

Abstract
The Fas (CD95) gene is among critical genetic factors in some autoimmune diseases, which are characterized by autoantibody (autoAb) productions. In mice, mutations in the Fas gene cause lymphoproliferation (lpr) which predominantly develops glomerulonephritis, whereas the mutations in human cause autoimmune lymphoproliferative syndrome (ALPS) characterized by autoimmune hemolytic anemia (AIHA) and thrombocytopenia. Although the mechanism of antinuclear Ab in Fas-deficient background has been well characterized, that of antierythrocyte Ab production in ALPS has been still unclear. To investigate this mechanism, we developed a mouse line by crossing the antierythrocyte antibody transgenic mice (H+L6 mice) and Fas-deficient mice. Although Fas deficiency did not break tolerance of autoreactive B-2 cells in H+L6 mice, autoreactive B-1 cells in Fas-deficient H+L6 homozygous mice became activated and differentiated into autoAb-producing cells in mesenteric lymph nodes and lamina propria of intestine, resulting in severe anemia. In addition, serum levels of interleukin (IL)-10 significantly increased in Fas−/− × H+L6 homozygous mice and administration of anti–IL-10 Ab prevented exacerbation of autoAb production and AIHA. These results suggest that activation of B-1 cells is responsible for induction of AIHA in Fas-deficient condition and that IL-10 plays a critical role in terminal differentiation of B-1 cells in these mice.
Keywords: autoantigen, autoantibody, B cell tolerance, autoimmune lymphoproliferative syndrome, peritoneal cavity
Introduction
Autoimmune diseases are the outcome of complicated inflammatory processes in which various cellular and humoral components are involved. In some autoimmune diseases such as myasthenia gravis, autoimmune hemolytic anemia (AIHA), and idiopathic thrombocytopenic purpura, pathogenic autoAbs produced by B cells primarily mediate the disorders (1–3). The pathogenic autoAbs directly bind to membrane-bound antigens and cause autoimmune diseases, whereas other disease-related autoAbs such as rheumatoid factor do not directly trigger clinical manifestations (4).
Fas (CD95) and Fas ligand (FasL) genes are among the critical genetic factors for autoAb production in mice and human (1, 5, 6). Fas is expressed in a variety of cell types including activated B cells and T cells, whereas FasL is only expressed in activated T cells and NK cells. Interaction of Fas and FasL induces rapid apoptosis and plays a critical role in homeostasis of peripheral lymphocytes and prevention of autoimmunity (5, 6). In mice, mutations in Fas and FasL genes cause lymphoproliferation (lpr) and generalized lymphoproliferative disease (gld), respectively (5, 6). MRL/lpr mice develop glomerulonephritis with lymphadenopathy and splenomegaly, caused by accumulation of CD4−CD8− T cells and production of autoAbs to intracellular antigens (5). The production of autoAbs was blocked by the specific expression of transgenic (Tg) Fas on B cells but not on T cells in lpr/lpr mice (7, 8), suggesting that Fas deficiency in B cells is responsible for autoAb productions. Several studies using Ig Tg/lpr mice have shown that central tolerance to soluble form of model antigens is partially impaired in Fas deficiency and that production of disease-associated autoAbs, such as anti-DNA Ab, rheumatoid factor, and anti-Sm Ab is markedly enhanced (9–12). However, it is not clear whether production of pathogenic autoAbs against membrane-bound antigens is affected in Fas deficiency.
In humans, mutations in Fas gene cause autoimmune lymphoproliferative syndrome (ALPS) (13). ALPS patients uniquely develop AIHA, idiopathic thrombocytopenic purpura, and autoimmune neutropenia, in addition to the common symptoms to MRL/lpr mice (13). Furthermore, ALPS patients but not MRL/lpr mice show increased numbers of CD5+ B cells and elevated levels of serum IL-10 (13–15). These results suggest that CD5+ B cells and IL-10 may play a role in production of pathogenic autoAbs to membrane-bound self-antigens.
We have generated several Tg mouse lines (H×L and H+L) in which almost all B cells have specificity for a membrane-bound antigen on self-RBC and cause AIHA (2). As autoreactive B cells are eliminated at the immature stages in the bone marrow, the number of mature B cells is markedly decreased in the periphery of these mice. In contrast, their peritoneal cavity (PerC) contains autoreactive B-1 cells, that can be activated to produce autoAb by IL-10, inducing AIHA (16, 17).
To assess whether production of pathogenic autoAb against membrane-bound self-antigens is affected in Fas deficiency, we crossed Fas-deficient mice and H+L6 mice. Autoreactive B-1 cells in Fas-deficient H+L6 homozygous mice were activated to induce severe anemia. In addition, serum levels of IL-10 significantly increased in these mice and administration of anti–IL-10 Ab blocked exacerbation of autoAb production and anemia. These results suggest that activation of B-1 cells, triggered by IL-10, is responsible for induction of AIHA in Fas-deficient condition.
Materials and Methods
Tg Mice.
We generated several lines of the anti-RBC mAb (4C8 mAb) Tg (H+L) mice which carried tandem joined H and L chain transgenes (18). By mating H+L6 heterozygous mice and Fas-deficient mice (19), we obtained Fas-deficient H+L6 homozygous mice. The genotype was determined by PCR of tail DNA. The PCR primers were described previously (18, 19). The homozygosity of the transgene was screened by Southern analysis. The mice were maintained under conventional conditions in our animal facility and were analyzed at 8–12 wk of age.
Detection of Anti-RBC AutoAb Production.
The amounts of autoAbs on RBCs were measured as described previously (20).
Preparation of Single Cell Suspensions.
Isolation of lamina propria (LP) lymphocytes from the small intestine and preparation of cells from bone marrow, mesenteric lymph nodes (MLNs), and PerC were done as described previously (20).
Flow Cytometry.
Flow cytometric analysis was performed by a FACSCalibur™ with CELLQuest™ software version 3.1 (Becton Dickinson) as described previously (20). After excluding dead cells by propidium iodide gating, cells present in the lymphocyte gate defined by forward and side light scatters were analyzed.
Enzyme-linked Immunospot Assay and Cytoplasmic Staining.
Enzyme-linked immunospot (ELISPOT) assay and cytoplasmic staining were performed on freshly isolated cells from lymphoid organs as described previously (20).
Cytokine ELISA.
The levels of serum IL-4, IL-5, IL-6, and IL-10 were assessed using the mouse IL-4–, IL-5–, IL-6–, and IL-10–ELISA systems (Amersham Pharmacia Biotech) according to the manufacturer's protocol.
Administration of Anti–Mouse IL-10 Ab.
Either rat anti–mouse IL-10 mAb (100 μg/injection; Genzyme) or control rat IgG (100 μg/injection; BD PharMingen) was injected intraperitoneally into 4-wk-old Fas-deficient H+L6 homozygous mice weekly for 4 wk.
Results
Fas Deficiency Markedly Enhances AutoAb Production and Anemia in H+L6 Homozygous Mice.
To assess how autoimmunity for membrane-bound autoantigens develops in Fas deficiency, we crossed H+L6 mice with Fas−/− mice and compared the phenotypes of H+L6 heterozygous or homozygous mice. In H+L6 mice, the size of Tg B-1 cell compartment in PerC is larger in homozygous than heterozygous as described previously (18). As bacterial infection and/or normal bacterial flora can induce autoAb production of B-1 cells in H×L mice (2) and conventional conditions represent the situations that ALPS patients are exposed in daily life, we bred H+L6 mice under conventional conditions.
First, we examined the amount of the autoAb bound to circulating RBCs by flow cytometry and the incidence of anemia by measuring hematocrit (Ht) values of the peripheral blood (Fig. 1). Neither the level of the autoAb nor the Ht values was changed between Fas+/− and Fas−/− H+L6 heterozygous mice. In contrast, a small amount of the autoAb was detected in Fas+/− × H+L6 homozygous mice and their Ht values were slightly reduced compared with Fas+/− × H+L6 heterozygous mice. More importantly, in Fas−/− × H+L6 homozygous mice, the amount of the autoAb was markedly increased and Ht values were drastically reduced. These results indicate that Fas deficiency enhances autoAb production and causes severe anemia in only H+L6 homozygous mice.
Figure 1.

Marked enhancement of autoAb production and anemia in H+L6 homozygous mice by Fas deficiency. (A) Amounts of anti-RBC autoAb bound to circulating RBCs. Peripheral blood RBCs were incubated with FITC-anti–mouse IgM antibody and analyzed by flow cytometry. Closed triangles indicate the mean fluorescence intensity of IgM on RBCs in individual mice. Horizontal long and short bars indicate the mean and SD of each group, respectively. (B) Hematocrit values of peripheral blood. Closed triangles indicate the hematocrit value of individual mice. Horizontal bars are as described in A. The Student's t test for unpaired data was used to compare the values between indicated groups.
Localization of Autoreactive B Cells and AutoAb-producing Cells in H+L6 Homozygous Mice Carrying Fas Deficiency.
As H+L6 homozygous mice contain a larger population of autoreactive B-1 cells than H+L6 heterozygous mice, it is possible that B-1 cells may be involved in the autoAb production in these mice. To examine the localization of autoreactive B cells, we stained cells in bone marrow, spleen, MLNs, PerC, and LP of the gut with the anti-IgM and anti-idiotype (S54) Abs and analyzed them by flow cytometry. As both anti-IgM and anti-idiotype Abs stain determinants on the same Ig molecule, the Tg B cells which express both Tg IgH and IgL on their surface were stained tight diagonal as described previously (10, 18, 21). Thus, this allows cleaner recognition and enumeration of truly autoreactive B cells.
A significant number of IgM+S54+ cells, expressing Mac-1 (data not shown), were observed in PerC of H+L6 homozygous mice, and their compartment was much larger in H+L6 homozygous than in heterozygous mice (Fig. 2 A). Importantly, although IgM+S54+ cells were slightly increased in PerC of Fas−/− × H+L6 heterozygous mice compared with the Fas+/− mice, the IgM+S54+ cells were significantly reduced in Fas−/− × H+L6 homozygous mice compared with the Fas+/− mice. Neither bone marrow or spleen contained distinct IgM+S54+ cells in H+L6 homozygous mice (Fig. 2 B), because of clonal deletion of autoreactive B-2 cells as described previously (18). In contrast, both MLNs and LP in Fas−/− × H+L6 homozygous mice had a small but significant number of IgM+S54+ cells. Taken together, the absence of B-2 cells in the periphery and the decrease of B-1 cells in PerC with concomitant appearance of IgM+S54+ cells in MLNs and LP are in parallel with the observation we described previously (20). Thus, these results strongly suggest that the autoreactive B-1 cells in PerC may have migrated to MLNs and LP and differentiated into plasma cells in Fas−/− × H+L6 homozygous mice. Staining of IgM+S54+ cells in MLNs and LP differs from that in PerC. This is probably because of lower surface Ig expression on Ig-secreting cells in MLNs and LP.
Figure 2.


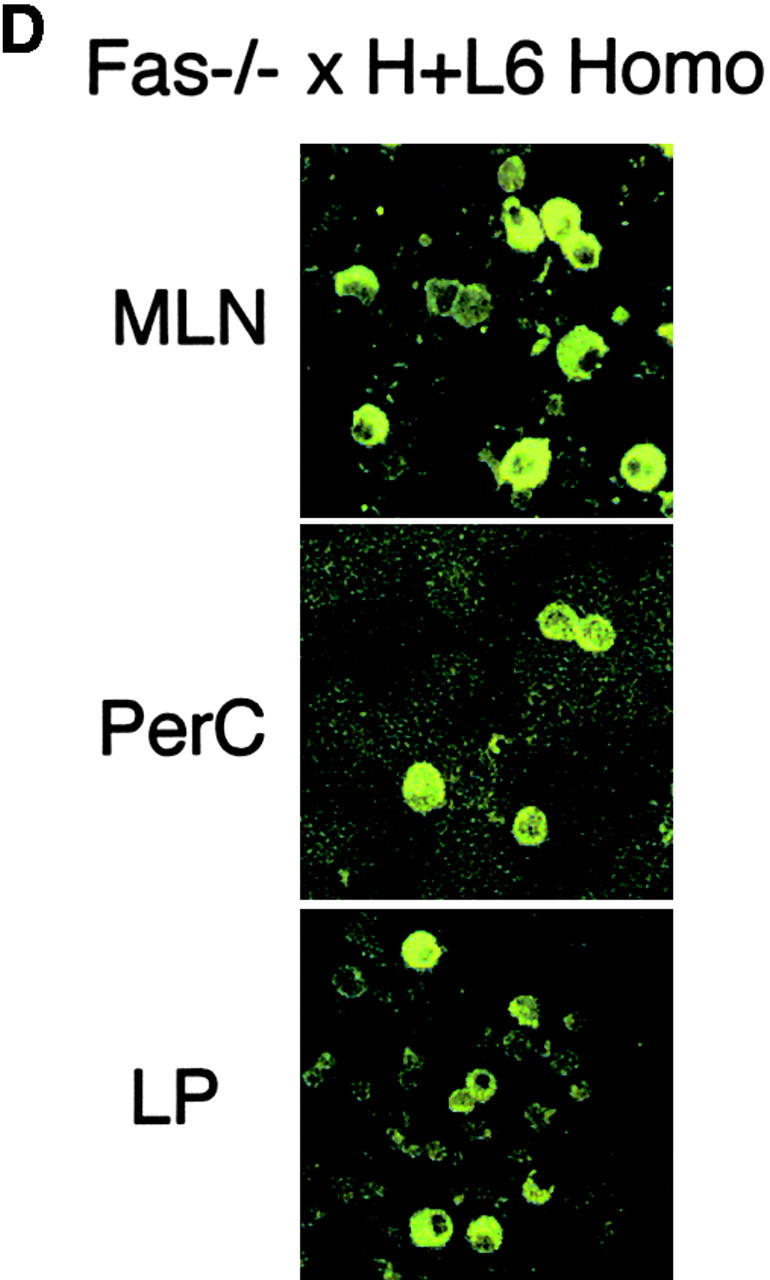
Localization of autoreactive B cells and autoAb-producing cells in Fas-deficient H+L6 homozygous mice. (A) Flow cytometry analysis of PerC in Fas+/− (left panels) or Fas−/− (right panels) × H+L6 homozygous, heterozygous, and non-Tg mice. Cells were stained with FITC–anti-IgM and biotin-conjugated anti-idiotype mAb followed by PE-conjugated SA. The percentages of the cells for a given phenotype in viable lymphocyte gate are shown. (B) Flow cytometry analysis of bone marrow (BM), spleen (SPL), MLN, and LP of Fas+/− (left panels) or Fas−/− (right panels) × H+L6 homozygous mice. (C) Numbers of IgM-producing cells of Fas+/− (white bars) or Fas−/− (black bars) × H+L6 homozygous mice in BM, SPL, MLN, PerC, and LP were measured by ELISPOT assay. The mean and SD of IgM-producing cells per 105 cells was calculated from at least three mice. (D) Cells were isolated from MLN, PerC, and LP of Fas−/− × H+L6 homozygous mice, and then 2 × 105 cells of each were fixed, and stained with FITC-anti–mouse IgM antibody. Preparations were examined and photographed by fluorescence microscope. Original magnifications: each panel ×400.
To show where the autoAb is produced in Fas−/− × H+L6 homozygous mice, we measured the numbers of autoAb-producing cells in the bone marrow, spleen, MLNs, PerC, and LP (Fig. 2 C). As H+L6 homozygosity strongly blocks the rearrangement of endogenous Ig genes, almost all IgM+ cells in these mice express Tg IgM (18). Therefore, we performed ELISPOT assay using anti-IgM Ab. Although only small numbers of IgM-producing cells were observed in Fas+/− × H+L6 homozygous mice, we detected large numbers of Ab-producing cells in MLNs and LP, but not in other organs, of Fas−/− × H+L6 homozygous mice.
As we found a difference in the distribution between surface Ig expressing cells and IgM-producing cells, we further confirmed staining pattern and morphology by cytoplasmic IgM staining. Almost all IgM+ cells in PerC of Fas−/− × H+L6 homozygous mice showed surface staining pattern of IgM (Fig. 2 D). In contrast, the majority of IgM+ cells in MLNs and LP had typical plasma cell morphology. These IgM+ plasma cells probably have lost some of surface Ig expression. Taken together, these results suggest that the autoreactive PerC B-1 cells may migrate and differentiate in MLNs and LP. They are also consistent with our previous observation that the autoAb production predominantly occurred in MLNs in recombination activating gene (RAG)2−/− × H×L mice with activated γδ T cells (20).
Serum Levels of IL-10 But Not IL-4, IL-5, and IL-6 Are Significantly Elevated in Fas-deficient H+L6 Homozygous Mice.
To examine which cytokine is involved in B-1 cell migration and terminal differentiation in Fas−/− × H+L6 homozygous mice, serum concentrations of IL-4, IL-5, IL-6, and IL-10 were assessed by ELISA. The levels of IL-10 but not IL-4, IL-5, and IL-6 were significantly elevated in Fas−/− × H+L6 homozygous mice compared with the other control mice (Fig. 3). Thus, these results suggest that the elevation of IL-10 may play a critical role in autoreactive B-1 cell migration and terminal differentiation in Fas−/− × H+L6 homozygous mice, as described previously (17, 20). In addition, although the levels of IL-10 in Fas−/− non-Tg mice were not increased (data not shown), the levels of IL-10 in Fas−/− × H+L6 heterozygous mice were slightly but significantly increased when compared with Fas+/− control mice. Therefore, the increase of serum IL-10 concentration closely correlated with the migration and terminal differentiation of autoreactive B-1 cells in Fas deficiency.
Figure 3.
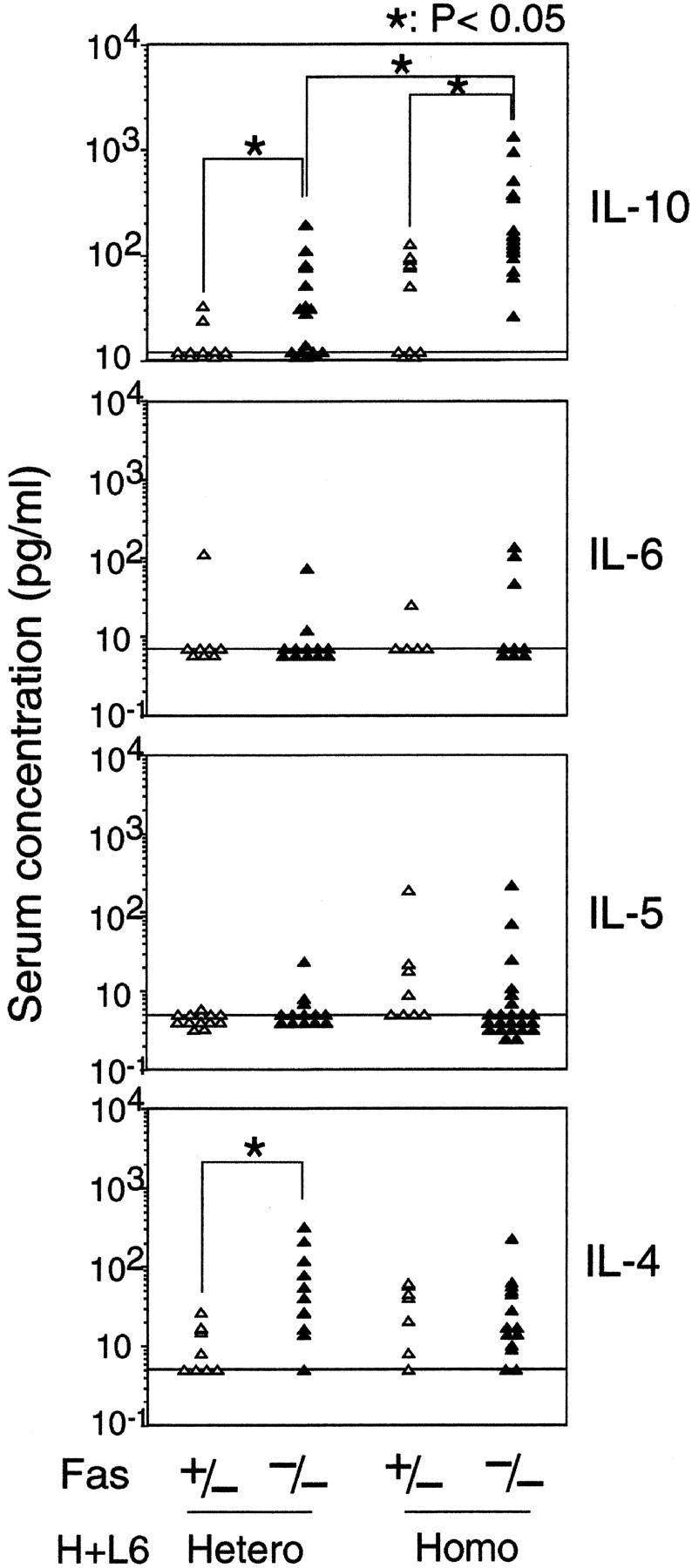
The serum levels of IL-10, but not IL-4, IL-5, and IL-6 were elevated in Fas-deficient H+L6 homozygous mice. Amounts of IL-4, IL-5, IL-6, and IL-10 in the serum were assessed in H+L6 heterozygous and homozygous mice with (closed triangles) or without (open triangles) Fas-deficient background by ELISA. Horizontal long bars indicate the minimum detectable levels of each cytokine. The Student's t test for unpaired data was used to compare the values between indicated groups.
Administration of Anti–IL-10 Ab Prevents Exacerbation of Anemia in Fas-deficient H+L6 Homozygous Mice.
To test whether the elevated IL-10 levels play an essential role in autoAb production in H+L6 homozygous mice, we injected the anti–IL-10 mAb into Fas-deficient H+L6 homozygous mice weekly for 4 wk. Injection of anti–IL-10 Ab blocked reduction of Ht values in Fas−/− × H+L6 homozygous mice compared with control rat IgG injection (Table I). IL-10 neutralization also suppressed increase in the amount of anti-RBC Ab and the numbers of autoAb-producing cells in the MLN of Fas−/− × H+L6 homozygous mice. These results clearly demonstrate that the elevation of serum IL-10 is responsible for the enhancement of autoAb production in Fas−/− × H+L6 homozygous mice.
Table I.
Administration of Anti–IL-10 mAb Blocks Exacerbation of Anemia in Fas-deficient H+L6 Homozygous Mice
| Treatments | Hematocrit values (%) |
Amounts of the autoAb bound to RBCs (mean fluorescence intensity) |
Ig-producing cells (per 105 cells) |
|---|---|---|---|
| Anti–IL-10 | 37.5 ± 2.5a | 494.4 ± 364.2b | 235.8 ± 173.1b |
| Control Ig | 31.5 ± 0.5 | 1,419.2 ± 497.2 | 660.6 ± 240.2 |
Either anti–IL-10 mAb or control rat IgG (100 μg/injection) was injected weekly into Fas-deficient H+L6 homozygous mice. 4 wk after the first injection, hematocrit values (%) were measured. Amounts of the autoAb bound to RBCs were assessed as mean fluorescence intensity of FITC-conjugated anti-IgM Ab bound to RBCs. Numbers of anti-RBC Ab-producing cells in MLN were measured by ELISPOT assay using anti-IgM Ab. Values represent the mean ± SD of five mice in each experimental group. Paired t test values calculated between anti–IL-10 and control rat IgG.
P < 0.01.
P < 0.02.
Discussion
In this study, we found marked autoAb production for RBC membrane Ag in only H+L6 homozygous but not in heterozygous mice under Fas-deficient background. Despite the same Ag specificity, higher levels of surface Tg immunoglobulin expression of H+L6 homozygous mice results in stronger clonal deletion of autoreactive B-2 cells from periphery compared with heterozygous mice. At the same time, the size of autoreactive B-1 cell compartment in PerC is larger in homozygous than in heterozygous mice (18). Thus, our results suggest that B-1 cells are responsible for autoAb production in Fas-deficient background. Indeed, in Fas−/− × H+L6 homozygous mice, autoreactive B-1 cells can efficiently migrate and differentiate into plasma cells (Fig. 2, A–D). In contrast, in H+L6 heterozygous mice, Fas deficiency did not alter the size of the B-1 cell population (data not shown, and Fig. 2 A).
Because Fas deficiency enhances migration and differentiation but not expansion of autoreactive B-1 cells, secondary genetic factors responsible for expansion of B-1 cells may be required for induction of AIHA. Indeed, genetic background plays a critical role for autoAb production and autoimmune diseases in lpr mice and activation of B-1 cells in PerC (22, 23). In human, additional genetic factors are also suggested to be involved in lymphoproliferation and autoimmunity in ALPS (24).
Although we have shown that the serum levels of IL-10 were significantly elevated in Fas−/− × H+L6 homozygous mice, we could not identify a particular cell population as the main source of IL-10 in vivo. Our preliminary experiments of intracellular cytokine staining suggested that not only B-1 cells but also aberrant T cells and macrophages in PerC of H+L6 homozygous mice can produce significant amount of IL-10 by ex vivo stimulation (unpublished data). Indeed, in ALPS patients, aberrant CD4−CD8− T cells with activated phenotypes accumulate in peripheral lymphoid organs and can produce large amounts of IL-10 (15). B-1 cells can also produce IL-10 (25, 26). In addition, we previously showed that both activated T cells and macrophages are involved in increased serum levels of IL-10 and in migration and differentiation of peritoneal B-1 cells (17, 20). Thus, aberrant T cells, autoreactive B-1 cells, and macrophages may cooperate in elevation of IL-10, resulting in activation of autoreactive B-1 cells in Fas-deficient condition.
How IL-10 induces activation of autoreactive B-1 cells is not yet clear. In vivo role of IL-10 in murine B cell function is limited (27). However, several previous studies suggest that IL-10 is important for B-1 cell expansion and activation (23, 28, 29). We have also shown that IL-10 elevation induced by LPS administration or IL-10 administration itself can induce autoAb production and anemia in H×L mice (16, 17). However, we could not detect direct effects of IL-10 for proliferation or differentiation of autoreactive B-1 cells (17). Furthermore, we did not find anti-apoptotic effect for B-1 cells in PerC against RBC injection in Fas−/− × H+L6 homozygous mice (data not shown). On the other hand, Fas-deficient background markedly enhances migration and differentiation of autoreactive B-1 cells to not only MLNs but also LP in H+L6 homozygous mice, whereas autoreactive B-1 cells in RAG2−/− × H×L mice with activated γδT cells predominantly migrate into MLNs but not LP (20). Thus, Fas deficiency may help autoreactive B-1 cells to terminally differentiate and migrate into LP, escaping from rapid apoptosis. Recently, Balabanian et al. have shown that IL-10 increases the effects of stromal cell–derived factor-1 on the proliferation and survival of B-1 cells in PerC and that IL-10 is chemokinetic for peritoneal B-1 cells, increasing their random mobility (30). Thus, these effects of IL-10 may play a role in indirect activation of autoreactive B-1 cells of Fas−/− × H+L6 homozygous mice.
In summary, we have shown that autoreactive B-1 cells in PerC migrate into MLNs and LP and differentiate into Ab-producing cells with Fas-deficient background. Elevated IL-10 levels are involved in the activation of autoreactive B-1 cells. These results are in parallel with the observation that ALPS patients with AIHA manifest increased numbers of CD5+ B cells and elevated serum levels of IL-10, implying that blocking of IL-10 signals may become a therapeutic approach to autoimmune diseases in ALPS patients.
Acknowledgments
We are grateful to Drs. Shigekazu Nagata and Hidehiro Fukuyama for their kind gift of Fas-deficient mice. We thank Dr. S. Fagarasan for critical discussion, and Ms. T. Taniuchi and Ms. M. Tanaka for their excellent technical assistance.
This work is supported by grants for Center of Excellence program from the Ministry of Education, Science, Sports and Culture of Japan.
References
- 1.Marrack, P., J. Kappler, and B.L. Kotzin. 2001. Autoimmune diseases: why and where it occurs. Nat. Med. 7:899–905. [DOI] [PubMed] [Google Scholar]
- 2.Fagarasan, S., N. Watanabe, and T. Honjo. 2002. Experimental models of autoimmune hemolytic anemia. The Molecular Pathology of Autoimmune Diseases, 2nd ed. A.N. Theofilopoulos and C.A. Bona, editors. Taylor and Francis, New York. 522–542.
- 3.Cines, D.B., and V.S. Blanchette. 2002. Immune thrombocytopenic purpura. N. Engl. J. Med. 346:995–1008. [DOI] [PubMed] [Google Scholar]
- 4.Lernmark, A. 2001. Autoimmune diseases: are markers ready for prediction? J. Clin. Invest. 108:1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 6.Nagata, S. 1997. Apoptosis by death factor. Cell. 88:355–365. [DOI] [PubMed] [Google Scholar]
- 7.Fukuyama, H., M. Adachi, S. Suematsu, K. Miwa, T. Suda, N. Yoshida, and S. Nagata. 1998. Transgenic expression of Fas in T cells blocks lymphoproliferation but not autoimmune disease in MRL-lpr mice. J. Immunol. 160:3805–3811. [PubMed] [Google Scholar]
- 8.Komano, H., Y. Ikegami, M. Yokoyama, R. Suzuki, S. Yonehara, Y. Yamasaki, and N. Shinohara. 1999. Severe impairment of B cell function in lpr/lpr mice expressing transgenic Fas selectively on B cells. Int. Immunol. 11:1035–1042. [DOI] [PubMed] [Google Scholar]
- 9.Elkon, K.B., and A. Marshak-Rothstein. 1996. B cells in systemic autoimmune disease: recent insights from Fas-deficient mice and men. Curr. Opin. Immunol. 8:852–859. [DOI] [PubMed] [Google Scholar]
- 10.Kench, J.A., D.M. Russell, and D. Nemazee. 1998. Efficient peripheral clonal elimination of B lymphocytes in MRL/lpr mice bearing autoantibody transgenes. J. Exp. Med. 188:909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, H., and M.J. Shlomchik. 1999. Autoantigen-specific B cell activation in Fas-deficient rheumatoid factor immunoglobulin transgenic mice. J. Exp. Med. 190:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santulli-Marotto, S., Y. Qian, S. Ferguson, and S.H. Clarke. 2001. Anti-Sm B cell differentiation in Ig transgenic MRL/Mp-lpr/lpr mice: Altered differentiation and an accelerated response. J. Immunol. 166:5292–5299. [DOI] [PubMed] [Google Scholar]
- 13.Straus, S.E., M.C. Sneller, M. Lenardo, J.M. Puck, and W. Strober. 1999. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann. Intern. Med. 130:591–601. [DOI] [PubMed] [Google Scholar]
- 14.Lim, M.S., S.E. Straus, J.K. Dale, T.A. Fleisher, M. Stetler-Stevenson, W. Strober, M.C. Sneller, J.M. Puck, M.J. Lenardo, K.S. Elenitoba-Johnson, et al. 1998. Pathological findings in human autoimmune lymphoproliferative syndrome. Am. J. Pathol. 153:1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopatin, U., X. Yao, R.K. Williams, J.J.H. Bleesing, J.K. Dale, D. Wong, J. Teruya-Feldstein, S. Fritz, M.R. Morrow, I. Fuss, et al. 2001. Increases in circulating and lymphoid tissue interleukin-10 in autoimmune lymphoproliferative syndrome are associated with disease expression. Blood. 97:3161–3170. [DOI] [PubMed] [Google Scholar]
- 16.Nisitani, S., T. Tsubata, M. Murakami, and T. Honjo. 1995. Administration of interleukin-5 or -10 activates peritoneal B-1 cells and induces autoimmune hemolytic anemia in anti-erythrocyte autoantibody-transgenic mice. Eur. J. Immunol. 25:3047–3052. [DOI] [PubMed] [Google Scholar]
- 17.Nisitani, S., T. Sakiyama, and T. Honjo. 1998. Involvement of IL-10 in induction of autoimmune hemolytic anemia in anti-erythrocyte Ig transgenic mice. Int. Immunol. 10:1039–1047. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe, N., S. Nisitani, K. Ikuta, M. Suzuki, T. Chiba, and T. Honjo. 1999. Expression levels of B cell surface immunoglobulin regulate efficiency of allelic exclusion and size of autoreactive B-1 cell compartment. J. Exp. Med. 190:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adachi, M., S. Suematsu, T. Kondo, J. Ogasawara, T. Tanaka, N. Yoshida, and S. Nagata. 1995. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat. Genet. 11:294–300. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe, N., K. Ikuta, S. Fagarasan, S. Yazumi, T. Chiba, and T. Honjo. 2000. Migration and differentiation of autoreactive B-1 cells induced by activated γ/δ T cells in antierythrocyte immunoglobulin transgenic mice. J. Exp. Med. 192:1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin, X.F., S. Schwers, W. Yu, F. Papavasiliou, H. Suh, A. Nussenzweig, K. Rajewsky, and M.C. Nussenzweig. 1999. Secondary V(D)J recombination in B-1 cells. Nature. 397:355–359. [DOI] [PubMed] [Google Scholar]
- 22.Vidal, S., D.H. Kono, and A.N. Theofilopoulos. 1998. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Fas lpr mice. J. Clin. Invest. 101:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velupillai, P., W.E. Secor, A.M. Horauf, and D.A. Harn. 1997. B-1 cell (CD5+B220+) outgrowth in murine schistosomiasis is genetically restricted and is largely due to activation by polylactosamine sugars. J. Immunol. 158:338–344. [PubMed] [Google Scholar]
- 24.Rieux-Laucat, F., S. Blachere, S. Danielan, J.P. de Villartay, M. Oleastro, E. Solary, B. Bader-Meunier, P. Arkwright, C. Pondare, F. Bernaudin, et al. 1999. Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations. Blood. 94:2575–2582. [PubMed] [Google Scholar]
- 25.O'Garra, A., G. Stapleton, V. Dhar, M. Pearce, J. Schumacher, H. Rugo, D. Barbis, A. Stall, J. Cupp, K. Moore, et al. 1990. Production of cytokines by mouse B cells: B lymphomas and normal B cells produce interleukin 10. Int. Immunol. 2:821–832. [DOI] [PubMed] [Google Scholar]
- 26.O'Garra, A., R. Chang, N. Go, R. Hastings, G. Haughton, and M. Howard. 1992. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur. J. Immunol. 22:711–717. [DOI] [PubMed] [Google Scholar]
- 27.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 28.Ramachandra, S., R.A. Metcalf, T. Fredrickson, G.E. Marti, and E. Raveche. 1996. Requirement for increased IL-10 in the development of B-1 lymphoproliferative disease in a murine model of CLL. J. Clin. Invest. 98:1788–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velupillai, P., J. Sypek, and D.A. Harn. 1996. Interleukin-12 and -10 and gamma interferon regulate polyclonal and ligand-specific expansion of murine B-1 cells. Infect. Immun. 64:4557–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balabanian, K., A. Foussat, L. Bouchet-Delbos, J. Couderc, R. Krzysiek, A. Amara, F. Baleux, A. Portier, P. Galanaud, and D. Emilie. 2002. Interleukin-10 modulates the sensitivity of peritoneal B lymphocytes to chemokines with opposite effects on stromal cell-derived factor-1 and B-lymphocyte chemoattractant. Blood. 99:427–436. [DOI] [PubMed] [Google Scholar]