

Abstract
The mechanisms by which CD8 effector populations interact with epithelial layers is a poorly defined aspect of adaptive immunity. Recognition that CD8 effectors have the capacity to express CD103, an integrin directed to the epithelial cell-specific ligand E-cadherin, potentially provides insight into such interactions. To assess the role of CD103 in promoting CD8-mediated destruction of epithelial layers, we herein examined the capacity of mice with targeted disruption of CD103 to reject pancreatic islet allografts. Wild-type hosts uniformly rejected islet allografts, concomitant with the appearance of CD8+CD103+ effectors at the graft site. In contrast, the majority of islet allografts transplanted into CD103−/− hosts survived indefinitely. Transfer of wild-type CD8 cells into CD103−/− hosts elicited prompt rejection of long-surviving islet allografts, whereas CD103−/− CD8 cells were completely ineffectual, demonstrating that the defect resides at the level of the CD8 cell. CD8 cells in CD103−/− hosts exhibited normal effector responses to donor alloantigens in vitro and trafficked normally to the graft site, but strikingly failed to infiltrate the islet allograft itself. These data establish a causal relationship between CD8+CD103+ effectors and destruction of graft epithelial elements and suggest that CD103 critically functions to promote intragraft migration of CD8 effectors into epithelial compartments.
Keywords: integrins, cytolytic T lymphocytes, epithelial cells, transplantation, graft infiltrating lymphocytes
Introduction
CD8 effector populations play important roles in host defense against intracellular parasites and tumors by direct destruction of cells expressing foreign MHC/peptide complexes, and it is widely accepted that donor-reactive CD8 effector populations play an analogous role in rejection of transplanted tissues (1, 2). After activation by MHC/peptide in the context of appropriate APC, naive, or memory CD8αβ+TCR-αβ+ cells (CD8 cells) differentiate into CD8 effector cells which efficiently eliminate cells expressing the target MHC class I/peptide alloantigen. Ubiquitous expression of class I molecules combined with loading of nascent class I molecules with peptides derived from endogenous proteins, assures that all donor cell types are susceptible to surveillance by CD8 effector populations. CD8 effectors possess a diverse and redundant armamentarium for destruction of target cells including induction of apoptosis by cytotoxic granule release or Fas–FasL pathways (3), and production of cytokines (4, 5) and chemokines (6) that regulate the effector activity of other leukocyte populations (7).
The mechanisms by which CD8 effectors interact with epithelial layers represents an important gap in our understanding of transplant immunity. The functional elements of most organ systems (i.e., renal tubules, pancreatic islets, hepatocytes, alveoli, etc.) are specialized epithelial layers, and specific attack of such structures by alloreactive CD8 effector populations constitutes a major component of organ allograft rejection (1, 2). Recent studies have provided detailed insight into the cellular and molecular interactions that promote trafficking of CD8 cells from the initial site of activation within peripheral lymphoid compartments to the graft site (8). However, little is known of the critical downstream events by which CD8 effectors gain access to and subsequently destroy graft epithelial compartments.
Previously, we have reported that alloreactive CD8 effector populations in both the mouse (9) and human (10) systems have the capacity to express the integrin heterodimer αE(CD103)/β7 (herein referred to as CD103). These observations potentially provide insight into CD8 effector–epithelial interactions because the principal ligand of CD103 is E-cadherin (11, 12), a tissue-restricted molecule selectively expressed by cells comprising epithelial layers (13). CD103 is poorly expressed by conventional CD8 effector populations (i.e., in vitro generated CTL populations; references 9 and 14), but is readily induced by exposure of such cells to bioactive TGF-β (9). The relevance of these data to transplant immunity is supported by the observation that CD103+CD8+ effectors accumulate in the kidney but not the spleen in a mouse model of GVHD (9), and are present at the site of clinical renal allograft rejection (10). CD8+CD103+ effectors that infiltrate rejecting renal allografts preferentially accumulate within graft epithelial compartments (10, 15), consistent with a key role for this effector subset in the rejection process.
CD103 was initially identified by its high level expression on the heterogeneous and poorly-defined T cell populations that reside within and adjacent to the mucosal epithelia of normal mice (16) and humans (17); intraepithelial and lamina propria lymphocytes, respectively (IEL and LPL)*. In contrast, CD103 is poorly expressed by T cells in lymphoid compartments such as the blood, spleen, and lymph nodes (16, 17). Consequently, previous studies of CD103 have focused almost exclusively on its potential role in retaining T cells at mucosal sites. Consistent with this possibility, mice with targeted disruption of CD103 exhibit a mild deficiency of T cells in the IEL and LPL compartments (18). It is important to note, however, that CD103-deficient mice are otherwise phenotypically normal, and that the deficiency in mucosal T cells is highly dependent on genetic background and environmental influences (18). Moreover, Lefrancois et al. (19) recently reported that CD103 expression is not required for long-term retention of antigen-specific CD8 cells in the intestinal epithelium. Thus, the functional significance of CD103 expression by T cells remains unclear.
The goal of this study was to determine if CD103 expression is required for destruction of graft epithelial elements by alloreactive CD8 cells. To test this hypothesis, we examined the capacity of mice with targeted disruption of the CD103-encoding gene (Itgae) (18) to reject pancreatic islet allografts. Pancreatic islets express both MHC class I and the CD103 ligand, E-cadherin (20), and thus should be susceptible to destruction by alloreactive CD103+CD8+ effectors. We report that CD103−/− hosts are strikingly deficient in the capacity to reject islet allografts, and provide evidence that the relevant defect resides at the level of intragraft migration of CD8 effector populations. These data are consistent with a critical role for CD103 in promoting entry and/or retention of antigen-specific CD8 effector populations in epithelial compartments.
Materials and Methods
Mice.
Female A/J and BALB/cJ mice (age 6 wk) were purchased from The Jackson Laboratory and maintained under pathogen-free housing conditions at the University of Maryland, Baltimore, MD. Breeding stocks of mice with targeted disruption of the CD103 (Itgae) gene (18) were bred and maintained (by C.M. Parker, Brigham and Women's Hospital, Boston, MA) under pathogen-free conditions at the University of Maryland animal facilities. Two different strains of CD103−/− mice were used in this study. Initially, CD103−/− mice backcrossed 10 generations to the BALB/c strain were used; wild-type BALB/cJ mice purchased from The Jackson Laboratory served as CD103+/+ controls. Subsequently, a CD103−/− strain rigorously selected to differ from BALB/c at only the Itgae locus were used for experiments; this congenic strain, designated C;129S2-Itgaetm1Cmp, has been deposited at The Jackson Laboratory. In this case, wild-type BALB/c mice bred in the University of Maryland animal facility in parallel with C; 129S2-Itgaetm1Cmp mice served as CD103+/+ controls. The two CD103−/− strains gave identical results compared with their respective wild-type controls with regard to all parameters examined herein. Breeding stocks for BALB/c-Thy1.1 congenic mice were provided by H. Levitsky (Johns Hopkins Medical School, Baltimore, MD). BALB/c hosts expressing both Thy1.1 and Thy1.2 were obtained by interbreeding BALB/c-Thy1.1 mice with BALB/cJ mice (Thy1.2+) to produce F1 hybrid mice.
Antibodies.
mAbs to mouse CD8 (3.155), CD103 (M290) (21), CD4 (GK1.5), and MHC class II (212.A1), were purified from hybridoma culture supernatants and conjugated to FITC by Bioexpress Cell Culture Services (West Lebanon, NH); mAbs to mouse CD4 (RL172.4), HSA, (J11D.B1), and H-2-IAd (MKD6) used for complement depletion were used as hybridoma culture supernatants. FITC- and CyChrome-conjugated mAbs to mouse CD8a, and PE-conjugated mAbs to mouse CD8b, CD11a, CD62L, CD44, TCR-αβ, CD8.2, Thy1.1, Thy1.2, and the respective species- and isotype-matched negative control mAbs were purchased from BD PharMingen.
Pancreatic Islet and Skin Transplantation.
Islets for transplantation were prepared as described by Gotoh et al. (22). In brief, HBSS (GIBCO BRL) containing 2 mg/ml collagenase (Sigma-Aldrich) was injected into the common bile duct of donor mice. The distended pancreas was removed and incubated at 37°C for 16 min, and the released islets were enriched by centrifugation on a discontinuous Ficoll (Sigma-Aldrich) gradient. Tissue fragments collected at the 21.5% interface were washed and resuspended in HBSS. Individual islets, free of attached acinar, vascular, and ductal tissue were selected under a dissecting microscope, yielding highly purified islets for transplantation. Recipients were rendered diabetic by injection of 220 mg/kg streptozotocin intraperitoneally 3–6 d before transplantation. An incision was made to expose the recipient's left kidney, and 500 fresh islets were injected into the renal subcapsular space. After recovery from anesthesia, recipients were maintained under pathogen-free conditions, and blood glucose levels were monitored on a daily basis. Rejection was defined as BG >300 mg/dL.
Skin transplantation was performed as described by Steinmuller (23). After 6 d, the bandages were removed and the grafts were assessed daily; rejection was defined as the day on which 100% graft scabbing was recorded.
FACS® Analyses.
Graft infiltrating lymphocytes (GILs) were isolated by mincing the graft and incubation for 30 min in media containing collagenase (Worthington Biochemical Corp.), soybean trypsin inhibitor (Sigma-Aldrich), and DNase (Boehringer Mannheim). Lymphocytes were isolated by centrifugation on Lympholyte-M (Cedarlane Laboratories Limited). For two-color FACS® analyses, cells were stained with PE-conjugated mAb to CD8b in combination with FITC-conjugated mAbs to markers of interest. For three-color analyses, cells were stained with anti-CD8a-CyChrome in combination with FITC- and PE-conjugated mAbs. Species and isotype-matched mAbs of irrelevant specificity were used as controls for nonspecific fluorescence. After staining, cells were fixed with 0.5% paraformaldehyde, and 3–6 × 104 cells were analyzed using a FACScan™ (Becton Dickinson). Intracellular cytokine staining for FACS® was performed using an intracellular cytokine staining kit (no. 2040KK) purchased from BD PharMingen. In brief, GILs isolated as described above were cultured in media containing PMA, ionomycin, and monensin for 4 h, then permeabilized and stained with anti-CD8a-FITC followed by PE-coupled mAbs to IFN-γ or TNF-α. Lymphocyte populations were gated by forward scatter/side scatter analysis to exclude monocytes and debris. WinMDI 2.8 software (downloaded from: http://facs.scripps.edu/software.html) developed by Joseph Trotter (Scripps Institute, La Jolla, CA) was used for analysis and graphic display of flow cytometry data. The percentage of positive cells for a given marker was based on cutoff points chosen to exclude >99% of the negative control population.
In Vitro Generation of CD8 Effectors.
CD8 effectors for 51[Cr] release assays and cytokine production assays were generated by coculturing 10 × 106 spleen cells (SCs) from CD103+/+ or CD103−/− donors with 10 × 106 irradiated (3,000 rads) A/J SCs in upright 25 cm2 culture flasks in 20 ml RPMI 1640 supplemented with sodium pyruvate, nonessential amino acids, 10 mM Hepes, 50 uM 2-ME, and 10% FCS (RPMI+). Red cells were removed from SC stimulators by treatment with RBC lysing buffer (Sigma-Aldrich). After 5 d, cultures were harvested and treated with a cocktail of mAbs to CD4 (RL172.4), HSA, (J11D.B1), and H-2-IAd (MKD6) followed by incubation in 1:10 Low-Tox M rabbit complement (Accurate) and centrifugation on Lympholyte-M. The resulting effector population contained >95% CD8+TCR-αβ+ cells as determined by FACS®.
T Cell Functional Assays.
51[Cr] release assays were performed as described previously (9). For targets, Con A blasts were prepared by culturing 25 × 106 red cell–depleted SCs from BALB/cJ or A/J donors for 3 d in 10 ml of RPMI+ containing 5 ug/ml Con A (Sigma-Aldrich). For cytokine production assays, 106 CD8 effectors were restimulated with 106 RBC-depleted A/J SCs (3,000 rads) in 2 ml of RPMI+. Supernatants from replicate cultures (n = 3) were harvested at various intervals and subjected to cytokine-specific ELISA by the University of Maryland Cytokine Core Laboratory to measure concentrations of IFN-γ and TNF-α.
Adoptive Transfer Models.
For adoptive transfer of CD8 cells into CD103−/− hosts with long-surviving A/J allografts, wild-type, or CD103-deficient mice (6-wk-old females) to be used as lymphocyte donors were injected intraperitoneally with 10 × 106 A/J SCs. On day 4, spleens and abdominal lymph nodes were harvested and centrifuged on Lympholyte-M (Cedarlane Laboratories Limited) to remove RBCs. The resulting cell suspension was enriched for CD8+ T cells by immunomagnetic depletion with mAbs to mouse class MHC class II (212.A1) and CD4 (GK1.5) followed by anti–rat IgG-, anti–mouse IgG-, and anti–mouse IgM-coupled magnetic beads (Polysciences). The negatively selected cell population comprised >60% CD8+ T cells with <0.1% CD4+ T cells. To assure that equivalent numbers of CD8 cells were transferred in different experiments, FACS® analyses using anti-CD8 and TCR-αβ was performed on each preparation to assess the degree of enrichment for CD8+ T cells. 50 × 106 CD8+ T cells were then injected into CD103−/− recipients with long-surviving A/J allografts by the tail vein, and blood glucose levels were monitored on a daily basis. In some experiments, CD8 cells were removed from the above “CD8” preparations by incubation with mAb to mouse CD8 (3.155) followed by complement. Such preparations contained >98% non-T cells (predominantly B cells) with <0.01% CD8+ T cells.
For adoptive transfer experiments to assess the relative efficiency with which CD103-deficient CD8 effectors traffic to the graft site, purified CD8 cells from naive CD103−/− (Thy1.1−/1.2+) and CD103+/+ (Thy1.1+/1.2−) donors were prepared as described above. 5 × 106 cells of each type were then injected intravenously into streptozotocin-treated (diabetic) BALB/c-Thy1.1 × BALB/cJ F1 hosts (Thy1.1+/Thy1.2+) which were transplanted with 500 A/J islets the following day. Blood glucose levels were monitored on a daily basis. At the time of early rejection (BG > 200 mg/dL), graft infiltrating lymphocytes were isolated and stained for three-color FACS® analyses with anti-Thy1.1-PE and anti-Thy1.2-FITC in combination with anti-CD8-CyChrome to discriminate host CD8 cells from each of the transferred CD8 populations.
Results
CD8+CD103+ Effectors Are Present at the Site of Islet Allograft Rejection in Wild-Type Hosts.
As shown in Fig. 1 A, A/J islet allografts (H-2a) transplanted into diabetic wild-type recipients (BALB/c, H-2d) were promptly rejected (n = 13, MST = 13.8 d). Recipient mice were hyperglycemic before transplant and transiently returned to a normoglycemic state 2–6 d after receiving A/J islet allografts (Fig. 1 A). In contrast, BALB/c recipients of islet isografts remained normoglycemic throughout the observation period (>300 d), confirming that the rapid return to a hyperglycemic state noted in allograft recipients reflects immunologic rejection (Fig. 1 A).
Figure 1.
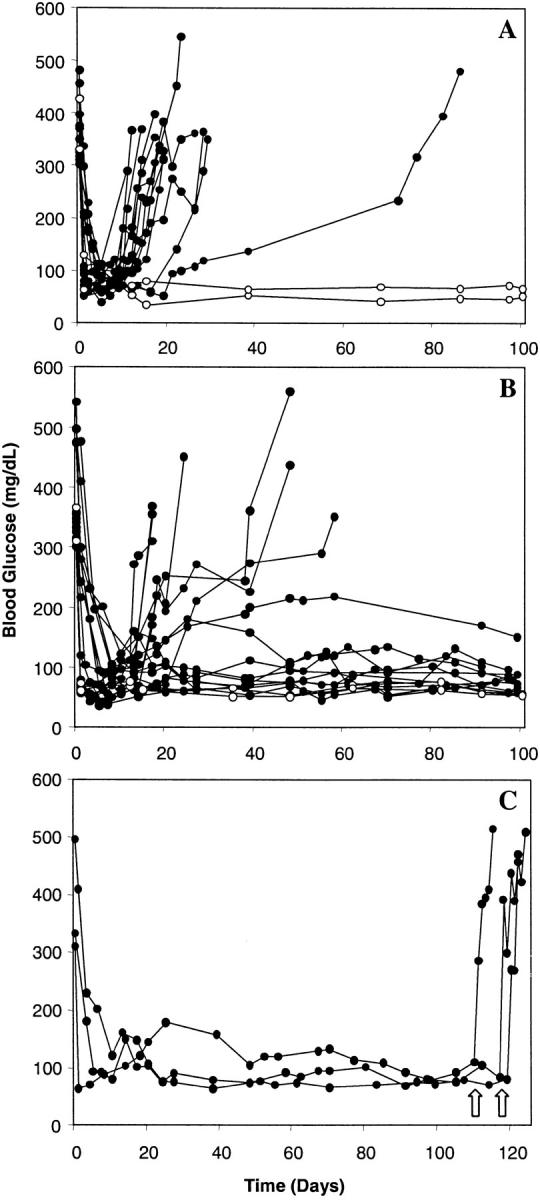
Survival of pancreatic islet allografts transplanted into CD103+/+ or CD103−/− hosts. Recipient mice were rendered diabetic by treatment with streptozotocin, then transplanted with 500 islets under the renal subcapsule; each line shown in A–C represents an individual recipient mouse. (A) Survival of A/J allografts (black circles, n = 13) or BALB/c isografts (white circles, n = 2) transplanted into CD103+/+ hosts. (B) Survival of A/J allografts (black circles, n = 17) or isografts (white circles, n = 2) transplanted into CD103−/− hosts. (C) Unilateral nephrectomy to remove the allograft (denoted by arrows) in CD103−/− recipients with long surviving allografts results in a prompt return to hyperglycemic state (n = 3).
FACS® analyses of GILs isolated from islet allografts at the time of rejection in wild-type hosts revealed a predominant population of CD8 cells (Fig. 2 A). Fig. 2 A further demonstrates that CD103 was expressed by a major subset of the CD8+ GIL population at the time of rejection. Note that CD103 expression by GILs was restricted almost exclusively to CD8+TCR-αβ+ cells. In four replicate experiments, 18.1 ± 3.0% of CD8+ GILs expressed CD103 at levels significantly above background staining. Fig. 2 B shows that CD8+ GILs were CD62L−CD44hiCD11ahi− TCR-αβ+ consistent with an effector phenotype. These data indicated that CD8+CD103+ effectors are present in the graft at the time of rejection, consistent with a role for these cells in the rejection process.
Figure 2.
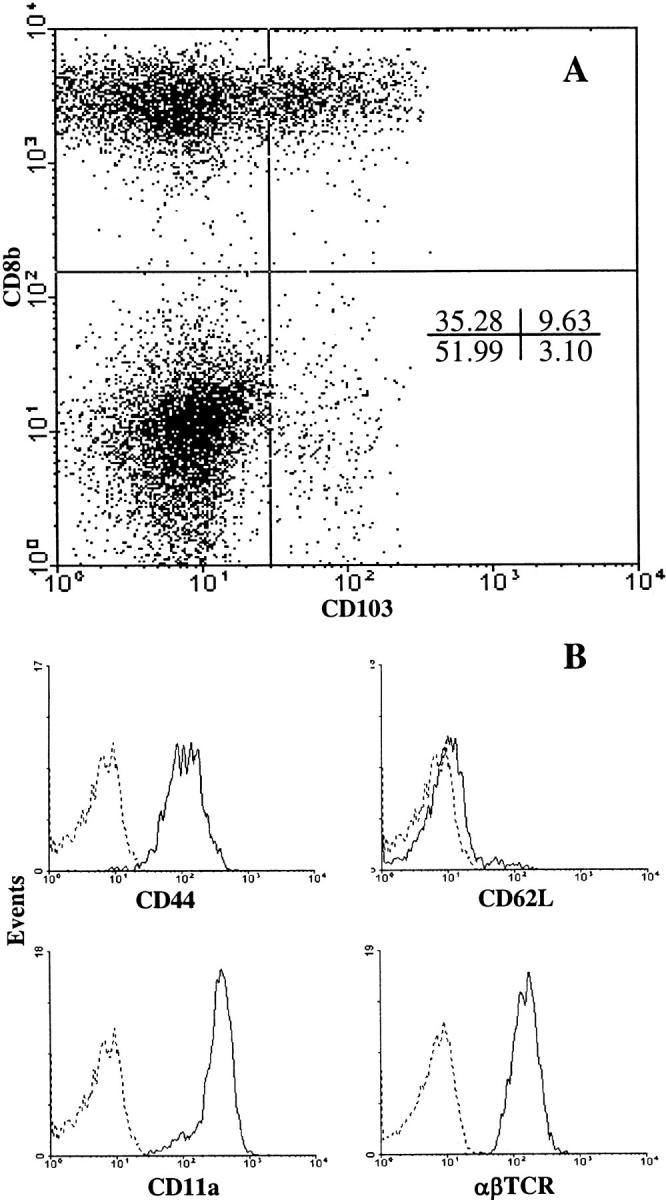
Phenotypic characterization of CD8+ T cells isolated from rejecting islet allografts. Wild-type recipients were transplanted with A/J islets as described in the legend to Fig. 1. At the time of rejection, lymphocytes were isolated from the graft and draining lymph node and subjected to two-color FACS® analyses. (A) Dot plot of CD103 versus CD8 expression by GILs. (B) Expression of CD44, CD11a, CD62L, and TCR-αβ by gated CD8+ GILs; isotype controls are shown by the dashed lines. Data are representative of four independent experiments.
Mice With Targeted Disruption of CD103 Exhibit a Specific Defect in the Capacity for Islet Allograft Rejection.
Mice with targeted disruption of the CD103-encoding gene (Itae) were used as recipients of islet allografts to directly assess the role of CD103 in islet allograft destruction. The CD103−/− mice used in this study were backcrossed 10 generations to the BALB/c strain; such mice completely fail to express the CD103 heterodimer (αEβ7) but are phenotypically indistinguishable from wild-type cohorts except for slightly reduced numbers of T cells diffusely distributed in the intestinal epithelium (18).
As shown in Fig. 1 B, CD103−/− hosts were highly deficient in their capacity to reject A/J islet allografts with the majority of allografts (11/17) surviving indefinitely. Importantly, removal of long-surviving allografts by unilateral nephrectomy (n = 3) resulted in a prompt return to hyperglycemia (Fig. 1 C), thereby confirming that the prolonged normoglycemic state was a reflection of stable graft function rather than the persistence of host islet function i.e., due to inadequate streptozotocin treatment.
The Defect in CD103−/− Hosts Resides at the Level of the CD8 Cell.
Adoptive transfer experiments were used to determine if the defect in CD103−/− mice resides at the level of the CD8 cell. This is a critical point because CD103 is also expressed by non-CD8 cells including dendritic cells (24), macrophages (25), and mast cells (26), which potentially could contribute to allograft rejection. In these experiments, purified CD8 cells from wild-type or CD103-deficient mice were adoptively transferred into CD103−/− hosts with long surviving A/J allografts. To circumvent the likely requirement for an anti-donor CD4 (Th) response, the CD8 population was primed to A/J alloantigens 4 d before CD8 purification and transfer.
As shown in Fig. 3 , transfer of highly-enriched CD8 cells into CD103−/− hosts elicited prompt rejection of long-surviving A/J allografts. In contrast, transfer of the same number of CD8 cells from CD103−/− donors completely failed to elicit rejection (Fig. 3). Moreover, treatment of the wild-type CD8 preparation with anti-CD8 mAb plus complement (non-CD8 cells in Fig. 3) completely abrogated its capacity to transfer rejection (Fig. 3). Thus, these data demonstrate that CD8 cells in CD103−/− hosts are deficient in their capacity to mediate rejection of islet allografts.
Figure 3.

Adoptive transfer experiments. 50 × 106 CD8+ T cells from CD103+/+ (black squares) or CD103−/− donors (white circles) were adoptively transferred into CD103−/− hosts with long surviving (>50 d) A/J islet allografts. The top and bottom panels show the results of two independent experiments. Donors were immunized with 107 A/J SCs intraperitoneally 4 d before cell isolation. A control group of CD103−/− hosts with long surviving A/J islet allografts received 25 × 106 purified non-CD8 cells from CD103+/+ donors (black circles). Each line represents blood glucose levels from a single recipient mouse. CD8 cells for these experiments were purified from combined spleens and lymph nodes of primed donors by immunomagnetic depletion with mAbs to CD4 and MHC class II, and comprised >60% CD8+ T cells with <0.1% CD4+ T cells. The non-CD8 preparation was obtained by treatment of the wild-type CD8 preparation with anti-CD8 mAb plus complement and comprised >97% non-T cells (splenic B cells) with <0.01% residual CD8+ cells.
CD103-deficient CD8 Cells Exhibit Normal Effector Responses to A/J Alloantigens and Efficiently Traffic to the Graft Site.
Despite their inability to mediate rejection of A/J islet allografts, CD8 cells in CD103−/− mice exhibited normal responsiveness to A/J alloantigens in conventional assays of CD8 activation and effector function. Fig. 4 shows that CD8 effectors derived from CD103−/− and wild-type mice mount comparable responses to A/J alloantigens both with respect to IFN-γ production (Fig. 4 A) and lytic activity toward lymphoblast targets (Fig. 4 B). CD103−/− and wild-type mice also exhibit comparable, albeit weak, proliferative responses to A/J alloantigens (unpublished data).
Figure 4.
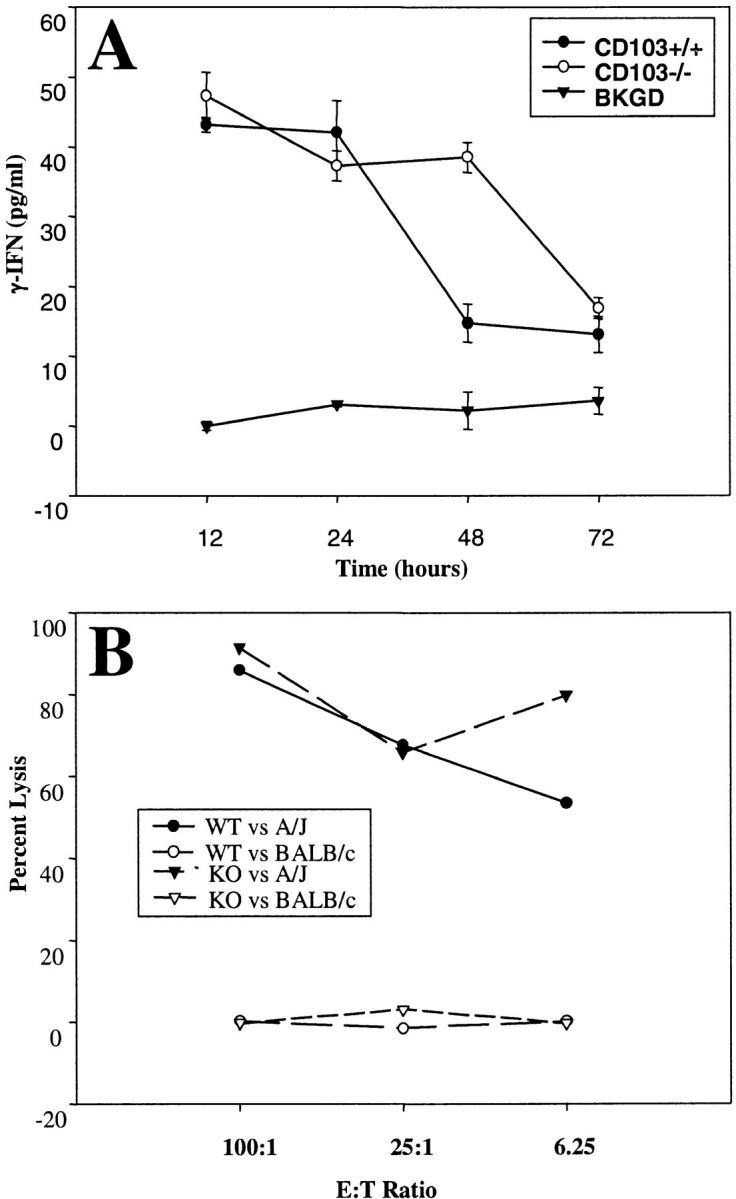
CD8 cells from CD103−/− mice exhibit normal responsiveness to A/J alloantigens in conventional assays of CD8 effector function. CD8 effectors were generated by coincubation of SCs from CD103+/+ or CD103−/− mice with A/J SCs in 5-d mixed lymphocyte cultures, and purified by negative selection with mAb plus complement. (A) IFN-γ production by CD8 effectors derived from CD103+/+ (black circles) or CD103−/− (white circles) in response to A/J SCs or media only (triangles). Data shown are means (±SE) of three replicate values. (B) Lytic activity of CD8 effectors derived from CD103+/+ (circles) or CD103−/− (triangles) to A/J (black symbols) and BALB/c (white symbols) lymphoblast targets in standard 4-h 51[Cr] release assays. Data shown are means of four replicate values; SD was <10% of mean values.
An adoptive transfer model was devised to assess the efficiency with which CD103-deficient CD8 effectors traffic to the graft site. In this model, equal numbers (5 × 106) of purified CD8 cells from CD103−/− (Thy1.1−/1.2+) and CD103+/+ (Thy1.1+/1.2−) donors were transferred into syngeneic wild-type (Thy1.1+/Thy1.2+) hosts which subsequently received A/J islet allografts. Three-color FACS® of GILs harvested at the time of early rejection (BG > 200 mg/dL) were then used to quantitate relative numbers of transferred CD8 populations which successfully trafficked to the graft site. As shown in Fig. 5 , CD103-deficient CD8 cells trafficked to the graft with efficiency comparable to that of wild-type CD8 cells. In three independent experiments, 18.7 ± 1.4% (mean ± SE) of the transferred cell population was of CD103−/− origin as compared with 20.6 ± 5.8% of CD103+/+ origin. Importantly, CD8 cells isolated from such grafts were devoid of naive (CD62LhiCD44lo) CD8 cells (unpublished data), thereby excluding the trivial possibility that the graft-associated CD8 cells derived from contaminating peripheral blood. Thus, these data demonstrate that CD103 expression is not required for effective activation of CD8 cells or trafficking of the resulting CD8 effectors to the general graft site.
Figure 5.

CD8 cells in CD103-deficient hosts traffic normally to the graft site. Equal numbers (5 × 106) of purified CD8 cells from CD103−/− (Thy1.1−/1.2+) and CD103+/+ (Thy1.1+/1.2−) donors were transferred into wild-type (Thy1.1+/Thy1.2+) hosts which were subsequently transplanted with A/J islet allografts. At the time of rejection, GILs were stained for three-color FACS® analyses with anti-Thy1.1-PE and anti-Thy1.2-FITC in combination with anti-CD8a-CyChrome. Data shown are for electronically gated CD8+ lymphocytes. The quadrant on right shows the relative positions of wild-type and CD103−/− cells in the density plot on left. Data shown are representative of three independent experiments.
Analyses of CD8 Cells that Infiltrate Islet Allografts in CD103-deficient Hosts.
Histological analyses of islet allografts removed from wild-type hosts at the time of rejection showed that the islet grafts were infiltrated with large numbers of lymphocytes of T cell morphology concomitant with loss of graft function (Fig. 6 A). In marked contrast, long surviving allografts removed from CD103−/− hosts (Fig. 6 B) were conspicuously free of infiltrating lymphocytes despite the presence of large numbers of lymphocytes in the subcapsular region. Similarly, islet allografts transplanted into CD103−/− hosts and removed at the time of rejection in wild-type hosts (day 14 after transplant) showed pristine islet allografts despite massive accumulation of lymphocytes in the subcapsular space (Fig. 6 C).
Figure 6.
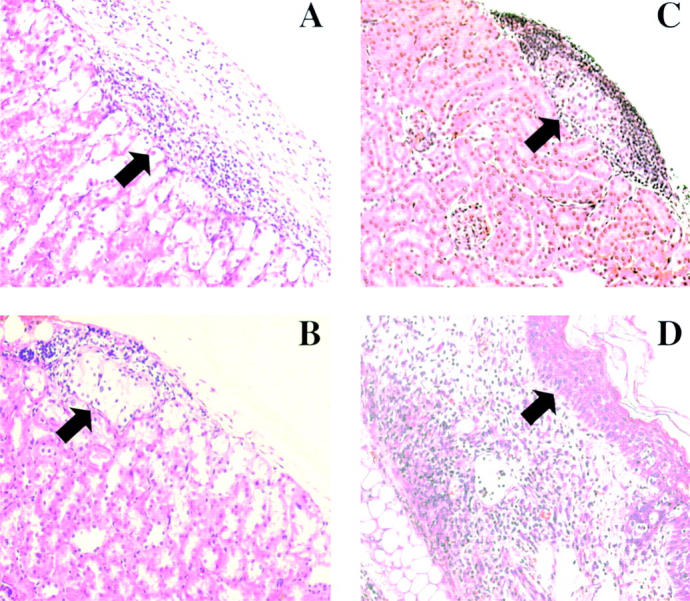
Graft histology. (A) Islet allograft transplanted into wild-type host harvested at the time of rejection (BG > 300 mg/dL). (B) Long surviving (>100 d) islet allograft removed from CD103−/− host as shown in Fig 1 C. (C) Islet allograft transplanted into CD103−/− host and harvested at the time of rejection in wild-type hosts (day 14 after transplant). (D) Skin allograft transplanted into wild-type host harvested at the time of rejection (day 8 after transplant). Arrows in A–C mark the position of islet allografts under the renal subcapsule; the arrow in D marks the postion of the graft epidermis. Data shown are H&E staining of paraffin-embedded sections.
FACS® analysis revealed that lymphocytes infiltrating the graft site in CD103−/− hosts (isolated at day 14 after transplant) were predominantly TCR-αβ+ cells (∼70%) of which ∼22% were CD8 cells (Fig. 7 A). Fig. 7 B shows that CD8 cells in such grafts exhibited an activated/memory phenotype. As shown in Fig. 7 C, the majority (>96%) of CD8 cells infiltrating such grafts were capable of rapid bursts of IFN-γ production after PMA/ionomycin stimulation, thereby confirming their effector status. Taken together with the above histologic analyses (Fig. 6, B and C), these data strongly suggest that deficient allograft rejection in CD103−/− hosts reflects a defect at the level of intragraft migration of CD8 effectors; i.e., CD103 is apparently required for efficient entry of CD8 effectors into islet allografts.
Figure 7.
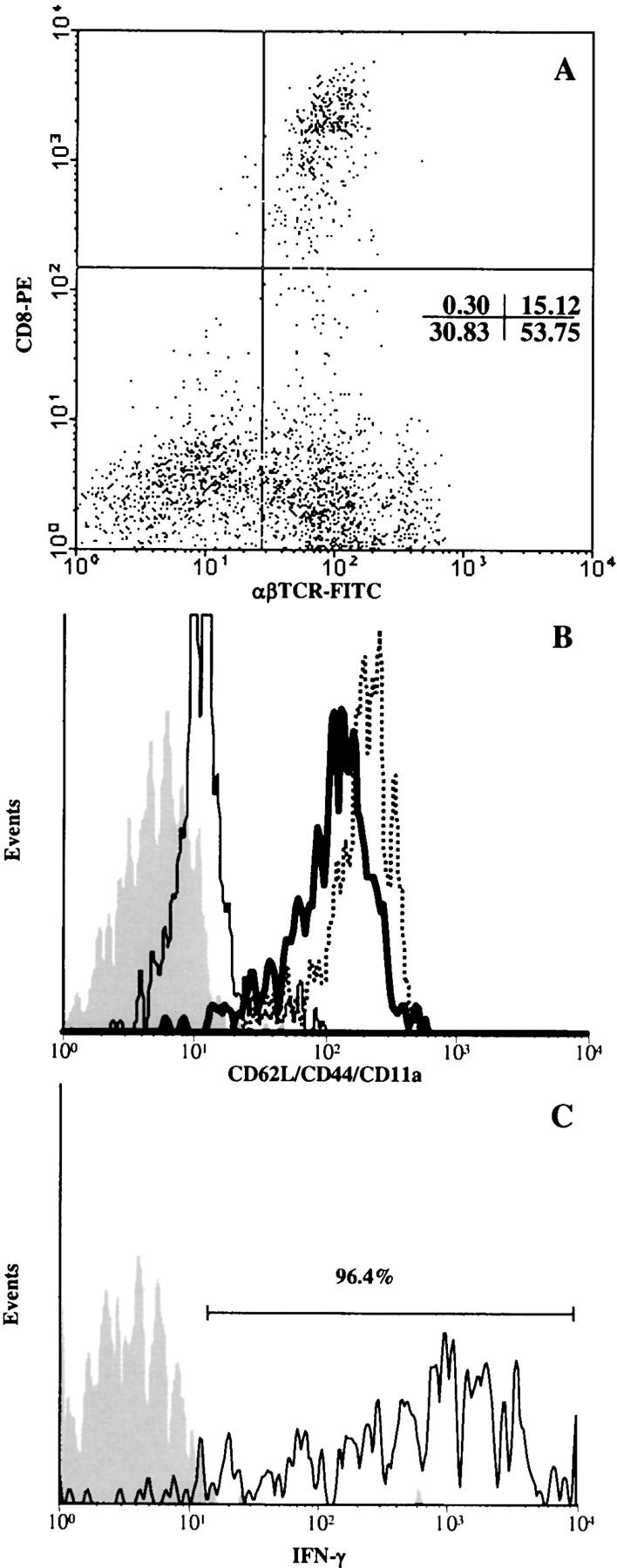
Phenotypic analysis of lymphocytes that infiltrate pancreatic islet allografts in CD103−/− hosts. CD103−/− recipients were transplanted with A/J islets as described in the legend to Fig. 1. At day 14 after transplant, GILs were isolated and subjected to FACS® analyses. (A) Dot plot of TCR-αβ versus CD8 expression by GILs. (B) Expression of CD62L (thin solid line), CD44 (thick solid line), and CD11a (dashed line) by electronically-gated CD8+ GILs. (C) IFN-γ production by gated CD8+ GILs after PMA/ionomycin activation; identical results were obtained for TNF-α production. Isotype control staining is shown by the gray peaks in B and C.
Normal Rejection of Skin Allografts in CD103−/− Hosts.
In contrast to their impaired capacity to reject A/J islet allografts, CD103−/− hosts were not deficient in the capacity to reject A/J skin allografts. CD103+/+ and CD103−/− hosts both rejected A/J skin allografts with similar kinetics: mean ± SEM = 7.67 ± 0.37 d (n = 10) for wild-type versus 8.22 ± 0.43 d (mean ± SEM) (n = 9) for CD103−/− hosts (P > 0.36). These data demonstrate that CD103−/− mice do not possess a global defect in allograft rejection.
Given that skin allografts contain important epithelial elements (epidermis), the lack of a requirement for CD103 in rejection of skin allografts was unexpected. To explore this issue more fully, histological analyses of A/J skin grafts transplanted onto CD103+/+ (Fig. 6 D) and CD103−/− recipients (unpublished data) were performed. Rejection of skin allografts in wild-type hosts is accompanied by massive infiltration of lymphocytes into the dermis and vascular bed of rejecting skin allografts yet the graft epidermis remains strikingly free of lymphocytic infiltration (see arrow, Fig. 6 D). Note that the epidermis in rejecting allografts remains intact except in focal areas with ischemic necrosis due to the vascular damage and secondary infiltration by neutrophils. Parallel analyses of skin allografts in CD103-deficient hosts revealed an identical histologic picture (unpublished data). Thus, in contrast to islet allograft rejection, skin allograft rejection in this strain combination apparently does not occur via direct destruction of epithelial compartments (epidermis) and, consequently, does not require CD103.
Discussion
The salient finding of this study is that expression of CD103 is required for efficient destruction of graft epithelial compartments by CD8 cells. This finding provides insight into the important clinical problem of organ allograft rejection. Similar to pancreatic islet allografts, the functional elements of most commonly transplanted organs express E-cadherin and are vulnerable to CD8-mediated rejection (1, 2). Robertson et al. (15) recently demonstrated that CD103+ T cells selectively accumulate within the graft tubular epithelium during clinical renal allograft rejection, and we have shown that such cells possess a classic CD8 effector phenotype (CD8αβ+TCR-αβ+CD11ahiperforin+; reference 27). This study provides compelling evidence that a causal relationship exists between the CD103+CD8+ effector subset and destruction of graft epithelial elements, and thus identifies CD103 as a novel target for therapeutic intervention in organ allograft rejection.
The critical role of CD8 cells in rejection of pancreatic islet allografts is well documented. CD8-deficient mice show delayed rejection of islet allografts (28), and treatment of wild-type mice with anti-CD8 mAb (29) or transplantation of MHC class I–deficient islets (30) leads to indefinite survival of islet allografts. This study extends these findings by demonstrating that CD103 expression by CD8 cells is required for efficient islet allograft destruction. Initially, we observed that CD8 effectors expressing high levels of CD103 are present at the site of islet allograft rejection (Fig. 2). Consistent with a role for such cells in the rejection process, islet allografts enjoyed strikingly prolonged survival in CD103−/− hosts compared with CD103+/+ hosts (Fig. 1). That the defect in CD103−/− mice resides at the level of the CD8 cell was established using adoptive transfer experiments. CD8 cells from wild-type mice promptly elicited rejection of long surviving allografts, whereas CD8 cells from CD103−/− mice were completely ineffectual in this regard (Fig. 3).
It remains to be determined how CD103 promotes destruction of islet allografts by CD8 cells. It is unlikely that the deficiency in CD103−/− mice reflects suboptimal activation or maturation of host CD8 cells because CD103-deficient CD8 effectors exhibit normal responsiveness to A/J (leukocyte) targets in conventional assays of CD8 activation and effector function (Fig. 4). Consistent with these in vitro observations, islet allografts in CD103−/− hosts are infiltrated with CD8 cells exhibiting the classic phenotypic and functional properties of CD8 effectors (Fig. 7). Adoptive transfer experiments confirmed that CD103 expression is not required for efficient trafficking of CD8 effectors to the graft site (Fig. 5). The latter data are consistent with the findings of Lefrancois et al. (19) who reported normal trafficking of CD103-deficient CD8 cells from peripheral lymphoid tissues to the lamina propria and IEL compartments. Thus, by process of elimination, these data strongly suggest that the inability of CD103-deficient CD8 cells to reject islet allografts reflects a defect at the level of intragraft migration or delivery of effector function.
Given that islet allografts express the CD103 ligand, E-cadherin, we favor the possibility that CD103 somehow targets donor-reactive CD8 effectors to graft epithelial cells, and thereby facilitates rejection. Importantly, combined histological and FACS® analyses of islet allografts in CD103−/− hosts (Figs. 6 and 7) revealed that CD8 effectors successfully extravasate into the general graft site (the renal subcapsule) but fail to accumulate within the islet allograft itself. These data suggest that CD103 critically functions to promote entry and/or retention of CD8 effectors into epithelial compartments. There is evidence that cytokine production (31), particularly IFN-γ (5), and not the perforin or Fas/FasL pathways (5, 32) is critical for islet allograft destruction by CD8 cells. Recent studies in cardiac (33, 34), skin (35), and tumor (36) rejection models indicate that IFN-γ produced by graft infiltrating CD8 effectors impacts graft survival by inducing intragraft expression of the CXC chemokines, IP-10 and Mig, which are potent chemoattractants for activated T cells. Based on these data, we postulate that the salient role of CD8+ CD103+ effectors in islet allograft rejection, by virtue of their unique capacity to interact with epithelial layers, may be to guide the migration of other inflammatory cells into critical areas of the graft (i.e., the graft functional elements). Consistent with this hypothesis, although islet allografts in wild-type hosts are thoroughly penetrated with infiltrating lymphocytes (Fig. 6 A), CD8+CD103+ cells comprise only a small fraction (<10%) of such cells (Fig. 2 A). Indeed, CD103− CD8+ (∼35%) and CD103−CD8− cells (∼50%, likely CD4 cells and B cells) comprise the vast majority of GILs (Fig. 2 A). These data suggest that CD103−CD8+ cells and other inflammatory cells are helped into the graft by a small subset of CD103−CD8+ cells which effectively penetrate the graft. Studies using chemokine/cytokine-deficient donors and hosts are underway to definitively test this hypothesis.
Studies of IEL populations provide precedent that CD103 can target T cells to epithelial layers. Consistent with a role for CD103 in promoting retention of T cells in the mucosal epithelium, the distribution of CD103+ IEL within the gut epithelium correlates with expression of E-cadherin, and CD103-deficient mice exhibit a specific deficiency (albeit mild) of TCR-αβ+ cells in the IEL compartment (18). Furthermore, CD103 has been shown to promote adhesion of intestinal IEL to epithelial cell lines in vitro (11, 12, 37), and there is evidence that CD103/E-cadherin adhesive interactions are enhanced by signaling through the TCR complex (37).
This data does not exclude a role for CD103 in promoting cytolysis of islet allografts and/or intragraft expansion of CD8 effectors subsequent to their migration into epithelial compartments. There is evidence that CD103-mediated signaling promotes both proliferation (38, 39) and cytolytic activity (21, 40) of IEL populations, and we have previously shown that anti-CD103 mAbs inhibit lysis of epithelial targets by allospecific (CD103+) CTL clones (14). The current dogma is that LFA-1 (CD11a/b1) plays a dominant role in delivery of CD8 effector function through recognition of its broadly expressed ligand, intracellular adhesion molecule (ICAM)-1 (41). CD103+CD8+ cells that infiltrate islet allografts express high levels of LFA-1 (CD11a, Fig. 2 B), but it remains unclear whether islet allografts express significant levels of ICAM-1. Although mouse islets can be induced to express ICAM-1 in vitro with inflammatory cytokines (42), in vivo studies indicate that ICAM-1 expression within inflamed islets is limited to infiltrating leukocyte populations (43); furthermore, treatment of islet allografts with ICAM-1 antisense oligodeoxynucleotide is ineffective in prolonging graft survival (44). Conversely, islets and other epithelial structures constitutively express the CD103 ligand, E-cadherin (20). Thus, CD103/E-cadherin interactions have the potential to impact CD8 effector function at multiple levels including not only migration into epithelial compartments but also in subsequent destruction of epithelial targets.
The concept that CD103 may promote migration of CD8 effectors into epithelial compartments is at odds with the findings of Lefrancois et al. (19), who used adoptive transfer of TCR-transgenic CD8 cells on a CD103−/− background to demonstrate that CD103 is not required for retention of CD8 cells within the intestinal IEL compartment. However, it is important to note that the Lefrancois study examined migration of OVA-specific CD8 cells into the gut following systemic immunization with OVA constructs; thus, it is not clear that OVA was expressed within the IEL compartment examined. Conversely, the CD8 response examined in the present study (BALB/c anti-A/J) is likely directed to the mismatched class I alloantigen (H-2Kk) which is well expressed by the epithelial compartment (islet allograft) in question. As discussed above, CD103/E-cadherin adhesive interactions are regulated by signaling through the TCR complex (37). Thus, the dramatic impact of CD103 on CD8 effector responses noted in the present study may depend on corecognition of MHC/peptide in the context of epithelial layers.
The present finding that CD103−/− hosts exhibit normal rejection of skin allografts was unexpected given that the skin contains important epithelial structures (keratinocytes comprising the epidermis). However, histologic analyses revealed that skin allograft rejection in the A/J to BALB/c strain combination does not involve direct T cell–mediated destruction of epithelial structures (epidermis) but rather reflects destruction of the underlying support structures leading to necrosis of the overlying epidermis (Fig. 6 D). These data are consistent with the findings of Wolman et al. (45) who reported that skin allograft rejection in BALB/c hosts reflects occlusion of blood vessels at the graft-host border with ischemia of the graft. Unlike islet allografts, skin allografts are highly vulnerable to CD4-mediated rejection (1, 2); moreover, A/J donors are fully mismatched from BALB/c at MHC class II alleles (I-Ak and I-Ek). Thus, destruction of skin allografts in this strain combination is likely mediated by CD4 effectors directed to class II alloantigens expressed by the graft vasculature, whereas islet rejection is likely mediated by CD8 effectors directed to class I alloantigens expressed by the graft functional elements (insulin-producing β cells). It is important to note, however, that this data does not exclude a role for CD103 in promoting CD8-mediated destruction of skin allografts in other strain combinations or transplant scenarios.
These findings are consistent with a general role for CD103 in facilitating immune surveillance of epithelial compartments by CD8 cells. The CD103 ligand, E-cadherin, functions at the level of adherent junctions (13) to promote homotypic interactions between adjacent epithelial cells and, consequently, is selectively yet ubiquitously expressed by cells comprising epithelial layers. Importantly, the initial site of attack by intracellular parasites is generally an epithelial layer (i.e., the mucosa of the skin, lungs, and gut), and there is abundant evidence that CD8-mediated destruction/neutralization of the infected epithelial cells is often required for clearance of such pathogens (46). Indeed, CD103+CD8αβ+TCR-αβ+ cells with potent cytolytic capability (albeit of unknown specificity) are abundantly present within the IEL and LPL compartments of normal individuals (47), where they are thought to function as an initial line of defense against frequent microbial attack at such sites. A role for CD103+CD8+ effectors in surveillance against tumors of epithelial origin (48), and as an effector mechanism in the epithelial lesions characteristic of GVHD (49) and autoimmune pathology (50) is also plausible. It will now be important to determine the extent to which these diverse sites are permissive for the induction/maintenance of CD103 expression by antigen-specific CD8 effectors, as this likely represents the salient factor limiting the role of CD103 in CD8-mediated immune responses.
Acknowledgments
We are indebted to Drs. Jan Cerny and Kamal Moudgil for critical reading of this manuscript, and to Dr. Anu Arya whose kind gift of CD103-deficient mice made this work possible.
This work was supported by a grant from the National Institutes of Health (AI36532) to G.A. Hadley.
Footnotes
Abbreviations used in this paper: GIL, graft infiltrating lymphocyte; ICAM, intracellular adhesion molecule; IEL, intraepithelial lymphocyte; LPL, lamina propria lymphocyte; SC, spleen cell.
References
- 1.Hall, B.M. 1991. Cells mediating allograft rejection. Transplantation. 51:1141–1151. [DOI] [PubMed] [Google Scholar]
- 2.Steinmuller, D. 1985. Which T cells mediate allograft rejection? Transplantation. 40:229–233. [DOI] [PubMed] [Google Scholar]
- 3.Topham, D.J., R.A. Tripp, and P.C. Doherty. 1997. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 159:5197–5200. [PubMed] [Google Scholar]
- 4.Barth, R.J., Jr., J.J. Mule, P.J. Spiess, and S.A. Rosenberg. 1991. Interferon γ and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J. Exp. Med. 173:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diamond, A.S., and R.G. Gill. 2000. An essential contribution by IFN-γ to CD8+ T cell-mediated rejection of pancreatic islet allografts. J. Immunol. 165:247–255. [DOI] [PubMed] [Google Scholar]
- 6.Cocchi, F., A.L. DeVico, A. Garzino-Demo, S.K. Arya, R.C. Gallo, and P. Lusso. 1995. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science. 270:1811–1815. [DOI] [PubMed] [Google Scholar]
- 7.Kim, J.J., L.K. Nottingham, J.I. Sin, A. Tsai, L. Morrison, J. Oh, K. Dang, Y. Hu, K. Kazahaya, M. Bennett, et al. 1998. CD8 positive T cells influence antigen-specific immune responses through the expression of chemokines. J. Clin. Invest. 102:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science. 272:60–66. [DOI] [PubMed] [Google Scholar]
- 9.Hadley, G.A., S.T. Bartlett, C.S. Via, E.A. Rostapshova, and S. Moainie. 1997. The epithelial cell-specific integrin, CD103 (α E integrin), defines a novel subset of alloreactive CD8+ CTL. J. Immunol. 159:3748–3756. [PubMed] [Google Scholar]
- 10.Hadley, G.A., E.A. Rostapshova, D.M. Gomolka, B.M. Taylor, S.T. Bartlett, C.I. Drachenberg, and M.R. Weir. 1999. Regulation of the epithelial cell-specific integrin, CD103, by human CD8+ cytolytic T lymphocytes. Transplantation. 67:1418–1425. [DOI] [PubMed] [Google Scholar]
- 11.Karecla, P.I., S.J. Bowden, S.J. Green, and P.J. Kilshaw. 1995. Recognition of E-cadherin on epithelial cells by the mucosal T cell integrin αM290 β 7 (αEβ7). Eur. J. Immunol. 25:852–856. [DOI] [PubMed] [Google Scholar]
- 12.Cepek, K.L., S.K. Shaw, C.M. Parker, G.J. Russell, J.S. Morrow, D.L. Rimm, and M.B. Brenner. 1994. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature. 372:190–193. [DOI] [PubMed] [Google Scholar]
- 13.Aberle, H., H. Schwartz, and R. Kemler. 1996. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell Biochem. 61:514–523. [DOI] [PubMed] [Google Scholar]
- 14.Rostapshova, E.A., J.M. Burns, S.T. Bartlett, and G.A. Hadley. 1998. Integrin-mediated interactions influence the tissue specificity of CD8+ cytolytic T lymphocytes. Eur. J. Immunol. 28:3031–3039. [DOI] [PubMed] [Google Scholar]
- 15.Robertson, H., W.K. Wong, D. Talbot, A.D. Burt, and J.A. Kirby. 2001. Tubulitis after renal transplantation: demonstration of an association between CD103+ T cells, transforming growth factor β1 expression and rejection grade. Transplantation. 71:306–313. [DOI] [PubMed] [Google Scholar]
- 16.Kilshaw, P.J., and S.J. Murant. 1990. A new surface antigen on intraepithelial lymphocytes in the intestine. Eur. J. Immunol. 20:2201–2207. [DOI] [PubMed] [Google Scholar]
- 17.Cerf-Bensussan, N., A. Jarry, N. Brousse, B. Lisowska-Grospierre, D. Guy-Grand, and C. Griscelli. 1987. A monoclonal antibody (HML-1) defining a novel membrane molecule present on human intestinal lymphocytes. Eur. J. Immunol. 17:1279–1285. [DOI] [PubMed] [Google Scholar]
- 18.Schon, M.P., A. Arya, E.A. Murphy, C.M. Adams, U.G. Strauch, W.W. Agace, J. Marsal, J.P. Donohue, H. Her, D.R. Beier, et al. 1999. Mucosal T lymphocyte numbers are selectively reduced in integrin αE (CD103)-deficient mice. J. Immunol. 162:6641–6649. [PubMed] [Google Scholar]
- 19.Lefrancois, L., C.M. Parker, S. Olson, W. Muller, N. Wagner, M.P. Schon, and L. Puddington. 1999. The role of β7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 189:1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cirulli, V., D. Baetens, U. Rutishauser, P.A. Halban, L. Orci, and D.G. Rouiller. 1994. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J. Cell Sci. 107:1429–1436. [DOI] [PubMed] [Google Scholar]
- 21.Lefrancois, L., T.A. Barrett, W.L. Havran, and L. Puddington. 1994. Developmental expression of the α IEL β7 integrin on T cell receptor γδ and T cell receptor αβ T cells. Eur. J. Immunol. 24:635–640. [DOI] [PubMed] [Google Scholar]
- 22.Gotoh, M., T. Maki, T. Kiyoizumi, S. Satomi, and A.P. Monaco. 1985. An improved method for isolation of mouse pancreatic islets. Transplantation. 40:437–438. [DOI] [PubMed] [Google Scholar]
- 23.Steinmuller, D. 1984. Skin grafting. Methods Enzymol. 108:20–28. [DOI] [PubMed] [Google Scholar]
- 24.Kilshaw, P.J. 1993. Expression of the mucosal T cell integrin αM290 β7 by a major subpopulation of dendritic cells in mice. Eur. J. Immunol. 23:3365–3368. [DOI] [PubMed] [Google Scholar]
- 25.Tiisala, S., T. Paavonen, and R. Renkonen. 1995. αEβ7 and α4β7 integrins associated with intraepithelial and mucosal homing, are expressed on macrophages. Eur. J. Immunol. 25:411–417. [DOI] [PubMed] [Google Scholar]
- 26.Smith, T.J., L.A. Ducharme, S.K. Shaw, C.M. Parker, M.B. Brenner, P.J. Kilshaw, and J.H. Weis. 1994. Murine M290 integrin expression modulated by mast cell activation. Immunity. 1:393–403. [DOI] [PubMed] [Google Scholar]
- 27.Hadley, G.A., C. Charandee, M.R. Weir, D. Wang, S.T. Bartlett, and C.B. Drachenberg. 2001. CD103+ CTL accumulate within the graft epithelium during clinical renal allograft rejection. Transplantation. 72:1546–1555. [DOI] [PubMed] [Google Scholar]
- 28.Desai, N.M., H. Bassiri, J. Kim, B.H. Koller, O. Smithies, C.F. Barker, A. Naji, and J.F. Markmann. 1993. Islet allograft, islet xenograft, and skin allograft survival in CD8+ T lymphocyte-deficient mice. Transplantation. 55:718–722. [DOI] [PubMed] [Google Scholar]
- 29.Simeonovic, C.J., D.J. Brown, M.J. Townsend, and J.D. Wilson. 1996. Differences in the contribution of CD4+ T cells to proislet and islet allograft rejection correlate with constitutive class II MHC alloantigen expression. Cell Transplant. 5:525–541. [DOI] [PubMed] [Google Scholar]
- 30.Markmann, J.F., H. Bassiri, N.M. Desai, J.S. Odorico, J.I. Kim, B.H. Koller, O. Smithies, and C.F. Barker. 1992. Indefinite survival of MHC class I-deficient murine pancreatic islet allografts. Transplantation. 54:1085–1089. [DOI] [PubMed] [Google Scholar]
- 31.Hodgkin, P.D., M. Agostino, K. Sellins, S.J. Prowse, D. Bellgrau, and K.J. Lafferty. 1985. T lymphocyte function in vivo. Ambivalence of the class I MHC antigen-reactive subset. Transplantation. 40:288–292. [PubMed] [Google Scholar]
- 32.Ahmed, K.R., T.B. Guo, and K.K. Gaal. 1997. Islet rejection in perforin-deficient mice: the role of perforin and Fas. Transplantation. 63:951–957. [DOI] [PubMed] [Google Scholar]
- 33.Kapoor, A., K. Morita, T.M. Engeman, S. Koga, E.M. Vapnek, M.G. Hobart, and R.L. Fairchild. 2000. Early expression of interferon-γ inducible protein 10 and monokine induced by interferon-γ in cardiac allografts is mediated by CD8+ T cells. Transplantation. 69:1147–1155. [DOI] [PubMed] [Google Scholar]
- 34.Miura, M., K. Morita, H. Kobayashi, T.A. Hamilton, M.D. Burdick, R.M. Strieter, and R.L. Fairchild. 2001. Monokine induced by IFN-γ is a dominant factor directing T cells into murine cardiac allografts during acute rejection. J. Immunol. 167:3494–3504. [DOI] [PubMed] [Google Scholar]
- 35.Watarai, Y., S. Koga, D.R. Paolone, T.M. Engeman, C. Tannenbaum, T.A. Hamilton, and R.L. Fairchild. 2000. Intraallograft chemokine RNA and protein during rejection of MHC-matched/multiple minor histocompatibility-disparate skin grafts. J. Immunol. 164:6027–6033. [DOI] [PubMed] [Google Scholar]
- 36.Dobrzanski, M.J., J.B. Reome, and R.W. Dutton. 2001. Immunopotentiating role of IFN-γ in early and late stages of type 1 CD8 effector cell-mediated tumor rejection. Clin. Immunol. 98:70–84. [DOI] [PubMed] [Google Scholar]
- 37.Higgins, J.M., D.A. Mandlebrot, S.K. Shaw, G.J. Russell, E.A. Murphy, Y.T. Chen, W.J. Nelson, C.M. Parker, and M.B. Brenner. 1998. Direct and regulated interaction of integrin αEβ7 with E-cadherin. J. Cell Biol. 140:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarnacki, S., B. Begue, H. Buc, F. Le Deist, and N. Cerf-Bensussan. 1992. Enhancement of CD3-induced activation of human intestinal intraepithelial lymphocytes by stimulation of the β7-containing integrin defined by HML-1 monoclonal antibody. Eur. J. Immunol. 22:2887–2892. [DOI] [PubMed] [Google Scholar]
- 39.Russell, G.J., C.M. Parker, K.L. Cepek, D.A. Mandelbrot, A. Sood, E. Mizoguchi, E.C. Ebert, M.B. Brenner, and A.K. Bhan. 1994. Distinct structural and functional epitopes of the αEβ7 integrin. Eur. J. Immunol. 24:2832–2841. [DOI] [PubMed] [Google Scholar]
- 40.Roberts, K., and P.J. Kilshaw. 1993. The mucosal T cell integrin αM290β7 recognizes a ligand on mucosal epithelial cell lines. Eur. J. Immunol. 23:1630–1635. [DOI] [PubMed] [Google Scholar]
- 41.Springer, T.A., M.L. Dustin, T.K. Kishimoto, and S.D. Marlin. 1987. The lymphocyte function-associated LFA-1, CD2, and LFA-3 molecules: cell adhesion receptors of the immune system. Annu. Rev. Immunol. 5:223–252. [DOI] [PubMed] [Google Scholar]
- 42.Prieto, J., E.E. Kaaya, L. Juntti-Berggren, P.O. Berggren, S. Sandler, P. Biberfeld, and M. Patarroyo. 1992. Induction of intercellular adhesion molecule-1 (CD54) on isolated mouse pancreatic β cells by inflammatory cytokines. Clin. Immunol. Immunopathol. 65:247–253. [DOI] [PubMed] [Google Scholar]
- 43.O'Reilly, L.A., P.R. Hutchings, P.R. Crocker, E. Simpson, T. Lund, D. Kioussis, F. Takei, J. Baird, and A. Cooke. 1991. Characterization of pancreatic islet cell infiltrates in NOD mice: effect of cell transfer and transgene expression. Eur. J. Immunol. 21:1171–1180. [DOI] [PubMed] [Google Scholar]
- 44.Katz, S.M., F. Bennett, K. Stecker, J.H. Clark, T. Pham, M.E. Wang, B.D. Kahan, and S.M. Stepkowski. 2000. ICAM-1 antisense oligodeoxynucleotide improves islet allograft survival and function. Cell Transplant. 9:817–828. [DOI] [PubMed] [Google Scholar]
- 45.Wolman, M., I. Bleiberg, and J. Leibovici. 1977. Histological criteria for immunological rejection of mouse skin homografts. J. Pathol. 122:1–7. [DOI] [PubMed] [Google Scholar]
- 46.Doherty, P.C. 1996. Cytotoxic T cell effector and memory function in viral immunity. Curr. Top. Microbiol. Immunol. 206:1–14. [DOI] [PubMed] [Google Scholar]
- 47.Hayday, A., E. Theodoridis, E. Ramsburg, and J. Shires. 2001. Intraepithelial lymphocytes: exploring the Third Way in immunology. Nat. Immunol. 2:997–1003. [DOI] [PubMed] [Google Scholar]
- 48.Svane, I.M., M. Boesen, and A.M. Engel. 1999. The role of cytotoxic T-lymphocytes in the prevention and immune surveillance of tumors–lessons from normal and immunodeficient mice. Med. Oncol. 16:223–238. [DOI] [PubMed] [Google Scholar]
- 49.Murphy, G.F., D. Whitaker, J. Sprent, and R. Korngold. 1991. Characterization of target injury of murine acute graft-versus-host disease directed to multiple minor histocompatibility antigens elicited by either CD4+ or CD8+ effector cells. Am. J. Pathol. 138:983–990. [PMC free article] [PubMed] [Google Scholar]
- 50.Amrani, A., J. Verdaguer, P. Serra, S. Tafuro, R. Tan, and P. Santamaria. 2000. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 406:739–742. [DOI] [PubMed] [Google Scholar]