

Abstract
Natural killer (NK) T cells initiate potent antitumor responses when stimulated by exogenous factors such as interleukin (IL)-12 or α-galactosylceramide (α-GalCer), however, it is not clear whether this reflects a physiological role for these cells in tumor immunity. Through adoptive transfer of NK T cells from wild-type to NK T cell–deficient (T cell receptor [TCR] Jα281−/−) mice, we demonstrate a critical role for NK T cells in immunosurveillance of methylcholanthrene (MCA)-induced fibrosarcomas, in the absence of exogenous stimulatory factors. Using the same approach with gene-targeted and/or antibody-depleted donor or recipient mice, we have shown that this effect depends on CD1d recognition and requires the additional involvement of both NK and CD8+ T cells. Interferon-γ production by both NK T cells and downstream, non-NK T cells, is essential for protection, and perforin production by effector cells, but not NK T cells, is also critical. The protective mechanisms in this more physiologically relevant system are distinct from those associated with α-GalCer–induced, NK T cell–mediated, tumor rejection. This study demonstrates that, in addition to their importance in tumor immunotherapy induced by IL-12 or α-GalCer, NK T cells can play a critical role in tumor immunosurveillance, at least against MCA-induced sarcomas, in the absence of exogenous stimulation.
Keywords: rodent, NK T cells, NK cells, tumor immunity, methylcholanthrene
Introduction
NK T cells are a specialized subset of T cells that, in mice, are commonly characterized by the coexpression of NK1.1, and a heavily biased αβ TCR, with the great majority expressing an invariant Vα14/Jα281 TCR α-chain coupled with Vβ8.2, 7 or 2 TCR-β chains (for a review, see references 1 and 2). This TCR specifically recognizes glycolipid-Ag in conjunction with the MHC class I–like molecule, CD1d (for a review, see references 2 and 3). Disruption of either the invariant TCR-α chain, or CD1d, results in a selective deficiency of NK T cells, while all other lymphocyte subsets remain intact (4–7). Upon TCR stimulation, NK T cells rapidly produce both proinflammatory cytokines, (e.g. IFN-γ and TNF-α) and antiinflammatory cytokines (e.g. IL-4, IL-10, IL-13), suggesting an important role for these cells in immunoregulation. Several studies have shown that NK T cells can suppress cell-mediated immunity and prevent self-tissue destruction. For example, NK T cells prevent pancreatic islet β cell destruction in NOD mice in an IL-4–/IL-10–dependent manner (8), mediate systemic suppression associated with anterior chamber-associated immune deviation via IL-10 production (9), induce allograft tolerance (10), and prevent graft-versus-host disease in mice (11). NK T cells have also been shown to suppress tumor antigen-specific, CTL-mediated tumor rejection in an IL-13–dependent manner (12).
In apparent contrast with the above studies, NK T cells promote potent tumor rejection in response to exogenous factors such as IL-12 (4, 13, 14) and α-galactosylceramide (α-GalCer)* (15–17). α-GalCer–induced NK T cell–dependent antitumor activity is dependent on IFN-γ production, and requires NK cells, (18–20), while the role for CD8+ T cells is controversial (19, 20). Despite the clear ability of NK T cells to initiate potent antitumor responses in response to exogenous immunotherapeutic stimuli, whether this represents a physiological role for NK T cells in tumor rejection remains unclear, with two recent studies suggesting an opposing role for NK T cells in tumor immunity. Terabe et al. (12) reported that NK T cells were responsible for incomplete tumor regression by IL-13–mediated inhibition of tumor-specific CTL, suggesting that NK T cells may normally inhibit tumor immunity, perhaps reflecting their immunosuppressive role in autoimmune disease. In contrast, we previously reported that NK T cell–deficient (TCR Jα281−/−) mice were more susceptible to methylcholanthrene (MCA)-induced fibrosarcoma, suggesting an aggressive role for NK T cells in preventing growth of these tumors (21). A disadvantage of the latter study was that it provided little insight into the role of NK T cells and NK T cell–derived factors in this model. It was not clear if NK T cells were directly killing the tumor in a perforin (pfp)-dependent fashion, as suggested by our in vitro studies, (21) or if they were acting indirectly via the production of factors which lead to the activation of downstream effectors such as NK cells as has been demonstrated in α-GalCer–induced systems (18–20). It was also unknown whether the NK T cell response, in the absence of α-GalCer, was dependent on an interaction between TCR and CD1d, which might suggest recognition of tumor-derived glycolipid antigen. In this study, using adoptive transfer of NK T cells from wild-type (WT) or various gene-targeted mice, we demonstrate unequivocally that NK T cells can play a critical role in immunosurveillance of MCA-induced fibrosarcomas, and reveal the mechanisms behind this process.
Materials and Methods
Mice.
Inbred C57BL/6 WT mice were purchased from The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia, and Clear Japan, Inc. The following gene-targeted mice were bred at the Peter MacCallum Cancer Institute: C57BL/6-TCRJα281-deficient (Jα281−/−) (provided by M. Taniguchi, Chiba University Graduate School of Medicine, Chiba, Japan and backcrossed to C57BL/6 for nine generations) (4); C57BL/6 pfp-deficient (pfp−/−) (from G. Karupiah, John Curtin School of Medical Research, Canberra, Australia and derived from C57BL/6 ES cells) (22); C57BL/6 IFN-γ deficient (IFN-γ−/−; from Genentech, Inc. and backcrossed to C57BL/6 for 10 generations) (23); C57BL/6 CD1d deficient (CD1d−/−; provided by L. van Kaer, Vanderbilt University School of Medicine, Nashville, TN and backcrossed to C57BL/6 for 10 generations) (6); and C57BL/6 recombination activation gene (RAG)-1 deficient (RAG-1−/−; from L. Corcoran, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia and backcrossed to C57BL/6 for 10 generations). Mice of 6–14 wk of age were used in all experiments that were performed according to animal experimental ethics committee guidelines.
Isolation of Liver Lymphocyte Subsets.
Lymphocytes were isolated from the liver as described previously (24). To avoid nonspecific binding of antibodies to FcR-γ, cells were preincubated with anti–mouse CD16/32 (2.4G2) mAb (grown in-house) before staining with FITC-conjugated anti-αβ TCR (clone H57–597) and PE-conjugated anti-NK1.1 (clone PK-136). All flow cytometry reagents were purchased from BD PharMingen, unless otherwise indicated. Cells were gated and sorted as described previously (24). After washing twice with PBS, the stained cells were analyzed on a FACStarPLUS™ (Becton Dickinson) and the data processed by the CELLQuest™ program (Becton Dickinson).
Tumor Models In Vivo.
MCA-1, -3, -4 and CD1.1 were derived from Jα281−/− and CD1d−/− mice, respectively, injected with 100 μg MCA. B16F10 mouse melanoma and MCA-induced sarcoma cell lines were maintained as described previously (20, 21). For growth of MCA-induced sarcoma lines, groups of WT and gene targeted and/or Ab-depleted mice were injected subcutaneously (right hind leg) with 105 sarcoma cells. On the same day, some mice received liver lymphocytes from WT or gene-targeted mice or FACS®-purified liver lymphocyte populations from WT mice, or 2% normal mouse serum (NMS) in PBS (2% NMS.PBS) via intravenous adoptive transfer. In the delayed transfer experiments, some mice received liver lymphocytes 7 d after tumor inoculation. All mice were observed every other day and tumor growth was measured with a caliper square as the product of two diameters. Results were recorded as the mean tumor size (cm2) ± SEM. Significant difference in the number of mice which remained susceptible to tumor development compared with PBS-treated Jα281−/− groups was determined using a Fisher's exact test (*P ≤ 0.05; **P ≤ 0.01). Significant difference in tumor growth rate, compared with PBS-treated control groups, was determined using a Mann-Whitney U test (n ≥ 4). In the B16F10 subcutaneous growth model, groups of WT mice were injected subcutaneously (hind leg) with 105 MCA-1 cells, and varying doses of B16F10 were injected subcutaneously within the same region at a distinct site. All mice were observed every other day and B16F10 subcutaneous tumor growth was measured with a caliper square as the product of two diameters. Results were recorded as the mean tumor size (cm2) ± SEM.
AsialoGM1 and CD8 Depletion.
NK cells were specifically depleted in Jα281−/− mice after intraperitoneal injection of 20 μg rabbit antiasialoGM1 Ab (Wako Chemicals) on days −1, 0, and 7 (day 0 being day of tumor inoculation) as described previously (25). CD8+ cells were specifically depleted using anti-CD8-depleting Ab (clone 53–6.7) (grown in-house) using the same approach.
Results
NK T Cell–deficient Mice Are Susceptible To Sarcoma Growth.
We have previously shown that NK T cell–deficient, TCR Jα281−/−, mice (Fig. 1 A) are more susceptible to MCA-induced fibrosarcomas and a fibrosarcoma line (MCA-1) when compared with WT mice, suggesting an important role for NK T cells in preventing the growth of these tumors (21). This phenotype can be demonstrated using several distinct fibrosarcoma lines (Fig. 1 B), including some that were generated from MCA-treated CD1d−/− mice. These data are consistent with previous observations that sarcomas derived from RAG-2−/− mice grow preferentially in RAG-2−/− mice over syngenic WT mice (26).
Figure 1.


NK T cell–deficient (Jα281−/−) mice are more susceptible to growth of MCA-induced sarcoma lines. (A) Liver lymphocytes were isolated from WT and Jα281−/− mice and labeled with CD1/α-GalCer tetramer and mAb specific for αβ TCR to identify NK T cells. (B) Groups of WT and Jα281−/− mice were injected subcutaneously (hind flank) with 105 MCA-1, -3, -4 or CD1.1 sarcoma cell lines. Results were recorded every other day as the mean tumor size (in cm2) ± SEM. Three mice per group were used. Significant difference from the PBS-treated Jα281−/− group was determined using a Mann-Whitney U test when n ≥ 4. *P ≤ 0.05.
Adoptive Transfer of NK T Cells into Jα281−/− Mice Restores Protection Against MCA-1 Sarcoma.
As the most obvious difference between WT and Jα281−/− mice is the presence of CD1d-restricted NK T cells (Fig. 1 A) (4, 27, 28), we sought to restore protection in Jα281−/− recipients treated with MCA-1 sarcoma by adoptive transfer of NK T cells. In initial experiments, total liver lymphocytes were transferred, since NK T cells are highly abundant within this population of cells, and we could avoid using mAb to engage cell surface molecules for purification. Over 80% of MCA-1-injected Jα281−/− mice treated with WT liver lymphocytes were able to completely inhibit the growth of MCA-1 (Fig. 2 A), while the rest had significantly smaller tumors (approximately one-fifth the size, measured in cm2; P ≤ 0.01) than PBS-injected controls (unpublished data). This was similar to the tumor incidence observed in WT mice, the majority of which remained tumor free. An additional control group of MCA-1–injected Jα281−/− mice were treated with Jα281−/−-derived liver lymphocytes, which have all populations except NK T cells. These mice remained susceptible to tumor growth and were indistinguishable from PBS-injected controls. Protection was also achieved when transferring WT liver lymphocytes into TCRJα281−/− mice that had been inoculated with either MCA-3 or MCA-4 tumor cells (unpublished data). This data strongly suggested that inhibition of growth was due to the transfer of CD1d-restricted NK T cells present among the WT liver lymphocytes.
Figure 2.


Adoptive transfer of NK T cells protects Jα281−/− mice against MCA-1 tumor growth. Groups of WT and Jα281−/− mice were inoculated subcutaneously (hind flank) with 105 MCA-1 cells. (A) Subgroups of Jα281−/− mice then received between 1 and 3 × 106 WT or 3 × 106 Jα281−/− liver lymphocytes, 200 μL 2% NMS.PBS, or (B) 2.5 × 105 purified NK1.1+αβ TCR+ (NK T), NK1.1+αβ TCR− (NK), NK1.1− αβ TCR+ (T) cells, or 200 μL 2% NMS.PBS, via intravenous adoptive transfer. NK T cells typically constituted between 30–35% of WT liver lymphocytes, and sorted populations were at least 97, 93, and 94% pure, respectively. Results represent pooled data from 10 (A) or 4 (B) independent experiments (with exception of Jα281−/− liver lymphocytes [A] and purified NK cell [B] transfers from one experiment only, and purified T cell transfers [B] from two experiments). Between three and six mice per group per experiment were used. Total number of mice in each group that developed tumors is shown in parentheses. Significant difference from the PBS-treated Jα281−/− groups was determined using a Fisher's exact test. *P ≤ 0.05; **P ≤ 0.01.
Having established that liver lymphocytes, probably NK T cells, could protect in this system in the absence of cell surface molecule engagement by mAb, we sought to formally define which liver lymphocyte subset was responsible for protection. NK1.1+ αβ TCR+ (NK T cells), NK1.1−αβ TCR+ (T cells), and NK1.1+αβ TCR− (NK cells) were purified by FACS® and adoptively transferred intravenously into MCA-1–injected Jα281−/− mice (Fig. 2 B). Mice that received NK T cells (2.5 × 105) were able to inhibit the growth of MCA-1, while those receiving the same number of T cells or NK cells could not, specifically defining NK T cells as the population responsible for restoring protection.
Dose Response for NK T Cell–mediated Protection Against MCA-1.
To determine the threshold number of NK T cells required to restore protection, several liver lymphocyte doses (3 × 106, 106, 0.33 × 106, and 0.11 × 106) (Fig. 3 A), and purified NK T cell doses (2.5 × 105, 105, and 5 × 104) (Fig. 3 B) were tested. Inhibition of tumor growth was clearly dose–dependent, with 106 liver lymphocytes (∼3.0 × 105 NK T cells) and 2.5 × 105 purified NK T cells required for optimal prevention of tumor growth. As the total number of NK T cells in the protective dose of liver lymphocytes was very similar to that of purified NK T cells, it was unlikely that mAb-binding altered the functional status of transferred NK T cells. Although lower doses of NK T cells failed to completely prevent tumor growth, partial protection was observed with smaller and slower growing tumors corresponding to the number of NK T cells injected.
Figure 3.
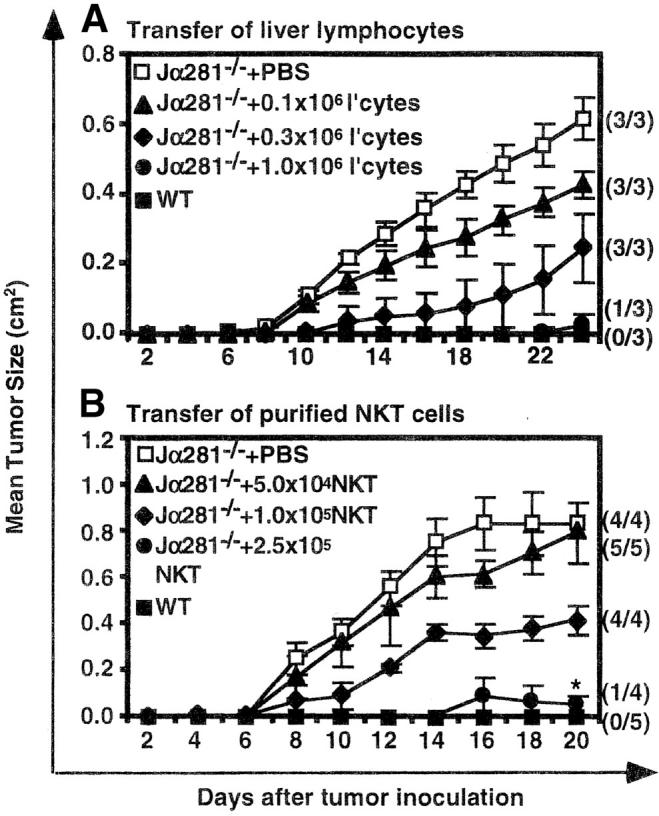
Dose response for NK T cell–mediated protection against MCA-1. Groups of WT and Jα281−/− mice were injected subcutaneously (hind flank) with 105 MCA-1 cells. (A) Subgroups of Jα281−/− mice then received either 3 × 106, 106, 0.33 × 106, 0.11 × 106 liver lymphocytes or 200 μL 2% NMS.PBS or (B) 2.5 × 105, 105, 5 × 104 purified NK1.1+αβ TCR+ (NK T) cells or 200 μL 2% NMS.PBS, via intravenous adoptive transfer. NK T cells constituted 31% of the liver lymphocyte population and sorted NK T cells were enriched to 98%. Results were recorded as the mean tumor size (cm2) ± SEM. Between three and five mice per group were used. Significant difference from the PBS-treated Jα281−/− groups was determined using a Mann-Whitney U test when n ≥ 4. *P ≤ 0.05.
Factors that Are Critical for NK T Cell–mediated Protection.
Having established that transfer of NK T cells effectively protected Jα281−/− mice from tumor growth, we sought to determine which NK T cell molecules were mediating this protection. When liver lymphocytes from IFN-γ−/− mice were used as a source of NK T cells, they were unable to prevent tumor growth (Fig. 4 A), despite the ability of endogenous cells in these Jα281−/− recipients to produce IFN-γ, indicating an essential role for NK T cell–derived IFN-γ. In contrast, pfp−/− liver lymphocytes retained the ability to inhibit tumor growth, indicating that direct, pfp-dependent, tumor lysis by NK T cells was not an essential mechanism in this model.
Figure 4.
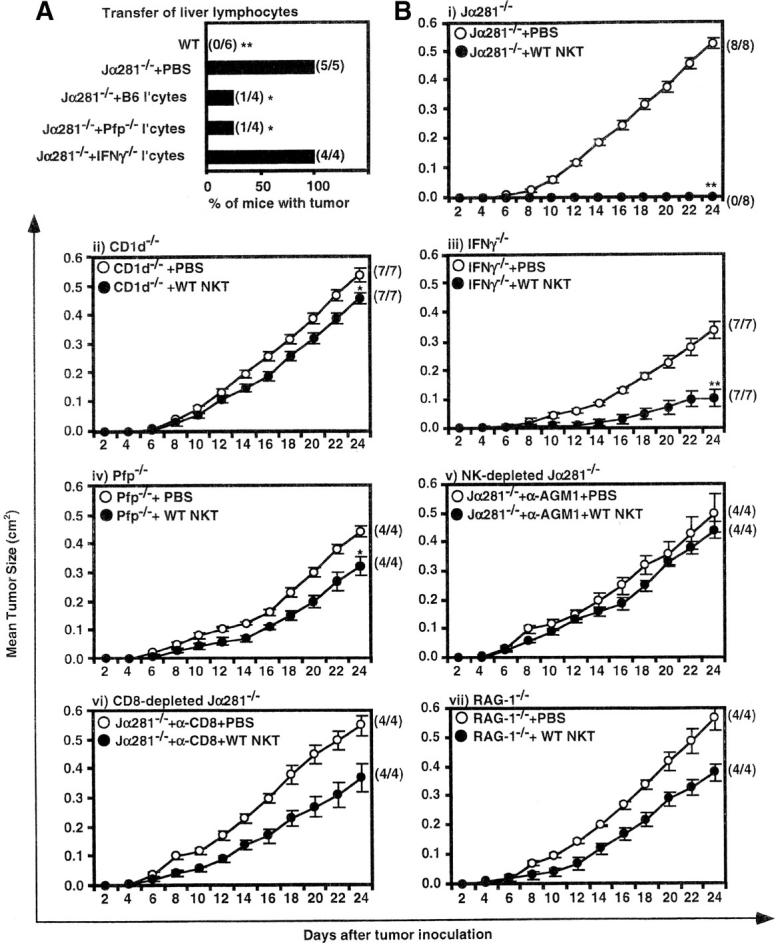
Factors that are critical for NK T cell–mediated protection. (A) WT or Jα281−/− mice were injected subcutaneously (hind flank) with 105 MCA-1 cells. Subgroups of Jα281−/− mice were simultaneously treated with either PBS, or 3 × 105 NKT cells, as part of a liver lymphocyte population, from either WT, IFN-γ−/−, or pfp−/− donor mice via intravenous adoptive transfer. Significant difference from the PBS-treated Jα281−/− group was determined by a Fisher's exact test. *P ≤ 0.05; **P ≤ 0.01. (B) Groups of (i) Jα281−/−, (ii) CD1d−/−, (iii) IFN-γ−/−, (iv) pfp−/−, (v) Jα281−/− mice treated with depleting anti-asGM1 Ab, (vi) Jα281−/− mice treated with depleting anti-CD8, and (vii) RAG-1−/− mice, were injected subcutaneously (hind flank) with 105 MCA-1 cells. One-half of each group of mice were simultaneously injected with 2.5 × 105 sorted WT NK T cells via intravenous adoptive transfer, while the other half were injected intravenously with 2% NMS.PBS. Sorted NK T cells were always at least 97% pure as determined by FACS®. (B) i, ii, and iii represent pooled data from two independent experiments, iv represents data from one of these experiments, v, vi, and vii represent data from one independent experiment with three to six mice per group per experiment. Significant difference from the PBS-treated control group of each experiment was determined using a Mann-Whitney U test. *P ≤ 0.05; **P ≤ 0.01.
As it was unlikely that NK T cells were acting alone, we further investigated what other cells and/or molecules were important downstream of, or in conjunction with, NK T cell activation. Purified WT NK T cells were transferred into various gene-targeted and/or mAb-depleted mice (Fig. 4 B i–vii). Similar to Jα281−/− mice (Fig. 4 B i), CD1d−/− mice were susceptible to MCA-1 growth (Fig. 4 B ii). However, in contrast to Jα281−/− recipients, NK T cell transfer failed to prevent tumor growth in CD1d−/− mice (Fig. 4 B ii), indicating that CD1d expression by cells other than the tumor, such as APCs, was critical for NK T cell–mediated immunosurveillance. CD1d expression by the tumor itself was not essential; MCA-1 is close to, if not completely, negative for CD1d expression (unpublished data) and, as shown above, a CD1d−/− tumor line grew in Jα281−/− but not WT mice (Fig. 1 B).
IFN-γ−/− recipients were also susceptible to MCA-1 growth, and transfer of (IFN-γ sufficient) WT NK T cells did not restore complete protection (Fig. 4 B ii), although growth was slower when compared with PBS-injected IFN-γ−/− mice. It is possible that transferred WT NK T cells may provide sufficient IFN-γ to confer initial protection, however, IFN-γ production by downstream non-NK T cells is clearly critical for effective eradication of the tumor. Pfp-deficient recipients were susceptible to MCA-1 growth (Fig. 4 B iv), and WT NK T cells were unable to protect in these mice, although again, the tumor grew at a slower rate when these cells were transferred. This indicated that, although NK T cell–derived pfp was not critical (Fig. 4 A), pfp-expressing NK T cells may be partially effective at controlling this tumor. However, pfp production by other cells, possibly NK or T cells, is clearly important for efficient NK T cell–mediated tumor immunosurveillance (Fig. 4 B iv). It was also noteworthy that both PBS-treated IFN-γ−/− and pfp−/− mice had smaller and slower growing tumors when compared with PBS-injected Jα281−/− mice (Fig. 4 B i, iii, and iv; P ≤ 0.01 and P ≤ 0.05, respectively). This suggests that NK T cells present within IFN-γ−/− and pfp−/− mice may still be able to use other, IFN-γ/pfp-independent, pathways/mechanisms to protect against MCA-1 tumor growth, although clearly, NK T cell–derived and non-NK T cell–derived IFN-γ, and pfp-expression by downstream effector cells appear to be critical in providing optimal protection against this tumor. Transfer of NK T cells into MCA-1–injected, NK or CD8-depleted Jα281−/− mice, or RAG-1−/− mice, revealed that NK, CD8+, and T cells are important for a clear protective response induced by NK T cells (Fig. 4 B v–vii). Tumor growth in PBS-injected, NK, or CD8-depleted Jα281−/−, or RAG-1−/− mice (Fig. 4 B v–vii) was not significantly different from that of PBS-injected Jα281−/− mice.
NK T Cells Are Required Early in the Response Against MCA-1.
To investigate whether NK T cells could also protect against established tumors, Jα281−/− mice were treated with WT liver lymphocytes 7 d after MCA-1 injection (Fig. 5). Delayed transfer of WT cells had no effect on tumor growth, indicating that NK T cells are required either earlier in the response, or in significantly greater numbers for effective inhibition of established tumors.
Figure 5.

NK T cells are required early in the response against MCA-1. Groups of WT and Jα281−/− mice were inoculated subcutaneously (hind flank) with 105 MCA-1 cells (d0, □, ▵, ▪). 7 d later, some Jα281−/− mice were injected with 106 WT liver lymphocytes via intravenous adoptive transfer (▵). As a control to show the WT liver lymphocytes (NK T cells) were protective, other Jα281−/− mice were inoculated with MCA-1 on the same day as lymphocyte transfer (•). NK T cells constituted 27% of the whole liver lymphocyte population as determined by FACS®. Results were recorded as the mean tumor size (in cm2) ± SEM. Four mice per group were used.
MCA-1 Does Not Induce α-GalCer–like Activation of NK T Cells.
We have previously shown that α-GalCer can activate NK T cells that have been adoptively transferred into B16F10-inoculated NK T cell–deficient mice, to initiate potent tumor rejection (20). To investigate whether MCA-1 tumor activates NK T cells in a manner similar to α-GalCer, possibly by providing CD1d-binding glycolipid antigens, WT mice were simultaneously injected with B16F10 and MCA-1. Mice were injected subcutaneously on the hind flank with varying doses of B16F10, and some mice were also injected on the same flank, in an adjacent region, with MCA-1 (Fig. 6). As expected, MCA-1 growth was inhibited, presumably due to the presence of NK T cells, however, this response had no effect on the adjacent growth of B16F10 tumor. Similar results were achieved when B16F10 was injected intravenously into WT mice, some of which were also injected subcutaneous with MCA-1 (unpublished data). Since B16F10 growth and metastasis is profoundly inhibited after α-GalCer treatment of Jα281−/− mice (20) that have been injected with WT NK T cells, these data suggest that MCA-1 sarcoma does not activate NK T cells to act in a manner similar to that following α-GalCer stimulation.
Figure 6.

MCA-1 does not induce α-GalCer–like activation of NK T cells. Groups of WT mice were injected subcutaneously (hind flank) with either 5 × 106, 106, or 5 × 105 B16F10 cells. Some mice were then injected on the same hind flank with 105 MCA-1 cells. Results were recorded as the mean tumor size (in cm2) ± SEM with five mice per group.
Discussion
It is well established that NK T cells can be induced by either IL-12 or α-GalCer to mediate potent antitumor effects both in vitro and in vivo (4, 15, 16), however, this does not necessarily indicate that NK T cells play a natural/physiological role in tumor recognition and immunity. We recently published that Ja281−/− mice are more susceptible to development of MCA-induced fibrosarcomas when compared with WT mice, suggesting that NK T cells might be important in the prevention of tumor growth in this model (21). Herein, using adoptive transfer techniques and gene-targeted mice, we have clearly demonstrated a critical role for NKT cells in immunosurveillance against MCA-induced fibrosarcomas, and have also identified the key cells and molecules that support this process.
Although NK T cell activation in this system does not require treatment with an exogenous CD1d-binding ligand such as α-GalCer, CD1d expression by recipient cells, possibly APCs, was nonetheless found to be important, indicating that NK T cells were responding to these tumors in a TCR-dependent fashion. What remains to be determined is whether NK T cell activation is caused by the presentation of tumor-derived Ag in the context of CD1d, or rather, whether the presence of CD1d is simply required in addition to other, CD1d-independent, tumor-induced ‘danger signals.’ It is conceivable that NK T cells were being activated by a CD1d-binding endogenous ligand, mimicking the effects of exogenously added α-GalCer, however, the downstream effector functions after NK T cell activation by MCA-1 were clearly distinct from those following NK T stimulation by α-GalCer. In contrast to α-GalCer–induced tumor rejection (18, 20), NK T cell–mediated tumor immunosurveillance in this model was pfp dependent, involved both NK and CD8+ T cells, and most importantly, did not mimic the effect of α-GalCer in its ability to invoke NK T cell–mediated B16F10 rejection.
We previously published that NK T cells can kill MCA-1 in vitro in a pfp-dependent manner when induced by IL-2 and IL-12 (21). This suggested that the critical contribution of NK T cells in vivo was direct tumor cell lysis. However, transferred pfp−/− NK T cells retained their ability to prevent MCA-1 growth, indicating NK T cell–derived pfp was clearly not critical for protection. WT NK T cells transferred into pfp−/− mice slowed the growth of MCA-1, indicating that, although not essential, NK T cell–derived pfp may make a minor contribution to MCA-1 rejection. On the other hand, pfp-expression by recipient cells was critical, supporting the idea that NK T cell–mediated tumor immunosurveillance relies on downstream pfp-expressing effector cells. These may include NK and CD8+ T cells, both of which were required for optimal prevention of sarcoma growth.
NK cells are known to be activated downstream of NK T cells after α-GalCer stimulation (29, 30). This is clearly necessary, although apparently not sufficient, for NK T cell–mediated immunosurveillance in this sarcoma model. These findings are in concert with our previous study showing that NK depletion by anti-asGM1 abrogates protection against primary MCA-induced sarcomas in WT mice (25). The involvement of both NK and CD1d-dependent NK T cells has also been shown to be important in the cyclophosphamide/IL-12–induced regression of a large MCA-induced sarcoma (31). In contrast the role for CD8+ T cells in α-GalCer–mediated tumor rejection is controversial (19, 20), although we have now shown that they play a critical role in immunosurveillance of MCA-induced tumor cell lines. Although the susceptibility of CD8-depleted mice to MCA-1 supports previous findings involving other MCA-induced sarcoma lines (32), our earlier findings did not reveal a role for CD8+ cells in NK T cell–mediated protection against primary MCA-induction of sarcomas (21). At least two possibilities may explain this. The role of CD8+ cells may differ in primary responses to MCA-induced sarcomas when compared with transplanted sarcoma lines; or, as the current experiments involve repopulation of NK T cell–deficient mice with relatively low numbers of NK T cells, it may be possible that in the absence of normal NK T cell numbers, the role for other cells such as CD8+ T cells increases.
IFN-γ is a critical factor in α-GalCer–mediated tumor rejection (18–20). This was also the case for the natural tumor surveillance model described herein. Importantly, similar to the α-GalCer model (20), both NK T cell–derived and non-NK T cell–derived IFN-γ was required, suggesting it may function at multiple stages in this process. This cytokine appears to be important for the activation of the downstream effector cells (29, 30, 33), which would explain why it must be initially produced by NK T cells. Additionally, IFN-γ production by recipient effector cells might act to amplify the immune response, or may have antitumor effects of its own (34–36).
The in vivo migratory and homing behavior of NK T cells is very poorly understood, as is the impact that a tumor may have on this process. Using immunohistology, we have failed to detect a significant population of NK T cells at the tumor site in WT mice (unpublished data), nor have we been able to detect these cells, at the doses used for protection, after adoptive transfer into tumor-treated, and nontumor-treated, Jα281−/− mice (unpublished data). This is probably due to the fact that we are transferring very low numbers of NK T cells, combined with the possibility that they die shortly after being activated in response to the tumor, as has been reported after α-GalCer stimulation (28, 30, 37, 38). The model we favor is that NK T cells are rapidly stimulated by the presence of the tumor cell glycolipids, leading to potent NK T cell–derived IFN-γ production and early activation of effector cells, including NK and CD8+ T cells. These effector cells then directly attack the tumor through direct pfp-dependent lysis and IFN-γ secretion. The early recognition of the tumor appears to be key, as delayed transfer of NK T cells provided no detectable protection.
A role for NK T cells in tumor rejection is usually only observed after stimulation with exogenous therapeutic factors such as IL-12 and α-GalCer. In the absence of these factors, some tumors such as B16F10 melanoma and 3LL lung carcinoma grow readily, despite the presence of NK T cells in these mice. Nonetheless, these earlier studies hinted that with the right environment, NK T cells had the potential to mediate tumor immunosurveillance. An obvious question relates to why NK T cells require exogenous stimulation to respond to some tumors, such as B16F10, but not for others, such as the sarcomas used in this study. This may be related to the type of CD1d-binding glycolipid Ag expressed by these tumors, which may be specific for sarcomas, MCA-induced tumors, or other as yet unidentified molecules, that might be differentially expressed by various tumors, such as activating ligands (e.g. NKG2D ligands; reference 39). Another possibility is that resistance of some tumors to NK T cells may relate to the length of time the tumor has been cultured in vitro. In support of this, we found that the MCA-1 line became more resistant to NK T cell–mediated tumor immunosurveillance with time in culture (unpublished data).
Clearly, the influence of NK T cells in tumor immunity may vary ranging from suppression (12) to promotion of tumor rejection (this study). Therefore, it will not only be important to determine the signals that activate NK T cells in response to various tumors, but also the signals that regulate the outcome of the NK T cell function (suppressive or aggressive; reference 40). These differential signals may be derived from different APCs (DC versus B cells, macrophages) or tumor cells themselves, in conjunction with distinct microenvironmental factors. These factors may include cytokines such as IL-12 (14, 41, 42) and IL-7 (43–45), which are thought to promote proinflammatory (via increased IFN-γ production), and antiinflammatory (via increased IL-4 and IL-13 production) responses, respectively.
An alternative possibility was that the tumor rejection observed in this model was due to minor transplantation antigens, expressed by the MCA-induced sarcoma lines, that may differ between the Jα281−/− donor mice from which they were originally derived, and the C57BL/6 recipients. We believe this to be highly unlikely for the following reasons. (a) The Jα281−/− mice (originally 129 strain) were backcrossed to C57BL/6 mice nine times before the induction of the sarcomas. Furthermore, we found a similar pattern of susceptibility in MCA-1–treated CD1d−/− mice, which had also backcrossed to C57BL/6 mice 10 times. Given the number of times each separate line had been backcrossed to C57BL/6 mice, CD1d−/− recipient mice would be unlikely to carry (and therefore be tolerant to) the same minor transplantation antigens that would be present on the Jα281−/− mouse-derived MCA-1 sarcoma line. Therefore, if the response was directed against minor transplantation antigens, we would have predicted that MCA-1 would have been rejected in CD1d−/− mice. (b) MCA-1, MCA-3, and MCA-4 tumor lines were also injected into (129 × C57BL/6)F1 recipient mice, which would express any of the potential minor transplantation antigens that may be expressed by the tumors. Whereas all these tumors grew as expected in Jα281−/− recipients, they were rejected in F1 mice in an identical fashion to that in C57BL/6 mice (unpublished data). (c) Adoptive transfer of purified, C57BL/6-derived, NK T cells was sufficient to mediate tumor rejection, whereas C57BL/6-derived, conventional NK1.1− T cells had no effect on tumor growth (Fig. 2 B). (d) The difference in tumor growth between C57BL/6 and Jα281−/− recipient mice was already apparent by day 3, which would represent a remarkably rapid response against a minor transplantation antigen. Taken together, these points indicate that this model involves a CD1d-dependent, NK T cell–mediated antitumor response, rather than a more conventional immune response against minor transplantation antigens.
In summary, this study clearly demonstrates a critical role for NK T cells in immunosurveillance of MCA-induced fibrosarcomas, in the absence of exogenous NK T cell stimulation. This is most likely mediated by their acting as an early warning system that initiates an antitumor response subsequently performed by dedicated effectors such as NK cells and/or CTLs. From a clinical viewpoint, this study suggests that maintenance and/or restoration of normal numbers of NK T cells, particularly after immune-depleting conditions, is an important consideration for the prevention of tumor growth and metastasis. Future research should now be aimed at determining whether this role for NK T cells in immunosurveillance extends to other tumor types, as well as gaining a more thorough understanding of the exact molecular mechanisms required for activation and regulation of NK T cell function in the absence of exogenous factors. In particular it will be important to attempt to isolate tumor-derived CD1d-binding ligands that may be key to this response.
Acknowledgments
The authors wish to thank Dr. Alan Baxter for critically reviewing the manuscript, Prof. Hideo Yagita for helpful discussions, Prof. Mitchell Kronenberg for provision of CD1d/α-GalCer tetramers, Prof. Masaru Tanigachi for provision of Jα281−/− mice, Prof. Luc Vankaer for provision of CD1d−/− mice, Dr. Phillip Darcy for assistance with tumor measurements, Daniel Pellicci and Konstantinos Kyparissoudis for technical assistance, Dr. Elise Randle-Barrett and Gerard Tarulli for operating the Flow Cytometric Cell Sorter, Christine Hall and the staff at Peter MacCallum Cancer Institute for their maintenance and care of the mice used in these studies, and Shayna Street for providing the Jα281−/−-derived MCA-induced sarcomas.
N.Y. Crowe is supported by an Australian Postgraduate Research Award. M.J. Smyth and D.I. Godfrey are supported by National Health and Medical Research Council of Australia (NHMRC) Research Fellowships and D.I. Godfrey was supported by an ADCORP/Diabetes Australia Fellowship. The project was supported by the Human Frontier Science Program and donations from Rothschild Australia.
M.J. Smyth and D.I. Godfrey are co-chief investigators.
Footnotes
Abbreviations used in this paper: α-GalCer, α-galactosylceramide; MCA, methylcholanthrene; NMS, normal mouse serum; pfp, perforin; RAG, recombination activation gene; WT, wild-type.
References
- 1.Bendelac, A., M.N. Rivera, S.H. Park, and J.H. Roark. 1997. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu. Rev. Immunol. 15:535–562. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey, D.I., K.J.L. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 3.Porcelli, S.A., B.W. Segelke, M. Sugita, I.A. Wilson, and M.B. Brenner. 1998. The CD1 family of lipid antigen-presenting molecules. Immunol. Today. 19:362–368. [DOI] [PubMed] [Google Scholar]
- 4.Cui, J., T. Shin, T. Kawano, H. Sato, E. Kondo, I. Toura, Y. Kaneko, H. Koseki, M. Kanno, and M. Taniguchi. 1997. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science. 278:1623–1626. [DOI] [PubMed] [Google Scholar]
- 5.Chen, Y.H., N.M. Chiu, M. Mandal, N. Wang, and C.R. Wang. 1997. Impaired NK1+ T cell development and early IL-4 production in CD1-deficient mice. Immunity. 6:459–467. [DOI] [PubMed] [Google Scholar]
- 6.Mendiratta, S.K., W.D. Martin, S. Hong, A. Boesteanu, S. Joyce, and L. Van Kaer. 1997. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 6:469–477. [DOI] [PubMed] [Google Scholar]
- 7.Smiley, S.T., M.H. Kaplan, and M.J. Grusby. 1997. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science. 275:977–979. [DOI] [PubMed] [Google Scholar]
- 8.Hammond, K.J.L., L.D. Poulton, L.J. Palmisano, P.A. Silveira, D.I. Godfrey, and A.G. Baxter. 1998. αβ-T cell receptor (TCR)+CD4−CD8− (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J. Exp. Med. 187:1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonoda, K.H., M. Exley, S. Snapper, S.P. Balk, and J. Stein-Streilein. 1999. CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J. Exp. Med. 190:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seino, K., K. Yanagisawa, H. Taniguchi, Y. Takada, K. Yuzawa, M. Otsuka, and K. Fukao. 2001. The effect of α-galactosylceramide upon allogenic rejection. Transpl. Proc. 33:437–438. [DOI] [PubMed] [Google Scholar]
- 11.Zeng, D., D. Lewis, S. Dejbakhsh-Jones, F.S. Lan, M. Garcia-Ojeda, R. Sibley, and S. Strober. 1999. Bone marrow NK1.1− and NK1.1+ T cells reciprocally regulate acute graft versus host disease. J. Exp. Med. 189:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terabe, M., S. Matsui, N. Noben-Trauth, H. Chen, C. Watson, D.D. Donaldson, D.P. Carbone, W.E. Paul, and J.A. Berzofsky. 2000. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 1:515–520. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto, W., K. Takeda, R. Anzai, K. Ogasawara, H. Sakihara, K. Sugiura, S. Seki, and K. Kumagai. 1995. Cytotoxic NK1.1 Ag+ αβ T cells with intermediate TCR induced in the liver of mice by IL-12. J. Immunol. 154:4333–4340. [PubMed] [Google Scholar]
- 14.Smyth, M.J., M. Taniguchi, and S.E. Street. 2000. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J. Immunol. 165:2665–2670. [DOI] [PubMed] [Google Scholar]
- 15.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, H. Sato, E. Kondo, M. Harada, H. Koseki, T. Nakayama, et al. 1998. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Vα14 NKT cells. Proc. Natl. Acad. Sci. USA. 95:5690–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa, R., K. Motoki, H. Ueno, R. Iijima, H. Nakamura, E. Kobayashi, A. Shimosaka, and Y. Koezuka. 1998. Treatment of hepatic metastasis of the Colon26 adenocarcinoma with an α-Galactosylceramide, KRN7000. Cancer Res. 58:1202–1207. [PubMed] [Google Scholar]
- 17.Toura, I., T. Kawano, Y. Akutsu, T. Nakayama, T. Ochiai, and M. Taniguchi. 1999. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with α-galactosylceramide. J. Immunol. 163:2387–2391. [PubMed] [Google Scholar]
- 18.Hayakawa, Y., K. Takeda, H. Yagita, S. Kakuta, Y. Iwakura, L. Van Kaer, I. Saiki, and K. Okumura. 2001. Critical contribution of IFN-γ and NK cells, but not perforin-mediated cytotoxicity, to anti-metastatic effect of α-galactosylceramide. Eur. J. Immunol. 31:1720–1727. [PubMed] [Google Scholar]
- 19.Nakagawa, R., I. Nagafune, Y. Tazunoki, H. Ehara, H. Tomura, R. Iijima, K. Motoki, M. Kamishohara, and S. Seki. 2001. Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by α-galactosylceramide in mice. J. Immunol. 166:6578–6584. [DOI] [PubMed] [Google Scholar]
- 20.Smyth, M.J., N.Y. Crowe, D.G. Pellicci, K. Kyparissoudis, J.M. Kelly, K. Takeda, H. Yagita, and D.I. Godfrey. 2002. Anti-metastatic effect of α-galactosylceramide is mediated by sequential production of IFN-γ by NKT and NK cells. Blood. 99:1259–1266. [DOI] [PubMed] [Google Scholar]
- 21.Smyth, M.J., K.Y. Thia, S.E. Street, E. Cretney, J.S. Trapani, M. Taniguchi, T. Kawano, S.B. Pelikan, N.Y. Crowe, and D.I. Godfrey. 2000. Differential tumour surveillance by natural killer (NK) and NK T cells. J. Exp. Med. 191:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagi, D., B. Lederman, K. Burki, P. Seiler, B. Odermatt, K.J. Olsen, E.R. Podack, R.M. Zinkernagel, and H. Hengartner. 1994. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 369:31–37. [DOI] [PubMed] [Google Scholar]
- 23.Dalton, D.K., S. Pitts-Meek, S. Keshav, I.S. Figari, A. Bradley, and T.A. Stewart. 1993. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 259:1739–1742. [DOI] [PubMed] [Google Scholar]
- 24.Hammond, K.J.L., S.B. Pelikan, N.Y. Crowe, E. Randle-Barrett, T. Nakayama, M. Taniguchi, M.J. Smyth, I.R. van Driel, R. Scollay, A.G. Baxter, et al. 1999. NKT cells are phenotypically and functionally diverse. Eur. J. Immunol. 29:3768–3781. [DOI] [PubMed] [Google Scholar]
- 25.Smyth, M.J., N.Y. Crowe, and D.I. Godfrey. 2001. NK and NKT cells collaborate in host protection from MCA-induced fibrosarcoma. Int. Immunol. 13:459–463. [DOI] [PubMed] [Google Scholar]
- 26.Shankaran, V., H. Ikeda, A.T. Bruce, J.M. White, P.E. Swanson, L.J. Old, and R.D. Screiber. 2001. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 410:1107–1111. [DOI] [PubMed] [Google Scholar]
- 27.Benlagha, K., A. Weiss, A. Beavis, L. Teyton, and A. Bendelac. 2000. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J. Exp. Med. 191:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnaud, C., D. Lee, O. Donnars, S.H. Park, A. Beavis, Y. Koezuka, and A. Bendelac. 1999. Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 163:4647–4650. [PubMed] [Google Scholar]
- 30.Eberl, G., and H.R. MacDonald. 2000. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur. J. Immunol. 30:985–992. [DOI] [PubMed] [Google Scholar]
- 31.Karnbach, C., M.R. Daws, E.C. Niemi, and M.C. Nakamura. 2001. Immune rejection of a large sarcoma following cyclophosphamide and IL-12 treatment requires both NK and NK T cells and is associated with the induction of a novel NK T cell population. J. Immunol. 167:2569–2576. [DOI] [PubMed] [Google Scholar]
- 32.Boeson, M., I.M. Svane, A.M. Engel, J. Rygaard, A.R. Thomsen, and O. Werdelin. 2000. CD8+ T cells are crucial for the ability of congenic normal mice to reject highly immunogenic sarcomas induced in nude mice with 3-methylcholanthrene. Clin. Exp. Immunol. 121:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eberl, G., P. Brawand, and H.R. MacDonald. 2000. Selective bystander proliferation of memory CD4+ and CD8+ T cells upon NK T or T cell activation. J. Immunol. 165:4305–4311. [DOI] [PubMed] [Google Scholar]
- 34.Gee, M.S., C.J. Koch, S.M. Evans, W.T. Jenkins, C.H. Pletcher, J.S. Moore, H.K. Koblish, J. Lee, E.M. Lord, G. Trinchieri, et al. 1999. Hypoxia-mediated apoptosis from angiogenesis inhibition underlies tumor control by recombinant interleukin 12. Cancer Res. 59:4882–4889. [PubMed] [Google Scholar]
- 35.Ruegg, C., A. Yilmaz, G. Bieler, J. Bamat, P. Chaubert, and F.J. Lejeune. 1998. Evidence for the involvement of endothelial cell integrin αVβ3 in the disruption of the tumor vasculature induced by TNF and IFN-γ. Nat. Med. 4:408–414. [DOI] [PubMed] [Google Scholar]
- 36.Yao, L., C. Sgadari, K. Furuke, E.T. Bloom, J. Teruya-Feldstein, and G. Tosato. 1999. Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood. 93:1612–1621. [PubMed] [Google Scholar]
- 37.Leite-De-Moraes, M.C., A. Herbelin, C. Gouarin, Y. Koezuka, E. Schneider, and M. Dy. 2000. Fas/Fas ligand interactions promote activation-induced cell death of NK T lymphocytes. J. Immunol. 165:4367–4371. [DOI] [PubMed] [Google Scholar]
- 38.Osman, Y., T. Kawamura, T. Naito, K. Takeda, L. Van Kaer, K. Okumura, and T. Abo. 2000. Activation of hepatic NKT cells and subsequent liver injury following administration of α-galactosylceramide. Eur. J. Immunol. 30:1919–1928. [DOI] [PubMed] [Google Scholar]
- 39.Wu, J., Y. Song, A.B. Bakker, S. Bauer, T. Spies, L.L. Lanier, and J.H. Phillips. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285:730–732. [DOI] [PubMed] [Google Scholar]
- 40.Smyth, M.J., and D.I. Godfrey. 2000. NKT cells and tumor immunity-a double-edged sword. Nat. Immunol. 1:459–460. [DOI] [PubMed] [Google Scholar]
- 41.Kawamura, T., K. Takeda, S.K. Mendiratta, H. Kawamura, L. Van Kaer, H. Yagita, T. Abo, and K. Okumura. 1998. Cutting edge: critical role of NK1+ T cells in IL-12-induced immune responses in vivo. J. Immunol. 160:16–19. [PubMed] [Google Scholar]
- 42.Leite de Moraes, M.C., G. Moreau, A. Arnould, F. Machavoine, C. Garcia, M. Papiernik, and M. Dy. 1998. IL-4-producing NK T cells are biased towards IFN-γ production by IL-12. Influence of the microenvironment on the functional capacities of NK T cells. Eur. J. Immunol. 28:1507–1515. [DOI] [PubMed] [Google Scholar]
- 43.Gombert, J.M., E. Tancrede-Bohin, A. Hameg, M.C. Leite de Moraes, A.P. Vicari, J.F. Bach, and A. Herbelin. 1996. IL-7 reverses NK1+ T cell-defective IL-4 production in the non-obese diabetic mouse. Int. Immunol. 8:1751–1758. [DOI] [PubMed] [Google Scholar]
- 44.Vicari, A.P., A. Herbelin, M.C. Leite-de-Moraes, U. Von Freeden-Jeffry, R. Murray, and A. Zlotnik. 1996. NK1.1+ T cells from IL-7-deficient mice have a normal distribution and selection but exhibit impaired cytokine production. Int. Immunol. 8:1759–1766. [DOI] [PubMed] [Google Scholar]
- 45.Hameg, A., C. Gouarin, J.M. Gombert, S. Hong, L. Van Kaer, J.F. Bach, and A. Herbelin. 1999. IL-7 up-regulates IL-4 production by splenic NK1.1+ and NK1.1− MHC class I-like/CD1-dependent CD4+ T cells. J. Immunol. 162:7067–7074. [PubMed] [Google Scholar]