCATCTTCTTCT 3′. To determine relative expression values, ΔCT (CT cytokine − CT cyclophilin) was used to derive an expression index (2ΔCT), which was then divided by the same index obtained with a reference sample of total spleen RNA.
Serum Transfer Protocol and Arthritis Scoring.
K/BxN serum pools were prepared from arthritic mice 60 d old. Arthritis was induced by intraperitoneal injection of 150–200 μl serum at days 0 and 2. A clinical index was evaluated over time (1 point for each affected paw; 0.5 points for a paw with only mild swelling/redness or only a few digits affected). Ankle thickness was measured by a caliper (6), with ankle thickening being defined as the difference in ankle thickness from the day 0 measure.
Histology.
Hind limbs were collected and the knee and ankle joints were separated mid-tibia. Specimens were dissected to remove skin and outer muscle, and subsequently fixed in 4% paraformaldehyde for a minimum of 12 h and demineralized for ∼2 wk in 14% EDTA, followed by paraffin embedding (model Citadel 1000; Shandon). For each specimen, twenty 4-μm sagittal serial sections were cut, and every fifth section was stained with hematoxylin and eosin (Sigma-Aldrich) for evaluation of inflammation, bone erosion, and cartilage destruction. An adjacent section was stained with toluidine blue (Sigma-Aldrich) for specific evaluation of proteoglycan. Histopathological scoring was performed as described previously (5, 23).
Results and Discussion
Kinetics of Inflammatory Cytokine Production.
Transfer of K/BxN serum into normal recipients induces rapid and synchronous development of arthritis, the first signs of joint inflammation appearing within 24 h in fully susceptible strains (9). To begin exploring the induction of various inflammatory cytokines in this model and their temporal relationships, we measured the expression of their mRNAs by quantitative real-time PCR. C57Bl/6 mice were injected with a single dose of K/BxN serum, RNA was prepared at different times thereafter from ankle tissue (pooled from two individuals), and real-time PCR was performed to quantitate spliced TNF-α, IL-1β, and IL-6 mRNA transcripts. A representative experiment is shown in Fig. 1.
Figure 1.
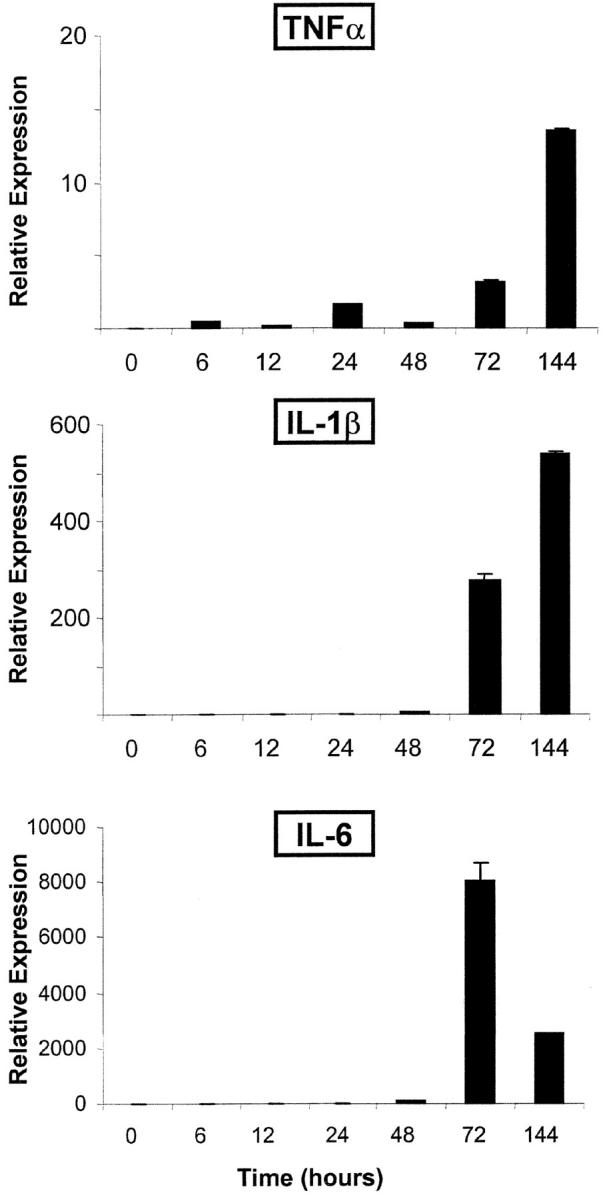
Kinetic of inflammatory cytokine expression. Arthritis was induced by injection of K/BxN serum into naive C57Bl/6 mice, and RNA was prepared from ankle joint cavities at various points thereafter. mRNA encoding inflammatory cytokines were quantitated by real-time PCR using cyclophilin mRNA as an internal standard. The results are presented as relative expression of the individual cytokine mRNAs standardized against a reference sample of total spleen RNA from a normal mouse. This is a representative experiment (of three), with two mice pooled for each point.
The first signs of induction were detectable a few hours after serum injection, with a modest but detectable rise from the baseline for all mRNAs at 6 h. TNFα message increased more substantially from 24 h onwards. IL-1 transcripts followed roughly the same pattern, but with a sharper induction at 48 h and far more extensive induction, reaching 13,000-fold at maximum. IL-6 showed a delay, with a maximum by 72 h followed by a decline at 144 h that was reproducibly observed in several experiments. These results are consistent with an early appearance of inflammatory cytokine transcripts (from cell recruitment, or from true induction of gene expression, or both), and a secondary, far more extensive, induction. The induction of IL-1 appears significantly more extensive than that of TNF-α.
No Role for IL-6.
The induction of arthritis by K/BxN serum transfer does not require any contribution from T or B cells (6). Thus, one can readily evaluate the role of inflammatory cytokines purely on the effector phase of the disease, unencumbered by their influences on the immunological induction phase. Such complications may have clouded results from collagen-induced arthritis (CIA) and antigen-induced arthritis models, where the known pleiotropic effects of such cytokines on the structure or responsiveness of the immune system complicate data interpretation. The K/BxN serum transfer system is applicable to a number of mouse strains (9), allowing one to investigate the effects of diverse natural and engineered mutations. This strategy was applied here, focusing on the contributions of IL-1, IL-6, and members of the TNF family, by transferring K/BxN serum into homozygous knockout mice lacking particular cytokines or cytokine receptors. Mice of matched genetic composition, bred in the same colony, were used as controls. In most cases, we preferred not to rely on injected cytokine inhibitors, such as anticytokine antibodies or soluble receptor molecules because negative results with such reagents can be difficult to interpret (sufficient dose or stability of the compound? completeness of the blockade?). This is particularly an issue in a context as aggressive as that of K/BxN arthritis.
We first investigated the importance of IL-6, a pleiotrophic cytokine expressed by a variety of cell types during inflammatory processes (24). IL-6 has complex pro- and antiinflammatory influences, with both local and systemic effects. For example, it promotes immune responses and plasma cell and macrophage differentiation (25), but also induces acute phase proteins, IL-1 receptor antagonist (26), and metalloproteinase inhibitors (27). Its role is variable in different inflammatory models (28). There have been conflicting reports of the requirement for IL-6 in animal models of arthritis: some investigators describe reduced disease in IL-6–deficient mice or after antibody blockade of its receptor (29, 30), whereas others report no such effect (31).
IL-6–deficient mice on the C57Bl/6 background (14) were transferred with serum from arthritic K/BxN mice, and arthritis development was monitored as described previously (6). The representative experiment in Fig. 2 A demonstrated a very similar arthritis course in IL-6–deficient and control mice. The initial onset of symptoms was the same, all distal joints were affected, and with a comparable degree of inflammation (measured as ankle thickness). These observations were confirmed by results from three individual experiments tabulated in Fig. 2 B. Histological examination of the ankle joints revealed the image of synovitis and joint infiltration typical of K/BxN mice (synovial thickening and infiltration, presence of neutrophils in the articular cavity, pannus formation, and cartilage destruction; Fig. 2 C; unpublished data). Furthermore, cartilage damage and proteoglycan loss was evident on toluidine blue–stained ankle sections from serum-injected mice at comparable levels for IL-6–deficient and control mice (unpublished data).
Figure 2.

No requirement for IL-6 in arthritis induced by K/BxN serum transfer. IL6-deficient and control mice (matched for gender/age and genetic background) were injected with 150 μl serum from arthritic K/BxN animals on days 0 and 2. Arthritis was evaluated by measuring clinical index and ankle thickening (Materials and Methods). (A) Data from a representative experiment, with each curve representing an individual mouse. (B) Tabulation of the results for 10 knockout mice and age/gender matched controls; MaxAT, maximum increase in ankle thickness in millimeters. MaxCI, maximum clinical index, sum of scores on each limb (for each limb: 0, no disease; 0.5, mild swelling of paw or of just a few digits; 1, clear joint inflammation in ankle or wrists); maximum score = 4. The histological score sums scores from knee, ankle, and tarsal joints (1, minimum synovial hyperplasia; 2, limited inflammatory infiltration; 3, massive infiltration; 4, massive infiltration with cartilage and bone destruction); maximum score = 12.
These data are in agreement with those of van den Berg and colleagues, who found little role for IL-6 in joint inflammation in CIA or zymosan-induced arthritis (31). They contrast with other reports showing an effect of IL-6 blockade in the CIA model (29, 30). The explanation for these discrepancies may lie in the positive impact of IL-6 on the immunological initiation phase of the CIA model: less intense immune responses were made to the collagen-II antigen in the absence of IL-6 function (29, 30). Together, then, the data are consistent with the notion that IL-6 does not play a major role in the inflammatory effector phase of arthritis.
An Essential Role for IL-1.
Although attempts at blocking the IL-1 pathway in RA patients in therapeutic trials have not met with as much success as those interfering with the activity of TNFα, there exists a substantial body of evidence implicating this inflammatory cytokine in several classic murine arthritis models, whether autoimmune in nature or induced by local microbial particles (32–36); similarly, high levels of IL-1 transcripts have been detected in RA synovium (4, 37).
We tested the susceptibility to serum-transferred arthritis of the IL-1R knockout strain (15), in which neither IL-1α nor IL-1β-mediated signals are possible. After K/BxN serum transfer, essentially no clinical signs of disease were observed in the IL-1R–deficient mice, except for a limited swelling of the digits and a slight flutter in the ankle-thickness curve (Fig. 3). To guard against possible influences of genetic background variability, we repeated the initial experiments performed in (B6 × 129)F2 mice in IL-IR–deficient mice thoroughly backcrossed onto the B6 background (our standard fully susceptible background; reference 11). Matched wild-type controls responded as usual. Histologically, no signs of joint inflammation were apparent in the four mice analyzed. Naturally, cartilage destruction and bone erosion were absent.
Figure 3.

Essential role of IL-1 ILIR–deficient and control mice (matched for gender/age and genetic background) were injected with 150 μl serum from arthritic K/BxN animals on days 0 and 2. Arthritis was evaluated by measuring clinical index and ankle thickening as in Fig. 2. (A) Data from a representative experiment in B6 recipients, with each curve representing an individual mouse. (B) Tabulation of the results for eight knockout mice and age/gender matched controls on either the standard (B6 × 129)F2 background or on an inbred B6 background. Scoring as described for Fig. 2; the star denotes a transient inflammation in the digits of one mouse.
These clear-cut results indicate that, in this serum-transfer model mediated by arthritogenic Igs, IL-1 plays a central role, critically required for disease progression. We have not been able to reproduce this effect by treatment with blocking anti–IL-1R mAb (unpublished data), likely because of the known difficulty to achieve complete blockade of IL-1 action with biomolecule inhibitors (for review see reference 4) The central importance of IL-1 in the K/BxN model is reminiscent of its requirement in CIA and other murine arthritis models (32, 33, 35). It is also consistent with the finding that intraarticular expression of IL-1β, alone, is sufficient to induce full-blown arthritis (38).
TNF Family Influences.
Members of the TNF family have received a great deal of attention in the context of inflammatory arthritis. This has ranged from the initial demonstration of TNF-α expression in arthritic synovium, to establishing the efficacy of TNF-α-/TNFR-blocking agents in animal models, to the successes of such reagents in therapeutic intervention in human RA (1, 4, 39–42). Aberrant expression of TNF-α is also sufficient to induce arthritis in transgenic animals (43). These results evoked models of arthritogenesis in which TNF-α plays a central and indispensable role (for review see 1). We tested the efficacy of K/BxN serum transfer in animals carrying knockout mutations of the genes encoding TNF-α or its close homologue, lymphotoxin (LT)-α (17–21). TNF-α and LT-α mediate their pleiotropic effects by binding to one of two known receptors, TNFR1 (p55) and TNFR2 (p75).
We also investigated the effect of knockout mutations of the genes encoding either or both of these molecules. The data, summarized in Table I, allow several conclusions. First, and most simply, LT-α seemed not to be required for the development of K/BxN serum-transferred arthritis. LT-α–deficient mice responded normally on all counts, in the kinetics and intensity of inflammation and in the appearance of histological lesions (proliferative synovitis, infiltration of the joint cavity by neutrophils, and formative of a destructive pannus).
Table I.
Arthritis Incidence in Mice Deficient in TNF and TNFR Families
| Strain | Arthritis | Days of onset | Max CI | |
|---|---|---|---|---|
| TNFR1 | +/+ | 8/8 | 4, 2, 2, 1, 1, 2, 2 | 3, 4, 4, 4, 4, 4, 4, 3 |
| (B6) | −/− | 8/8 | 4, 2, 2, 3, 2, 3, 5, 2 | 4, 3, 4, 3, 4, 3.5, 2.5, 2.5 |
| TNFR2 | +/+ | 8/8 | 4, 2, 2, 1, 1, 1, 2, 2 | 3, 4, 4, 4, 4, 4, 4, 3 |
| (B6) | −/− | 8/8 | 1, 4, 1, 1, 1, 1, 1, 2 | 4, 4, 4, 4, 4, 4, 4, 3.5 |
| TNFR1/2 | +/+ | 6/6 | 2, 2, 1, 4, 2, 2 | 4, 4, 4, 1.5, 3.5, 3 |
| (B6x129F2) | −/− | 6/6 | 3, 7, 2, 2, 2, 2 | 2.5, 0.5,4, 4, 4, 4 |
| Ltα | +/+ | 8/8 | 2, 2, 2, 2, 2, 1, 4, 2 | 4, 4, 4, 4, 4, 2, 4, 3 |
| (B6x129F2) | −/− | 8/8 | 2, 2, 5, 3, 2, 2, 2, 2 | 2, 2, 3, 3, 4, 4, 4, 4 |
| TNFα | +/+ | 9/9 | 2, 2, 2, 3, 5, 4, 2, 3, 3 | 2.5, 4, 4, 4, 2, 4, 4, 2.5, 3.5 |
| (B6x129F2) | −/− | 4/14 | -, -, -, -, 8, 19, -, -, - | 0, 0, 0, 0, 1.5, 1, 0, 0, 0 |
| 2, 5, -, -, -, 1 | 4, 1.5, 0.5, 0.5, 0.5 |
Second, the absence of TNF-α had a marked impact on arthritogenesis. Many TNF-α–deficient mice developed no disease whatsoever upon transfer of K/BxN serum, either clinically or histologically (Table I). However, a number of such animals did develop joint inflammation, overall in 9/23 examined over the course of this study. This finding is illustrated for representative cohorts in Fig. 4. The presence of responder TNFα−/− mice was not restricted to one or two experimental groups, but was observed in a number of independent experiments. In contrast, a certain degree of clustering was observed, some experimental groups showing a high incidence of arthritis development (see below). When disease did develop, the time of onset was quite variable, usually delayed by several days relative to wild-type controls, and the degree of inflammation always remained below the maximum attainable. Histological analysis also revealed significant signs of inflammation in those mice with clinically detectable arthritis.
Figure 4.

Variability of arthritis in TNFα-deficient mice. TNFα-deficient (left) and control mice (right; matched for gender/age and genetic background) were injected with 150 μl serum from arthritic K/BxN animals on days 0 and 2. Arthritis was evaluated by measuring ankle thickening as in Fig. 2. The data are pooled from six different experiments. All mice originated from the Jackson Laboratory.
Third, joint inflammation developed normally in both the TNFR1- and TNFR2-deficient mice, as well as in TNFR1/TNFR2 double–deficient animals (Table I; the genotypes of the mice were reconfirmed at the end of the experiment). Clinical and histological parameters were essentially indistinguishable from normal controls. This observation was quite unexpected, as TNFR1 and TNFR2 are the only known receptors for TNF-α, with no reported indication of another possible receptor in spite of the broad attention that TNF-α has received (44). As both the cytokine and cytokine receptor mutations were on a susceptible (B6 × 129) F2 background, one would have expected that they have the same phenotype in both deficient strains.
These conflicting results prompted us to question the effect of the TNF-α mutation: was the poor responsiveness in TNF-α–deficient mice truly due to the absence of the cytokine, or instead to some independent factor (a linked gene effect, quite plausible given the genomic localization of the TNFα locus; an independent mutation; protective genes segregating by chance, etc.)? If the former were true, it should be possible to complement the deficiency by TNF-α replacement, e.g., by triggering TNFR1 with an agonistic mAb. To test this prediction, we injected cohorts of TNFα-deficient mice with K/BxN serum, selected those mice that remained free of arthritis after 7 d, and administered the agonistic anti-TNFR1 Ab 55R-593 (45). As shown in Fig. 5, the Ab had a marked effect, provoking arthritis in all the TNF-α–deficient mice that had previously received K/BxN serum. No arthritis was observed when 55R-593 was injected without serum pretreatment (unpublished data). Several control mAbs were used in parallel to rule out trivial explanations for this observation: an isotype-matched control Ab, anti-TNFR1 mAbs with blocking or antagonist activity (55R-170, 55R-286). None of these reagents induced arthritis (Fig. 5 B), at least not beyond the minority of TNF-α–deficient mice one might expect to eventually progress spontaneously to arthritis on the basis of the results presented in Fig. 4. Thus, results from these experiments confirmed that TNF-α is indeed the element missing in TNF-α–deficient mice that is required for robust development of arthritis.
Figure 5.

Triggering the TNF receptor complements TNFα deficiency. TNFα-deficient mice were injected with 150 μl serum from arthritic K/BxN animals on days 0 and 2. Animals not presenting any signs of disease by day 7 were injected at days 7, 11, and 15 with anti-TNFR1 mAb 55R-293, which has significant agonist activity (A) or with control mAbs (B). These controls included anti-TNFR1 reagents devoid of agonist activity or an irrelevant mAb. (C) Anti-TNFR mAbs were injected without K/BxN serum. Arthritis was evaluated by measuring ankle thickening as above. The data are pooled from four different experiments. All mice originated from the Jackson Laboratory.
Further experiments were performed to address the cause of the variable effect of the TNF-α deficiency. It could be explained by genetic, epigenetic, or environmental variation controlling the activity of TNF-α–independent pathways; stochastic threshold effects could also be involved, arthritogenesis requiring a certain degree of local inflammatory insult, only seldom reached in the absence of TNF-α. As the knockout mutation was carried on a mixed (129xB6) F2 background, we reasoned that modifier alleles at other loci, able to complement the TNF deficiency, might segregate randomly in the F2 knockout mice. To test this hypothesis, several crosses were set up between combinations of resistant or susceptible TNF-α–deficient mice. Should alleles at independent loci be segregating, there should be heritable transmission of these traits to the progeny. As shown in Fig. 6 A, this was not the case. A cross between two resistant mice yielded a dominant proportion of responder mice; the transmission of a recessive susceptibility allele in this family would be very unlikely to yield such a pattern (P < 0.001). Thus, the variability does not stem from Mendelian genetic elements. Epigenetic variation could perhaps account for these results. However, we observed a clear correlation between the origin and life history of the mice and their responses to K/BxN serum (Fig. 6 B). Those mice bred at the Jackson Laboratory and shipped to Boston 7–15 d before challenge showed mainly a resistant phenotype, whereas those bred in Boston and tested there were mainly susceptible (P < 0.003). In both cases, the barrier facilities have SPF status, free of major mouse pathogens, but minor bacterial flora varies. Thus, the segregation of responses is more consistent with an environmental explanation than with an epigenetic one.
Figure 6.

Environmental, not genetic, influences on TNF-independent arthritis. (A) TNFα-deficient mice from the Jackson Laboratory were tested by transfer of K/BxN serum, and animals of different phenotypes were crossed (white symbols, resistant mice; black symbols, susceptible mice, where resistance and susceptibility are defined as the presence of clear arthritis [grade > 1]) in the first 10 d after serum transfer. Their progeny was similarly tested when 4–5 wk old. (B) A compilation of results of challenge of TNFα-deficient mice with K/BxN serum, either from mice purchased from the Jackson Laboratory or bred in our Boston colony; χ2, P = 0.003.
Together, these experiments point to a distinct involvement of TNF-α in Ab-induced arthritis, but one that is not absolutely essential. This conclusion differs from that reached by Kyburz et al. (13), who found no effect of anti-TNF-α therapy in arthritis development in straight K/BxN transgenic mice. We have also made similar observations, injecting several different anti-TNF-α reagents into young K/BxN mice (unpublished data). However, we interpret these negative results with caution because of the very aggressive nature of the disease that develops in the transgenic mice and uncertainties concerning the efficiency of Ab-mediated blockade. On the other hand, the present results do concur with reports of robust development of CIA in TNF-α–deficient mice (46). Although it is conceivable that the cytokine network adapts somewhat in TNF-α–deficient animals, other compensatory cytokines being more active than usual, the results do show that TNF-α is not the indispensable cytokine for the development of Ab-induced arthritis.
The significant mouse-to-mouse variability we observed with TNF-α–deficient animals is, in a sense, reminiscent of the variability in the response of RA patients to TNF-α/TNFR blockade (1). The results of Fig. 6 make it perhaps more plausible that environmental effects are at play, the degree of TNF-α involvement being dependent on the general inflammatory state of the individual. It should be worthwhile trying to pinpoint what these influences might be, in both mice and humans, and the present system does provide a handle.
There are several potential interpretations for the strong arthritis that develops in TNFR1/2-deficient mice. The most straightforward is that other receptors can compensate and mediate TNF-α signals. Although the existence of such a receptor has not been reported to date, the breadth of the TNFR family makes it quite possible that other receptors will be found to bind TNF-α. Whether these are indeed the primary receptors mediating arthritis, or whether they only come into play when the primary TNFR1/2 receptors are absent, will need to be explored. Alternatively, one might propose that TNF-α–independent arthritis pathways are particularly active when TNFR1/2 are missing, perhaps by commandeering downstream signal transduction adaptors. For example, the absence of TNFR1 might free TRADD, FADD, or TRAF molecules for more efficient interaction with other receptors.
Bone Destruction and Formation.
There is some debate about the role of inflammatory cytokines in promoting focal bone erosion in the course of arthritic diseases. Osteoclasts are essential to the process, and essentially no focal destruction of the bone occurs in their absence. Resistance to bone erosion was previously demonstrated in mice deficient in the TNF family member receptor activator of NF-KB ligand (RANKL) that had received K/BxN serum, as in the CIA model after blockade of RANKL by osteoprotegerin treatment (23, 47). This finding is consistent with the fact that RANK/RANKL axis is required for the generation of osteoclasts and also plays a role in their activation (for review see reference 48). In contrast, it is also possible that other inflammatory cytokines play a role. IL-1α can activate osteoclasts, and promotes bone resorption in vitro (49, 50). TNF promotes osteoclast differentiation in the presence of RANKL (51, 52), and there are indications that TNF/TNFR blockade can retard bone destruction in RA patients, even when the effect on the inflammatory component is limited (53). Thus, we asked whether bone destruction could be seen in the absence of these cytokines. As described previously, obvious instances of focal bone destruction were seen in normal mice injected with K/BxN serum; similar images were also observed in LTα-deficient mice (Fig. 7, A and B). For TNF-α, we focused in particular on those mice that showed significant joint inflammation. In these instances, clear evidence of focal bone destruction was also observed (Fig. 7 C). Although impossible to truly quantitate, given the variability of inflammation in the TNF-deficient animals, the extent of the erosive lesions in the absence of TNF-α was largely on par with the extent of inflammation.
Figure 7.
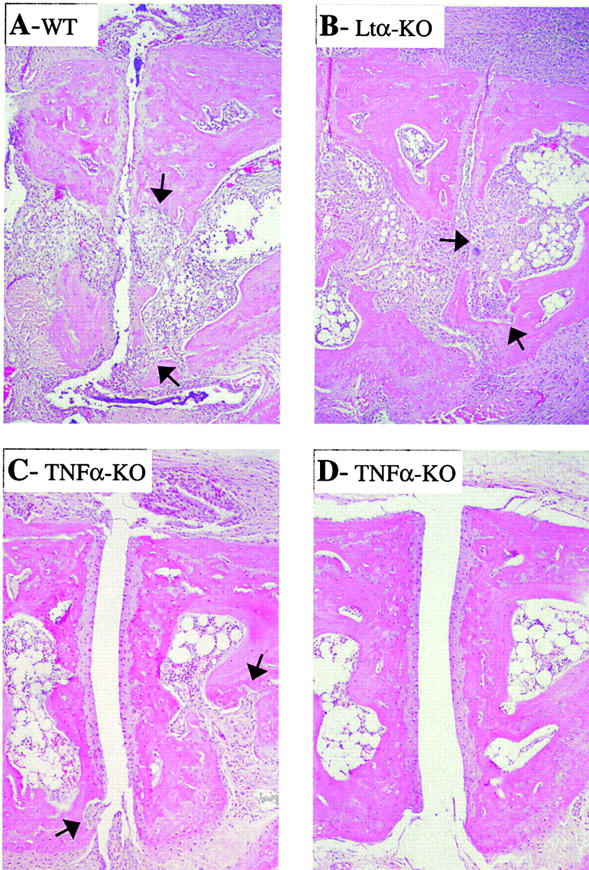
Bone destruction in control and KO mice. Bone erosion was assessed in hematoxylin and eosin–stained ankle sections from TNFα- and LTα-deficient mice and control mice after transfer of K/BxN serum. (A) Control mouse, full inflammation and areas of focal bone destruction (arrows). (B) Arthritic LTα-deficient mouse with inflammation and focal bone erosion. (C) TNFα-deficient mouse with clinical manifestations, showing inflammation and focal bone erosion. (D) TNFα-deficient mouse with no clinically detectable symptoms, showing minimal inflammation (matched with A).
We could not draw any conclusion on the role of IL-1 in bone destruction, as the upstream inflammatory phase did not develop in its absence. However, our results are not consistent with the view that TNF-α plays an obligate role in promoting bone destruction: synovitis and joint inflammation could still lead to extensive destruction in its absence.
Synthesis: Intersection of IL-1 and TNF Pathways.
There has been quite some debate as to the relative roles and importance of IL-1 and TNF-α in arthritogenesis. In animal models where the function of these cytokines has been tested, their importance varies somewhat with the disease-eliciting agent, although IL-1 may play a dominant role in the cartilage and bone destruction that ultimately ensues (for review see reference 4). For Ab-mediated arthritis that the K/BxN disease may typify, our results point to a more crucial function for IL-1. These roles, and the slightly different kinetics of induction of cytokine transcription in the joint during arthritis initiation, are consistent with a model in which the point of action of TNF-α would be upstream of that of IL-1 (1). TNF-α–independent pathways, perhaps relying on other members of the TNF family, may also trigger IL-1 independently. This view is consistent with the importance of TNF-α in promoting IL-1 production by synoviocytes from RA patients (54), or with the fact that IL-1 blockade prevents the arthritis induced by transgene-encoded TNF-α misexpression (34). It should also be pointed out that the experiments shown in Fig. 1 only detect transcriptionally induced TNF-α production. However, it is likely that even earlier release of TNF-α occurs in the first minutes or hours of the disease, released from intracellular stores of synoviocytes or mast cells upon triggering by C5a or FcγRIII. These molecules constitute two essential links between the anti-GPI Abs and the inflammatory manifestations of K/BxN arthritis (11), and both pathways are known to precipitate rapid TNF-α release.
The relevance of the Ab-mediated arthritis model that K/BxN mice present to human arthritic diseases had been questioned, in part, because it does not fit well with the paradigm in which autoreactive T cells within the joint provoke local TNF-α release, a model bolstered by the successes of anti–TNF-α therapy. The present results show that arthritis induced by Ab complexes in the joint also end up with the production of TNF-α and IL-1, and is highly dependent on these cytokines.
Acknowledgments
We would like to thank Drs. R. Schreiber for the generous gift of mAbs, and J. Hergueux, S. Johnson, and Q.M. Pham for excellently managing the KRN colony.
This work was supported by grants from the Association pour la Recherche contre la Polyarthrite and the National Institutes of Health (1R01 AR/AI46580-01) to D. Mathis and C. Benoist, and an Arthritis Foundation Biomedical Science grant to E. Gravallese. K. Ohmura received a fellowship from the Uehara Memorial Foundation, and A. Pettit is supported by the National Health and Medical Research Council of Australia, and by the Arthritis Foundation.
The present address of H. Ji is Serono Pharmaceutical Research Institute, 14 Chemin des Aulx, 1228, Plan-les-Ouates, Geneva, Switzerland.
Footnotes
Abbreviations used in this paper: CIA, collagen-induced arthritis; GPI, glucose-6-phosphate isomerase; LT, lymphotoxin; RA, rheumatoid arthritis.
References
- 1.Feldmann, M., and R.N. Maini. 2001. Anti-TNFα therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol. 19:163–196. [DOI] [PubMed] [Google Scholar]
- 2.Aarvak, T., and J.B. Natvig. 2001. Cell-cell interactions in synovitis: antigen presenting cells and T cell interaction in rheumatoid arthritis. Arthritis Res. 3:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Firestein, G.S., and N.J. Zvaifler. 1990. How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 33:768–773. [DOI] [PubMed] [Google Scholar]
- 4.van den Berg, W.B. 2001. Anti-cytokine therapy in chronic destructive arthritis. Arthritis Res. 3:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kouskoff, V., A.-S. Korganow, V. Duchatelle, C. Degott, C. Benoist, and D. Mathis. 1996. Organ-specific disease provoked by systemic autoreactivity. Cell. 87:811–822. [DOI] [PubMed] [Google Scholar]
- 6.Korganow, A.-S., H. Ji, S. Mangialaio, V. Duchatelle, R. Pelanda, T. Martin, C. Degott, H. Kikutani, K. Rajewsky, J.-L. Pasquali, et al. 1999. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 10:451–461. [DOI] [PubMed] [Google Scholar]
- 7.Mangialaio, S., A. Staub, C. Benoist, and D. Mathis. 1999. The arthritogenic T cell receptor and its ligand in a spontaneous model of arthritis. Arthritis Rheum. 42:2517–2523. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto, I., A. Staub, C. Benoist, and D. Mathis. 1999. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 286:1732–1735. [DOI] [PubMed] [Google Scholar]
- 9.Ji, H., D. Gauguier, K. Ohmura, A. Gonzalez, V. Duchatelle, P. Danoy, H.J. Garchon, C. Degott, M. Lathrop, C. Benoist, and D. Mathis. 2001. Genetic influences on the end-stage effector phase of arthritis. J. Exp. Med. 194:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto, I., M. Maccioni, D.M. Lee, M. Maurice, B. Simmons, M. Brenner, D. Mathis, and C. Benoist. 2002. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat. Immunol. 3:360–365. [DOI] [PubMed] [Google Scholar]
- 11.Ji, H., K. Ohmura, U. Mahmood, D.M. Lee, F.M.A. Hofhuis, S.A. Boackle, V.M. Holers, M. Walport, C. Gerard, A. Ezekowitz, et al. 2002. Arthritis critically dependent on innate immune system players. Immunity. 16:157–168. [DOI] [PubMed] [Google Scholar]
- 12.Schaller, M., D.R. Burton, and H. Ditzel. 2001. Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nat. Immunol. 2:746–753. [DOI] [PubMed] [Google Scholar]
- 13.Kyburz, D., D.A. Carson, and M. Corr. 2000. The role of CD40 ligand and tumor necrosis factor alpha signaling in the transgenic K/BxN mouse model of rheumatoid arthritis. Arthritis Rheum. 43:2571–2577. [DOI] [PubMed] [Google Scholar]
- 14.Kopf, M., H. Baumann, G. Freer, M. Freudenberg, M. Lamers, T. Kishimoto, R. Zinkernagel, H. Bluethmann, and G. Kohler. 1994. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 368:339–342. [DOI] [PubMed] [Google Scholar]
- 15.Glaccum, M.B., K.L. Stocking, K. Charrier, J.L. Smith, C.R. Willis, C. Maliszewski, D.J. Livingston, J.J. Peschon, and P.J. Morrissey. 1997. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 159:3364–3371. [PubMed] [Google Scholar]
- 16.Labow, M., D. Shuster, D. Zetterstrom, P. Nunes, R. Terry, E.B. Cullinan, T. Bartfai, C. Solorzano, L.L. Moldawer, R. Chizzonite, and K.W. McIntyre. 1997. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J. Immunol. 159:2452–2461. [PubMed] [Google Scholar]
- 17.Pasparakis, M., L. Alexopoulou, V. Episkopou, and G. Kollias. 1996. Immune and inflammatory responses in TNF-α-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Togni, P., J. Goellner, N.H. Ruddle, P.R. Streeter, A. Fick, S. Mariathasan, S.C. Smith, R. Carlson, L.P. Shornick, J. Strauss-Schoenberger, et al. 1994. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 264:667–669. [DOI] [PubMed] [Google Scholar]
- 19.Pfeffer, K., T. Matsuyama, T.M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P.S. Ohashi, M. Kronke, and T.W. Mak. 1993. Mice deficient for the 55 kD tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73:457–467. [DOI] [PubMed] [Google Scholar]
- 20.Erickson, S.L., F.J. de Sauvage, K. Kikly, K. Carver-Moore, S. Pitts-Meek, N. Gillett, K.C. Sheehan, R.D. Schreiber, D.V. Goeddel, and M.W. Moore. 1994. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 372:560–563. [DOI] [PubMed] [Google Scholar]
- 21.Peschon, J.J., D.S. Torrance, K.L. Stocking, M.B. Glaccum, C. Otten, C.R. Willis, K. Charrier, P.J. Morrissey, C.B. Ware, and K.M. Mohler. 1998. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 160:943–952. [PubMed] [Google Scholar]
- 22.Auffray, C., and F. Rougeon. 1980. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur. J. Biochem. 107:303–314. [DOI] [PubMed] [Google Scholar]
- 23.Pettit, A.R., H. Ji, D. von Stechow, R. Muller, S.R. Goldring, Y. Choi, C. Benoist, and E.M. Gravallese. 2001. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159:1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taga, T., and T. Kishimoto. 1997. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 15:797–819. [DOI] [PubMed] [Google Scholar]
- 25.Hirano, T., K. Yasukawa, H. Harada, T. Taga, Y. Watanabe, T. Matsuda, S. Kashiwamura, K. Nakajima, K. Koyama, A. Iwamatsu, et al. 1986. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 324:73–76. [DOI] [PubMed] [Google Scholar]
- 26.Tilg, H., E. Trehu, M.B. Atkins, C.A. Dinarello, and J.W. Mier. 1994. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 83:113–118. [PubMed] [Google Scholar]
- 27.Silacci, P., J.M. Dayer, A. Desgeorges, R. Peter, C. Manueddu, and P.A. Guerne. 1998. Interleukin (IL)-6 and its soluble receptor induce TIMP-1 expression in synoviocytes and chondrocytes, and block IL-1-induced collagenolytic activity. J. Biol. Chem. 273:13625–13629. [DOI] [PubMed] [Google Scholar]
- 28.Fattori, E., M. Cappelletti, P. Costa, C. Sellitto, L. Cantoni, M. Carelli, R. Faggioni, G. Fantuzzi, P. Ghezzi, and V. Poli. 1994. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 180:1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohshima, S., Y. Saeki, T. Mima, M. Sasai, K. Nishioka, S. Nomura, M. Kopf, Y. Katada, T. Tanaka, M. Suemura, and T. Kishimoto. 1998. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA. 95:8222–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takagi, N., M. Mihara, Y. Moriya, N. Nishimoto, K. Yoshizaki, T. Kishimoto, Y. Takeda, and Y. Ohsugi. 1998. Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 41:2117–2121. [DOI] [PubMed] [Google Scholar]
- 31.van de Loo, F.A., S. Kuiper, F.H. van Enckevort, O.J. Arntz, and W.B. van den Berg. 1997. Interleukin-6 reduces cartilage destruction during experimental arthritis. A study in interleukin-6-deficient mice. Am. J. Pathol. 151:177–191. [PMC free article] [PubMed] [Google Scholar]
- 32.Wooley, P.H., J.D. Whalen, D.L. Chapman, A.E. Berger, K.A. Richard, D.G. Aspar, and N.D. Staite. 1993. The effect of an interleukin-1 receptor antagonist protein on type II collagen-induced arthritis and antigen-induced arthritis in mice. Arthritis Rheum. 36:1305–1314. [DOI] [PubMed] [Google Scholar]
- 33.van den Berg, W.B., L. Joosten, M.M.A. Helsen, and A.A.J. van de Loo. 1994. Amelioration of established murine collagen induced arthritis with anti-IL-1 treatment. Clin. Exp. Immunol. 95:237–243. 8306498 [Google Scholar]
- 34.Probert, L., D. Plows, G. Kontogeorgos, and G. Kollias. 1995. The type I interleukin-1 receptor acts in series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur. J. Immunol. 25:1794–1797. [DOI] [PubMed] [Google Scholar]
- 35.Joosten, L., M.M.A. Helsen, F. van de Loo, and W.B. van den Berg. 1996. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: a comparative study using anti-TNF-α, anti-IL-Iα/β, and IL-1ra. Arthritis Rheum. 39:797–809. [DOI] [PubMed] [Google Scholar]
- 36.Joosten, L.A., M.M. Helsen, T. Saxne, F.A. van de Loo, D. Heinegard, and W.B. van den Berg. 1999. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J. Immunol. 163:5049–5055. [PubMed] [Google Scholar]
- 37.Feldmann, M., F.M. Brennan, and R.N. Maini. 1996. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 14:397–440. [DOI] [PubMed] [Google Scholar]
- 38.Ghivizzani, S.C., R. Kang, H.I. Georgescu, E.R. Lechman, D. Jaffurs, J.M. Engle, S.C. Watkins, M.H. Tindal, M.K. Suchanek, L.R. McKenzie, et al. 1997. Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J. Immunol. 159:3604–3612. [PubMed] [Google Scholar]
- 39.Thorbecke, G.J., R. Shah, C.H. Leu, A.P. Kuruvilla, A.M. Hardison, and M.A. Palladino. 1992. Involvement of endogenous tumor necrosis factor alpha and transforming growth factor beta during induction of collagen type II arthritis in mice. Proc. Natl. Acad. Sci. USA. 89:7375–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piguet, P.F., G.E. Grau, C. Vesin, H. Loetscher, R. Gentz, and W. Lesslauer. 1992. Evolution of collagen arthritis in mice is arrested by treatment with anti-tumour necrosis factor (TNF) antibody or a recombinant soluble TNF receptor. Immunology. 77:510–514. [PMC free article] [PubMed] [Google Scholar]
- 41.Williams, R.O., M. Feldmann, and R.N. Maini. 1992. Anti-TNF ameliorates joint disease in murine collagen induced arthritis. Proc. Natl. Acad. Sci. USA. 89:9784–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wooley, P.H., J. Dutcher, M.B. Widmer, and S. Gillis. 1993. Influence of a recombinant human soluble TNF receptor Fc fusion protein on type II collagen induced arthritis in mice. J. Immunol. 151:6602–6607. [PubMed] [Google Scholar]
- 43.Keffer, J., L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis, and G. Kollias. 1991. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krakauer, T., J. Vilcek, and J.J. Oppenheim. 1999. Proinflammatory cytokines: TNF and IL-1 families, chemokines, TGF-β, and others. Fundamental Immunology. W.E. Paul, editor. Lippincott-Raven Publishers, Philadelphia. 775.
- 45.Sheehan, K.C., J.K. Pinckard, C.D. Arthur, L.P. Dehner, D.V. Goeddel, and R.D. Schreiber. 1995. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J. Exp. Med. 181:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell, I.K., K. O'Donnell, K.E. Lawlor, and I.P. Wicks. 2001. Severe inflammatory arthritis and lymphadenopathy in the absence of TNF. J. Clin. Invest. 107:1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong, Y.Y., U. Feige, I. Sarosi, B. Bolon, A. Tafuri, S. Morony, C. Capparelli, J. Li, R. Elliott, S. McCabe, et al. 1999. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 402:304–309. [DOI] [PubMed] [Google Scholar]
- 48.Gravallese, E.M., D.L. Galson, S.R. Goldring, and P.E. Auron. 2001. The role of TNF-receptor family members and other TRAF-dependent receptors in bone resorption. Arthritis Res. 3:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi, K., N. Takahashi, E. Jimi, N. Udagawa, M. Takami, S. Kotake, N. Nakagawa, M. Kinosaki, K. Yamaguchi, N. Shima, et al. 2000. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 191:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimi, E., I. Nakamura, L.T. Duong, T. Ikebe, N. Takahashi, G.A. Rodan, and T. Suda. 1999. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp. Cell Res. 247:84–93. [DOI] [PubMed] [Google Scholar]
- 51.Abu-Amer, Y., J. Erdmann, L. Alexopoulou, G. Kollias, F.P. Ross, and S.L. Teitelbaum. 2000. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 275:27307–27310. [DOI] [PubMed] [Google Scholar]
- 52.Azuma, Y., K. Kaji, R. Katogi, S. Takeshita, and A. Kudo. 2000. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 275:4858–4864. [DOI] [PubMed] [Google Scholar]
- 53.Lipsky, P.E., D.M. van der Heijde, E.W. St. Clair, D.E. Furst, F.C. Breedveld, J.R. Kalden, J.S. Smolen, M. Weisman, P. Emery, M. Feldmann, et al. 2000. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study group. N. Engl. J. Med. 343:1594–1602. [DOI] [PubMed] [Google Scholar]
- 54.Brennan, F.M., D. Chantry, A. Jackson, R. Maini, and M. Feldmann. 1989. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 2:244–247. [DOI] [PubMed] [Google Scholar]