

Abstract
Pseudomonas aeruginosa is an important opportunistic human pathogen. Certain strains can transmigrate across epithelial cells, and their invasive phenotype is correlated with capacity to cause invasive human disease and fatal septicemia in mice. Four multidrug efflux systems have been described in P. aeruginosa, however, their contribution to virulence is unclear. To clarify the role of efflux systems in invasiveness, P. aeruginosa PAO1 wild-type (WT) and its efflux mutants were evaluated in a Madin-Darby canine kidney (MDCK) epithelial cell monolayer system and in a murine model of endogenous septicemia. All efflux mutants except a ΔmexCD-oprJ deletion demonstrated significantly reduced invasiveness compared with WT. In particular, a ΔmexAB-oprM deletion strain was compromised in its capacity to invade or transmigrate across MDCK cells, and could not kill mice, in contrast to WT which was highly invasive (P < 0.0006) and caused fatal infection (P < 0.0001). The other mutants, including ΔmexB and ΔmexXY mutants, were intermediate between WT and the ΔmexAB-oprM mutant in invasiveness and murine virulence. Invasiveness was restored to the ΔmexAB-oprM mutant by complementation with mexAB-oprM or by addition of culture supernatant from MDCK cells infected with WT. We conclude that the P. aeruginosa MexAB-OprM efflux system exports virulence determinants that contribute to bacterial virulence.
Keywords: Pseudomonas aeruginosa, bacterial invasion, multidrug efflux system, outer membrane protein, endogenous bacteremia
Introduction
Pseudomonas aeruginosa is a versatile Gram-negative bacterium that is an important opportunistic human pathogen. Although this organism does not cause bloodstream infections in patients with chronic respiratory infections despite long-term colonization, it can be invasive in other patients and cause bacteremia associated with high mortality rates, particularly in those with neutropenia. We have reported that most clinical blood isolates cause lethal endogenous bacteremia in leukopenic mice, whereas human respiratory isolates do not cause bacteremia (1, 2). We have also reported that clinical blood isolates of P. aeruginosa penetrate human intestinal Caco-2 and Madin-Darby canine kidney (MDCK)* epithelial cell monolayers to a greater extent than do respiratory isolates (3, 4). In the MDCK epithelial cell monolayer system, PAO1 and most blood isolates are invasive but lack exoU, which encodes a 70-kD cytotoxic protein ExoU (5, 6), and showed high penetration ability without severe epithelial damage. In contrast, PA103 and ∼8% of clinical isolates are cytotoxic, possess exoU, and pass through the monolayer after epithelial cell death (4).
P. aeruginosa is intrinsically resistant to conventional penicillins and cephems due to its low outer membrane (OM) permeability coupled to the production of an inducible chromosomal β-lactamase, which hydrolyzes these β-lactams (7) and can become mutationally resistant to even newly-developed antipseudomonal agents (8). In addition to the recent emergence of metallo-β-lactamase producing P. aeruginosa (9), multidrug resistant (MDR) efflux systems are becoming recognized as important antimicrobial resistance mechanisms for this organism. To date, four resistance-nodulation-division (RND) MDR efflux systems have been described in P. aeruginosa: MexAB-OprM (10–13); MexCD-OprJ (14); MexEF-OprN (15); and MexXY-OprM (16, 17, for a review, see references 18–21). The genes encoding these systems are arranged as operons, with the first gene encoding a linker protein that is associated with the cytoplasmic membrane (MexA, MexC, MexE, and MexX). The second gene encodes an efflux pump that exports substrates across the inner membrane (MexB, MexD, MexF, and MexY). In three cases, a third gene encodes an OM protein, which facilitates passage of the substrate across the OM (OprM, OprJ, and OprN). OprM is also involved in the MexXY-OprM system (16, 22). The three components form a channel that traverses the inner membrane, periplasm, and OM and allows the substrate to be effluxed directly from the cytoplasm or cytoplasmic membrane to the extracellular environment (18). Among these four efflux systems, only MexAB-OprM is expressed constitutively in wild-type (WT) P. aeruginosa. It was the first P. aeruginosa MDR efflux system to be described, and contributes to intrinsic resistance to a number of antimicrobials, such as fluoroquinolones, β-lactams, tetracycline, macrolides, chloramphenicol, novobiocin, trimethoprim, and sulphonamides (18). In contrast, other systems have different substrate spectra and are not expressed during normal laboratory growth (18). In addition to medically relevant antibiotics, MexAB-OprM also exports a variety of dyes, detergents, inhibitors of fatty acid biosynthesis, and organic solvents (18). Hence, this and other RND efflux systems are generally thought to expel harmful substances from bacteria.
Recently, certain studies have suggested a relationship between efflux and virulence in P. aeruginosa. For instance, it has been reported that MexAB-OprM exports homoserine lactones which are involved in quorum sensing (cell-to-cell signaling; references 23–25) and consequent regulation of the expression of a variety of virulence determinants. Thus, nalB mutants which hyperexpress the MexAB-OprM efflux system produce reduced levels of several extracellular virulence factors (25). Other studies have demonstrated that, compared with WT, nfxC type mutants, overexpressing the MexEF-OprN efflux system, produce lower levels of pyocyanin, elastase, and rhamnolipids, which are controlled by the las and rhl quorum-sensing systems (26, 27). It has also been suggested that MexAB-OprM contributes to the antibiotic resistance of P. aeruginosa biofilms (28), while other studies have reported the obverse indicating that the four characterized efflux systems described above did not play a significant role in the resistance of P. aeruginosa biofilm to antibiotics (29). However, the role of efflux systems in specific clinical pathogenesis of P. aeruginosa, including invasiveness, has not been determined.
To gain a better understanding the role of efflux in bacterial pathogenesis, the invasiveness of P. aeruginosa PAO1 and its efflux mutants was evaluated using in vitro MDCK epithelial cell monolayer penetration (4, 30, 31) and gentamicin survival (31, 32) assays, and by electron microscopy. Virulence was also investigated in a murine model of endogenous P. aeruginosa bacteremia, closely mimicking the pathophysiology of septicemia in humans (2, 33, 34).
Materials and Methods
Bacterial Strains and Construction of Mutants.
Bacterial strains and plasmids used in this study are summarized in the Table I. P. aeruginosa PAO1 strain K767 (35) was used as the WT, which is invasive and penetrates MDCK cell monolayers by 3 h after infection (4). Strain K767 lacks exoU and did not show any cytotoxicity to MDCK cells within 6 h after infection. To construct P. aeruginosa deletion mutants, the deletions were first made in sacB-containing vectors and were subsequently introduced into the chromosome of K767 by gene replacement (36). The mexAB-oprM deletion strain K1119 (37) and the mexCD-oprJ deletion strain K1521 (29) were constructed using the mexAB-oprM deletion construct pRSP21 and the mexCD-oprJ deletion construct pRSP05, respectively, using the strategy reported previously (38). In these instances, however, the selections of transconjugants carrying a copy of pRSP21 or pRSP05 were on Luria-Bertani (LB) agar containing 1.5 mg of kanamycin per ml. The mexXY deletion strain K1525 was constructed as described previously (29). To construct the mexB deletion strain K1523, a 4.6–kb KpnI-HindIII fragment from pRSP19 (39), containing mexA and part of mexB, was first cloned into pEX18Tc to generate pRSP77. Plasmid pRSP77 was digested with BamHI and MluI at two unique sites within mexB, 1.8-kb apart, and the 9.1-kb fragment containing the vector was isolated. This fragment was treated with Klenow (New England Biolabs) to facilitate back filling of the 3′ recessed ends and ligated to yield pRSP81. The internal deletion in mexB created in this manner was confirmed to be in-frame by sequencing pRSP81 with primer bacmexbf1 (5′-ATG TCG AAG TTT TTC ATT GAT AGG-3′). Plasmid pRSP81 was mobilized into P. aeruginosa strain K767 and a chromosomal mexB deletion selected using the protocol described previously for the mexXY deletion (29).
Table I.
Bacterial Strains and Plasmids Used in this Study
| Strain or plasmid | Relevant genotype or phenotype | Source or reference |
|---|---|---|
| P. aeruginosa strains | ||
| K767 | WT PAO1 | 35 |
| K1119 | K767 ΔmexAB-oprM; MexAB-OprM deletion strain | 37 |
| K1521 | K767 ΔmexCD-oprJ; MexCD-OprJ deletion strain | 29 |
| K1523 | K767 ΔmexB; MexB deletion strain | this study |
| K1525 | K767 ΔmexXY; MexXY deletion strain | 29 |
| OCR1 | K767 nalB; MexAB-OprM overproducing strain | 35 |
| K1536 | K767 nfxB; MexCD-OprJ overproducing strain | this study |
| KG4521 | K767 oprM:Sm; oprM knockout mutant | this study |
| Plasmids | ||
| pRK415 | Broad-host-range cloning vector, tetracycline-resistant | 40 |
| pRSP17 | pRK415:mexAB-oprM; mexAB-oprM expression plasmid | 39 |
| pKMM128 | oprM expression plasmid, carbenicillin-resistant | 45 |
The chromosomal deletion of mexB was confirmed by PCR using primers rspoligo3 (5′-CAG CAG CTC TAC CAG ATC GAC-3′) and rspoligo4 (5′-GTG TCC TTG GTC AGC TGC AAC-3′). The nfxB strain K1536 was obtained after plating strain K767 on LB plates containing 0.4 μg ciprofloxacin (Sigma-Aldrich) per ml. Those overexpressing MexCD-OprJ were confirmed by immunoblotting of cell envelopes with antibodies specific to OprJ as described previously (38). The nalB strain OCR1 has been described previously (35). Plasmids pRK415 (40) and its derivative pRSP17 (39), which carries mexAB-oprM were maintained in strain K1119 (ΔmexAB-oprM) by inclusion of tetracycline (10 μg/ml) in the growth media. To constructe a oprM knockout mutant derived from K767 (WT), PCR primers for amplification of the 1.5-kb fragment were synthesized based on nucleotide sequences of the Pseudomonas genome sequencing project database. After amplifying a 1.5-kb region including oprM gene (11) on strain K767 (WT) genomic DNA as a template using #oprM4 (5′-TCATAAGCTTATGAAAACGGTCCTTCCTTTCC-3′) and #oprM3 (5′-TTGAGAATTCTCAAGCCTGGGGATCTTCCTT-3′), a primer pair containing a newly added cutting site (underlined) for restriction nucleases, the region was ligated into the HindIII-EcoRI site in a multicloning site of pMT5059 (41) to yield pMT5059M. The SalI fragment encompassing ΩSm gene from pS1918 (42) was ligated into the SalI site in the oprM gene on pMT5059M to yield pMT5059MSM. Then, the Mob cassette from pMT5071 (43) was cloned into the NotI site, on pMT5059 (41) to yield pMK1. The resulting plasmid was mobilized from the E. coli strain S17–1 to K767 (WT) to introduce the streptomycin-resistant determinant into the oprM gene on the recipient chromosomes to yield KG4521. Disruption of the oprM gene of KG4521 was confirmed by PCR and Western immunoblot analysis using the anti-OprM antibody (44) (unpublished data). Plasmid pKMM128 (45) carrying oprM was maintained in KG4521 (ΔoprM) in the presence of 10 μg/ml of carbenicillin. All mutants derived from K767 were motile, resistant to 10% pooled fresh normal human serum, noncytotoxic, and grew in LB broth and MEM as well as their parent strain K767.
MDCK Cell Monolayer Penetration Assay.
The assay was performed as reported previously (4, 30, 31). In brief, strain 1 MDCK cells in MEM with 10% FBS were seeded at 1.5 × 105 cells per well in Transwell filter units (Costar) containing 0.33-cm2 porous filter membranes (3.0-μm pores). Monolayers were incubated at 37°C in 5% CO2 for 4 d until the transmonolayer electrical resistance (TER) reached the proper range (900–1,200 Ωcm2), as measured with a Millicell-ESR apparatus (Millipore). Monolayers were infected with bacteria by adding 5 μl (3.5 × 106 CFU) freshly grown bacteria cultured in LB broth overnight at 37°C with shaking at 150 rpm. In some experiments, TER was monitored at several time intervals after infection to assess damage to monolayers. The assay was performed in triplicate, and results are expressed as an average ± SD. Each assay was repeated at least three times to confirm the reproducibility.
Bacterial Association and Invasion Assay.
Gentamicin survival assays were performed to quantify the bacteria which invaded MDCK cells (31, 32). After a 3-h infection, monolayers were washed six times with PBS and then incubated a further 2 h in fresh medium containing 200 μg/ml of gentamicin to kill extracellular bacteria only. Monolayers were washed three times with PBS and then lysed with a 1% Triton X-100 (Sigma-Aldrich) for 15 min. Appropriate dilutions were spread onto agar plates, incubated at 37°C overnight, and CFUs counted to quantify bacteria surviving intracellularly. Associated bacteria (including both adherent and invading bacteria) were measured by the addition of a 1% Triton X-100 to the monolayers 3 h after infection followed by six washes without gentamicin treatment. As a control, filter units with medium only and without MDCK cells were inoculated with bacteria to determine baseline adherence to plastic. The baseline values were subtracted from those obtained by incubation with epithelial cells as described above.
Electron Microscopy.
For transmission electron microscopy (TEM), monolayers infected with bacteria were washed seven times with PBS at 37°C and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate containing 2 mM CaCl2 (cacodylate buffer) for 60 min. After washing with cacodylate buffer, samples were postfixed in cold 1% OsO4 in the same buffer for 30 min, and then stained with cold 2% uranyl acetate for 30 min. Samples were dehydrated in a series of ethanol and embedded in EPON 812 resin, and were sectioned and stained with uranyl acetate and lead citrate before examination in a JEOL JEM-1210 transmission electron microscope. For scanning electron microscopy (SEM), the monolayers were fixed as described above, dehydrated in a critical point apparatus, and were examined with a JEOL JSM-35C/LaB6 III–A scanning electron microscope, after a gold sputter coating.
Evaluation of Virulence of the Efflux Mutants in a Murine Model of Endogenous Bacteremia.
The animal studies were approved by the Animal Care and Use Committee of Nagasaki University, and were conducted in accordance to the Guidelines for Animal Experimentation, Nagasaki University. Specific-pathogen-free male BALB/c mice (Japan S.L.C. Co., Ltd.) weighing 20–24 g were used in these experiments. Fecal specimens were cultured before the study to ensure the absence of P. aeruginosa. Endogenous bacteremia was induced as described previously (2, 33, 34). In brief, a suspension of 107 CFU/ml P. aeruginosa in sterile 0.45% saline was given in the drinking water between days 1 and 4. The normal intestinal flora of the mice was disturbed by administration of sodium ampicillin (Meiji Seika Kaisha, Ltd.) by daily intraperitoneal injection during the same period, to assist in the colonization by P. aeruginosa. Mice were then given 200 mg/kg cyclophosphamide (Shionogi & Co.) by intraperitoneal injection on days 5, 7, and 9. These doses of cyclophosphamide induce leukopenia (<1,000/mm3) in mice from days 8 to 14 without lethality in the absence of oral bacterial inoculation (33, 34).
Effect of Monolayer Supernates on Penetration of P. aeruginosa Strains.
The supernates of monolayers infected with strains K767 (WT) or K1119 (ΔmexAB-oprM) were collected 3 h or overnight after infection of MDCK cells, and diluted appropriately. The medium of the MDCK cell monolayers on day 4 was replaced with these supernates and strains K767 or K1119 were added 1 h later as described above. As controls, the supernates from MDCK cell-free MEM or LB broth inoculated with the strains were also tested.
Statistics.
Analysis of variance (ANOVA) was used for the comparisons among three or more groups. Student's t test was used to compare means between the two groups. Chi-square test was used to compare survival rates. A level of 5% was accepted as statistically significant.
Results
Penetration of Strain K767 (WT) and Its Efflux Mutants through MDCK Cell Monolayers.
Both the parent WT strain K767 and strain K1521 (ΔmexCD-oprJ) penetrated MDCK monolayers by 3 h, whereas K1119 (ΔmexAB-oprM) was not detected in the basolateral medium until 6 h after infection (Fig. 1 A). At all time points, the penetration of both K767 (WT) and K1521 (ΔmexCD-oprJ) was significantly greater than that for K1119 (ΔmexAB-oprM) (P < 0.0006, ANOVA). Compared with the WT, relative capacities of the mutants to penetrate monolayers at 3 and 6 h after infection are summarized in Fig. 1 B. Strains K1523 (ΔmexB) and K1525 (ΔmexXY) were also compromised for invasion, but to lesser extents than the mutant with total deletion of mexAB-oprM (K1119). The invasive capacities of strains OCR1 (nalB) and K1536 (nfxB), hyperexpressing efflux systems, were also intermediate between strains K767 (WT) and K1119 (ΔmexAB-oprM).
Figure 1.


Penetration of P. aeruginosa strain K767 (WT) and its efflux mutants. Bacteria were inoculated at 3.5 × 106 CFU per well to the apical surfaces of MDCK cell monolayers. (A) The assay was performed in triplicate, and results are expressed as mean ± SD. (B) The values are expressed as percentages of values obtained with strain K767 at 3 and 6 h.
Fig. 2 shows the time course of changes in TER of the MDCK cell monolayers infected with strain K767 (WT) and its efflux mutants. TER of monolayers infected with strains K767 (WT) or K1521 (ΔmexCD-oprJ) decreased in a time-dependent manner. The drops in TER for these strains were slower than that for cytotoxic strains, which induced a steep drop (4). When monolayers were infected with strain K1119 (ΔmexAB-oprM), TER decreased more slowly over time than for strains K767 (WT) and K1521 (ΔmexCD-oprJ). The values of TER for strains K1523 (ΔmexB) and K1525 (ΔmexXY) were always intermediate between those of strains K767 (WT) and K1119 (ΔmexAB-oprM).
Figure 2.

Changes of TER after infection with P. aeruginosa strain K767 (WT of PAO1) and its efflux mutants. TER was measured in triplicate, and results are expressed as an average ± SD of area multiplied by resistance.
Association and Invasion of Strains K767 (WT) and K1119 (ΔmexAB-oprM).
Strain K1119 (ΔmexAB-oprM) adhered to MDCK cells, as well as K767 (WT) (5.25 ± 1.79 × 105 vs. 4.78 ± 1.21 × 105 CFU, not significant, Fig. 3 A). However, the gentamicin survival assay revealed that the efflux mutant K1119 (ΔmexAB-oprM) showed a significantly decreased level of invasion (86.7 ± 9.6 vs. 1.18 ± 0.15 × 104 CFU, P = 0.0002, Fig. 3 B). Fig. 3 C shows the time courses of invasion for the strains K767 (WT) and K1119 (ΔmexAB-oprM) by the gentamicin survival assay. Significantly greater numbers of K767 (WT) than K1119 (ΔmexAB-oprM) invaded the epithelial cells (P < 0.0065, at each time point). Strain K767 (WT), which penetrated monolayers by 3 h (Fig. 1 A), entered the cells within 30 min, and the number of intracellular bacteria per monolayer increased in a time-dependent manner. The small number of K1119 (ΔmexAB-oprM) which had transversed the monolayers by 6 h (Fig. 1 A), was reflected in by the low level of invasion by 60 min. The number of intracellular K1119 (ΔmexAB-oprM) bacteria also increased in a time-dependent manner, but the value at 360 min was comparable to K767 (WT) only 60 min after infection (200 ± 45 vs. 130 ± 17 CFU, P = 0.0653).
Figure 3.

Bacterial association with (A) and invasion into (B and C) MDCK epithelial cells of P. aeruginosa strain K767 (WT of PAO1) and its efflux mutant K1119 (ΔmexAB-oprM). Bacterial association was evaluated by lysis of the epithelial cells with Triton X-100, 3 h after infection. Bacterial invasion was assessed at indicated times similarly but after the treatment with 200 μg/ml of gentamicin for 2 h to kill extracellular bacteria. The assay was performed in triplicate, and results are expressed as mean ± SD.
Electron Microscopy.
When observed by scanning electron microscopy, most of the bacteria, including strain K767 (WT) and its efflux mutants, adhered near the edge (cell-to-cell junctions) of MDCK cells (Fig. 4 A), whereas K767 (WT) showed an additional adherence pattern, adherence around the center of the epithelial cells and seemed to be in the process of invading (Fig. 4 B). The microvilli of MDCK cells, to which P. aeruginosa isolates adhered, were grossly unaltered. Transmission electron microscopy showed that K767 (WT) contacted with the epithelial cell surface (Fig. 4 C) and a bacterium was internalized (Fig. 4 D) 15 min after infection. At later time points bacteria invaded into the cells and reached the layer of the polycarbonate filter within 3 h after infection, as reported elsewhere (46, 47) but without appearance of cell injury. In contrast, K1119 (ΔmexAB-oprM) did not penetrate at all by 30 min, and only a small number of bacteria invaded cells by 3 h after infection (data not shown).
Figure 4.
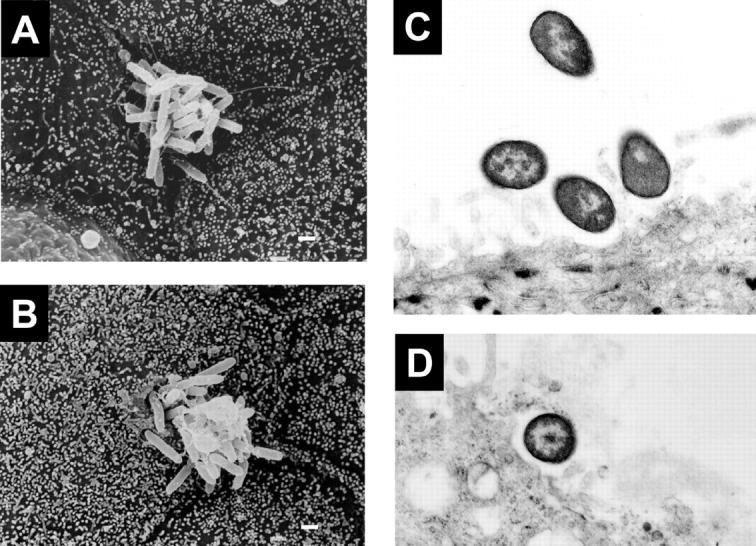
Scanning (A and B) and transmission (C and D) electron micrographs of MDCK epithelial cell monolayers infected with P. aeruginosa strain K767 (WT of PAO1) and its efflux mutants K1119 (ΔmexAB-oprM). Bacteria adhere near the edge of the epithelial cells without loss of microvilli (A; K1119, 3 h after infection). Bacteria adhere around the center of the epithelial cells and some bacteria appear to be invading into the MDCK cells, without loss of microvilli (B; K767, 1 h after infection). Bacteria are contacting the epithelial cell surface (C; K767) and a bacterium is internalized into a MDCK cell (D; K767) 15 min after infection. Original magnifications, 4,800× (A and B) and 10,000× (C and D). Bar, 1 μm.
Evaluation of the Virulence of the Efflux Mutants in a Murine Model.
The animal model used in this study reflects the differences in the important pathophysiological steps in authentic human infections, including bacterial colonization and invasion. All strains of P. aeruginosa tested survived equally well in the drinking water; all were recovered at equal titer at the end of the oral bacterial challenge. Furthermore, on day 8, equivalent numbers of P. aeruginosa were recovered from the stools of animals challenged with each different bacterial strain. Strains K767 (WT) and K1521 (ΔmexCD-oprJ) induced lethal endogenous septicemia in mice, whereas strain K1119 (ΔmexAB-oprM) failed to kill any mice (P < 0.0001, Fig. 5 A). The mortality rates of mice given K1523 (ΔmexB, 20% mortality rate, P < 0.0001 compared with K767), K1525 (ΔmexXY, 55%, P = 0.00066), OCR1 (nalB, 80%, P = 0.035), and K1536 (nfxB, 70%, P = 0.0079) were intermediate between those for strains K767 (100%) and K1119 (0%) (Fig. 5 B).
Figure 5.


Survival kinetics of leukopenic mice given P. aeruginosa strain K767 (WT) and its efflux mutants K1119 (ΔmexAB-oprM) and K1521 (ΔmexCD-oprJ) (A) and mortality rates of mice given P. aeruginosa WT and its efflux mutants (B). Mice were given 200 mg of ampicillin (A) and 200 mg of cyclophosphamide (CY) per kg on the indicated days. Bacteria were given orally in drinking water in sterile 0.45% saline at the concentration of 107 CFU/ml between days 1 and 4. The numbers of mice per group are indicated in the figure. (B) Data are expressed as the mortality rate for each group given a different P. aeruginosa strain.
Influence of Complementation of K1119 (ΔmexAB-oprM) with mexAB-oprM on Its Invasiveness.
To confirm the role of mexAB-oprM in the expression of invasiveness, the ΔmexAB-oprM deletion strain K1119 was complemented with mexAB-oprM by introducing plasmid pRSP17 carrying the genes and examined with strains K767 (WT), K1119 (ΔmexAB-oprM), and K1119/pRK415 (plasmid control). K1119/pRSP17 (mexAB-oprM complemented strain) showed the invasiveness equivalent to WT in both in vitro (Fig. 6 A) and the animal model (Fig. 6 B), while plasmid control strain K1119/pRK415 had the same phenotype, which was compromised in its capacity to penetrate MDCK monolayer (Fig. 6 A, P < 0.0001) and to kill leukopenic mice (Fig. 6 B, P < 0.0001) as K1119 (ΔmexAB-oprM).
Figure 6.


Penetration of P. aeruginosa strain K767 (WT), K1119 (ΔmexAB-oprM), mexAB-oprM complemented strain, and oprM knockout mutant (A) and mortality rates of leukemic mice given the P. aeruginosa strains (B). Strains K1119/pRSP17 and K1119/pRK415 are K1119 (ΔmexAB-oprM), complemented with mexAB-oprM and its plasmid control, respectively. KG4521 and KG4521/pKMM128 are oprM knockout mutant from K767 and oprM complemented mutant of KG4521, respectively. The assay was performed in triplicate, and results are expressed as mean ± SD (A). Data are expressed as the mortality rate for each group (at least 10 mice per group) given a different P. aeruginosa strain (B).
Influence of a Single Mutation in oprM on the Invasiveness of P. aeruginosa.
A oprM knockout mutant KG4521 was examined with K767 (WT) and KG4521/pKMM128 (oprM complemented strain) to clarify the role of OprM in the loss of virulence in P. aeruginosa. Strain KG4521 (oprM knockout) was significantly less virulent in the MDCK epithelial monolayer system (Fig. 6 A, P = 0.0466) and in the animal model (Fig. 6 B, P = 0.02703). However, the loss of virulence was modest compared with the complete mexAB-oprM deletion mutant K1119. Complementation of KG4521 with oprM restored the virulence entirely (Fig. 6).
Effect of Monolayer Supernates on Penetration of P. aeruginosa Strains.
Experiments were performed to determine if soluble factors enhanced the penetration of P. aeruginosa across MDCK cells. Neither supernates of K767 (WT) or K1119 (ΔmexAB-oprM) cultured overnight in cell-free LB broth, nor K1119 (ΔmexAB-oprM) cultured overnight in MEM in the presence of MDCK cells, when readded to MDCK cells, influenced the penetration of K767 (WT). On the other hand, the supernate from MDCK cells incubated overnight together with K767 (WT), caused a slight but significant enhancement of the penetration of strain K767 (WT) itself into MDCK monolayers (P = 0.0475, ANOVA). Supernates from MDCK cell monolayers infected overnight with K767 (WT) dramatically and significantly enhanced the penetration of K1119 (ΔmexAB-oprM) into MDCK cells (P < 0.0001, ANOVA). The K1119 (ΔmexAB-oprM) supernate also significantly enhanced the penetration of K1119 (ΔmexAB-oprM) (P < 0.05), but the effect was nearly 10-fold less than that observed with the K767 (WT) supernate (245 ± 128 vs. 2320 ± 385, P = 0.0081) (Fig. 7 A). The supernates of K767 (WT) incubated in MEM in the absence of MDCK cells showed a very low level of enhancement of invasion of the K1119 (ΔmexAB-oprM) cells (unpublished data). The supernates of strain K767 (WT) incubated with MDCK cells overnight, enhanced the penetration of strain K1119 (ΔmexAB-oprM) through monolayers in a concentration-dependent manner (Fig. 7 B). In contrast, the supernates obtained 3 h after infection demonstrated a relatively weak enhancement effect compared with those from overnight cultures.
Figure 7.


Influence of bacterial culture supernates on penetration of P. aeruginosa strain K767 (WT of PAO1) and its efflux mutant K1119 (ΔmexAB-oprM) (A) and the effect of concentration of supernates (B). MDCK cell monolayers were infected with K767 or K1119 overnight and the supernates were collected. Medium of MDCK cell monolayers were replaced 1 h before infection with the supernates obtained in the different condition as described. Fresh MEM was used as a control. The assay was performed in triplicate, and results are expressed as mean ± SD. Legend: sup in LB, bacterial culture supernate from LB broth; sup in MEM/MDCK, bacterial culture supernate from MDCK cell monolayer infected with bacteria;. sup 3 h, bacterial culture supernate from MDCK cell monolayer infected for 3 h; sup O/N, bacterial culture supernate from MDCK cell monolayer infected overnight.
Discussion
This study using MDCK epithelial cell monolayer system demonstrates an association between bacterial efflux mechanisms and invasion of eukaryotic cells. The ΔmexAB-oprM deletion strain K1119 was the least invasive among the efflux mutants tested, while K1521, the mexCD-oprJ deletion strain, was as invasive as WT. The loss of only mexB (K1523) or mexXY (K1525) decreased invasion but in a less dramatic fashion than for the deletion of the entire mexAB-oprM operon (K1119). Data from gentamicin survival assays, electron microscopy studies, and the animal studies supported these findings. The current in vitro and in vivo animal models represent intestinal epithelium and gut-derived endogenous bacteremia, respectively, but not respiratory epithelium nor pneumonia models. Mex and OM proteins may influence the bacterial susceptibility to antibody independent complement killing or opsonic killing by neutrophils; however, all mutants in this study were serum-resistant as the WT, suggesting that the mutations did not affect their susceptibility to antibody-independent complement killing. This study did not include phagocytes in the in vitro system and our animal model was leukopenic. Therefore, we do not believe that the changes we observed in the mutants influenced their interactions with phagocytic cells.
Our data suggest that deletion of all three genes in the MexAB-OprM efflux system is necessary to yield the maximum loss of invasiveness. The complementation of ΔmexAB-oprM deletion strain K1119 with mexAB-oprM restored the invasiveness to the level of WT, and consequently confirmed that the genes, mexAB-oprM, actually played an important role in the loss of invasiveness. The MexXY-OprM and MexAB-OprM systems share OprM as a channel-tunnel (16, 17), and only the deletion of genes in these two efflux systems decreased the level of invasiveness modestly. However, the mexB deletion strain K1523 displayed a more similar phenotype to the complete mexAB-oprM mutant K1119 than the mexXY mutant K1525. The difference in virulence between the ΔmexB mutant and the ΔmexXY mutant may depend on the expression of each efflux system; MexAB-OprM is expressed constitutively. We expected that OprM is a critical determinant of invasiveness in P. aeruginosa and that the invasive determinant(s) are exported through OprM only; however, the loss of invasiveness in the oprM knockout mutant, KG4521 was modest, suggesting the possibility that OM proteins other than OprM also could work with MexAB.
The efflux system-overproducing strains such as nalB and nfxB, also demonstrated somewhat reduced invasiveness, akin to the efflux deletion strains (Fig. 1 B). These paradoxical findings may be explained by recent reports, which showed that nalB type mutants, hyperexpressing the MexAB-OprM (23–25) and nfxC type mutants, overexpressing the MexEF-OprN (26, 27), produce lower level of virulence factors because of their influence on cell-to-cell signaling regulated by quorum-sensing systems. Therefore, it appears that normal efflux levels are necessary for P. aeruginosa to express maximal invasiveness.
Data from gentamicin survival assays (Fig. 3 A) and electron microscopy studies (Fig. 4 A) also confirmed that the differences between WT and the ΔmexAB-oprM mutant in bacterial penetration through MDCK epithelial cell monolayers, were correlated with differences in their invasiveness; the strains adhered to the epithelial cells equally well. Electron microscopy studies showed a very similar invasion pattern to that reported in the study using tracheal epithelial cells (48). Interestingly, in contrast to the observation of microvilli loss when MDCK cells are infected with Salmonella isolates for at least 2 h (31), microvilli appeared to remain intact when such cells were infected with P. aeruginosa strains for at least 3 h. The difference between the two bacterial species is consistent with the observation that P. aeruginosa frequently colonizes the intestine of normal human subjects (49) without producing any symptoms, and shares characteristics with normal enteric microbial flora. Conversely, Salmonella species are not part of the normal flora and have the capacity to cause invasive disease in normal hosts. The molecular mechanisms accounting for differences in the pathology of disease in the gastrointestinal tract remains unexplained in spite of the reported use of the cystic fibrosis transmembrane conductance regulator by both Salmonella typhi and P. aeruginosa to enter epithelial cells (50–52). Recently, it has been reported that S. typhi LPS is altered by contact with epithelial cells, exposing the bacterial ligand for cystic fibrosis transmembrane conductance regulator on the LPS (53). Similar findings in P. aeruginosa–epithelial cell interaction could further clarify the molecular mechanisms of invasion for this bacterial species.
Our previous study demonstrated that noncytotoxic isolates of P. aeruginosa, including PAO1, expressed minimal epithelial cell damage; PAO1 did not induce LDH release from MDCK cells or the formation of plaques as detected by staining with trypan blue at 6 h after infection; cytotoxic isolates showed clear evidence of cell damage within 3 h (4). In this study, changes in TER of monolayers infected with the efflux mutants, in particular the mexAB-oprM deletion strain, were substantially less than for the WT (Fig. 2). These findings may mean that bacterial invasion may disrupt the tight junctions between epithelial cells even though they do not induce demonstrable cytotoxicity.
The supernates from MDCK cells infected overnight with WT restored the capacity of the ΔmexAB-oprM strain K1119 to penetrate monolayers in a concentration-dependent manner (Fig. 7). When the WT strain was added from the basolateral side, neither the basolateral nor apical supernates showed any enhancing effect (data not shown). The enhancement of invasion by the K1119 (ΔmexAB-oprM) supernate was modest, and much less than by the WT supernate, suggesting that other efflux systems may export virulence determinants, albeit to a lesser degree than for MexAB-OprM. These findings strongly indicate that WT exports invasion determinant(s) into the extracellular environment, using predominantly MexAB-OprM (with lesser contributions by MexXY-OprM or other undetermined systems). It remains to be determined if the putative determinant(s) of invasiveness act directly as invasive factor(s) or as regulatory factor(s). Although it is still possible that mammalian products could be involved in the findings, studies are currently underway to purify and determine the molecular nature of the determinant(s) in the bacterial supernate which confer invasiveness.
The recovery of invasiveness of the mexAB-oprM deletion strain, mediated by the supernate of WT, required cell-to-cell contact between the bacterium and MDCK cells (Fig. 7). These data suggest that the type III protein secretion system (for a review, see reference 54) may be involved in the expression of invasiveness in P. aeruginosa. Type III secretion systems require close contact between bacterium and eukaryotic cell in order to deliver virulent bacterial proteins directly into the cytoplasm of the cell. ExoU and some other exoenzymes are recognized type III effectors in P. aeruginosa (55). In other bacterial species such as Salmonella, the bacteria modulate actin organization to induce their own uptake by nonphagocytic cells (56, 57). Efflux protein homologues can secrete proteins since type I secretion systems are RND homologues (58). However, the relation of efflux systems to the type III protein secretion system has not been demonstrated to date, and proteins secreted by the type III systems are likely too large for export via MDR efflux systems. Therefore, it is possible that contact of bacteria with the epithelial cells may be a trigger to stimulate the efflux of invasiveness determinant(s).
Our findings in this study strongly suggest that invasion determinant(s) are predominantly exported by P. aeruginosa via MexAB-OprM. Hence, MDR efflux systems in P. aeruginosa might be critical for the efflux of virulence factors, in addition to their established role of exporting harmful substances such as antibiotics or detergents. It seems practical for bacteria to utilize efflux systems to export virulence determinants and physiological products, as a physiological process. In fact, recent reports suggest that E. coli expels bacterial cellular metabolites such as indole and bile acids using the efflux systems AcrEF (59) and AcrAB (60), respectively. Our findings are consistent with a novel physiological role for such “multidrug” efflux systems, namely in enhancing bacterial invasiveness in P. aeruginosa infections. Furthermore, efflux inhibitors, which are under development to depress MDR by efflux systems (61), may also serve to reduce bacterial invasiveness in P. aeruginosa infections.
Acknowledgments
We are grateful to B. Brett Finlay for helpful suggestions. We thank Shadi Neshat and Takeshi Murata for the construction of mutants; Kelly MacDonald for serum sensitivity testing; Manjeet Bains for technical expertise and discussions; and Elaine Humphrey and Ashka Patel for helping in the sample preparation for electron microscopy.
Y. Hirakata was supported by Uehara Memorial Foundation, Canadian Cystic Fibrosis Foundation (CCFF), and British Columbia Lung Association, and by a Grant-in-Aid for Scientific Research in 2001 (13877042) from Japan Society for the Promotion of Science. R. Srikumar was a recipient of a Postdoctoral Fellowship from the CCFF. K. Poole and D.P. Speert were supported by grants from the CCFF. K. Poole is a CCFF scholar. R.E.W. Hancock is a Canada Research Chair in Microbiology and his research was supported by the Canadian Institutes of Health Research.
Footnotes
Abbreviations used in this paper: ANOVA, analysis of variance; LB, Luria-Bertani; MDCK, Madin-Darby canine kidney; MDR, multidrug resistant; OM, outer membrane; RND, resistance-nodulation-division; TER, transmonolayer electrical resistance; WT, wild-type.
References
- 1.Hirakata, Y., K. Poole, R. Srikumar, S. Kamihira, S. Kohno, B.B. Finlay, R.E.W. Hancock, and D.P. Speert. 2001. MexAB-OprM and MexXY-OprM systems play an important role in the invasiveness of Pseudomonas aeruginosa 101st General Meeting. Am. Soc. Microbiol. 276:D5 (Abstr. [Google Scholar]
- 2.Furuya, N., Y. Hirakata, K. Tomono, T. Matsumoto, K. Tateda, M. Kaku, and K. Yamaguchi. 1993. Mortality rates amongst mice with endogenous septicaemia caused by Pseudomonas aeruginosa isolates from various clinical sources. J. Med. Microbiol. 39:141–146. [DOI] [PubMed] [Google Scholar]
- 3.Hirakata, Y., K. Izumikawa, T. Yamaguchi, S. Igimi, N. Furuya, S. Maesaki, K. Tomono, Y. Yamada, S. Kohno, K. Yamaguchi, and S. Kamihira. 1998. Adherence to and penetration of human intestinal Caco-2 epithelial cell monolayers by Pseudomonas aeruginosa. Infect. Immun. 66:1748–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirakata, Y., B.B. Finlay, D.A. Simpson, S. Kohno, S. Kamihira, and D.P. Speert. 2000. Penetration of clinical isolates of Pseudomonas aeruginosa through MDCK epithelial cell monolayers. J. Infect. Dis. 181:765–769. [DOI] [PubMed] [Google Scholar]
- 5.Finck-Barbancon, V., J. Goranson, L. Zhu, T. Sawa, J.P. Wiener-Kronish, S.M. Fleiszig, C. Wu, L. Mende-Mueller, and D.W. Frank. 1997. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 25:547–557. [DOI] [PubMed] [Google Scholar]
- 6.Hauser, A.R., P.J. Kang, and J.N. Engel. 1998. PepA, a secreted protein of Pseudomonas aeruginosa, is necessary for cytotoxicity and virulence. Mol. Microbiol. 27:807–818. [DOI] [PubMed] [Google Scholar]
- 7.Hancock, R.E.W. 1998. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 27:S93–S99. [DOI] [PubMed] [Google Scholar]
- 8.Hancock, R.E.W., and D.P. Speert. 2000. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist. Update. 3:247–255. [DOI] [PubMed] [Google Scholar]
- 9.Hirakata, Y., K. Izumikawa, T. Yamaguchi, H. Takemura, H. Tanaka, R. Yoshida, J. Matsuda, M. Nakano, K. Tomono, S. Maesaki, et al. 1998. Rapid detection and evaluation of clinical characteristics of emerging multiple-drug-resistant gram-negative rods carrying metallo-β-lactamase gene bla IMP. Antimicrob. Agents Chemother. 42:2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poole, K., D.E. Heinrichs, and S. Neshat. 1993. Cloning and sequence analysis of an EnvCD homologue in Pseudomonas aeruginosa: regulation by iron and possible involvement in the secretion of the siderophore pyoverdine. Mol. Microbiol. 10:529–544. [DOI] [PubMed] [Google Scholar]
- 11.Poole, K., K. Krebes, C. McNally, and S. Neshat. 1993. Multiple antibiotic resistance in Pseudomonas aeruginosa: evidence for involvement of an efflux operon. J. Bacteriol. 175:7363–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotoh, N., H. Tsujimoto, K. Poole, J. Yamagishi, and T. Nishino. 1995. The outer membrane protein OprM of Pseudomonas aeruginosa is encoded by oprK of the mexA-mexB-oprK multidrug resistance operon. Antimicrob. Agents Chemother. 39:2567–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, X.Z., H. Nikaido, and K. Poole. 1995. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39:1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole, K., N. Gotoh, H. Tsujimoto, Q. Zhao, A. Wada, T. Yamasaki, S. Neshat, J. Yamagishi, X.Z. Li, and T. Nishino. 1996. Overexpression of the mexC-mexD-oprJ efflux operon in nfxB-type multidrug-resistant strains of Pseudomonas aeruginosa. Mol. Microbiol. 21:713–724. [DOI] [PubMed] [Google Scholar]
- 15.Kohler, T., M. Michea-Hamzehpour, U. Henze, N. Gotoh, L.K. Curty, and J.C. Pechere. 1997. Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23:345–354. [DOI] [PubMed] [Google Scholar]
- 16.Mine, T., Y. Morita, A. Kataoka, T. Mizushima, and T. Tsuchiya. 1999. Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43:415–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westbrock-Wadman, S., D.R. Sherman, M.J. Hickey, S.N. Coulter, Y.Q. Zhu, P. Warrener, L.Y. Nguyen, R.M. Shawar, K.R. Folger, and C.K. Stover. 1999. Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob. Agents Chemother. 43:2975–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole, K. 2001. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 3:255–264. [PubMed] [Google Scholar]
- 19.Poole, K. 2000. Efflux-mediated resistance to fluoroquinolones in gram-negative bacteria. Antimicrob. Agents Chemother. 44:2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikaido, H. 1998. Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin. Infect. Dis. 27:S32–S41. [DOI] [PubMed] [Google Scholar]
- 21.Zgurskaya, H.I., and H. Nikaido. 2000. Multidrug resistance mechanisms: drug efflux across two membranes. Mol. Microbiol. 37:219–225. [DOI] [PubMed] [Google Scholar]
- 22.Aires, J.R., T. Kohler, H. Nikaido, and P. Plesiat. 1999. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 43:2624–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans, K., L. Passador, R. Srikumar, E. Tsang, J. Nezezon, and K. Poole. 1998. Influence of the MexAB-OprM multidrug efflux system on quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 180:5443–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans, K., and K. Poole. 1999. The MexA-MexB-OprM multidrug efflux system of Pseudomonas aeruginosa is growth-phase regulated. FEMS Microbiol. Lett. 173:35–39. [DOI] [PubMed] [Google Scholar]
- 25.Pearson, J.P., C. Van Delden, and B.H. Iglewski. 1999. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 181:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohler, T., M. Michea-Hamzehpour, U. Henze, N. Gotoh, L.K. Curty, and J.C. Pechere. 1977. Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa Mol. Microbiol. 23:345–354. [DOI] [PubMed] [Google Scholar]
- 27.Kohler, T., C. van Delden, L.K. Curty, M. Michea-Hamzehpour, and J.C. Pechere. 2001. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183:5213–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooun, A., S. Liu, and K. Lewis. 2000. A dose–response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44:640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Kievit, T.R., M.D. Parkins, R.J. Gillis, R. Srikumar, H. Ceri, K. Poole, B.H. Iglewski, and D.G. Storey. 2001. Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 45:1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finlay, B.B., M.N. Starnbach, C.L. Francis, B.A. Stocker, S. Chatfield, G. Dougan, and S. Falkow. 1988. Identification and characterization of TnphoA mutants of Salmonella that are unable to pass through a polarized MDCK epithelial cell monolayer. Mol. Microbiol. 2:757–766. [DOI] [PubMed] [Google Scholar]
- 31.Finlay, B.B., B. Gumbiner, and S. Falkow. 1988. Penetration of Salmonella through a polarized Madin-Darby canine kidney epithelial cell monolayer. J. Cell Biol. 107:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleiszig, S.M., T.S. Zaidi, M.J. Preston, M. Grout, D.J. Evans, and G.B. Pier. 1996. Relationship between cytotoxicity and corneal epithelial cell invasion by clinical isolates of Pseudomonas aeruginosa. Infect. Immun. 64:2288–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirakata, Y., M. Kaku, K. Tomono, K. Tateda, N. Furuya, T. Matsumoto, R. Araki, and K. Yamaguchi. 1992. Efficacy of erythromycin lactobionate for treating Pseudomonas aeruginosa bacteremia in mice. Antimicrob. Agents Chemother. 36:1198–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirakata, Y., N. Furuya, K. Tateda, M. Kaku, and K. Yamaguchi. 1993. In vivo production of exotoxin A and its role in endogenous Pseudomonas aeruginosa septicemia in mice. Infect. Immun. 61:2468–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masuda, N., and S. Ohya. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36:1847–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer, A., A. Tauch, W. Jager, J. Kalinowski, G. Thierbach, and A. Puhler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 145:69–73. [DOI] [PubMed] [Google Scholar]
- 37.Li, X.Z., L. Zhang, R. Srikumar, and K. Poole. 1998. β-lactamase inhibitors are substrates for the multidrug efflux pumps of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srikumar, R., X.Z. Li, and K. Poole. 1997. Inner membrane efflux components are responsible for β-lactam specificity of multidrug efflux pumps in Pseudomonas aeruginosa. J. Bacteriol. 179:7875–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srikumar, R., T. Kon, N. Gotoh, and K. Poole. 1998. Expression of Pseudomonas aeruginosa multidrug efflux pumps MexA-MexB-OprM and MexC-MexD-OprJ in a multidrug-sensitive Escherichia coli strain. Antimicrob. Agents Chemother. 42:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keen, N.T., S. Tamaki, D. Kobayashi, and D. Trollinger. 1988. Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene. 70:191–197. [DOI] [PubMed] [Google Scholar]
- 41.Tsuda, M., H. Miyazaki, and T. Nakazawa. 1995. Genetic and physical mapping of genes involved in pyoverdine production in Pseudomonas aeruginosa PAO. J. Bacteriol. 177:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prentki, P., A. Binda, and A. Epstein. 1991. Plasmid vectors for selecting IS1-promoted deletions in cloned DNA: sequence analysis of the omega interposon. Gene. 103:17–23. [DOI] [PubMed] [Google Scholar]
- 43.Tsuda, M. 1998. Use of a transposon-encoded site-specific resolution system for construction of large and defined deletion mutations in bacterial chromosome. Gene. 207:33–41. [DOI] [PubMed] [Google Scholar]
- 44.Gotoh, N., H. Tsujimoto, M. Tsuda, K. Okamoto, A. Nomura, T. Wada, M. Nakahashi, and T. Nishino. 1998. Characterization of the MexC-MexD-OprJ multidrug efflux system in mexA-mexB-oprM mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42:1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gotoh, N., H. Tsujimoto, A. Nomura, K. Okamoto, M. Tsuda, and T. Nishino. 1998. Functional replacement of OprJ by OprM in the MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 165:21–27. [DOI] [PubMed] [Google Scholar]
- 46.Apodaca, G., M. Bomsel, R. Lindstedt, J. Engel, D. Frank, K.E. Mostov, and J. Wiener-Kronish. 1995. Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation-defective host cells are resistant to bacterial killing. Infect. Immun. 63:1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleiszig, S.M., J.P. Wiener-Kronish, H. Miyazaki, V. Vallas, K.E. Mostov, D. Kanada, T. Sawa, T.S. Yen, and D.W. Frank. 1997. Pseudomonas aeruginosa-mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzyme S. Infect. Immun. 65:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroeder, T.H., N. Reiniger, G. Meluleni, M. Grout, F.T. Coleman, and G.B. Pier. 2001. Transgenic cystic fibrosis mice exhibit reduced early clearance of Pseudomonas aeruginosa from the respiratory tract. J. Immunol. 166:7410–7418. [DOI] [PubMed] [Google Scholar]
- 49.Speert, D.P., M.E. Campbell, A.G. Davidson, and L.T. Wong. 1993. Pseudomonas aeruginosa colonization of the gastrointestinal tract in patients with cystic fibrosis. J. Infect. Dis. 167:226–229. [DOI] [PubMed] [Google Scholar]
- 50.Pier, G.B. 2000. Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc. Natl. Acad. Sci. USA. 97:8822–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pier, G.B., M. Grout, T. Zaidi, G. Meluleni, S.S. Mueschenborn, G. Banting, R. Ratcliff, M.J. Evans, and W.H. Colledge. 1998. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 393:79–82. [DOI] [PubMed] [Google Scholar]
- 52.Pier, G.B., M. Grout, T.S. Zaidi, J.C. Olsen, L.G. Johnson, J.R. Yankaskas, and J.B. Goldberg. 1996. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 271:64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyczak, J.B., T.S. Zaidi, M. Grout, M. Bittner, I. Contreras, and G.B. Pier. 2001. Epithelial cell contact-induced alterations in Salmonella enterica serovar Typhi lipopolysaccharide are critical for bacterial internalization. Cell. Microbiol. 3:763–772. [DOI] [PubMed] [Google Scholar]
- 54.Hueck, C.J. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dacheux, D., B. Toussaint, M. Richard, G. Brochier, J. Croize, and I. Attree. 2000. Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect. Immun. 68:2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galan, J.E., and A. Collmer. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 284:1322–1328. [DOI] [PubMed] [Google Scholar]
- 57.Galan, J.E. 1999. Interaction of Salmonella with host cells through the centisome 63 type III secretion system. Curr. Opin. Microbiol. 2:46–50. [DOI] [PubMed] [Google Scholar]
- 58.Doung, F., A. Lazdunski, B. Cami, and M. Murgier. 1992. Sequence of a cluster of genes controlling synthesis and secretion of alkaline protease in Pseudomonas aeruginosa: relationships to other secretory pathways. Gene. 121:47–54. [DOI] [PubMed] [Google Scholar]
- 59.Kawamura-Sato, K., K. Shibayama, T. Horii, Y. Iimuma, Y. Arakawa, and M. Ohta. 1999. Role of multiple efflux pumps in Escherichia coli in indole expulsion. FEMS Microbiol. Lett. 179:345–352. [DOI] [PubMed] [Google Scholar]
- 60.Thanassi, D.G., L.W. Cheng, and H. Nikaido. 1997. Active efflux of bile salts by Escherichia coli. J. Bacteriol. 179:2512–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lomovskaya, O., M.S. Warren, A. Lee, J. Galazzo, R. Fronko, M. Lee, J. Blais, D. Cho, S. Chamberland, T. Renau, et al. 2001. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]