

Abstract
The contribution of platelets to the process of atherosclerosis remains unclear. Here, we show in vivo that platelets adhere to the vascular endothelium of the carotid artery in ApoE − / − mice before the development of manifest atherosclerotic lesions. Platelet–endothelial cell interaction involved both platelet glycoprotein (GP)Ibα and GPIIb-IIIa. Platelet adhesion to the endothelium coincides with inflammatory gene expression and preceded atherosclerotic plaque invasion by leukocytes. Prolonged blockade of platelet adhesion in ApoE − / − mice profoundly reduced leukocyte accumulation in the arterial intima and attenuated atherosclerotic lesion formation in the carotid artery bifurcation, the aortic sinus, and the coronary arteries. These findings establish the platelet as a major player in initiation of the atherogenetic process.
Keywords: platelets, endothelium, atherosclerosis, integrin, glycoprotein Ib
Introduction
Atherosclerosis is a systemic inflammatory disease characterized by the accumulation of monocytes/macrophages and lymphocytes in the intima of large arteries (1, 2). Rupture or erosion of the advanced lesion initiates platelet activation and aggregation on the surface of the disrupted atherosclerotic plaque. Thrombotic vascular occlusion is associated with ischemic episodes, including acute coronary syndromes or cerebral infarction. While it is widely accepted that platelets play a significant role in thromboembolic complications of advanced atherosclerotic lesions, their involvement in the initiation of the atherosclerotic process has received scant attention.
Several indications suggest that platelets might significantly contribute to the inflammatory processes that promote atherosclerotic lesion formation. Platelets can adhere directly to the intact endothelial monolayer even in the absence of endothelial disruption (3–6). During adhesion platelets are activated and release proinflammatory cytokines and chemoattractants (e.g., IL-1β, or RANTES) and surface express CD40 ligand (CD40L) (7–9). In vitro, interaction of platelets with endothelial cells triggers the secretion of chemokines and the expression of adhesion molecules and promotes the adhesion of leukocytes (9–11). In this manner, the adhesion of platelets to the endothelial surface might generate signals for recruitment and extravasation of monocytes during atherosclerotic plaque formation, a process of paramount importance for atherogenesis (12). However, the relevance of platelets for initiation of atherosclerosis in vivo has not been established.
The development of ApoE-deficient (ApoE −/−) mice has provided an opportunity to directly assess the dynamics of platelet-vessel wall interactions in the process of atherosclerosis and to evaluate the significance of platelet adhesion for the initiation of atherosclerotic plaque formation. ApoE −/− mice acquire widespread arterial lesions with a pathomorphology similar to humans that progress from simple fatty streaks to complex fibrous plaques (13). Using intravital microscopy we assessed platelet–endothelial cell interactions in the carotid artery of ApoE −/− mice in the early and advanced stages of atherosclerosis in vivo and evaluated the significance of platelet–endothelial cell interactions for plaque formation and plaque progression. Our results substantiate a critical role for platelets in the initiation of atherosclerotic lesion development.
Materials and Methods
Animals.
4-wk-old male ApoE −/− (C57BL/6J-ApoE tm1Unc) mice (The Jackson Laboratory) consumed a 0.25% cholesterol diet (Harlan Research diets, 0% cholate) for another 2, 4, 6, 8, 12, or 18 wk (n = 20 per group). C57BL/6J wild-type mice (Charles River Laboratories) served as controls (n = 20).
Assessment of Vascular Resistance Index by Duplex Color Sonography.
The animals were anaesthetized and the common carotid artery was visualized by duplex color sonography. Using a Acuson Sequoia 512 with a 15 MHz transducer (Acuson) the maximum systolic Vsys and the end-diastolic flow velocity Vdia were determined. The resistance index of the common carotid artery was calculated as the difference between Vsys and Vdia divided by Vsys.
Determination of Platelet and Leukocyte Adhesion.
Platelet and leukocyte adhesion dynamics in the process of atherosclerosis were monitored in vivo by use of video fluorescence microscopy. In brief, mice were anaesthetized by intraperitoneal injection of a solution of midazolame (5 mg/kg body weight; Ratiopharm), medetomidine (0.5 mg/kg body weight; Pfizer), and fentanyl (0.05 mg/kg body weight; CuraMed Pharma GmbH). The right common carotid artery was carefully exposed at a distance of ∼3 mm distal and 7-mm proximal of the carotid bifurcation. The exposed tissue was continuously superfused with a thermostated bicarbonate-buffered saline solution equilibrated with 5% CO2 in nitrogen to maintain physiological pH. Polyethylene catheters were implanted into the right jugular vein for injection of fluorescent dyes.
Murine Platelets Isolated from Subsets of ApoE−/−.
Mice from each group or human platelets (normal or glycoprotein (GP)* IIb-IIIa deficient; reference 6) were labeled with CFSE (DCF). The DCF-labeled platelet suspension was adjusted to a final concentration of 50 × 106 platelets per 250 μl and infused intravenously. Where indicated, fluorescent wild-type platelets were preincubated with 50 μg/ml anti-GPIbα (p0p/B-Fab) or anti-GPIIb-IIIa (JON/A-F(ab)2 (14) for 10 min before infusion. Leukocytes were stained in vivo by intravenous injection of 100 μl 0.02% rhodamine 6G (Molecular Probes). Subsequently, the carotid artery was visualized at low (250-fold) magnification using a Zeiss Axiotech microscope (water immersion objective: 20×, W 20×/0.5; Zeiss) with a 100 W HBO mercury lamp for epi-illumination. As illustrated in Fig. 1 A, two perpendicular axes were dropped through the origin of the internal and the external common carotid artery. A third line was drawn connecting the two perpendicular axes, where they crossed the vessel wall. Platelet and leukocyte adhesion were determined at high magnification (500-fold) in a 200 μm × 100 μm area adjacent to the third line. This implicates that platelet recruitment was assessed exactly at the carotid bifurcation, which is a predilection site for the development of atherosclerotic lesions.
Figure 1.

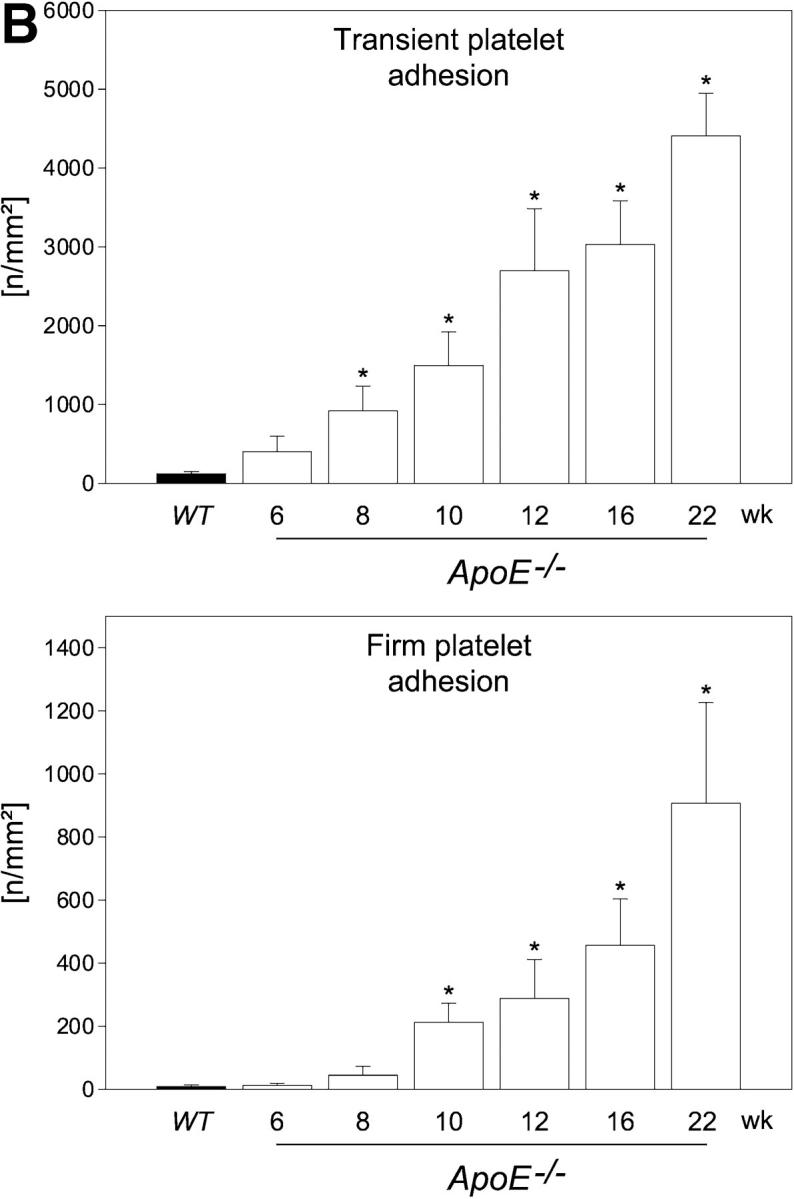


Platelet adhesion to the endothelium of the common carotid artery in ApoE −/− mice in vivo. (A) Assessment of platelet adhesion at the carotid bifurcation (lesion-prone-site, A) and within the proximal portion of the common carotid artery. For in vivo microscopy, two imaginary perpendicular axes were dropped through the origin of the internal and the external common carotid artery. Platelet and leukocyte adhesion were determined at high magnification (500-fold) in a 200 × 100 μm area adjacent to a third line, connecting the perpendicular axes at their intersection with the vessel wall (lesion-prone site, A). In a subset of experiments, platelet adhesion was also determined in the proximal portion of the common carotid artery 500 μm upstream of the carotid bifurcation (nonlesion-prone site, B). ACC, common carotid artery; ACE, external carotid artery; ACI, internal carotid artery. (B) Platelet–endothelial cell interactions were investigated in 6-, 8-, 10-, 12-, 16-, and 22-wk-old ApoE −/− mice by in vivo fluorescence microscopy of the common carotid artery in situ. Wild-type animals served as controls. The top and bottom panels summarize transient and firm platelet adhesion, respectively, of 10 experiments per group. Platelets were classified according to their interaction with the endothelial cell lining as described previously (reference 5) and are given per mm2 of vessel surface. (C) The microphotographs show representative in vivo fluorescence microscopy images at distinct time points. White arrows indicate adherent platelets, and the black arrow indicates the area of a fatty streak. The small inserts demonstrate histological sections of the corresponding carotid artery under investigation (original magnification: 200-fold). Underneath, the in vivo microscopic images representative en face carotid arteries, stained with Sudan III, are given. (D) Assessment of platelet adhesion to lesion-prone (white bars) and nonlesion-prone sites (black bars). Platelet adhesion was quantified in the proximal (nonlesion-prone) carotid artery and adjacent to the carotid bifurcation (lesion-prone) in both, young (10-wk-old) and old (22-wk-old) ApoE −/− mice (left). The photomicrograph in the middle shows a representative carotid artery, stained with Sudan III. Atherosclerotic plaque formation occurs preferentially at the carotid bifurcation. Representative histological sections of the carotid bifurcation and the proximal common carotid artery are presented in the right panels (original magnification: 200-fold).
All images were videotaped and evaluated off-line using a computer-assisted image analysis program (Cap Image 7.1; Dr. H. Zeintl, Ingenieurbüro Dr. Zeintl, Heidelberg, Germany) (references 5 and 6). Transiently adherent platelets were defined as cells crossing an imaginary perpendicular through the vessel at a velocity significantly lower than the centerline velocity; their numbers are given as cells per mm2 endothelial surface. The number of adherent cells (leukocytes or platelets) was assessed by counting the cells that did not move or detach from the endothelial surface within 20 s.
In a subset of experiments, we determined whether or not platelet adhesion is restricted to lesion-prone sites within the carotid bifurcation or rather occurs throughout the entire carotid artery. To address this issue, platelet adhesion was analyzed at the carotid bifurcation (lesion-prone site) and in the proximal portion of the common carotid artery, ∼500 μm upstream of the carotid bifurcation (nonlesion-prone site; see Fig. 1 A) in young (10 wk, n = 8) and old ApoE −/− mice (n = 8, 22 wk).
Characterization of Intimal Inflammation and Atheroprogression.
After intravital videofluorescence microscopy, the animals were killed for collection of the arterial tree. The common carotid arteries and the descending aorta were shock-frozen for EMSA (15). To determine monocyte recruitment into the intima of the carotid artery and aorta, shock-frozen tissue samples were additionally evaluated for CD14 and CD11b mRNA expression by RT-PCR. The primer pairs were: CD11b (forward) 5′-CTG GTC CAA AGC TTG GTT TT-3′, CD11b (reverse) 5′-AGC CAT CCA TTG TGA GAT CC-3′, CD14 (forward) 5′-TAC CGA CCA TGG AGC GTG TG-3′ and CD14 (reverse) 5′-GCC GGT TAC CTC GAG ATT TT-3′ CD14. The 18s ribosomal RNA was used as amplification control (forward: 5′-CGG CTA CCA CAT CCA AGG AA-3′ and reverse: 5′-GCT GGA ATT ACC GCG GCT-3′). The remaining carotid arteries were embedded in paraffin and cut into a series of sections (5 μm in thickness). 20 sections downstream of the carotid bifurcation were used for assessment of plaque formation at the lesion-prone site. All sections were stained with H&E. The remaining carotid arteries of each group were opened longitudinally, mounted on slides, and stained with Sudan III to assess plaque formation. For immunolabeling studies cryoblock (medite, Medizintechnik GmbH) embedded aortae were cut into 5-μm sections and stained with anti–vascular cell adhesion molecule (VCAM)-1 (rabbit polyclonal IgG), or anti–MCP-1 (goat polyclonal IgG), purchased from Santa Cruz Biotechnology.
Chronic Inhibition of Platelet Adhesion.
To determine the relevance of platelet adhesion for atherosclerosis, 6-wk-old ApoE −/− mice were randomly assigned to receive either an irrelevant rat Ig (50 μg, n = 4) or a rat anti–mouse GPIbα (50 μg, mAb p0p/B, n = 6) for another 12 wk twice a week by intraperitoneal injection. Untreated ApoE −/− mice served as controls (n = 6). 3 d after the last mAb injection, the animals were anaesthetized, and the vascular resistance index was determined by duplex sonography (see above). Thereafter, leukocyte adhesion to the carotid bifurcation was assessed by in vivo microscopy as described above. Subsequently, blood samples were drawn from all mice for determination of circulating rat IgG by ELISA. Finally, all mice were killed and the carotid arteries, hearts, and ascending aortae were processed for histomorphometry according to a standardized protocol as follows.
In brief, one carotid artery per animal was excised. Carotid arteries were opened longitudinally, stained with Sudan III, and mounted on slides using glycerol gelatine. Plaque extension was quantified as μm2 Sudan III-stained surface area by an image analysis program (Cap Image 7.4; Dr. H. Zeintl). In addition, shock-frozen tissue samples were evaluated for CD14 and CD11b mRNA expression using RT-PCR, to determine monocyte recruitment into the intima.
To assess cross-sectional plaque area, the remaining arterial tree was perfusion-fixed in situ with 4% paraformaldehyde, pH 7.0). Thereafter, the carotid arteries and hearts were excised and further fixed with 4% paraformaldehyde for 24 h. The carotid arteries were embedded in paraffin blocks and cut into 5-μm sections from the distal to the proximal end. 20 sections downstream of the carotid bifurcation were used for quantification of plaque formation. The hearts and ascending aortae were embedded in paraffin, and cross-sectioned. The sections were discarded until reaching the junction of the heart muscle and aorta, where the valve cusps become visible. Once this area was localized, 20 5-μm sections were collected on glass slides and stained with elastica van Giesson reagent. The area of lesions in the aortic sinus was assessed by a computerized image analysis program as described previously (16, 17). In all sections, cross-sections of the proximal coronary arteries were identified and cross-sectional plaque area in the proximal coronary arteries was determined using an image analysis software.
Statistical Analysis.
Comparisons between group means were performed using Kruskal-Wallis One Way Analysis of Variance on Ranks. Data represent mean ±SEM. A value of P < 0.05 was regarded as significant.
Results and Discussion
To address the dynamic processes of platelet accumulation during atherogenesis, we used in vivo fluorescence microscopy to directly visualize and quantify platelet–vessel wall interactions (5, 6, 18) in the common carotid artery of ApoE −/− mice in situ (Fig. 1). ApoE −/− mice reveal consistent plaque formation with a pathomorphology similar to humans (13, 19, 20). Already at the age of 6 wk, a substantial increase in platelet adhesion to the carotid endothelium was detectable in ApoE −/− animals (Fig. 1 B). In contrast, platelet–endothelial cell interactions were virtually absent in wild-type mice. While the number of adherent platelets constantly increased with age, the occurrence of platelet–endothelial cell interactions strictly preceded the development of manifest atherosclerotic lesions. In fact, the number of platelets adherent to the carotid endothelium was significantly increased (Fig. 1, B and C) even in the complete absence of early vascular alterations, as assessed ex vivo by histomorphology (Fig. 1 C) and in vivo by color duplex sonography (unpublished data).
Correspondingly, 10-wk-old ApoE −/− mice showed complete absence of atherosclerotic lesions in the carotid artery, whereas both transient and firm platelet adhesion were increased 12- and 24-fold, respectively, as compared with wild-type mice (P < 0.05). Notably, platelet recruitment occurred preferentially at lesion-prone sites in young and old ApoE −/−mice, since the number of adherent platelets was significantly higher at the carotid bifurcation compared with the proximal carotid artery (nonlesion-prone site) in both 10- and 22-wk-old ApoE −/− mice (Fig. 1 D).
Platelet adhesion to endothelial cells was followed by activation of NF-κB (unpublished data) and NF-κB–regulated genes, e.g., VCAM-1 (Fig. 2 A) or MCP-1 (unpublished data), that play a pivotal role for chemotaxis and endothelial transmigration of monocytes (10–12, 15, 21) and contribute substantially to atherosclerotic lesion formation (22, 23). Importantly, we did not observe leukocyte adhesion in ApoE −/− mice before the development of initial vascular lesions as verified by in vivo microscopy (Fig. 2 B). In addition, CD14 or CD11b mRNA in the arterial tree of 6-, 8-, or 10-wk-old ApoE −/− mice did not differ from wild-type animals (unpublished data). Thus, platelet adhesion clearly preceded the recruitment of leukocytes to the arterial wall. However, significant leukocyte recruitment was encountered in mice with overt early atherosclerotic lesions detectable at 12, 16, or 22 wk of age (Fig. 2 B). In line with our findings, Eriksson et al. did not observe leukocyte–endothelium interactions in the aorta of ApoE −/− mice in the absence of atherosclerotic lesions, but demonstrated significant leukocyte adhesion in animals with diseased arteries (24, 25).
Figure 2.


Platelet adhesion precedes leukocyte chemotaxis. (A) Photomicrographs show frozen sections immunostained for VCAM-1 in the time course of atherogenesis (original magnification: 200-fold). (B) Leukocyte adhesion to the endothelium of the common carotid artery was assessed by fluorescence microscopy as described previously (reference 5). The right panel shows representative in vivo fluorescence microscopy images at distinct time points; the data are summarized in the left panel. White arrows indicate adherent leukocytes. Mean ± SEM, n = 10 each group. *P < 0.05.
Next, we addressed the molecular determinants underlying platelet adhesion in the early stage of atherogenesis before the development of manifest vascular lesions. Platelet GPIbα and GPIIb-IIIa have been demonstrated to largely contribute to endothelial platelet adhesion in vitro (26) making them good candidates for inhibition. Therefore, 10-wk-old ApoE −/− mice received fluorescence-tagged syngeneic platelets preincubated with noncytotoxic derivatives of function blocking mAbs against GPIbα (p0p/B) or GPIIb-IIIa (JON/A). Inhibition of GPIbα by p0p/B-Fab reduced both transient and firm adhesion to the vascular surface of the common carotid artery by 85 and 99%, respectively (P < 0.05) (Fig. 3 A). In contrast, inhibition of GPIIb-IIIa by JON/A-F(ab)2 (14) had only partial effects on transient platelet adhesion but almost completely prevented firm attachment to endothelial cells in vivo (by 95%, Fig. 3 A). A similar reduction in transient and firm adhesion to the vascular surface of the common carotid artery in ApoE −/− mice was observed with human platelets isolated from a patient with Glanzmann's thrombasthenia that are deficient in functional GPIIb-IIIa (6) when compared with normal human platelets (Fig. 3 B). Hence, we have identified two distinct mechanisms in vivo that act in concert to initiate recruitment of flowing platelets to the vascular surface in the very initial stage of atherogenesis. Similar to platelet adhesion to subendothelial matrix (27), GPIbα is mandatory for the initial capturing process, whereas GPIIb-IIIa is critical for firm adhesion of platelets to the endothelium.
Figure 3.


Platelet membrane glycoproteins GPIbα and GPIIb-IIIa mediate platelet–endothelium adhesion in the common carotid artery of ApoE −/− mice. (A) Platelets were preincubated with blocking mAbs (50 μg/ml) directed against GPIbα (p0p/B-Fab) or GPIIb-IIIa (JON/A-F(ab)2 before they were administered into ApoE −/− mice (10-wk-old). Platelet–endothelial cell interactions were investigated by in vivo fluorescence microscopy (see Materials and Methods). The top panel shows representative photomicrographs, the bottom panel summarizes the data (mean and SEM) of n = 4 experiments. *P < 0.05. (B) Platelets isolated from a Glanzmann's patient with a functional defect in the GPIIb-IIIa receptor (reference 6) or from a healthy control individual were evaluated for platelet–endothelium adhesion in ApoE −/− mice. n = 4, mean ± SEM. *P < 0.05.
Next, we examined whether inhibition of platelet adhesion mitigates atheroprogression in ApoE −/− mice. Therefore, 6-wk-old ApoE −/− mice were treated with the anti-GPIbα mAb p0p/B over a period of 12 wk (twice a week 50 μg) by intraperitoneal injection. The intact IgG of p0p/B was chosen for this long-term treatment because of its higher avidity and significantly prolonged half-life in vivo as compared with the Fab fragments of the mAb. P0p/B inhibits binding of von Willebrand factor to GPIbα (unpublished data) and prevents platelet adhesion in vivo (Fig. 3 A). Furthermore, the intact IgG of p0p/B significantly reduces the platelet count in vivo by Fc-independent mechanisms (28). As reported earlier by Mach et al. (29), free circulating antibody was detectable in both groups of rat IgG-treated mice after 12 wk of treatment (control IgG: 1.75 ± 0.73 μg/ml; anti-GPIbα: 1.20 ± 0.90 μg/ml), demonstrating that the rat Ig was not cleared from the circulation.
Strikingly, inhibition of platelet adhesion (for 12 wk) significantly reduced the increase in vascular resistance index in 18-wk-old ApoE −/− mice, when compared with untreated 18-wk-old animals (Fig. 4 A, P < 0.05). Furthermore, the anti-GPIbα treatment dramatically limited atherosclerotic lesion formation as compared with control animals. Quantitative computer-assisted image analysis showed an ∼90% reduction in the extent of fatty streak formation in 18-wk-old ApoE −/− mice treated with anti-GPIbα mAb for 12 wk, as assessed by Sudan III staining (Fig. 4 B). Moreover, anti–GPIbα-treated ApoE −/− mice showed substantial decreases in cross-sectional carotid plaque area (by 81%, P < 0.05) (Fig. 4 C). In contrast, in ApoE −/− mice, treated with isotype-matched control rat IgG, atheroprogression in the carotid artery was neither accelerated nor retarded, when compared with untreated ApoE −/− mice.
Figure 4.



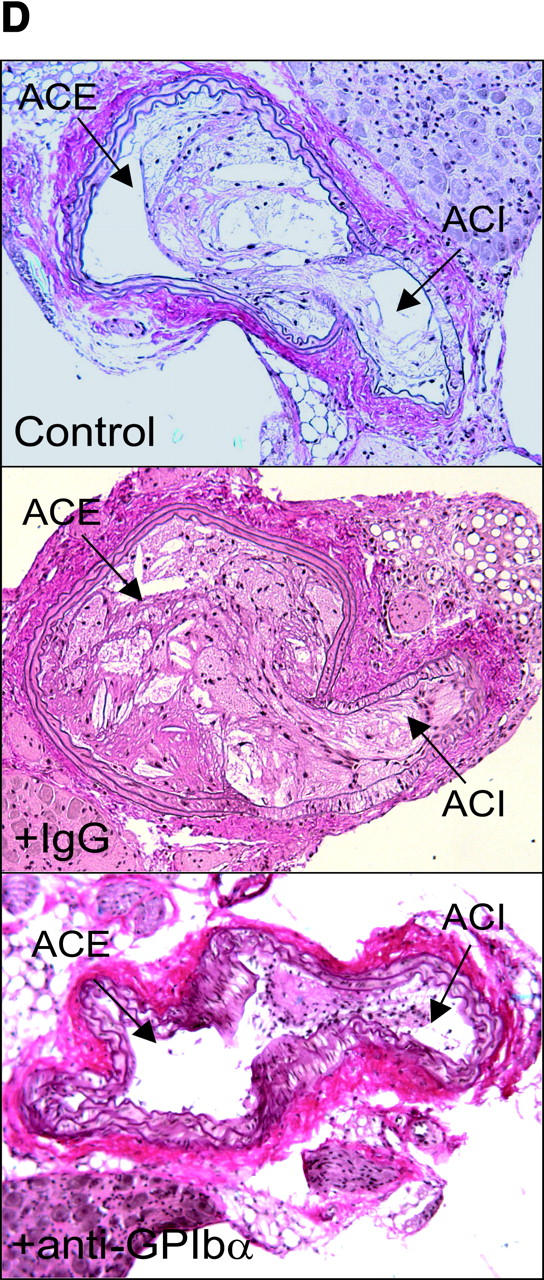
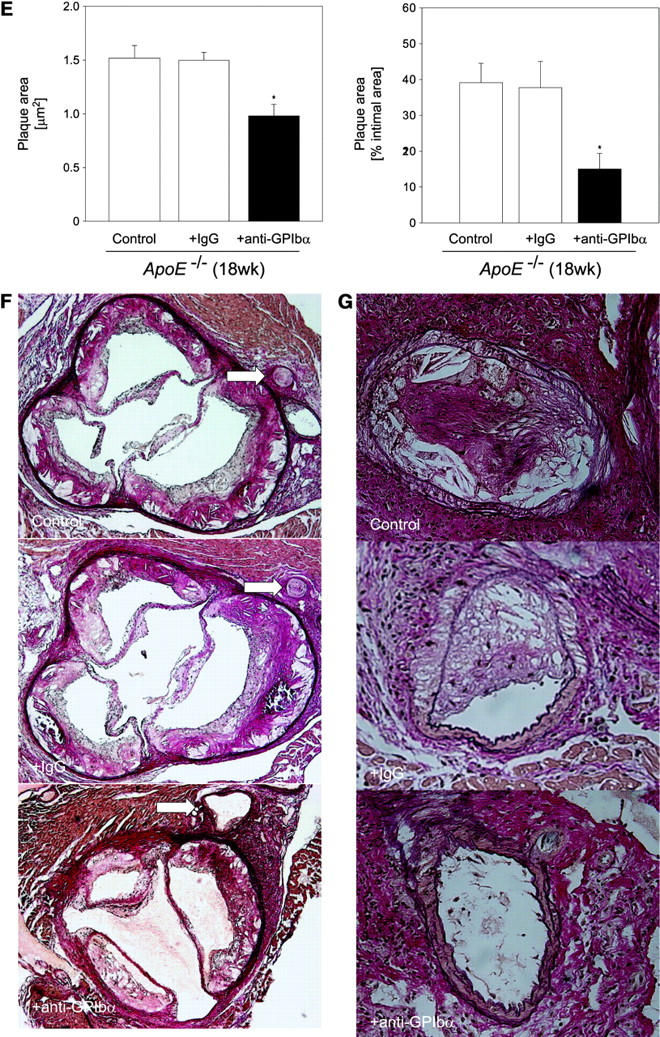
Reduction of atherosclerotic lesion formation after inhibition of platelet adhesion by anti-GPIbα treatment. (A) Vascular resistance index (RI) was analyzed by color duplex sonography. Mean ± SEM of 4–6 experiments per group; *P < 0.05. (B) The extension of fatty streaks (μm2) was quantified in common carotid of ApoE −/− mice by Sudan III staining in anti–GPIbα-treated mice and untreated or rat IgG-treated control animals. Data represent mean values with SEM (4–6 experiments per group) of 18-wk-old ApoE −/− mice treated 12 wk with anti-GPIbα mAb. *P < 0.05. (C) Cross-sectional plaque area was assessed on 20 serial section of the common carotid artery adjacent to the carotid bifurcation and was evaluated for each animal as the difference between the area, delimited by the internal elastic lamina, and the lumen area. The results were normalized to total vessel cross-sectional area to eliminate variations due to vessel size. Data represent mean values with SEM (4–6 experiments per group) of ApoE −/− mice treated for 12 wk with anti-GPIbα mAb. *P < 0.05. Representative sections (original magnification: 200-fold) stained with elastica van Giesson reagent are presented in D. ACC, common carotid artery; ACE, external carotid artery, ACI, internal carotid artery. (E) Role of platelet adhesion for atherosclerotic lesion formation in the aortic sinus (top panel) and the right and left main coronary arteries (bottom panel, plaque area is presented as percentage of total cross-sectional intimal area). 18-wk-old ApoE −/− mice were treated with vehicle (Control), irrelevant rat IgG, or anti-GPIbα mAb for 12 wk. Atherosclerotic lesion formation was assessed in the aortic sinus and the proximal coronary arteries. Inhibition of platelet adhesion induced a significant reduction in atherosclerotic lesion formation in the aortic sinus and the proximal coronary arteries. Aortic plaque area is presented in μm2; coronary plaque area is given as percentage of the area delimited by the internal elastic lamina (intimal area). Mean ± SEM, P < 0.05 versus Control. Representative histological sections of the aortic sinus and the coronary arteries stained with elastica van Giesson reagent are presented in F and G, respectively (original magnification: 200-fold).
To exclude that the effects of chronic GPIbα-inhibition on atherosclerotic lesion formation were restricted to the carotid artery, we evaluated lesion formation in other vascular beds including the aortic sinus and the left and right proximal coronary artery. As demonstrated in Fig. 4, E–G, chronic inhibition of GPIbα, but not administration of irrelevant rat IgG, significantly reduced atherosclerotic lesion formation in both the aortic sinus (by ∼35%, P < 0.05) and the coronary artery (by 63%, P < 0.05), compared with untreated controls.
To further address the pathophysiological role of platelets in the inflammatory process of atherosclerosis, we assessed leukocyte recruitment to the carotid wall in 18-wk-old ApoE-deficient mice, treated with anti-GPIbα− or irrelevant rat IgG. Leukocyte recruitment was assessed in vivo by intravital fluorescence microscopy of the carotid bifurcation (Fig. 5 A). In addition, to determine monocyte recruitment into the intima, shock-frozen tissue samples were evaluated for CD14 and CD11b mRNA expression using RT-PCR (Fig. 5 B). As illustrated in Fig. 5, anti–GPIb-α but not control IgG-treated mice showed reduced leukocyte adhesion to the vascular endothelium compared with untreated control animals by ∼75% (P < 0.05), as determined by in vivo fluorescence microscopy. Furthermore, intimal monocyte recruitment was significantly attenuated as assessed by CD11b and CD14 mRNA expression (Fig. 5 B). Together, the present data demonstrate that inhibition of platelet adhesion (to the vessel wall) at the very initial stage of atherogenesis in ApoE −/− mice attenuates intimal inflammatory cell accumulation and results in a dramatic reduction of early atherosclerotic lesions. These findings provide strong evidence for a critical role of platelets and platelet–endothelium adhesion in atheroma formation in response to elevated cholesterol, a common risk factor for cardiovascular diseases in humans.
Figure 5.


Leukocyte recruitment to the vascular wall of ApoE-deficient mice, treated with anti-GPIbα mAb or irrelevant control IgG. Leukocyte accumulation was assessed by in vivo microscopy (A) (4–6 experiments per group) or by CD14 and CD11b mRNA expression in the arterial wall (B). The presence of 18s RNA served as loading control.
Previous studies (3, 5, 6) and the present work show that platelets adhere to the arterial wall in vivo in the absence of endothelial cell denudation. GPIbα mediates platelet adhesion under high shear rates (30) and binds to vWF and P-selectin both of which are found on activated endothelial cells (31, 32). Ligation of the GPIb-IX-V complex during platelet tethering may lead to activation of platelet integrin GPIIb-IIIa and shear-resistant platelet deposition (33). Subsequent “outside-in” GPIIb-IIIa signaling increases cytoplasmic Ca2+ leading to secretion of granules and local release of preformed potent inflammatory mediators in areas around adherent platelets. Previously, two cytokines, CD40L and IL-1β, have been identified in platelets that alter the chemotactic and adhesive properties of endothelium by enhancing endothelial surface expression of adhesion receptors including VCAM-1 as well as the release of chemokines such as MCP-1 (7, 9, 10). Both VCAM-1 and MCP-1 initiate monocyte recruitment and subsequent transmigration across the endothelial monolayer into the intima (12, 34) and have been shown to play a central role in atherosclerotic lesion formation (22, 23). Interestingly, inhibition of CD40L (29) or the lack of CD40L expression (20) substantially reduced atherosclerotic lesion formation in hypercholesterolemic mice. Here, we show for the first time that platelet adhesion plays a critical role in the initiation of atherosclerosis. These data extend our knowledge of the early events in the atherogenetic process and point to the importance of platelets as a target for novel anti-atherosclerotic therapies.
Acknowledgments
Histomorphometry was performed with the skillful help of Katrin Hemler and Julia Kersting. We thank Dr. R. Brandl, Marion Weber, and Caroline Nothdurfter for their valuable technical advice. The authors are indebted to S.W. for kind donation of thrombasthenic platelets. We further acknowledge the excellent technical assistance of Antje Wallmuth and Renate Hegenloh.
The study was supported by a grant from the Bayerische Forschungsstiftung. Dr. Brandl was supported by the Deutsche Forschungsgemeinschaft (Br 1583/1-2).
Footnotes
Abbreviations used in this paper: GP, glycoprotein; VCAM, vascular cell adhesion molecule.
References
- 1.Ross, R. 1999. Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 2.Lusis, A.J. 2000. Atherosclerosis. Nature. 407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frenette, P.S., R.C. Johnson, R.O. Hynes, and D.D. Wagner. 1995. Platelets roll on stimulated endothelium in vivo: an interaction mediated by endothelial P-selectin. Proc. Natl. Acad. Sci. USA. 92:7450–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frenette, P.S., T.N. Mayadas, H. Rayburn, R.O. Hynes, and D.D. Wagner. 1996. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 84:563–574. [DOI] [PubMed] [Google Scholar]
- 5.Massberg, S., G. Enders, R. Leiderer, S. Eisenmenger, D. Vestweber, F. Krombach, and K. Messmer. 1998. Platelet-endothelial cell interactions during ischemia/reperfusion: the role of P-selectin. Blood. 92:507–515. [PubMed] [Google Scholar]
- 6.Massberg, S., G. Enders, F.C. Matos, L.I. Tomic, R. Leiderer, S. Eisenmenger, K. Messmer, and F. Krombach. 1999. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood. 94:3829–3838. [PubMed] [Google Scholar]
- 7.Lindemann, S., N.D. Tolley, D.A. Dixon, T.M. McIntyre, S.M. Prescott, G.A. Zimmerman, and A.S. Weyrich. 2001. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 154:485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEver, R. 2001. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb. Haemost. 86:746–756. [PubMed] [Google Scholar]
- 9.Henn, V., J.R. Slupsky, M. Grafe, I. Anagnostopoulos, R. Forster, G. Müller-Berghaus, and R.A. Kroczek. 1998. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 391:591–594. [DOI] [PubMed] [Google Scholar]
- 10.Gawaz, M., F.J. Neumann, T. Dickfeld, W. Koch, K.L. Laugwitz, H. Adelsberger, K. Langenbrink, S. Page, D. Neumeier, A. Schömig, and K. Brand. 1998. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation. 98:1164–1171. [DOI] [PubMed] [Google Scholar]
- 11.Gawaz, M., K. Brand, T. Dickfeld, G. Pogatsa-Murray, S. Page, C. Bogner, W. Koch, A. Schömig, and F. Neumann. 2000. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis. 148:75–85. [DOI] [PubMed] [Google Scholar]
- 12.Gu, L., Y. Okada, S.K. Clinton, C. Gerard, G.K. Sukhova, P. Libby, and B.J. Rollins. 1998. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 2:275–281. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, S.H., R.L. Reddick, J.A. Piedrahita, and N. Maeda. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 258:468–471. [DOI] [PubMed] [Google Scholar]
- 14.Nieswandt, B., V. Schulte, W. Bergmeier, R. Mokhtari-Nejad, K. Rackebrandt, J.P. Cazenave, P. Ohlmann, C. Gachet, and H. Zirngibl. 2001. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 193:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brand, K., S. Page, G. Rogler, A. Bartsch, R. Brandl, R. Knuechel, M. Page, C. Kaltschmidt, P.A. Baeuerle, and D. Neumeier. 1996. Activated transcription factor nuclear factor-κB is present in the atherosclerotic lesion. J. Clin. Invest. 97:1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson, R.C., S.M. Chapman, Z.M. Dong, J.M. Ordovas, T.N. Mayadas, J. Herz, R.O. Hynes, E.J. Schaefer, and D.D. Wagner. 1997. Absence of P-selectin delays fatty streak formation in mice. J. Clin. Invest. 99:1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Methia, N., P. Andre, C.V. Denis, M. Economopoulos, and D.D. Wagner. 2001. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 98:1424–1428. [DOI] [PubMed] [Google Scholar]
- 18.Massberg, S., M. Sausbier, P. Klatt, M. Bauer, A. Pfeifer, W. Siess, R. Fässler, P. Ruth, F. Krombach, and F. Hofmann. 1999. Prevention of platelet adhesion and intravascular platelet aggregation by cGMP-kinase I during ischemia/reperfusion in vivo. J. Exp. Med. 189:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Celletti, F.L., J.M. Waugh, P.G. Amabile, A. Brendolan, P.R. Hilfiker, and M.D. Dake. 2001. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat. Med. 7:425–429. [DOI] [PubMed] [Google Scholar]
- 20.Lutgens, E., L. Gorelik, M.J. Daemen, E.D. de Muinck, I.S. Grewal, V.E. Koteliansky, and R.A. Flavell. 1999. Requirement for CD154 in the progression of atherosclerosis. Nat. Med. 5:1313–1316. [DOI] [PubMed] [Google Scholar]
- 21.Collins, T., and M.I. Cybulsky. 2001. NF-κB: pivotal mediator or innocent bystander in atherogenesis? J. Clin. Invest. 107:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boring, L., J. Gosling, M. Cleary, and I.F. Charo. 1998. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 394:894–897. [DOI] [PubMed] [Google Scholar]
- 23.Cybulsky, M.I., K. Iiyama, H. Li, S. Zhu, M. Chen, M. Iiyama, V. Davis, J.C. Gutierrez-Ramos, P.W. Connelly, and D.S. Milstone. 2001. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eriksson, E.E., X. Xie, J. Werr, P. Thoren, and L. Lindbom. 2001. Direct viewing of atherosclerosis in vivo: plaque invasion by leukocytes is initiated by the endothelial selectins. FASEB J. 15:1149–1157. [DOI] [PubMed] [Google Scholar]
- 25.Eriksson, E.E., X. Xie, J. Werr, P. Thoren, and L. Lindbom. 2001. Importance of primary capture and l-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J. Exp. Med. 194:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bombeli, T., B.R. Schwartz, and J.M. Harlan. 1998. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), αωβ3 integrin, and GPIbα. J. Exp. Med. 187:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savage, B., E. Saldivar, and Z.M. Ruggeri. 1996. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 84:289–297. [DOI] [PubMed] [Google Scholar]
- 28.Bergmeier, W., K. Rackebrandt, W. Schroder, H. Zirngibl, and B. Nieswandt. 2000. Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood. 95:886–893. [PubMed] [Google Scholar]
- 29.Mach, F., U. Schonbeck, G.K. Sukhova, E. Atkinson, and P. Libby. 1998. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 394:200–203. [DOI] [PubMed] [Google Scholar]
- 30.Ruggeri, Z.M. 1997. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 78:611–616. [PubMed] [Google Scholar]
- 31.Andre, P., C.V. Denis, J. Ware, S. Saffaripour, R.O. Hynes, Z.M. Ruggeri, and D.D. Wagner. 2000. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood. 96:3322–3328. [PubMed] [Google Scholar]
- 32.Romo, G.M., J.-F. Dong, A. Schade, E.E. Gardiner, G.S. Kansas, C.Q. Li, L.V. McIntire, M.C. Berndt, and J.A. López. 1999. The glykoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J. Exp. Med. 190:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasirer-Friede, A., J. Ware, L. Leng, P. Marchese, Z.M. Ruggeri, and S.J. Shattil. 2002. Lateral clustering of platelet GP Ib-IX complexes leads to up-regulation of the adhesive function of Integrin αIIbβ3. J. Biol. Chem. 277:11949–11956. [DOI] [PubMed] [Google Scholar]
- 34.Ramos, C.L., Y. Huo, U. Jung, S. Ghosh, D.R. Manka, I.J. Sarembock, and K. Ley. 1999. Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ. Res. 84:1237–1244. [DOI] [PubMed] [Google Scholar]