

Abstract
The number of circulating follicular B lymphocytes is normally kept within a precise range despite their dispersion through the body and daily overproduction of precursors in the bone marrow. By establishing a genome wide recessive mutation screen in C57BL/6 mice to identify critical components of immune system regulation, we identified a mutant strain with selective deficiency in recirculating B cells but not immature or peritoneal B1 cells. Analysis of mixed bone marrow chimeras established that the mutation affects a cell autonomous process within B cells that is required for their accumulation after emigrating to peripheral lymphoid organs. The defect is caused by a point mutation in the gene encoding transcription factor nuclear factor (NF)-κB2, terminating the encoded protein within the DNA-binding domain. These findings establish the feasibility of analyzing immune regulation by genome wide mutant screens and demonstrates an intrinsic requirement for NF-κB2 in regulating circulating follicular B cell numbers.
Keywords: Rel/NF-κB, B lymphocytes, follicular, ethylnitrosourea, mutagenesis
Introduction
Abnormal numbers of circulating lymphocytes are associated with leukemia/lymphoma, immunodeficiency, and autoimmunity. Despite the importance of processes that regulate peripheral lymphocyte numbers, details on the genetics and pathways involved remain to be elucidated. To investigate the components of pathways involved in immune system regulation we have explored the possibility of using the potent germline mutagen ethylnitrosourea (ENU) to generate libraries of C57BL/6 mice with random point mutations throughout their genomes. ENU was originally defined as a supermutagen for mouse spermatogonial stem cells, by treating outbred male mice and then breeding with tester females homozygous for seven visible recessive traits (1). This one generation “specific locus test” revealed new recessive loss-of-function alleles induced by ENU on average once in every 700 gametes. Several potential barriers may nevertheless block extension of this strategy to the entire genome, to discover recessive mutations affecting the immune system of adult mice in an unbiased manner. First, when inbreeding strategies are used to bring recessive ENU traits to homozygosity across an entire mutagenized genome, the frequency of embryonic or neonatal lethal mutations may prevent sufficient animals reaching adulthood. Second, the burden of homozygozed mutations may result in complex, multigenic interactions so that the majority of immunological traits may fail to breed true and be difficult to pinpoint. Third, genome wide screens impose the difficulty of mapping each mutation to a chromosomal interval and identifying the mutated gene. On the other hand, it would be useful if new immunological mutants were induced in the C57BL/6 strain, since a large number of congenic and mutant strains are available for immunobiological analyses in this strain background. Here, we demonstrate the feasibility and utility of genome wide mutagenesis and screening for recessive immunological traits. By isolating and analyzing a mutant strain with diminished numbers of circulating B cells, we confirm earlier studies on nuclear factor (NF)-κB2–deficient mice (2–4) and extend these findings to show that the transcription factor NF-κB2 plays an essential, B cell–intrinsic role in maintaining the numbers of mature B2 and marginal zone (MZ) B cells, while B1 cell numbers do not require this factor.
Materials and Methods
ENU Mutagenesis.
Male C57BL/6 mice 8–15 wk old were treated three or four times, 1 wk apart, with 85 mg/kg or 100 mg/kg N-ethyl-N-nitrosourea (Sigma-Aldrich) freshly dissolved in 10% ethanol in citrate buffer, pH 5.0, as described previously (1). The xander strain arose from a G0 treated with the dose of 4 × 85 mg/kg ENU.
Flow Cytometry.
Single cell suspensions were stained with FITC-conjugated anti-IgM, anti-B220, and anti-Ly5a and PE-conjugated anti-Thy1.2, anti-IgD, anti-CD23, and anti-B220 (BD PharMingen). Biotinylated anti-CD21 and anti-Ly5a were secondary stained with streptavidin cychrome (BD PharMingen). Samples were collected on a FACScan™ (Becton Dickinson) and data was analyzed using Flowjo™ software (Tree Star).
Immunohistochemistry.
5-μm sections of frozen spleen were cut and fixed in cold acetone, air-dried, and stored at –20°C. Sections were immunostained with mAbs anti-IgM (Serotec and The Binding Site), anti-CD3 (Serotec), anti-IgD (Southern Biotechnology, Inc.), anti-CD21 (BD PharMingen), and MOMA1 (Serotec). HRP-conjugated antibodies were developed with 3,3′-diaminobenzidine tablets (Sigma-Aldrich). Biotinylated antibodies were stained with streptavidin alkaline phosphatase and visualized using alkaline phosphatase substrate kit (Vector Laboratories).
Bone Marrow Chimeras.
Recipient mice were irradiated with two doses of 5.5 Gy 3 h apart. The next day, recipient mice were injected intravenously with a total of 2 × 106 bone marrow (BM) cells. Mice were killed and analyzed 8 wk after reconstitution.
Mapping.
The xdr mutation was out crossed to the NOD. H-2k strain by breeding male xdr/xdr mice on C57BL/6 background to NOD.H-2k females to yield F1 intercross mice. F1 mice were intercrossed to generate F2 intercross mice that were screened to identify mutant individuals. Analysis of F2 intercross DNA was performed by amplifying regions of the genome with defined simple sequence length polymorphism (SSLP) markers (5). C57BL/6 and NOD.H-2k DNA amplification products from each SSLP were resolved by agarose gel electrophoresis.
Sequencing and Protein Expression.
Spleen cDNA was amplified by PCR and sequenced using 16–20 bp NF-κB2 complementary primers (sequences available on request). Cell lysates were prepared in RIPA buffer from splenocytes and fractionated by SDS-PAGE using standard protocols. Membranes were probed with a rabbit polyclonal antibody against NF-κB2 (Santa Cruz Biotechnology, Inc.), followed by HRP-conjugated goat anti–rabbit IgG (Santa Cruz Biotechnology, Inc.) and developed with enhanced chemiluminescence (NEN Life Sciences).
Results
Generation of ENU Mutant Strain ‘xander’.
We generated a library of recessive mutant mice by treating male C57BL/6 mice with ENU. To observe the effect of recessive mutations, which are the most frequent class of loss-of-function mutation, we adopted a strategy of inbreeding mice to homozygozity by constructing three generation pedigrees (Fig. 1 A). ENU treated male mice (generation 0; G0) were mated with wild-type (WT) C57BL/6 females yielding G1 offspring. Each G1 mouse is heterozygous for a different constellation of point mutations inherited on the paternal chromosomes. To homozygoze these mutations, a pedigree is founded by a single G1 male mated with a WT C57BL/6 female and then with 4–6 of his G2 daughters to yield G3 mice. Since half of the G2 daughters will inherit any particular mutation carried by the G1, one in eight of the G3 mice are expected to be homozygous for this mutation. A target of 24 G3 mice was bred in each pedigree and reared to adulthood, and their blood lymphocyte subsets screened by flow cytometry. In pedigree number 3, several G3 mice were identified with fewer circulating B cells in the blood (Fig. 1 B). The B cell deficiency was inherited as a simple recessive Mendelian trait both during intercrossing with siblings on the C57BL/6 background and in an outcross with NOD.H-2k followed by an F2 intercross. The mutant strain thus established was named ‘xander’ (xdr).
Figure 1.

An ENU-induced recessive mutation causing selective deficiency of peripheral B cells. (A) Initial generations of ‘xander’ pedigree. Note that apparent fluctuations from expected Mendelian frequency in the small numbers shown are not significant, and that the expected frequencies were observed in large crosses. Black squares and circles, affected individuals; white squares and circles, unaffected; cross, not typed by blood. (B) Reduced numbers of B cells in blood of xdr/xdr mice compared with heterozygous and WT siblings. Representative FACS® plots of B220 and Thy1.2 expression on blood from C57BL/6 and xdr/xdr mice, and the percent of each cell subset within the lymphocyte gate. Graph shows the percentage of B220+ and Thy1.2+ cells for xdr/xdr, xdr/+ and WT siblings.
xdr/xdr Mice Have a Selective Deficiency in the Mature B2 Cell Population.
Analysis of splenic lymphocyte populations in xdr/xdr mice reveals a reduction in the number of B cells, but not T cells compared with WT mice (Fig. 2 A). After B cell formation in the BM, B cell maturation in the spleen proceeds from transitional type 1 (T1) cells that have recently emigrated from the BM to transitional type 2 (T2) cells, mature recirculating (follicular) B cells and MZ B cells. These subsets can be distinguished by cell surface expression of IgM, IgD, and CD21 (6). Analysis of B cell subsets in the spleens shows that xdr/xdr mice have significantly reduced numbers of T2, mature, and MZ B cells compared with WT mice (Fig. 2 B). Recirculating B cells were also dramatically reduced in blood (Fig. 1) and all peripheral lymph nodes (unpublished data). There was no significant reduction in numbers of T1 B cells, suggesting that B cell export from the BM was normal. Consistent with normal BM B cell production, there was no difference in the percent of pro-B/pre-B cells or immature B cells in the BM of WT and xdr/xdr mice, but recirculating mature B cells were again reduced (Fig. 2 C). These results are consistent with a defect in the accumulation of mature B cells in xdr/xdr mice.
Figure 2.

Decreased numbers of mature B2 cells in spleen, but normal B cell production in BM. (A) Peripheral B cell deficiency, but normal T cell numbers in xdr/xdr mice. Graphs show the mean number of cells ± SD (n = 3). Gray bars, C57BL/6; white bars, xdr/xdr. (B) Splenic B cell subsets. T2 cells are IgMhigh IgD+ and mature B cells are IgMlow and IgD+. Histograms of CD21 expression, after gating on IgMhigh IgD− cells, resolves T1 cells (CD21−) from MZ B cells (CD21high). Significant differences as determined by Student's t test are represented as *P < 0.05, **P < 0.01, and ***P < 0.001. Results shown are representative of several independent experiments. (C) BM pro- and pre-B cells (B220low IgM−), immature B cells (B220low IgM+ IgD−), and recirculating mature B cells (B220high IgM+ IgD+). (D) Numbers of peritoneal B1 cells (IgM+ CD23−) and B2 cells (IgM+ CD23+). Equivalent results were obtained using B220 and CD5 to resolve these subsets.
The B1 subset of mature peripheral B cells differs from recirculating B2 cells in its anatomical localization, restricted Ig repertoire, cell surface markers, and capacity for self-renewal (7). B1 cells were enumerated in the peritoneal cavity of xdr/xdr and WT mice. In contrast to the deficiency of mature B cells in other lymphoid compartments, xdr/xdr mice had a sevenfold increase in the number of B1 cells in the peritoneal cavity (Fig. 2 D). Interestingly, the number of B2 cells in the peritoneal cavity is comparable for WT and xdr/xdr mice (Fig. 2 D).
xdr/xdr Mice Have Abnormalities in Secondary Lymphoid Tissues.
In addition to the deficiency in recirculating B cells, xdr/xdr mice have abnormalities in secondary lymphoid tissues including small peripheral lymph nodes and disturbed splenic microarchitecture. In spleen sections from xdr/xdr mice, the T cell zone is normal in appearance, but B cell follicles are reduced to a small ring around the T zone and MZ B cells are visible although fewer in number (Fig. 3, A and B) . Marginal metallophilic macrophages (MMM) detected with MOMA-1 staining form a continuous ring around the white pulp in spleens of WT animals whereas there are few MOMA1+ cells in xdr/xdr spleens (Fig. 3 C). Follicular dendritic cells (FDC) detected by high expression of CD21 form prominent networks within the B cell follicles in WT spleen sections, whereas these FDC clusters are not visible in spleen sections from xdr/xdr mice (Fig. 3 D).
Figure 3.
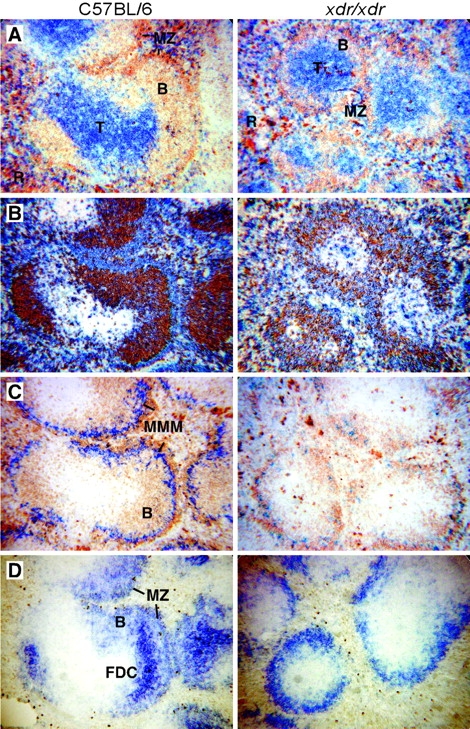
Abnormal splenic microarchitecture in xdr/xdr mice. Spleen sections from WT and xdr/xdr mice were immunostained for expression of (A) IgM (brown) and CD3 (blue), (B) IgD (brown) and IgM (blue), (C) IgM (brown) and MOMA1 (blue) and (D) CD21. T, T cell zone; B, B cell follicle; R, red pulp; MMM, marginal metallophilic macrophages.
B Cell Deficiency in xdr/xdr Mice Is Due to a B Cell Intrinsic Defect.
To determine if the circulating B cell deficiency was secondary to these FDC and macrophage supporting cell abnormalities or due to a B cell intrinsic defect, BM chimeras were constructed with mixtures of normal and mutant BM that could be distinguished by alleles of the cell surface marker Ly5/CD45 (Fig. 4) . In the test group (xdr versus WT chimeras), irradiated B6.Ly5a recipient mice were reconstituted with a mixture containing equal numbers of xdr/xdr B6.Ly5b and WT B6.Ly5a BM cells. As controls, B6.Ly5a recipients received equal numbers of B6.Ly5b and B6.Ly5a BM cells (WT versus WT chimeras). After reconstitution, the control (WT versus WT) chimeric animals contained equivalent proportions of Ly5a and Ly5b pre-B cells, immature, and mature B cells in their BM and periphery, indicating equal reconstitution with the two sources of stem cells and equal accumulation of mature B cell progeny (Fig. 4). The xdr versus WT chimeras were also equally reconstituted with the two donor marrows, and formed similar numbers of mutant and WT pre-B cells and immature B cells in their BM (Fig. 4). The numbers of IgM+IgD− splenic cells, containing the recent BM emigrants, was also not significantly different, but there was a dramatic and selective deficiency of more mature B cells of xdr/xdr origin such that only a few percent of mature B cells in xdr versus WT recipients were of xdr/xdr origin and there were almost no xdr/xdr-derived MZ B cells (Fig. 4). Since WT B cells accumulated normally in the xdr versus WT chimeras, the selective deficit in mutant B cell accumulation establishes that the mutation disrupts a cell autonomous step within B cells rather than any defect in other supporting hemopoietic cells.
Figure 4.

Intrinsic defect in peripheral B cells in xdr/xdr mice. Lethally irradiated B6.Ly5a recipient mice were reconstituted with a mixture of 106 B6.Ly5b and 106 B6.Ly5a marrow (WT versus WT) or 106 B6.Ly5b xdr/xdr and 106 B6.Ly5a marrow (xdr versus WT). Chimeric mice were killed 8 wk after reconstitution and B cell subsets derived from the two donors were distinguished by staining for cell surface marker Ly5a. There was no significant difference in the absolute number of spleen cells: 6.68 × 107 ± 2.76 × 107 for WT versus WT; 5.89 × 107 ± 1.4 × 107. Graphs show the mean ± SD (n = 4). Gray bars, Ly5a; white bars, Ly5b. Significant differences as determined by Student's t test are represented as **P < 0.01 and ***P < 0.001. Data is representative of several experiments.
xdr/xdr Mice Have a Point Mutation in Gene Encoding Transcription Factor NF-κB2.
An F2 intercross between B6 xdr/xdr and NOD.H-2k mice was used to locate the mutated gene (Fig. 1 A). From 9 pairs of F2 breeders, 108 offspring were typed for blood B cell deficiency. 32 offspring were affected (30%) confirming a simple, fully penetrant recessive trait. Pooled DNA from affected individuals was typed using a genome wide panel of SSLP markers (5), revealing linkage to chromosome 19. Typing individual animals with additional SSLP markers localized the gene between D19Mit53 and D19Mit70 (Fig. 5 A). The Nf-κb2 gene within this interval was a strong candidate because like xdr/xdr mice, Nf-κb2 knockout animals exhibit B cell defects and lack FDC networks and MOMA1+ MZ macrophages in the spleen (2–4).
Figure 5.

Intronic point mutation in NF-κB2 results in aberrant mRNA splicing and premature stop codons. (A) Chromosomal mapping of xdr/xdr mutation. Haplotypes of individual xdr/xdr affected mice are shown. Black squares indicate C57BL/6 homozygote; white squares indicate C57BL/6/NOD heterozygote. (B) Amplification of NF-κB2 cDNA reveals multiple aberrant spliced products between exons 5–8 in xdr/xdr mice. Spleen cDNA from WT and xdr/xdr mice was amplified by PCR using the indicated primer pairs. Primer pair 204F and 550R amplifies exons 2–5, 457F and 872R amplifies exons 5–8 and 791F and 3013R amplify exons 8–23. (C) The PCR products resulting from amplification of exons 5–8 from WT and xdr/xdr mice were cloned and sequenced. Intronic base number 3163 mutated in xdr/xdr mice shown as bold A. (D) Absence of NFκB2 protein in xdr/xdr mice. Cell lysates were prepared from the spleens of WT and xdr/xdr mice and probed with a polyclonal antibody against NF-κB2. (E) T to A conversion of Nf-κb2 intronic base 3163 in amplified genomic DNA in xdr/xdr mice. (F) Schematic representation of the effect of the ENU-induced point mutation in Nf-κb2.
NF-κB2 mRNA was amplified by RT-PCR from the spleens of WT and xdr/xdr mice. Amplification of exons 5–8 yielded multiple aberrant sized products from xdr/xdr mice, none of which correspond to the WT species (Fig. 5 B). cDNA from the amplification of exons 5–8 was cloned and sequenced (Fig. 5 C). In xdr/xdr mice no correctly spliced products were found and five different aberrant transcript types were revealed. Two classes retained an unspliced intron between exon 6 and 7. In the others, exon 6 was spliced to cryptic splice acceptor sites within exon 7 resulting in the absence of the first 34 or 37 bases of exon 7. All of the aberrant products generate in frame-stop codons that would prematurely truncate NF-κB2 in the middle of the DNA-binding domain (8) so that no functional protein could be made. The absence of full-length NF-κB2 protein in xdr/xdr mice was confirmed by Western blot analysis (Fig. 5 D).
Sequencing of genomic DNA revealed the ENU-induced point mutation in Nf-κb2 as an intronic T to A conversion of base 3163 between the parental B6 and xdr mutant, in the conserved polypyrimidine tract of the exon 7 splice acceptor site (Fig. 5 E; reference 8). This base change, thus prevents mRNA splicing of exon 6 to the correct exon 7 acceptor site, causing an accumulation of unspliced intermediates and aberrant splicing to sequences within exon 7 that resemble splice acceptor sites. The effect of the ENU-induced point mutation on mRNA splicing and protein expression is shown schematically in Fig. 5 F.
Discussion
This study establishes the feasibility and utility of analyzing immune regulation by genome wide screens for recessive immune abnormalities induced in C57BL/6 mice by ENU, and further defines the role of NF-κB2 in regulating B cell numbers. The mutation identified, in the gene encoding the transcription factor NF-κB2, lies in an intron and causes aberrant mRNA splicing so that the protein reading frame is shifted and the protein coding region terminated prematurely within the DNA-binding domain. Thus, no functional NF-κB2 protein is predicted, consistent with the absence of p100 in Western blot analysis. The aberrant splicing resulting from a point mutation is consistent with previous analyses of ENU-induced mutations, where ∼25% result from aberrant mRNA splicing (9).
Previous analyses of mice with a targeted null mutation in NF-κB2 found reduced numbers of B cells in the spleen and absence of FDCs and MMM paralleling the findings here (2–4). Our analysis extends these findings by showing that the B cell deficiency first appears after the T1 stage of B cell maturation in the spleen, severely reducing the follicular and MZ populations in the spleen, lymph node, and blood. The mixed chimera analysis establishes that the failure to accumulate these mature B cell subsets reflects a primary, cell intrinsic function of NF-κB2 in these B cells or their immediate T1/T2 precursors, rather than being secondary to NF-κB2 defects in other hemopoietic or stromal cells. We also show for the first time that NF-κB2 is not required to establish normal numbers of B2 and B1 cells in the peritoneum, indeed the numbers of B1 cells are markedly increased in the xander mice. Alymphoplasia (aly) mice, a strain with a point mutation in NF-κB-inducing kinase (NIK), exhibit multiple defects including reduced numbers of mature B cells in their peripheral lymphoid organs (10). Interestingly, like xdr/xdr mice, aly/aly mice have normal numbers of B2 cells and increased B1 cells in the peritoneal cavity possibly arising from a migration defect in aly/aly peritoneal B cells (10). The possible role of NF-κB2 downstream of NIK and chemokine receptor signaling is yet to be explored.
The specific pattern of B cell subsets deficient in the xander mutant mice raises the question of what B cell survival signaling pathway this transcription factor is likely to serve as an essential intermediate. Two cell surface receptor systems have recently emerged as regulating peripheral B cell numbers. Mature B cells require a basal stimulation through the B cell receptor (BCR) to survive and accumulate (11, 12). Unlike the selective deficiency of recirculating and MZ B cells in Nf-κb2 xdr mice, however, peripheral B cell deficiencies due to defects in the BCR signaling pathway are most marked within the peritoneal B1 subsets (13–19). By contrast, the selective deficiency of splenic T2, recirculating and MZ cells but not peritoneal B cells in Nf-κb2 xdr mice closely resembles the B cell phenotype of A/WySnJ mice, in which the BAFF-R is naturally mutated (20–22), and in mice deficient for BAFF (23, 24), or NIK (10, 25). Thus it is reasonable to consider that NF-κB2 plays a major role in B2 cells transducing the survival signals from BAFF/BAFF-R.
The demonstration here that NF-κB2 is required within follicular B cells for their accumulation, at precisely the developmental stages and locations that BAFF is required, is interesting given that activating mutations in this transcription factor or its partner, Bcl-3, occur in B cell lymphoma (for a review, see reference 26). This signaling system may normally regulate B cell numbers by requiring that circulating B cells regularly pass through limiting sites of BAFF production such as the macrophage-rich zones of spleen and lymph nodes (27). Mutations that activate this pathway may thus allow follicular lymphoma cells to survive and accumulate independently of this trophic support. It will be important in future work to better define the follicular B cell intrinsic pathway served by NF-κB2 and the downstream target genes that it regulates in these cells.
Acknowledgments
We thank L. Wilson and S. Chaudhry, I. Whiting, L. Foster, and staff of the Medical Genome Centre for the expert care and breeding of mutant mice, and J. Cyster, S. Kay, C. Vinuesa, and S. Martin for comments on the manuscript.
M. Blery is supported by a Human Frontier Science Program Organization long-term fellowship.
References
- 1.Hitotsumachi, S., D.A. Carpenter, and W.L. Russell. 1985. Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc. Natl. Acad. Sci. USA. 82:6619–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franzoso, G., L. Carlson, L. Poljak, E.W. Shores, S. Epstein, A. Leonardi, A. Grinberg, T. Tran, T. Scharton-Kersten, M. Anver, et al. 1998. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 187:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caamano, J.H., C.A. Rizzo, S.K. Durham, D.S. Barton, C. Raventos-Suarez, C.M. Snapper, and R. Bravo. 1998. Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med. 187:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poljak, L., L. Carlson, K. Cunningham, M.H. Kosco-Vilbois, and U. Siebenlist. 1999. Distinct activities of p52/NF-κB required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J. Immunol. 163:6581–6588. [PubMed] [Google Scholar]
- 5.Dietrich, W.F., J. Miller, R. Steen, M.A. Merchant, D. Damron-Boles, Z. Husain, R. Dredge, M.J. Daly, K.A. Ingalls, T.J. O'Connor, et al. 1996. A comprehensive genetic map of the mouse genome. Nature. 380:149–152. [DOI] [PubMed] [Google Scholar]
- 6.Loder, F., B. Mutschler, R.J. Ray, C.J. Paige, P. Sideras, R. Torres, M.C. Lamers, and R. Carsetti. 1999. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy, R.R., and K. Hayakawa. 2001. B cell development pathways. Annu. Rev. Immunol. 19:595–621. [DOI] [PubMed] [Google Scholar]
- 8.Paxian, S., S. Liptay, G. Adler, H. Hameister, and R.M. Schmid. 1999. Genomic organization and chromosomal mapping of mouse nuclear factor κB 2 (NFKB2). Immunogenetics. 49:743–750. [DOI] [PubMed] [Google Scholar]
- 9.Justice, M.J. 2000. Capitalizing on large-scale mouse mutagenesis screens. Nat. Rev. Genet. 1:109–115. [DOI] [PubMed] [Google Scholar]
- 10.Fagarasan, S., R. Shinkura, T. Kamata, F. Nogaki, K. Ikuta, K. Tashiro, and T. Honjo. 2000. Alymphoplasia (aly)-type nuclear factor κB-inducing kinase (NIK) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med. 191:1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyster, J.G., J.I. Healy, K. Kishihara, T.W. Mak, M.L. Thomas, and C.C. Goodnow. 1996. Regulation of B-lymphocyte negative and positive selection by tyrosine phosphatase CD45. Nature. 381:325–328. [DOI] [PubMed] [Google Scholar]
- 12.Lam, K.P., R. Kuhn, and K. Rajewsky. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 90:1073–1083. [DOI] [PubMed] [Google Scholar]
- 13.Fruman, D.A., S.B. Snapper, C.M. Yballe, L. Davidson, J.Y. Yu, F.W. Alt, and L.C. Cantley. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science. 283:393–397. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki, H., Y. Terauchi, M. Fujiwara, S. Aizawa, Y. Yazaki, T. Kadowaki, and S. Koyasu. 1999. Xid-like immunodeficiency in mice with disruption of the p85α subunit of phosphoinositide 3-kinase. Science. 283:390–392. [DOI] [PubMed] [Google Scholar]
- 15.Thomas, J.D., P. Sideras, C.I. Smith, I. Vorechovsky, V. Chapman, and W.E. Paul. 1993. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 261:355–358. [DOI] [PubMed] [Google Scholar]
- 16.Khan, W.N., F.W. Alt, R.M. Gerstein, B.A. Malynn, I. Larsson, G. Rathbun, L. Davidson, S. Muller, A.B. Kantor, L.A. Herzenberg, et al. 1995. Defective B cell development and function in Btk-deficient mice. Immunity. 3:283–299. [DOI] [PubMed] [Google Scholar]
- 17.Cariappa, A., T.J. Kim, and S. Pillai. 1999. Accelerated emigration of B lymphocytes in the Xid mouse. J. Immunol. 162:4417–4423. [PubMed] [Google Scholar]
- 18.Hashimoto, A., K. Takeda, M. Inaba, M. Sekimata, T. Kaisho, S. Ikehara, Y. Homma, S. Akira, and T. Kurosaki. 2000. Cutting edge: essential role of phospholipase C-γ 2 in B cell development and function. J. Immunol. 165:1738–1742. [DOI] [PubMed] [Google Scholar]
- 19.Wang, D., J. Feng, R. Wen, J.C. Marine, M.Y. Sangster, E. Parganas, A. Hoffmeyer, C.W. Jackson, J.L. Cleveland, P.J. Murray, and J.N. Ihle. 2000. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity. 13:25–35. [DOI] [PubMed] [Google Scholar]
- 20.Lentz, V.M., C.E. Hayes, and M.P. Cancro. 1998. Bcmd decreases the life span of B-2 but not B-1 cells in A/WySnJ mice. J. Immunol. 160:3743–3747. [PubMed] [Google Scholar]
- 21.Yan, M., J.R. Brady, B. Chan, W.P. Lee, B. Hsu, S. Harless, M. Cancro, I.S. Grewal, and V.M. Dixit. 2001. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr. Biol. 11:1547–1552. [DOI] [PubMed] [Google Scholar]
- 22.Thompson, J.S., S.A. Bixler, F. Qian, K. Vora, M.L. Scott, T.G. Cachero, C. Hession, P. Schneider, I.D. Sizing, C. Mullen, et al. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 293:2108–2111. [DOI] [PubMed] [Google Scholar]
- 23.Schiemann, B., J.L. Gommerman, K. Vora, T.G. Cachero, S. Shulga-Morskaya, M. Dobles, E. Frew, and M.L. Scott. 2001. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 293:2111–2114. [DOI] [PubMed] [Google Scholar]
- 24.Gross, J.A., S.R. Dillon, S. Mudri, J. Johnston, A. Littau, R. Roque, M. Rixon, O. Schou, K.P. Foley, H. Haugen, et al. 2001. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. Impaired B cell maturation in mice lacking BLyS. Immunity. 15:289–302. [DOI] [PubMed] [Google Scholar]
- 25.Yamada, T., T. Mitani, K. Yorita, D. Uchida, A. Matsushima, K. Iwamasa, S. Fujita, and M. Matsumoto. 2000. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-κB-inducing kinase. J. Immunol. 165:804–812. [DOI] [PubMed] [Google Scholar]
- 26.Rayet, B., and C. Gelinas. 1999. Aberrant rel/nfκb genes and activity in human cancer. Oncogene. 18:6938–6947. [DOI] [PubMed] [Google Scholar]
- 27.Goodnow, C.C., J.G. Cyster, S.B. Hartley, S.E. Bell, M.P. Cooke, J.I. Healy, S. Akkaraju, J.C. Rathmell, S.L. Pogue, and K.P. Shokat. 1995. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 59:279–368. [DOI] [PubMed] [Google Scholar]