

Abstract
Accumulating evidence has shown the importance of Stat6-mediated signaling in allergic diseases. In this study, we show a novel regulatory mechanism of Stat6-mediated signaling in mast cells. When Stat6 is activated by interleukin (IL)-4 and translocated to the nucleus, Stat6 is cleaved by a nucleus-associated protease in mast cells. The cleaved 65-kD Stat6 lacks the COOH-terminal transactivation domain and functions as a dominant-negative molecule to Stat6-mediated transcription. The retrovirus-mediated expression of cleavage-resistant Stat6 mutants prolongs the nuclear accumulation of Stat6 upon IL-4 stimulation and enhances IL-4–induced gene expression and growth inhibition in mast cells. These results indicate that the proteolytic processing of Stat6 functions as a lineage-specific negative regulator of Stat6-dependent signaling in mast cells, and thus suggest that it may account for the limited role of Stat6 in IL-4 signaling in mast cells.
Keywords: Stat6, short isoform, proteolysis, mast cells, apoptosis
Introduction
IL-4 is a multifunctional cytokine that plays a critical role in the regulation of immune responses (1). Its effects depend upon binding to and signaling through a receptor complex consisting of the IL-4Rα chain and either the common cytokine receptor γ chain (γc) or IL-13Rα 1 chain, resulting in a series of phosphorylation events mediated by receptor-associated Janus kinases (JAKs;* reference 2). Among downstream molecules of JAKs, Stat6 acts as a direct connection between the cytokine receptor and the transcription apparatus (3). Stat6 is critical in the enhanced expression of many IL-4–responsive genes, including class II major histocompatibility molecules, low affinity IgE receptor (CD23), and IL-4Rα chain (2). Recent studies with Stat6-deficient (Stat6−/−) mice also show that the Stat6 pathway is the principal signaling pathway involved in the commitment of CD4+ T cells to the Th2 phenotype and IgE isotype switching in B cells (4–6). Although the role and the positive regulatory mechanisms of Stat6 activation are well documented, little is known about the mechanisms involved in the down-regulation of Stat6-dependent signaling.
Mast cells are recognized as the major effector cells of type I hypersensitivity reactions and play a pivotal role in allergic diseases such as asthma, allergic rhinitis, and atopic dermatitis (7). IL-4 is a potent growth factor of mast cells in human (8) and mouse (9), although in some situations IL-4 enhances the apoptosis of mast cells (10). Stat6 is not essential for IL-4–induced proliferation of bone marrow–derived mast cells (BMMCs; reference 11), whereas Stat6 is essential for IL-4–induced proliferation of T cells (4–6). On the other hand, it has recently been shown, especially in the presence of IL-10, that IL-4 induces the apoptosis of IL-3–dependent mast cells by Stat6-dependent signaling (10). In addition, intestinal mastcytosis induced by parasitic infection is enhanced in Stat6−/− mice (12). Recently, we and others have shown that a short isoform of Stat6 (65-kD Stat6) is expressed in mast cells (13, 14). However, the mechanism of 65-kD Stat6 production as well as the role of 65-kD Stat6 in mast cells remains to be determined.
In this study, we show a novel regulatory mechanism of Stat6-mediated signaling in mast cells. When Stat6 is activated by IL-4 and translocated to the nucleus, Stat6 is cleaved at the COOH terminus by a nucleus-associated protease in mast cells. The cleaved 65-kD Stat6 lacks the transcriptional activity and acts as a dominant-negative molecule to conventional Stat6-mediated transcription. The Stat6 protease activity is sensitive to serine protease inhibitors, but different from previously described Stat5 protease in myeloid progenitors (15–18). Moreover, the retrovirus-mediated expression of cleavage-resistant Stat6 mutants prolongs the nuclear accumulation of Stat6 upon IL-4 stimulation and enhances IL-4–induced gene expression and growth inhibition in mast cells. Our results indicate that the proteolytic processing of Stat6 functions as a lineage-specific negative regulator of Stat6-dependent signaling in mast cells.
Materials and Methods
Mice.
Stat6-deficient (Stat6−/−) (4) and C57BL/6 mice (Japan SLC) were housed in microisolator cages under pathogen-free conditions. All experiments were performed according to the guidelines of Chiba University (Chiba, Japan).
Constructs.
Flag epitope–tagged Stat6 at the COOH terminus (C-Flag Stat6) was constructed as previously described (19). COOH-terminal–truncated mutants at amino acid (aa) 673 (673 Stat6), aa 695 (695 Stat6), or aa 715 (715 Stat6) were generated by changing each aa to a stop codon by using a PCR-based, site-directed mutagenesis kit (Stratagene). Alanine substitutions of Stat6 on serine at aa 683 (S683A), serine at aa 684 (S684A), aspartic acid at aa 685 (D685A), or methionine at aa 686 (M686A) were generated by using a PCR-based, site-directed mutagenesis kit (Stratagene). All mutations were confirmed by DNA sequencing.
Culture of BMMCs.
Primary culture of IL-3–dependent BMMCs was prepared from 8–12-wk-old C57BL/6 or Stat6−/− mice and maintained as previously described (13). BMMCs obtained after 4 wk of culture were >99% mast cells.
Luciferase Assay.
Stat6-dependent reporter construct, TPU474, was previously described (20). For Stat6-dependent luciferase assays, COS7 cells were transiently transfected with TPU474 (provided by U. Schindler, Tularik Inc., San Francisco, CA) and pRL-TK in the presence of various Stat6 expression vectors alone or in their combinations using FuGENE6 transfection reagents (Roche Diagnostics Co.). 20 h after transfection, cells were stimulated with IL-4 (human IL-4, 10 ng/ml; Genzyme) at 37°C for an additional 12 h and the luciferase activity was measured by the dual luciferase assay system (Promega). Firefly luciferase activity of TPU474 was normalized by Renilla luciferase activity of pRL-TK.
Preparation of Cell Extracts.
Cell fractionation was performed as follows. All buffers were added to these reagents: 1 mM Na3VO4, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 2 μg/ml pepstatin, and 0.5 mM PMSF. For the preparation of whole cell extracts, cells were resuspended in lysis buffer W (1% NP-40, 20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 4 mM EGTA, 10 mM NaF, and 10% glycerol), incubated on ice for 30 min, and then the supernatant was collected by centrifugation. Nuclear or cytoplasmic fraction of cells was prepared as previously described (21). In brief, after 107 cells were washed with PBS, cells were resuspended in 100 μl hypotonic lysis buffer A (0.1% NP-40, 10 mM Hepes-KOH, pH 7.9, 10 mM KCl, 1 mM DTT, 0.1 mM EDTA, 2 mM MgCl2, and 0.5 M sucrose) and lysed by 10 strokes in a glass Dounce homogenizer. The supernatant was collected as a cytoplasmic fraction by centrifugation. Subsequently, nuclear fraction was prepared by resuspending nuclear pellets with 15 μl high salt lysis buffer C (20 mM Hepes-KOH, pH 7.9, 420 mM NaCl, 1 mM DTT, 0.2 mM EDTA, 1.5 mM MgCl2, and 5% glycerol). The nuclear fraction was then mixed with 22.5 μl of a low salt buffer (20 mM Hepes-KOH, pH 7.9, 50 mM KCl, 1 mM DTT, 0.2 mM EDTA, and 20% glycerol). The purity of the extracts was evaluated by measuring lactate dehydrogenase activity and we found that the nuclear extract contained <5% of contamination (unpublished data). In addition, NaCl concentration of cytoplasmic extract as well as nuclear extract was adjusted to 150 mM before Stat6 cleaving assay. In some experiments, splenocytes or BMMCs were stimulated with IL-4 (10 ng/ml) at 37°C for 20 min before the preparation of cell extracts. In other experiments, BMMCs were starved from IL-3 for 5 h and then stimulated with IL-3 (10 ng/ml; Genzyme) at 37°C for 20 min before preparing cell extracts.
Stat6 Cleaving Assay.
Stat6 protein from either wild-type (WT) splenocytes or murine Stat6 expression vector-transfected COS7 cells (provided by J.N. Ihle, St. Jude Children's Hospital, Memphis, TN) was incubated with cell extracts from Stat6−/− BMMCs at 37°C for 20 min. Where indicated, p-chloromercuribenzoate (PCMB; 0.1 mM; Wako Pure Chemical Industries), matrix metalloproteinase (MMP) inhibitor I (4-Abz-Gly-Pro-D-Leu-D-Ala-OH; 0.1 mM; Calbiochem-Novabiochem), lactacystin (clasto-lactacystin-β-lactone; 1 μM; Boston Biochem), z-VAD (z-VAD-FMK; 0.1 mM; Calbiochem-Novabiochem), ICE inhibitor III (z-Asp-2,6-dichlorobenzoyloxymethylketone; 0.1 mM; Funakoshi), ALLN calpain inhibitor I (N-acetyl-Leu-Leu-norleucinal; 0.1 mM; Calbiochem-Novabiochem), or 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF; 0.3–16 mM; Roche Diagnostics Co.) was added during the incubation. The reaction was separated on 10% SDS polyacrylamide gels and subjected to Western blotting as described below.
Stat5 Cleaving Assay.
After WT BMMCs or FDC-P1 cells (provided by T. Kitamura, University of Tokyo, Tokyo, Japan) were starved from IL-3 for 5 h, nuclear extracts were prepared as described above. COS7 cells were transfected with murine Stat5a expression vector (provided by L. Hennighausen, National Institutes of Health [NIH], Bethesda, MD) and the extract was incubated with nuclear extract from either WT BMMCs or FDC-P1 cells at 37°C for 20 min. The reaction was terminated by SDS sample buffer, separated on 6% polyacrylamide gels, and subjected to Western blotting as described below.
Western Blotting.
Immunoblotting was performed as previously described (13) using antisera to mouse Stat6 (M20; Santa Cruz Biotechnology Inc.), mouse Stat6 (M200; Santa Cruz Biotechnology Inc.), phospho-Stat6 (New England Biolabs, Inc.), mouse Stat5 (Transduction Laboratories), phospho-Stat5 (New England Biolabs, Inc.), human Stat3 (Upstate Biotechnology), or Flag-epitope tag (M2; Sigma-Aldrich).
Retrovirus-mediated Gene Expression in BMMCs.
To overcome the limited efficiency of transfection on BMMCs, we used a bicistronic retrovirus system, in which infected cells were identified by coexpressed green fluorescent protein (GFP; pMX-internal ribosome entry site [IRES]-GFP vector; reference 22) or coexpressed Thy1.1 (murine stem cell virus [MSCV]-IRES-Thy1.1 vector; reference 23). Retrovirus vectors, pMX-WT Stat6-IRES-GFP, pMX-D685A Stat6-IRES-GFP, pMX-673 Stat6-IRES-GFP, pMX-IRES-GFP (as a negative control; provided by T. Kitamura), MSCV-C-Flag WT Stat6-IRES-Thy1.1, MSCV-C-Flag D685A Stat6-IRES-Thy1.1, and MSCV-IRES-Thy1.1 (as a negative control; provided by P. Marrack, University of Colorado Medical School, Denver, CO) were transfected to a transient retrovirus packaging cell line PlatE (provided by T. Kitamura; reference 24) using FuGENE6 transfection reagents. 48 h after transfection, retrovirus was collected by centrifugation and stored at −80°C until use. For infection on BMMCs, cells were incubated twice with 1,000 μl of the retrovirus for 4 h at a 48-h interval in the presence of 10 ng/ml murine IL-3, 100 ng/ml murine stem cell factor (provided by Kirin Brewery Co., Takasaki, Japan), and 10 μg/ml polybrene (Sigma-Aldrich). After the cells were washed with PBS, they were allowed to grow in fresh medium containing IL-3. Under these conditions, the efficiency of infection to BMMCs was 5–10% in all viruses as assessed by GFP+ or Thy1.1+ cells by FACS®.
Retrovirus-mediated Gene Expression in CFTL-15 Cells.
CFTL-15 cells (provided by M.A. Brown [Williams College, Williamstown, MA] and W.E. Paul [NIH, Bethesda, MD]), a murine mast cell line that expresses 65-kD Stat6 (25), were cultured in RPMI 1640 medium containing murine IL-3. CFTL-15 cells were infected with retroviruses for 4 h in the presence of 10 ng/ml IL-3 and 10 μg/ml polybrene. The efficiency of infection to CFTL-15 cells was ∼30%.
MACS Sorting of Thy1.1+ Cells.
5 d after the retrovirus infection, cells were incubated with biotin-conjugated anti-Thy1.1 antibody (OX-7; BD PharMingen), washed three times with PBS, and then incubated with streptavidin magnetic microbeads (Miltenyi Biotec). After washing, infected (Thy1.1+) cells were positively collected using MACS RS+ column (Miltenyi Biotec). The purity of Thy1.1+ cells was >98%. Cytoplasmic extracts of Thy1.1+ cells were subjected to Western blotting with anti-Flag antibody to evaluate the levels of retrovirus-mediated Stat6 expression in the infected cells.
Immunohistochemistry.
CFTL-15 cells were infected with either MSCV-C-Flag WT Stat6-IRES-Thy1.1 retrovirus or MSCV-C-Flag D685A Stat6-IRES-Thy1.1 retrovirus and infected (Thy1.1+) cells were positively collected as described above. As controls, COS7 cells were transiently transfected with either C-Flag WT Stat6 or C-Flag D685A Stat6 expression plasmids using FuGENE6 transfection reagents. CFTL-15 cells and COS7 cells were stimulated with 10 ng/ml murine IL-4 and 10 ng/ml human IL-4, respectively, at 37°C for 15 min, washed with PBS, and then cultured with IL-4–free medium for an additional 45 or 165 min. Where indicated, to block nuclear export, cells were incubated with 10 ng/ml leptomycin B (Sigma-Aldrich) at 37°C from 60 min before IL-4 stimulation until harvest as previously described (26). At the indicated times after IL-4 stimulation, CFTL-15 cells were placed on glass microscope slides using cytospin. Cells were fixed in methanol at −20°C for 10 min and then in acetone at −20°C for 1 min. After washing with PBS, cells were incubated with PBS containing 3% bovine serum albumin for 30 min and then with PBS containing 5% donkey serum for 30 min at room temperature to block nonspecific binding. Cells were then washed three times with PBS, incubated with Cy3-conjugated anti-Flag antibody (M2; Sigma-Aldrich) at room temperature for 1 h, and then washed an additional three times with PBS. Their nuclei were stained with DAPI (4′-6-diamidino-2-phenylindole; Sigma-Aldrich) for 15 min at room temperature.
Detection of IgE Receptors on BMMCs.
The expression of IgE receptors on BMMCs was analyzed by FACS® using mouse anti–DNP IgE and anti-IgE antibody as previously described (13).
Data Analysis.
Data are summarized as mean ± SD. The statistical analysis of the results was performed by the unpaired t test. P values <0.05 were considered significant.
Results
A 65-kD Isoform of Stat6 Is Produced by Proteolytic Processing.
In previous reports, we and others have shown that a 65-kD isoform of Stat6 (65-kD Stat6) is expressed in BMMCs (13, 14). The 65-kD Stat6 in BMMCs is detected by anti-Stat6 (M200) antibody, which recognizes the middle portion of Stat6 (aa 280–480), but not by anti-Stat6 (M20) antibody, which recognizes the COOH terminus of Stat6 (13, 14). In addition, when BMMCs are stimulated with IL-4, the phosphorylated form of Stat6 is detected at 65 kD by anti-phospho Stat6 antibody, which recognizes the tyrosine residue at aa 641 (Y641) of Stat6 (13). These findings indicate that the 65-kD Stat6 lacks the COOH terminus but contains the Y641, which is essential for the homodimerization of Stat6 (3). To determine whether the 65-kD Stat6 is a product of protein processing, we first performed the coincubation assay in which the conventional 94-kD Stat6 from splenocytes was incubated with cell extracts of BMMCs and analyzed for the size of Stat6 protein by anti-Stat6 Western blotting. To eliminate the influence of endogenous Stat6 expression in BMMCs, we prepared whole cell extracts from BMMCs in Stat6−/− mice (Stat6−/− BMMCs) as a possible source of the protease(s). Interestingly, when conventional Stat6 (94-kD Stat6) was incubated with Stat6−/− BMMC extract, the 94-kD Stat6 was cleaved to 65 kD (Fig. 1 A, compare lanes 3 and 4). The cleaved Stat6 was detected by anti-Stat6 (M200) antibody (Fig. 1 A, top) but not by anti-Stat6 (M20) antibody (Fig. 1 A, bottom), suggesting that the cleaved Stat6 also lacks the COOH terminus. These results indicate that the 65-kD Stat6 is produced by the cleavage of the 94-kD Stat6 in BMMCs.
Figure 1.

A 65-kD isoform of Stat6 is produced by proteolytic processing. (A) Cell extracts from WT splenocytes were incubated with cell extracts of BMMCs from Stat6−/− mice at 37°C for 20 min and analyzed by Western blotting with anti-Stat6 (M200) antibody (top) or anti-Stat6 (M20) antibody (bottom). As controls, cell extracts from WT BMMCs and Stat6−/− BMMCs were blotted with anti-Stat6 antibodies. Representative blots from four independent experiments are shown. (B) COS7 cells were transfected with Stat6 expression vector and their cell extracts were used as a source of Stat6 protein. Transfected Stat6 was incubated with cell extracts of thymocytes, splenocytes, or BMMCs from Stat6−/− mice at 37°C for 20 min and analyzed by Western blotting with anti-Stat6 (M200) antibody. A representative blot from four independent experiments is shown.
To further analyze the Stat6 protease activity, we developed the Stat6 cleaving assay using transfected Stat6 as a substrate of the protease (Fig. 1 B). COS7 cells were transfected with Stat6 expression vector and the cell extracts of these cells were incubated with Stat6−/− BMMC extract and subjected to Western blotting using anti-Stat6 (M200) antibody. Consistent with the above findings (Fig. 1 A), incubation of the 94-kD Stat6 with Stat6−/− BMMC extract decreased the size of Stat6 to 65 kD (Fig. 1 B, lane 7). In contrast, incubation with cell extracts from either Stat6−/− thymocytes or Stat6−/− splenocytes did not change the size of the 94-kD Stat6 (Fig. 1 B), indicating that Stat6 protease activity is absent in thymocytes and splenocytes.
Stat6 Protease Activity Is Localized in the Nucleus.
Next, we examined the subcellular localization of Stat6 protease activity in BMMCs. Cell extracts were prepared from the cytoplasmic or nuclear fraction of Stat6−/− BMMCs and then incubated with 94-kD Stat6. Interestingly, 94-kD Stat6 was cleaved to 65-kD Stat6 by the incubation with nuclear extract but not with the cytoplasmic extract from Stat6−/− BMMCs (Fig. 2 A). To exclude the possibility that the protease is normally in a protected cellular compartment that is detergent or high salt soluble, we added NP-40 or NaCl to the cytoplasmic fraction to the levels that we used for whole cell or nuclear extract preparation (1% NP-40 or 420 mM NaCl), and then examined the Stat6 protease activity. However, there was still no detectable Stat6 protease activity in the cytoplasmic fraction of BMMCs (Fig. 2 A). These results indicate that Stat6 protease activity is localized in the nucleus.
Figure 2.
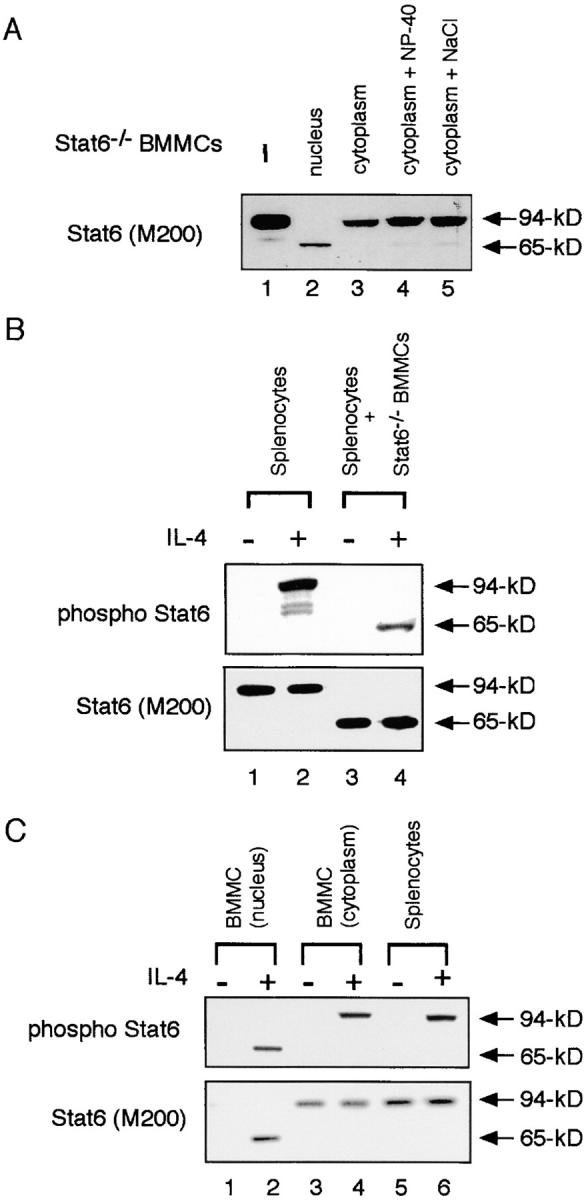
Stat6 protease activity is localized in the nucleus. (A) Subfraction of cell extracts (nuclear or cytoplasmic) was prepared from Stat6−/− BMMCs. Where indicated, 1% NP-40 or 420 mM NaCl was added to the cytoplasmic fraction. These extracts were then incubated with transfected Stat6 at 37°C for 20 min and subjected to Western blotting with anti-Stat6 (M200) antibody. (B) Cell extracts from IL-4–stimulated or –unstimulated WT splenocytes were incubated with Stat6−/− BMMC extract and blotted with anti–phospho-Stat6 antibody (top) or anti-Stat6 (M200) antibody (bottom). (C) Subfraction of cell extracts was prepared from either IL-4–stimulated or –unstimulated WT BMMCs and blotted with anti–phospho-Stat6 antibody (top) or anti-Stat6 (M200) antibody (bottom). As a control, cell extracts from IL-4–stimulated or –unstimulated WT splenocytes were blotted with these antibodies.
Stat6 has been shown to translocate to the nucleus only after Stat6 is phosphorylated by JAK kinases (2, 3). Therefore, we tested whether phosphorylated Stat6 was a substrate of the Stat6 protease. When phosphorylated Stat6 in IL-4–stimulated splenocytes was incubated with Stat6−/− BMMC extract, the phosphorylated form of Stat6 was efficiently cleaved to 65-kD Stat6 (Fig. 2 B, compare lanes 2 and 4). In addition, the unphosphorylated form of Stat6 was also cleaved to 65-kD Stat6 by Stat6−/− BMMC extract (Fig. 2 B, compare lanes 1 and 3). Thus, both phosphorylated and unphosphorylated forms of Stat6 are sensitive to the Stat6 protease activity. However, the in vivo substrate of Stat6 protease should be the phosphorylated form of Stat6 because of the nuclear localization of Stat6 protease activity.
Because Stat6 protease activity is restricted in the nucleus, it is possible that the conventional 94-kD Stat6 is expressed in the cytoplasm of WT BMMCs. Indeed, 94-kD Stat6 but not 65-kD Stat6 was found in the cytoplasm of WT BMMCs (Fig. 2 C, lanes 3 and 4). In contrast to the cytoplasm, Stat6 was detected at 65 kD in the nucleus only after IL-4 stimulation (Fig. 2 C, lane 2). These results indicate that Stat6 is normally produced as the conventional 94-kD form and that after translocation to the nucleus, 94-kD Stat6 is cleaved to 65-kD Stat6 by a nucleus-associated protease.
Stat6 Protease Activity Is Inhibited by Serine Protease Inhibitors.
We next sought to classify the Stat6 protease in BMMCs. Although a battery of protease inhibitors (Materials and Methods) was routinely added to all buffers used in the preparation and analysis of cell extracts of BMMCs, none of these effectively inhibited the observed proteolytic activity to Stat6. Thus, we examined the effect of other protease inhibitors on the Stat6 cleaving activity in BMMCs. As shown in Fig. 3 A, PCMB, MMP inhibitor I, lactacystin, z-VAD-FMK (z-VAD), ICE inhibitor III, nor calpain inhibitor I inhibited the Stat6 protease activity of Stat6−/− BMMC extract. Interestingly, however, Stat6 protease activity was significantly inhibited by AEBSF, a serine protease inhibitor. The Stat6 protease activity was inhibited in part by 4 mM AEBSF and completely by 16 mM AEBSF (Fig. 3 B). These results suggest that the Stat6 protease belongs to the serine protease family. In addition, the Stat6 protease activity was also inhibited by a high concentration (5 mM) of PMSF (unpublished data), another serine protease inhibitor, although 0.5 mM PMSF did not significantly inhibit the Stat6 protease activity.
Figure 3.

Stat6 protease activity is inhibited by serine protease inhibitors. (A) Similar to Fig. 1 B, transfected Stat6 was incubated with Stat6−/− BMMC extract. Where indicated, PCMB, MMP inhibitor I, lactacystin, z-VAD, ICE inhibitor III, or calpain inhibitor I was added during the incubation. All cell extracts were prepared in the presence of EGTA, aprotinin, leupeptin, pepstatin, and 0.5 mM PMSF. A representative blot from four independent experiments is shown. (B) Similarly, the indicated concentrations of AEBSF were added during the incubation. (C) Nuclear extract was prepared from IL-4–stimulated WT BMMCs in the presence (lane 3) or absence (lane 2) of 16 mM AEBSF. Nuclear extract was also prepared by directly resuspending the nuclear pellets of IL-4–stimulated WT BMMCs with preheated SDS sample buffer (lane 4). These extracts were then subjected to Western blotting with anti-phospho Stat6 antibody.
Stat6 Is Cleaved in the Nucleus of Living Mast Cells.
To further show that IL-4–activated Stat6 is physiologically cleaved by the protease in the nucleus of living mast cells, the nuclear extract of IL-4–stimulated WT BMMCs was prepared either in the presence of 16 mM AEBSF or in preheated SDS sample buffer. These extracts were then subjected to immunoblotting with anti-phospho Stat6 antibody. As shown in Fig. 3 C, 94-kD Stat6 was significantly increased in these extracts, which indicates that a part of Stat6 proteolysis occurred during the preparation of the extracts. However, 65-kD Stat6 was still observed in the nuclear extracts of IL-4–stimulated BMMCs. These results indicate that Stat6 is cleaved by the Stat6 protease in the nucleus of IL-4–stimulated living mast cells.
BMMC Extract Does Not Cleave Stat5.
It has recently been shown that Stat5 is cleaved by an undefined Stat5 protease in myeloid progenitors (15–18). Although the characteristics of Stat5 protease are still largely unknown, it has been reported that Stat5 protease activity is inhibited by serine protease inhibitors (15–17). To determine whether Stat6 protease is identical to Stat5 protease, we first analyzed the size of Stat5 proteins (Stat5a and Stat5b) in whole cell extracts of BMMCs. As shown in Fig. 4 A, BMMCs expressed only conventional forms of Stat5a and Stat5b. In addition, when BMMCs were stimulated with IL-3, phosphorylated Stat5 was detected at ∼94 kD by anti-phospho Stat5a antibody (Fig. 4 A, top). Moreover, using FDC-P1 cells as a source of Stat5 protease (15–17), we examined the substrate specificity of Stat6 and Stat5 protease. As shown in Fig. 4 B, the nuclear extract from FDC-P1 cells cleaved 94-kD murine Stat5a to 80 kD (Fig. 4 B, lane 5, top), whereas the FDC-P1 extract did not cleave Stat6 (Fig. 4 B, lane 5, bottom). In contrast, the nuclear extract from WT BMMCs cleaved Stat6 but not Stat5 (Fig. 4 B, lane 4). In addition, Stat6 was expressed as 94-kD Stat6 in whole cell extracts of FDC-P1 cells (unpublished data). Taken together, although both Stat6 and Stat5 protease have not been cloned yet, these results indicate that Stat6 differs from Stat5 protease.
Figure 4.

BMMC extract cleaves Stat6 but not Stat5. (A) Cell extracts from IL-3–stimulated WT BMMCs were blotted with either anti–phospho-Stat5 (top) or anti–pan-Stat5 (bottom) antibody. (B) Either murine Stat5a (top) or murine Stat6 (bottom) from transfected COS7 cells was incubated with nuclear extract from either WT BMMCs or FDC-P1 cells. Reactions were separated on 6 or 10% polyacrylamide gels and subjected to Western blotting with anti-pan Stat5 antibody (top) or anti-Stat6 (M200) antibody (bottom).
Stat6 Is Cleaved between aa 673 and 695.
To determine the cleavage site of Stat6, we prepared several COOH-terminal–truncated mutants of Stat6, including 715 Stat6, 695 Stat6, and 673 Stat6. These mutants were expressed in COS7 cells and the extracts were subjected to coincubation assay with Stat6−/− BMMC extract. As shown in Fig. 5 A, when 715 Stat6 was incubated with Stat6−/− BMMC extract, two bands were detected with anti-Stat6 (M200) antibody (lane 2). The upper band exhibited the same mobility to the 715 Stat6 and the lower band exhibited the same mobility to 65-kD Stat6. Similarly, the incubation of 695 Stat6 with Stat6−/− BMMC extract resulted in two bands of Stat6, the original 695 Stat6 and the 65-kD Stat6 (Fig. 5 A, lane 4). In contrast, the size of 673 Stat6 was smaller than the 65-kD Stat6 and the 673 Stat6 was not cleaved by the Stat6−/− BMMC extract (Fig. 5 A). These results indicate that the cleavage site is located between aa 673 and 695 of Stat6.
Figure 5.

Stat6 is cleaved between aa 673 and 695 by the Stat6 protease and the resultant Stat6 functions as a dominant-negative molecule. (A) COOH-terminal–truncated mutants of Stat6 (715, 695, or 673 Stat6) were transfected to COS7 cells. Cell extracts from these cells were incubated with or without Stat6−/− BMMC extract and blotted with anti-Stat6 (M200) antibody. A representative blot from four independent experiments is shown. (B) COS7 cells were transfected with 695, 673, or WT Stat6 (as a control) in the presence of a Stat6-responsive reporter construct, TPU474. 20 h after transfection, cells were stimulated with or without IL-4 for an additional 12 h and the luciferase activity of TPU474 was measured by the dual luciferase reporter system. Data are means ± SD for four experiments. (C and D) Similar to B, COS7 cells were transfected with WT Stat6 in the presence of the indicated amounts of either (C) 695 or (D) 673 Stat6. Data are means ± SD for four experiments.
65-kD Stat6 Functions as a Dominant-negative Molecule to Stat6-mediated Transcription.
To investigate the functional property of 65-kD Stat6, we first examined the transcriptional activity of 695 and 673 Stat6. COS7 cells were transfected with 695 or 673 Stat6 in the presence of a Stat6-dependent reporter construct (TPU474; reference 20) and the luciferase activity was measured in the presence or absence of IL-4 stimulation. As a control, COS7 cells were transfected with WT Stat6 and stimulated with IL-4. In cells expressing WT Stat6, IL-4 enhanced the transcription of TPU474 (Fig. 5 B). In contrast, the expression of 695 or 673 Stat6 did not enhance the IL-4–induced transcription (Fig. 5 B). Moreover, when 695 (Fig. 5 C) or 673 Stat6 (Fig. 5 D) was coexpressed with WT Stat6, these mutants inhibited the Stat6-dependent transcription of WT Stat6 in a dose-dependent fashion. These results indicate that 695 and 673 Stat6 function as dominant-negative regulators to Stat6-dependent transcription and thus suggest that 65-kD Stat6 may also act as a dominant-negative molecule.
D685A and M686A Stat6 Are Resistant to the Stat6 Protease Activity.
To address the role of Stat6 protease activity in mast cells, we tried to identify a mutant Stat6 that was resistant to the protease activity. Because Stat6 was cleaved between aa 673 and 695 and the mobility of 65-kD Stat6 was approximately in the middle of 695 and 673 Stat6 (Fig. 5 A), we prepared a series of point mutants of Stat6 by substituting each residues to alanine. The sensitivity of these mutants to the Stat6 protease activity was then examined by the coincubation assay. Interestingly, two of these mutants, D685A (aspartic acid at aa 685 to alanine) and M686A Stat6 (methionine at aa 686 to alanine), were resistant to the Stat6 protease activity (Fig. 6, A and B). In contrast, S683A and S684A Stat6 were as sensitive to the protease activity as WT Stat6 (Fig. 6 A).
Figure 6.

D685A and M686A Stat6 are resistant to the Stat6 protease activity. (A) Alanine substitutions of Stat6 (S683A, S684A, D685A, or M686A Stat6) were transfected to COS7 cells. Cell extracts were then incubated with or without Stat6−/− BMMC extract and blotted with anti-Stat6 (M200) antibody. A representative blot from four independent experiments is shown. (B) Relevant aa sequence of the putative cleavage site of Stat6 is shown in comparison with a reported cleavage site of Stat5a and Stat5b (17). (C) Similar to Fig. 5 B, COS7 cells were transfected with D685A, M686A, or WT Stat6 and the luciferase activity of TPU474 was measured in the presence or absence of IL-4. Data are means ± SD for four experiments.
We next examined the transcriptional activity of cleavage-resistant D685A and M686A Stat6. As shown in Fig. 6 C, D685A and M686A Stat6 exhibited a comparable transcriptional activity upon IL-4 stimulation to that of WT Stat6 in COS7 cells.
Retrovirus-mediated Expression of Cleavage-resistant D685A Stat6 Prolongs the Nuclear Accumulation of Stat6 in IL-4–stimulated Mast Cells.
D685A Stat6 is resistant to the Stat6 protease activity (Fig. 6 A) but normal in its transcriptional activity in COS7 cells lacking Stat6 protease activity (Fig. 6 C). Thus, we now have a tool to investigate the in vivo role of the Stat6 protease activity in mast cells. First, we examined the kinetics of nuclear accumulation of D685A Stat6 upon IL-4 stimulation in CFTL-15 cells, a murine mast cell line that expresses 65-kD Stat6 (25). CFTL-15 cells were infected with either MSCV-C-Flag WT Stat6-IRES-Thy1.1 or MSCV-C-Flag D685A Stat6-IRES-Thy1.1 retrovirus and before and after IL-4 stimulation, subcellular localization of transfected Stat6 was visualized with anti-Flag antibody. In the absence of IL-4 stimulation, both WT and D685A Stat6 localized in the cytoplasm (Fig. 7 A). When stimulated with IL-4 for 15 min, both WT and D685A Stat6 similarly accumulated in the nucleus (Fig. 7 A). Interestingly, although WT Stat6 disappeared from the nucleus 60 min after IL-4 stimulation, D685A Stat6 still accumulated in the nucleus (Fig. 7 A).
Figure 7.

Stat6 protease regulates the nuclear accumulation of Stat6 in mast cells. (A) CFTL-15 cells were infected with either MSCV-C-Flag WT Stat6-IRES-Thy1.1 retrovirus or MSCV-C-Flag D685A Stat6-IRES-Thy1.1 retrovirus and infected (Thy1.1+) cells were positively collected by magnetic cell sorting. Before and after IL-4 stimulation, subcellular localization of transfected Stat6 was visualized by Cy3-conjugated anti-Flag antibody. Their nuclei were stained with 4′-6-Diamidino-2-phenylindole. Where indicated, 10 ng/ml leptomycin B was added to the culture to block nuclear export. DAPI, 4′-6-Diamidino-2-phenylindole; LMB, leptomycin B. (B) COS7 cells were transiently transfected with either C-Flag WT Stat6 or C-Flag D685A Stat6 expression plasmids. Before and after IL-4 stimulation, subcellular localization of transfected Stat6 was visualized by Cy3-conjugated anti-Flag antibody.
To exclude the possibility that the difference between WT and D685A Stat6 in their kinetics of nuclear accumulation results from the nuclear export rather than the sensitivity to the proteolytic processing, the analogous experiments were performed in the presence of a nuclear export inhibitor, leptomycin B. Even in the presence of leptomycin B, WT Stat6 disappeared from the nucleus of CFTL-15 cells much faster than D685A Stat6 did (Fig. 7 A, bottom panels). In contrast, in COS7 cells that lacked Stat6 protease activity, WT and D685A Stat6 exhibited indistinguishable kinetics of subcellular distribution upon IL-4 stimulation, and WT Stat6 still accumulated in the nucleus of COS7 cells, even at 60 min (Fig. 7 B). As expected, leptomycin B similarly prolonged the nuclear accumulation of WT and D685A Stat6 in COS7 cells (Fig. 7 B). Taken together, these results suggest that proteolytic processing of Stat6 by the Stat6 protease is involved in the control of nuclear accumulation of Stat6 in living mast cells.
Retrovirus-mediated Expression of D685A Stat6 Enhances IL-4–induced Apoptosis and Growth Inhibition in Mast Cells.
We next investigated the physiological significance of the Stat6 protease activity in mast cells. When CFTL-15 cells were infected with control retrovirus (MSCV-IRES-Thy1.1) and then cultured with IL-3 alone or IL-3 plus IL-4, the number of apoptotic cells (annexin V+ cells) was significantly increased in cells cultured with IL-3 plus IL-4 than in those cultured with IL-3 alone (IL-3, 2.7 ± 0.4% vs. IL-3 plus IL-4, 8.7 ± 2.1% annexin V+ cells in Thy1.1+ cells; mean ± SD; n = 5 experiments; P < 0.005; Fig. 8 A). These results are consistent with a previous finding that IL-4 increases the apoptosis of IL-3–dependent mast cells (10). Interestingly, IL-4–induced apoptosis of CFTL-15 cells was significantly increased by the expression of D685A but not WT Stat6 (D685A Stat6, 16.2 ± 2.8% vs. WT Stat6 8.1 ± 1.9%; n = 5; P < 0.005; Fig. 8 A). To exclude the possibility that the difference resulted from the expression levels of D685A and WT Stat6, we examined the expression of retrovirus-mediated D685A and WT Stat6 by blotting cytoplasmic extracts from infected (Thy1.1+) cells with anti-Flag antibody. As shown in Fig. 8 B, the expression levels of D685A and WT Stat6 were comparable in infected CFTL-15 cells. These results suggest that the cleavage-resistant D685A Stat6 behaves as hyperfunctional Stat6 in CFTL-15 cells and the proteolytic processing of Stat6 is involved in the negative regulation of Stat6-mediated signaling in CFTL-15 cells.
Figure 8.

Retrovirus-mediated expression of the cleavage-resistant Stat6 increases IL-4–induced apoptosis in CFTL-15 cells. (A) CFTL-15 cells were infected with MSCV-C-Flag WT Stat6-IRES-Thy1.1, MSCV-C-Flag D685A Stat6-IRES-Thy1.1, or MSCV-IRES-Thy1.1 (as a control) retroviruses. Cells were cultured in the presence of IL-3 for 2 d and then cultured with IL-3 alone or IL-3 plus IL-4 for an additional 3 d. The percentage of annexin V+ cells in Thy1.1+ cells was then evaluated by FACS®. Data are mean ± SD for five experiments. *, Significantly different from the mean value of MSCV-C-Flag WT Stat6-IRES-Thy1.1 infected cells; P < 0.005. (B) Anti-Flag and anti-Stat3 (as a control) blottings of sorted Thy1.1+ cells.
Next, we examined the effect of D685A Stat6 on the cell growth of IL-3–dependent BMMCs using bicistronic retroviruses that coexpress GFP. Stat6−/− BMMCs were infected with D685A Stat6 retrovirus, WT Stat6 retrovirus, or control retrovirus, and then cultured with either IL-3 alone or IL-3 plus IL-4. The number of live infected mast cells (GFP+ c-kit+ PI− cells) was then determined 3 and 7 d later. As shown in Fig. 9 A (left), in the absence of IL-4 stimulation, no significant difference was observed in IL-3–induced proliferation of BMMCs among these viruses. In Stat6−/− BMMCs expressing control retrovirus, IL-4 increased the number of IL-3–dependent BMMCs (Fig. 9 A). In contrast, IL-4 decreased the number of Stat6−/− BMMCs when these cells were infected with WT Stat6 retrovirus (Fig. 9 A). Retrovirus-mediated expression of 673 Stat6 did not affect the IL-4–induced growth inhibition of Stat6−/− BMMCs (unpublished data). Interestingly, IL-4–mediated growth inhibition was significantly enhanced in Stat6−/− BMMCs infected with D685A Stat6 retrovirus compared with those infected with WT Stat6 retrovirus (D685A Stat6, 32.4 ± 15.7% vs. WT Stat6, 90.2 ± 20.4% of percent cell growth at day 7; n = 5 experiments; P < 0.001; Fig. 9 A). Consistent with this finding, apoptotic cells (annexin V+ cells) were increased in D685A Stat6–expressing BMMCs than WT Stat6–expressing BMMCs (D685A Stat6, 12.5 ± 3.1% vs. WT Stat6, 6.8 ± 2.5%; n = 5; P < 0.01). These results suggest that the Stat6 protease activity is involved in the regulation of Stat6-dependent cell death pathway in mast cells.
Figure 9.
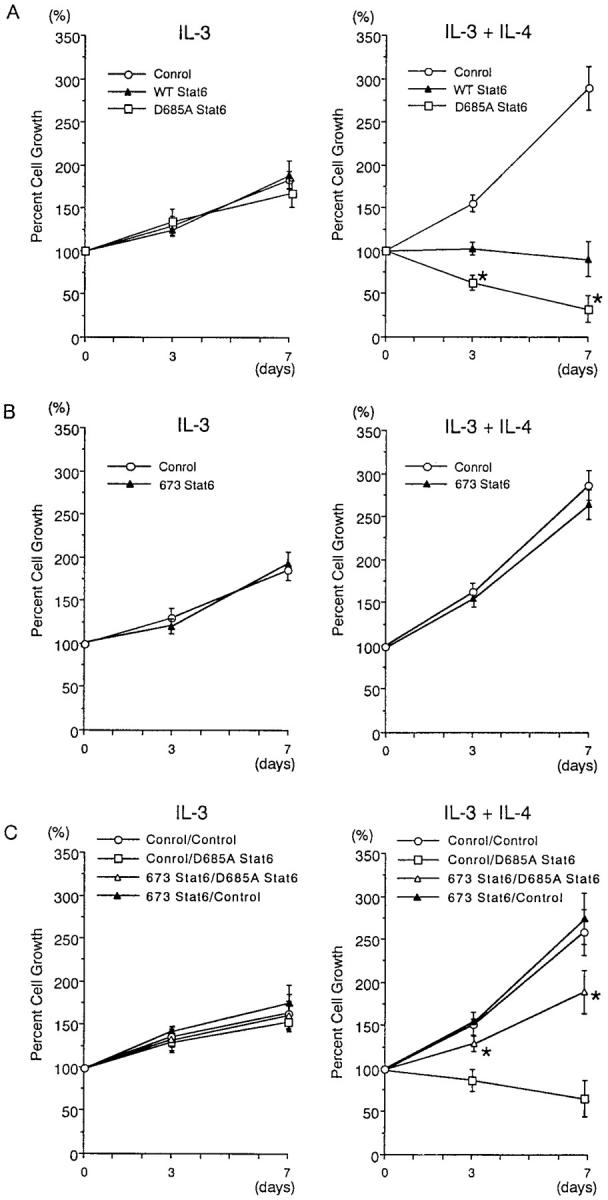
Retrovirus-mediated expression of D685A Stat6 enhances IL-4–induced growth inhibition of Stat6−/− BMMCs. (A) Stat6−/− BMMCs were infected with retroviruses of pMX-WT Stat6-IRES-GFP, pMX-D685A Stat6-IRES-GFP, or pMX-IRES-GFP, and then cultured with IL-3 alone (left) or IL-3 plus IL-4 (right). The number of live infected BMMCs (GFP+ c-kit+ PI− cells) was determined at days 0, 3, and 7 using a hemocytometer and FACS®. Data are means ± SD for five experiments. *, significantly different from the mean value of pMX-WT Stat6-IRES-GFP infected cells; P < 0.001. (B) WT BMMCs were infected with retroviruses of pMX-673 Stat6-IRES-GFP or pMX-IRES-GFP and then cultured with IL-3 alone (left) or IL-3 plus IL-4 (right). The number of live infected BMMCs was determined as described above. (C) Stat6−/− BMMCs were doubly infected with GFP-labeled retroviruses (either pMX-673 Stat6-IRES-GFP or pMX-IRES-GFP) and Thy1.1-labeled retroviruses (either MSCV-C-Flag D685A Stat6-IRES-Thy1.1 or MSCV-IRES-Thy1.1), and then cultured with IL-3 alone (left) or IL-3 plus IL-4 (right). The number of live doubly infected BMMCs (GFP+ Thy1.1+ PI− cells) was determined at day 0, 3, and 7 using a hemocytometer and FACS®. Data are means ± SD for four experiments. *, Significantly different from the mean value of pMX-IRES-GFP and/or MSCV-C-Flag D685A Stat6-IRES-Thy1.1 infected cells; P < 0.01.
65-kD Stat6 Functions as a Dominant-negative Regulator in Mast Cells.
To determine whether 65-kD Stat6 functions as a dominant-negative regulator in mast cells, we examined the effect of retrovirus-mediated expression of 673 Stat6 on IL-4–induced growth inhibition of BMMCs. In contrast to its apparent dominant-negative effect in COS7 cells (Fig. 5 D), the dominant-negative effect of 673 Stat6 was not obvious in WT BMMCs (Fig. 9 B). However, because WT BMMCs express abundant 65-kD Stat6 in the nucleus after IL-4 stimulation (Fig. 3 C), it is possible that the endogenously produced 65-kD Stat6 masks the effect of 673 Stat6 in this situation. To eliminate the interference by the endogenously produced 65-kD Stat6, we examined the effect of retrovirus-mediated expression of 673 Stat6 on D685A Stat6–expressing Stat6−/− BMMCs (Fig. 9 C). When 673 Stat6 retrovirus was coinfected with D685A Stat6 retrovirus in Stat6−/− BMMCs, 673 Stat6 reversed the effect of D685A Stat6 on IL-4–induced growth inhibition of Stat6−/− BMMCs (Fig. 9 C). These results suggest that 673 Stat6 functions as a dominant-negative regulator in mast cells. Taken together with the data shown in Fig. 9 B, these results also suggest that the endogenously produced 65-kD Stat6 may function as a dominant-negative regulator on full-length Stat6 in mast cells.
Retrovirus-mediated Expression of D685A Stat6 Enhances IL-4–induced Down-regulation of IgE Receptors on Mast Cells.
It has recently been shown that IL-4 decreases the expression of IgE receptors on mast cells in a Stat6-dependent manner (11). Therefore, we examined the effect of retrovirus-mediated expression of D685A Stat6 on the levels of IgE receptors on BMMCs. Stat6−/− BMMCs were infected with WT Stat6 retrovirus, D685A Stat6 retrovirus, 673 Stat6 retrovirus, or control retrovirus, allowed to grow for 3 d in the presence of IL-3, and then cultured with either IL-3 alone or IL-3 plus IL-4 for 24 h. The expression of IgE receptors on infected (GFP+) BMMCs was analyzed by flow cytometry. As shown in Fig. 10, in the absence of IL-4 stimulation, no significant differences were observed in the expression of IgE receptors among these retrovirus-infected BMMCs. Consistent with the previous report (11), IL-4 decreased the expression of IgE receptors in WT Stat6 virus–infected Stat6−/− BMMCs (IgE-bindinglow cells, 25.3 ± 5.5%; n = 4; Fig. 10). Interestingly, IL-4–mediated down-regulation of IgE receptors was significantly enhanced when Stat6−/− BMMCs were infected with D685A Stat6 virus (IgE-bindinglow cells, 53.5 ± 6.8%; P < 0.01), which suggests that D685A Stat6 functions as hyperfunctional Stat6 in IL-4–induced down-regulation of IgE receptors on BMMCs. In contrast, IL-4 did not decrease the expression of IgE receptors on Stat6−/− BMMCs that were infected with 673 Stat6 or control virus (Fig. 10), indicating that IL-4–induced down-regulation of IgE receptors requires the transcriptional activity of Stat6.
Figure 10.

D685A Stat6 enhances IL-4–induced down-regulation of IgE receptors on Stat6−/− BMMCs. Stat6−/− BMMCs were infected with retroviruses of pMX-WT Stat6-IRES-GFP, pMX-D685A Stat6-IRES-GFP, pMX-673 Stat6-IRES-GFP, or pMX-IRES-GFP, and then cultured with IL-3 alone or IL-3 plus IL-4 for 24 h. The expression of IgE receptors on infected (GFP+) BMMCs was determined by FACS®. Representative FACS® profiles from four independent experiments are shown.
Discussion
In this study, we show a novel regulatory mechanism of Stat6-mediated signaling in murine mast cells. We found that when Stat6 was phosphorylated on IL-4 stimulation and translocated to the nucleus, phosphorylated Stat6 was cleaved by a nucleus-associated protease in BMMCs (Figs. 1 and 2). We also found that the cleaved 65-kD Stat6 lacked the COOH-terminal transactivation domain (Fig. 1) and functioned as a dominant-negative molecule to Stat6-mediated transcription (Fig. 5). Moreover, the retrovirus-mediated expression of cleavage-resistant D685A Stat6 prolonged the nuclear accumulation of Stat6 upon IL-4 stimulation in mast cell line CFTL-15 cells but not in COS7 cells that lack the protease activity (Fig. 7). Furthermore, the expression of D685A Stat6 enhanced IL-4–induced apoptosis and growth inhibition in mast cells (Figs. 8 and 9). D685A Stat6 also enhanced IL-4–induced down-regulation of IgE receptors in BMMCs (Fig. 10). Taken together, these results indicate that the proteolytic processing of Stat6 is a lineage-specific negative regulatory mechanism of Stat6-dependent signaling in mast cells.
We show that phosphorylated Stat6 is cleaved by the Stat6 protease in the nucleus of mast cells and that the resultant 65-kD Stat6 lacks the transcriptional activity and functions as a dominant-negative molecule to Stat6-mediated signaling. Stat6 was cleaved by the protease between aa 673 and 695 of Stat6 (Fig. 5 A). Furthermore, we found that alanine substitution of aspartic acid at 685 or methionine at 686 of Stat6 was resistant to the Stat6 protease activity (Fig. 6 A), which suggests that Stat6 is cleaved around this region. Thus, the 65-kD Stat6 is apparently different from the previously reported Stat6 isoforms in human fibroblast, Stat6b and Stat6c, which encode an NH2-terminal truncation and an SH2 domain deletion, respectively, and result from alternative splicing (27). Regarding the functional property of 65-kD Stat6, we found that COOH-terminal–truncated mutants of Stat6 at either aa 673 or 695 lost the transcriptional activity in a Stat6-dependent reporter assay (Fig. 5). Moreover, the coexpression of these truncated mutants with full-length Stat6 inhibited the Stat6-mediated transcription (Fig. 5) and growth inhibition of mast cells (Fig. 9 C), suggesting that 65-kD Stat6 functions as a dominant-negative regulator to Stat6-mediated signaling. This observation is in agreement with the previous finding that COOH-terminal–truncated mutants of Stat6 function as dominant-negative molecules in Stat6-mediated transcription (20).
It has recently been shown that a ubiquitin-dependent proteasome pathway regulates the turnover of Stat5 and Stat6 in lymphoid cells (28). In the case of Stat5, Wang et al. (28) clearly demonstrated that a relatively small, potentially amphipathic α-helical region in the carboxyl-terminal of Stat5 played an important role in the rapid turnover of Stat5 protein. However, the analogous region was not found in Stat6 (28). In addition, we found that Stat6 cleaving activity in mast cells was not inhibited by a proteasome inhibitor, lactacystin (Fig. 3 A). Thus, the Stat6 cleaving activity in mast cells seems independent of proteasome pathway.
On the other hand, we found that the Stat6 protease activity in mast cells was inhibited by a serine protease inhibitor, AEBSF (Fig. 3 B) or PMSF (unpublished data), suggesting that the protease may belong to serine protease family. In addition, we found that the Stat6 protease activity was localized in the nucleus (Fig. 2 A). Interestingly, these two properties are shared with the recently described Stat5 protease in myeloid progenitors (15–17). It has also been shown that Stat5 protease cleaves both Stat5a and Stat5b at the COOH terminus (15–18). Although a number of properties are shared between the Stat6 protease activity and the Stat5 protease activity, we believe that the Stat6 protease activity is not identical to the Stat5 protease activity for the following reasons. First, both Stat5a and Stat5b were expressed as conventional forms in BMMCs (Fig. 4 A). Second, Stat6 was expressed as 94-kD Stat6 in FDC-P1 cells (unpublished data). Third, FDC-P1 cell extract cleaved Stat5 but not Stat6, whereas BMMC extract cleaved Stat6 but not Stat5 (Fig. 4 B). Finally, the recognition sequence of Stat5 protease, ATYMDQA (17), is not found in Stat6 (Fig. 6 B). Taken together, these results indicate that the Stat6 protease activity differs from the Stat5 protease activity, although these two proteases may belong to the same family. Thus, proteolytic inactivation of Stat proteins by cell type–specific Stat proteases may be a general mechanism in lineage-specific down-regulation of Stat-mediated signaling.
At present, the mechanism underlying the restricted expression of Stat proteases and the regulation of their activities in a given cell type are unknown. Stat6 and Stat5 protease activity are restricted in mast cells and myeloid progenitors, respectively (Fig. 1 and references 13–17). In addition, we found that Stat6 protease activity was detected in the nucleus of BMMCs (Fig. 2) and CFTL-15 cells (unpublished data) in the absence of IL-4 stimulation. Moreover, Stat6 protease activity was even found in Stat6−/− BMMCs (Fig. 2). These results indicate that the Stat6 protease is in an active form in the nucleus of mast cells and not dependent on the presence of its substrate. However, because homeostatic regulation of the proteolytic processing by serine proteases is achieved, in part, through the interaction between a protease and a corresponding endogenous serine protease inhibitor (serpin; reference 29), the loss of inhibitory activity of serpin may result in an imbalance between proteases and their inhibitors. Thus, it is still possible that mast cell–specific proteolytic processing of Stat6 may be achieved by the specific loss of an inhibitor to the Stat6 protease activity in the nucleus of mast cells.
It is clear from recent experiments using Stat6−/− mice that Stat6 is not essential for IL-4–mediated proliferation of mast cells (11). On the other hand, in some situations, Stat6-mediated signaling has been shown to be essential for the death-promoting effect of IL-4 on IL-3–dependent mast cells by an undefined mechanism(s) (10). In agreement with this observation, we found that the enforced expression of WT Stat6 in Stat6−/− BMMCs diminished the IL-4–induced cell growth (Fig. 9). Moreover, the Stat6-mediated growth inhibition was significantly enhanced when cleavage-resistant D685A Stat6 was expressed in Stat6−/− BMMCs (Fig. 9). Therefore, our results suggest that the proteolytic inactivation of Stat6 by the Stat6 protease may prevent Stat6-mediated cell death of mast cells.
Our results also indicate that Stat6-mediated gene expression is regulated in part by the Stat6 protease activity in mast cells. Stat6 has been shown to be involved in the expression of a number of important genes in various cell types (2). We found that IL-4–induced down-regulation of IgE receptors on BMMCs was enhanced by the expression of D685A Stat6 (Fig. 10). We also found that IL-4–induced expression of CD23 was enhanced by the expression of D685A Stat6 in BMMCs (unpublished data). Therefore, our results suggest that in addition to the survival, the proteolytic processing of Stat6 may also be involved in Stat6-dependent gene regulation in mast cells.
Mast cells are not only important effector cells in acute IgE-associated allergic reaction, but also contribute significantly to the protection from bacterial and parasitic infection (30). It has recently been shown that the activation of mast cells by IL-4 is essential to expel gastrointestinal nematode, Trichinella spiralis (31), suggesting that mast cells should survive in IL-4–rich environments to expel parasites. Therefore, Stat6 protease system may play a role in the prolongation of mast cell survival in this situation. The further role of the Stat6 protease will be determined when the protease is cloned and mice lacking the protease are analyzed.
In conclusion, we have shown that the proteolytic processing of Stat6 functions as a lineage-specific negative regulator of Stat6-dependent signaling in mast cells. Currently, the mechanisms underlying the selective expression of Stat6 protease activity in mast cells are unclear. The protease activity in mast cells appears to be independent of IL-4– and/or Stat6-mediated signaling. In this regard, this regulatory system differs from the SOCS/CIS/SSI system, in which expression is induced in response to cytokine-induced Stat activation (32). Thus, the Stat6 protease system may be more potent than the SOCS/CIS/SSI system in abolishing the early phase of Stat6-mediated signaling. It is suggested that the Stat6 protease system might be applicable for the treatment of allergic diseases by inducing the Stat6 protease activity in T or B cells.
Acknowledgments
This work was supported in part by grants from the Japanese Ministry of Education, Science and Culture, Health Science Research Grants of Japan, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Japan Research Foundation for Clinical Pharmacology. K. Suzuki was supported in part by the Japan Society for the Promotion of Science Research Fellowships for Young Scientists.
K. Suzuki and H. Nakajima contributed equally to this work.
Footnotes
Abbreviations used in this paper: aa, amino acid; AEBSF, 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride; BMMC, bone marrow–derived mast cell; GFP, green fluorescent protein; IRES, internal ribosome entry site; JAK, Janus kinase; MMP, matrix metalloproteinase; MSCV, murine stem cell virus; PCMB, p-chloromercuribenzoate; WT, wild-type.
References
- 1.Seder, R.A., and W.E. Paul. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673. [DOI] [PubMed] [Google Scholar]
- 2.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 3.Wurster, A.L., T. Tanaka, and M.J. Grusby. 2000. The biology of Stat4 and Stat6. Oncogene. 19:2577–2584. [DOI] [PubMed] [Google Scholar]
- 4.Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role of Stat6 in IL-4 signalling. Nature. 380:627–630. [DOI] [PubMed] [Google Scholar]
- 5.Shimoda, K., J. van Deursen, M.Y. Sangster, S.R. Sarawar, R.T. Carson, R.A. Tripp, C. Chu, F.W. Quelle, T. Nosaka, D.A.A. Vignali, et al. 1996. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 380:630–633. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan, M.H., U. Schindler, S.T. Smiley, and M.J. Grusby. 1996. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 4:313–319. [DOI] [PubMed] [Google Scholar]
- 7.Metcalfe, D.D., D. Baram, and Y.A. Mekori. 1997. Mast cells. Physiol. Rev. 77:1033–1079. [DOI] [PubMed] [Google Scholar]
- 8.Yanagida, M., H. Fukamachi, K. Ohgami, T. Kuwaki, H. Ishii, H. Uzumaki, K. Amano, T. Tokiwa, H. Mitsui, and H. Saito. 1995. Effects of T-helper 2-type cytokines, IL-3, IL-4, IL-5, and IL-6 on the survival of cultured human mast cells. Blood. 86:3705–3714. [PubMed] [Google Scholar]
- 9.Tepper, R.I., D.A. Levinson, B.Z. Stanger, J. Campos-Torres, A.K. Abbas, and P. Leder. 1990. IL-4 induces allergic-like inflammatory disease and alters T cell development in transgenic mice. Cell. 62:457–467. [DOI] [PubMed] [Google Scholar]
- 10.Yeatman, C.F., II, S.M. Jacobs-Helber, P. Mirmonsef, S.R. Gillespie, L.A. Bouton, H.A. Collins, S.T. Sawyer, C.P. Shelburne, and J.J. Ryan. 2000. Combined stimulation with the T helper cell type 2 cytokines interleukin (IL)-4 and IL-10 induces mouse mast cell apoptosis. J. Exp. Med. 192:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryan, J.J., S. DeSimone, G. Klisch, C. Shelburne, L.J. McReynolds, K. Han, R. Kovacs, P. Mirmonsef, and T.F. Huff. 1998. IL-4 inhibits mouse mast cell FcɛRI expression through a STAT6-dependent mechanism. J. Immunol. 161:6915–6923. [PubMed] [Google Scholar]
- 12.Urban, J.F., Jr., N. Noben-Trauth, D.D. Donaldson, K.B. Madden, S.C. Morris, M. Collins, and F.D. Finkelman. 1998. IL-13, IL-4Rα, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 8:255–264. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki, K., H. Nakajima, N. Watanabe, S. Kagami, A. Suto, Y. Saito, T. Saito, and I. Iwamoto. 2000. Role of common cytokine receptor γ chain (γc)- and Jak3-dependent signaling in the proliferation and survival of murine mast cells. Blood. 96:2172–2180. [PubMed] [Google Scholar]
- 14.Sherman, M.A., V.H. Secor, and M.A. Brown. 1999. IL-4 preferentially activates a novel STAT6 isoform in mast cells. J. Immunol. 162:2703–2708. [PubMed] [Google Scholar]
- 15.Azam, M., C. Lee, I. Strehlow, and C. Schindler. 1997. Functionally distinct isoforms of STAT5 are generated by protein processing. Immunity. 6:691–701. [DOI] [PubMed] [Google Scholar]
- 16.Meyer, J., M. Jucker, W. Ostertag, and C. Stocking. 1998. Carboxyl-truncated STAT5β is generated by a nucleus-associated serine protease in early hematopoietic progenitors. Blood. 91:1901–1908. [PubMed] [Google Scholar]
- 17.Lee, C., F. Piazza, S. Brutsaert, J. Valens, I. Strehlow, M. Jarosinski, C. Saris, and C. Schindler. 1999. Characterization of the Stat5 protease. J. Biol. Chem. 274:26767–26775. [DOI] [PubMed] [Google Scholar]
- 18.Piazza, F., J. Valens, E. Lagasse, and C. Schindler. 2000. Myeloid differentiation of FdCP1 cells is dependent on Stat5 processing. Blood. 96:1358–1365. [PubMed] [Google Scholar]
- 19.Noguchi, M., A. Sarin, M.J. Aman, H. Nakajima, E.W. Shores, P.A. Henkart, and W.J. Leonard. 1997. Functional cleavage of the common cytokine receptor γ chain (γc) by calpain. Proc. Natl. Acad. Sci. USA. 94:11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikita, T., D. Campbell, P. Wu, K. Williamson, and U. Schindler. 1996. Requirements for interleukin-4-induced gene expression and functional characterization of Stat6. Mol. Cell. Biol. 16:5811–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schtze, S., K. Potthoff, T. Machleidt, D. Berkovic, K. Wiegmann, and M. Kronke. 1992. TNF activates NF-κB by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell. 71:765–776. [DOI] [PubMed] [Google Scholar]
- 22.Nosaka, T., T. Kawashima, K. Misawa, K. Ikuta, A.L. Mui, and T. Kitamura. 1999. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 18:4754–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell, T.C., D. Hildeman, R.M. Kedl, T.K. Teague, B.C. Schaefer, J. White, Y. Zhu, J. Kappler, and P. Marrack. 2001. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat. Immunol. 2:397–402. [DOI] [PubMed] [Google Scholar]
- 24.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066. [DOI] [PubMed] [Google Scholar]
- 25.Sherman, M.A. 2001. The role of STAT6 in mast cell IL-4 production. Immunol. Rev. 179:48–56. [DOI] [PubMed] [Google Scholar]
- 26.Begitt, A., T. Meyer, M. van Rossum, and U. Vinkemeier. 2000. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl. Acad. Sci. USA. 97:10418–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel, B.K.R., J.H. Pierce, and W.J. LaRochelle. 1998. Regulation of interleukin 4-mediated signaling by naturally occurring dominant negative and attenuated forms of human Stat6. Proc. Natl. Acad. Sci. USA. 95:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang, D., R. Moriggl, D. Stravopodis, N. Carpino, J.-C. Marine, S. Teglund, J. Feng, and J.N. Ihle. 2000. A small amphipathic α-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. EMBO J. 19:392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janciauskiene, S. 2001. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim. Biophys. Acta. 1535:221–235. [DOI] [PubMed] [Google Scholar]
- 30.Wedemeyer, J., M. Tsai, and S.J. Galli. 2000. Roles of mast cells and basophils in innate and acquired immunity. Curr. Opin. Immunol. 12:624–631. [DOI] [PubMed] [Google Scholar]
- 31.Urban, J.F., Jr., L. Schopf, S.C. Morris, T. Orekhova, K.B. Madden, C.J. Betts, H.R. Gamble, C. Byrd, D. Donaldson, K. Else, et al. 2000. Stat6 signaling promotes protective immunity against Trichinella spiralis through a mast cell- and T cell-dependent mechanism. J. Immunol. 164:2046–2052. [DOI] [PubMed] [Google Scholar]
- 32.Yasukawa, H., A. Sasaki, and A. Yoshimura. 2000. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 18:143–164. [DOI] [PubMed] [Google Scholar]