

Abstract
High risk human papillomaviruses (HPVs) are central to the development of cervical cancer and the deregulated expression of high risk HPV oncogenes is a critical event in this process. Here, we find that the cell protein nucleolin binds in a sequence-specific manner to the HPV18 enhancer. The DNA binding activity of nucleolin is primarily S phase specific, much like the transcription of the E6 and E7 oncoproteins of HPV18 in cervical cancer cells. Antisense inactivation of nucleolin blocks E6 and E7 oncogene transcription and selectively decreases HPV18+ cervical cancer cell growth. Furthermore, nucleolin controls the chromatin structure of the HPV18 enhancer. In contrast, HPV16 oncogene transcription and proliferation rates of HPV16+ SiHa cervical cancer cells are independent of nucleolin activity. Moreover, nucleolin expression is altered in HPV18+ precancerous and cancerous tissue from the cervix uteri. Whereas nucleolin was homogeneously distributed in the nuclei of normal epithelial cells, it showed a speckled nuclear phenotype in HPV18+ carcinomas. Thus, the host cell protein nucleolin is directly linked to HPV18-induced cervical carcinogenesis.
Keywords: tumor virus, carcinogenesis, cell cycle, proliferation, chromatin
Introduction
High risk human papillomaviruses (HPVs)*play a central etiologic role in the development of cervical cancer (1), which represents the second most common malignancy in women with an annual incidence of ∼500,000 new cases (2). A recent report demonstrated that 99.7% of all cervical cancers contain high risk HPV types (3). Of the oncogenic HPV types, HPV16 and HPV18 are found with the highest frequency in invasive cervical carcinomas in most populations (3). In adenocarcinomas, however, HPV18 is the most prevalent type (4). Accumulating clinical and experimental research suggests that HPV18 infection is associated with a more aggressive form of cervical neoplasia than HPV16 infection (5–8) and it has been shown by a cell transformation assay that its upstream regulatory region (URR) has a 10–50-fold higher transforming activity than HPV16 (9). Papillomaviruses initially infect the basal cell layer of the cervical squamous epithelium and persist as a latent infection (for review see reference 10). In high grade squamous intraepithelial lesions (HSILs), squamous cell carcinomas and adenocarcinomas, the HPV genomes are usually integrated into the host cell genome and the E6 and E7 oncogenes are highly expressed (11, 12). Most recently it was reported that within the host cell genome no preferential integration sites for HPV18 and HPV16 genomes could be identified (13). These results indicate that target-directed insertional mutagenesis or the activation of nearby oncogenes apparently plays a minor role for the development of HPV-associated cervical cancers. In contrast, the E6 and E7 oncoproteins exert their carcinogenic potential by inactivation of the tumor suppressors pRB and p53 (14–16). The papillomavirus oncogenes cause progressive squamous intraepithelial neoplasias (17). Thus, deregulated high risk HPV oncogene expression is a critical event in papillomavirus-associated carcinoma development. Therefore, it is essential to work out the mechanisms of HPV oncogene expression to understand cervical cancer development at a molecular level. The HPV18 enhancer (18) has been studied extensively by DNase I footprinting, site directed mutagenesis, and transient reporter gene assays, and it was shown that in cervical carcinoma cells E6/E7 oncogene expression is dependent not on viral, but on cellular transcription regulators (19–26). It is, however, not completely understood how HPV18 oncogene expression is controlled in the process of human squamous cell carcinoma and adenocarcinoma development. Here, we report a previously unrecognized involvement of the cellular protein nucleolin in HPV18-associated cervical carcinogenesis. Nucleolin is a phosphoprotein whose synthesis positively correlates with cell division rates (27). Nucleolin is a multifunctional protein and is a downstream target of several signal transduction pathways (for review see references 28 and 29). For example, fibroblast growth factor 2 stimulates the activity of CK2 toward nucleolin (30), nucleolin phosphorylation is controlled by androgens (31), and nucleolin expression is activated by IL-2 in T cells (32). Here, we show that nucleolin is a key activator of HPV18 oncogene transcription involved in chromatin structure regulation and we demonstrate that nucleolin is not only expressed in HPV18+ squamous cell carcinomas and adenocarcinomas of the cervix uteri, but also shows an abnormal intranuclear localization compared with normal cervical epithelial cells. Thus, we have identified nucleolin as a cellular protein with oncogenic potential.
Materials and Methods
Preparation of Nuclear Extracts, Protein Purification, and Sequencing.
HeLa cells were purchased from the 4C Biotech and nuclear extracts were prepared as previously described (33). Protein purification on DNA affinity columns was performed according to a published protocol (34). In brief, nuclear extracts were loaded onto heparin-Sepharose in a buffer containing 20 mM Tris, pH 8.0, 60 mM NaCl, 1 mM EDTA, 1 mM 1,4-dithio-DL-threitol, 5% glycerol, and protease inhibitors. The fraction eluted from the column with loading buffer containing 500 mM NaCl was dialyzed and applied to a DNA affinity column, which was prepared by coupling the double stranded enhancer fragment corresponding to nucleotide numbers 7634–7671 (35), termed here RP3, to CNBr-activated Sepharose. Proteins eluted from the RP3 oligonucleotide DNA affinity column with 500 mM NaCl were analyzed by SDS-PAGE. The final protein preparation was apparently homogeneous, as judged by 2D electrophoresis. Internal peptides were generated from the corresponding gel band by in situ digestion with trypsin (36) and peptides were separated on a reverse phase column using a SMART System (Amersham Biosciences). Alternatively, the protein was transferred to a high retention polyvinylidene difluoride membrane (Bio-Rad Laboratories) using 10 mM Caps buffer, pH 11.0, containing 10% methanol (v/v), 0.07% SDS, for 2.5 h at 1 mA/cm2 and the blotted protein and selected fractions from the HPLC run of internal peptides were subjected to N-terminal Edman microsequencing using the Procise® sequencer (Applied Biosystems).
Generation of Polyclonal Nucleolin Peptide Antibodies.
A conjugate of the NH2-terminal peptide of nucleolin, VKLAKAGKNQGDPKKM, coupled to KLH was used to generate a polyclonal rabbit Ab specific for nucleolin. The Ab was affinity purified on a peptide column according to standard protocols. Affinity-purified Ab specific for nucleolin was used throughout this study.
Cell Culture and Synchronization.
Hela cells, HeLa-fibroblast hybrid cell line 444 (37), SW756 cells (38), SiHa cells, and HaCaT and SiHa × HaCaT hybrids (39) were maintained in DME supplemented with 10% FCS. Detailed cell synchronization conditions and the necessary controls have been described (33). In brief, G1 phase cells were prepared by 20 μM lovastatin treatment for 24 h, S phase cells by 3 μM aphidicolin treatment for 24 h followed by a 2-h release in fresh medium, and G2 phase cells by 3 μM aphidicolin treatment for 24 h followed by a 6-h release in fresh medium. Mitotic cells were prepared by 10 μg/ml nocodazole treatment for 18 h and thereafter the shake-off cells were collected. G0 phase cells were prepared by a 48-h cultivation in serum-free medium. Synchronization efficiencies were monitored by FACS® analysis. In addition, S phase synchronization was controlled by the detection of histone H3 mRNA, and G2 phase synchronization was monitored by immunoblotting with a cyclin B1 Ab. To assess G0 synchronization, the expression of Ki67-Ag was determined by immunoblot analysis.
Antisense Inhibition of Nucleolin Expression and Proliferation Assays.
The nucleolin phosphorothioate-antisense oligonucleotide 5′-TCACCATGATGGCGGCGG-3′ is complementary to the 5′ end of the mRNA of human nucleolin encompassing the translation initiation region. For antisense inhibition experiments, phosphorothioate-modified oligonucleotides were added in concentrations indicated in the figure legends. After 60 h, the cells were harvested and used for the preparation of nuclei, nuclear extracts, and RNA. Proliferation assays were performed using the bromodeoxuridine (BrdUrd) proliferation ELISA kit (Roche Diagnostics). Nucleolin antisense inhibition in proliferation assays was performed using 1 μM phosphorothioate-modified antisense and sense oligonucleotides, and oligonucleotides were applied to growing cells using DOTAP reagent (Roche Diagnostics) to enhance transfection efficiency. After an initial incubation for 15 h, the culture medium was changed, and the cells were incubated for an additional 48 h. BrdUrd was added for 9 h and the BrdUrd incorporation was determined as described in the manufacturer's protocol.
Electrophoretic Mobility Shift Assay (EMSA), Immunoshift, and Immunoblot Analysis.
The conditions for DNA binding and EMSA have been described (33). For immunoshift assays, nuclear protein extracts were preincubated with the nucleolin Ab for 30 min before processing the reactions for EMSA. As specificity control, the Ab specific for nucleolin was presaturated with the immunizing nucleolin peptide for 15 min at room temperature.
The glutathione S-transferase (GST)–nucleolin fusion protein was generated by cloning the cDNA fragment corresponding to amino acid residues 289–709 of the nucleolin cDNA (40) in frame with GST cDNA into vector pGEX2T. The fusion protein was produced in Escherichia coli and purified by affinity chromatography using glutathione-Sepharose (Amersham Biosciences) as described in the manufacturer's protocol. To detect DNA binding, 50 ng purified GST–nucleolin protein or GST protein was incubated with 50 ng poly (dIdC)(dIdC) and labeled, double stranded RP3 or AP1 oligonucleotides for 15 min at room temperature, and analyzed by EMSA.
Immunoblot analysis was performed according to standard procedures. The following are antibodies and their dilutions: Ab specific for nucleolin, 1:10,000; Ab specific for Sp1 (PEP2; Santa Cruz Biotechnology, Inc.), 1:2,000; mAb specific for proliferating cell nuclear antigen (PCNA; Santa Cruz Biotechnology, Inc.), 1:2,000; Ab specific for Cdk4 (provided by M. Pagano, New York University School of Medicine, New York, NY), 1:2,000; and mAb specific for β actin (Sigma-Aldrich), 1:2,000.
Preparation of RNA and Northern Blot Analysis.
The preparation of total cellular RNA and Northern blot analysis was performed according to previously published procedures (41, 42) with minor modifications. For the preparation of nuclear RNA, the cells were first lysed with a buffer containing 140 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl, pH 8.0, 0.5% NP-40, 1 mM 1,4-dithio-DL-threitol. Subsequently, nuclei were collected by low speed centrifugation and RNA was isolated according to the protocol described above. For Northern blot analysis of HPV18 mRNA, total or nuclear RNA (5 μg) was fractionated on 1.2% agarose-formaldehyde gels, transferred to charged nylon membranes (Zeta-Probe; Bio-Rad Laboratories), and hybridized overnight at 47°C in the presence of 50% formamide. HPV18 DNA probes were radioactively labeled to a specific activity of 107 cpm/50 ng DNA using a nick translation kit (Roche Diagnostics). For the detection of HPV16 mRNA in SiHa and SiHa × HaCaT hybrid cells by Northern hybridization, we used a HPV16 unit length DNA (provided by M. Dürst, University of Jena, Jena, Germany; reference 43). Here, we used 0.5 μg poly A–selected RNA per sample and RNA was separated on a 1% agarose gel in the presence of ethidium bromide under nondenaturing conditions (44) and transferred to GeneScreen Plus membranes (NEN Life Science Products). The filter was hybridized under stringent conditions with specific probes labeled with [32P]dCTP by random priming (45).
Transient Transfections and Chloramphenicol Acetyltransferase Reporter Gene (CAT) Assays.
We have reported the creation of an HPV16+ cervical carcinoma cell line (SiHa), where contiguous HPV18 URR fused to the CAT is stably integrated, and HaCaT hybrid cells thereof (39). To determine the activity of the integrated HPV18 URR-CAT construct in SiHa and SiHa × HaCaT hybrid cells we used a CAT ELISA kit (Roche Diagnostics). The results obtained by CAT ELISA were normalized to protein concentration according to the manufacturer's specifications. For transient transfections, a cytomegalovirus promoter-driven nucleolin expression construct was used (46). The DNA was purified using a plasmid purification system (QIAGEN) and cells were transiently transfected according to standard procedures. HPV18 URR-CAT activity in transiently transfected cells was measured by a CAT-ELISA kit (Roche Diagnostics).
Detection of a DNase I Hypersensitive (HS) Site in the HPV18 Enhancer.
For the detection of the DNase I HS site in the HPV18 enhancer chromatin, we followed our published procedure (47). In brief, cells were lysed with 0.6% NP-40 and the nuclei were washed twice with isotonic Tris buffer and resuspended in DNase I digestion buffer (10 mM Tris-HCl, pH 7.5, 30 mM NaCl, 3 mM MgCl2). The nuclear DNA concentration was determined and adjusted to 1 mg/ml, and then nuclei were digested with DNase I (Roche Diagnostics) for 10 min at 37°C. The concentrations of DNase I are indicated in the figure legends. The reactions were terminated by the addition of an equal volume of a stop mixture containing 2% SDS and 40 mM EDTA. After proteinase K digestion (50 μg/ml for 2 h at 37°C), the DNA was phenol extracted, ethanol precipitated, and dissolved in 1× Tris-EDTA buffer. DNA was then digested with EcoR I (30 U EcoR I/5 μg DNA) for 5 h at 37°C, treated with 10 μg/ml RNase A, and size fractionated in 0.8% agarose gels. After the transfer to Zeta-Probe membranes (Bio-Rad Laboratories), the filters were hybridized overnight at 47°C in the presence of 50% formamide according to the manufacturer's specifications. A DNA fragment corresponding to the open reading frames of the E6-E7 oncogenes of HPV18 was used as a probe for indirect end labeling of the HPV18-containing EcoR I fragment on the Southern blot (47). The DNA probe was labeled to a specific activity of 107 cpm/50 ng DNA using a nick translation kit (Roche Diagnostics) and [32P]-labeled dNTPs.
Immunohistochemistry and Detection of HPV18 by PCR.
Paraffin sections of human cervix specimens were immunostained with the Ab specific for nucleolin after microwave treatment. For immunohistochemistry, the polyclonal Ab specific for nucleolin was used at a dilution of 1:200. As second Ab, a swine anti–rabbit IgG was used at a 1:300 dilution. Specimens were then incubated with an avidin biotin complex at a 1:200 dilution and then developed by an incubation with 3,3′ diaminobenzidine tetrachloride for 3 min and stained with hematoxylin for 30 s. For HPV detection, DNA was prepared from formalin-fixed, paraffin-embedded tissue by standard procedures. HPV DNA was detected by GP5+/6+ PCR enzyme immunoassay and typing was performed by reverse line blot hybridization according to our published protocol (48).
Results
Nucleolin is an S Phase–specific HPV18 Enhancer Binding Protein.
To gain insight into the control of HPV18 oncogene transcription, we analyzed whether this process would be subject to cell cycle–dependent regulation. 444 cells were biochemically synchronized according to cell cycle stage and HPV18 oncogene transcription was measured by Northern blot analysis using nuclear RNA. Cell cycle synchronization efficiencies were controlled as previously described (33). E6/E7 oncogene transcription was just detectable in G1, G2, and M phase, however, HPV18 oncogenes were strongly transcribed in S phase (Fig. 1 A, top). To monitor S phase synchronization, histone H3 mRNA was detected (Fig. 1 A, bottom). Similar results were obtained using cells synchronized by serum starvation followed by serum induction (not depicted). We have previously obtained evidence for a cell cycle–regulated protein that binds to the HPV18 enhancer in S phase of the cell cycle (33). Here, this HPV18 enhancer binding protein was purified by DNA affinity chromatography with the use of the HPV18 enhancer fragment corresponding to nucleotide numbers 7634–7671 (35) and termed RP3 (33). A protein fraction eluted from the RP3 oligonucleotide DNA affinity column with 500 mM sodium chloride was analyzed by SDS-PAGE and 2D gel electrophoresis, and the amino acid sequence of the predominant polypeptide was determined by protein microsequencing. The first 20 NH2-terminal amino acids as well as 5 internal peptides, amino acids 295–309, 347–361, 410–419, 486–500, and 577–591, corresponded to predicted nucleolin residues (40). Next, we analyzed the binding of nucleolin to the HPV18 enhancer. Nuclear extracts of 444 cells synchronized in S phase were analyzed by EMSA (Fig. 1 B) using the labeled RP3 oligonucleotide. A retarded DNA–protein complex was detected, and this complex formation was abolished by the addition of an Ab specific for nucleolin (Fig. 1 B, lane 2). After presaturating the Ab with the immunizing nucleolin peptide, a reappearance of retarded nucleolin–DNA complex was observed (Fig. 1 B, lanes 3 and 4).
Figure 1.




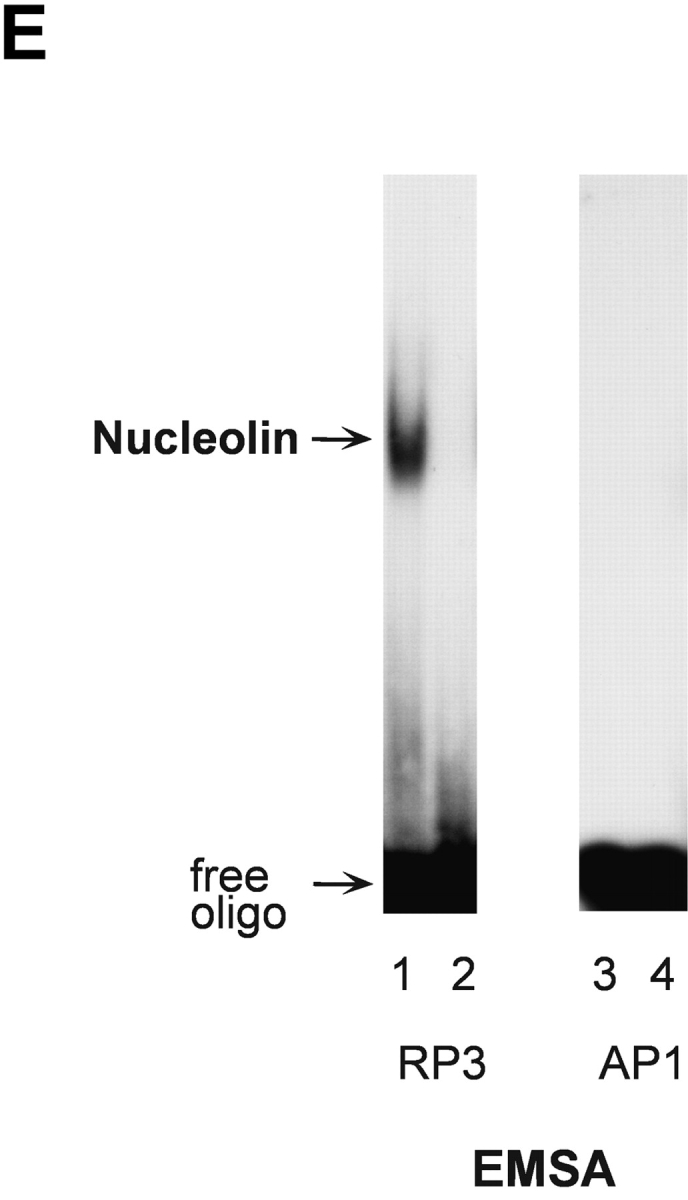
S phase– and sequence-specific binding of nucleolin to the HPV18 enhancer. (A) HPV18 oncogene transcription was examined by Northern blot analysis using nuclear RNA and an E6-E7 cDNA probe (top). S phase synchronization was controlled by hybridization with a histone H3 probe (bottom) and RNA concentrations were verified by hybridization with a β actin probe (middle). The differentially spliced HPV18 early gene transcripts are indicated by arrows (reference 62). (B) Nucleolin binds to the HPV18 enhancer. The labeled HPV18 enhancer oligonucleotide RP3 and a nuclear extract from S phase synchronized cells were used in EMSA. Lane 1, S phase nuclear extract; lane 2, nucleolin Ab was added to the DNA binding reaction; lanes 3 and 4, nucleolin Ab was presaturated with increasing amounts of the immunizing peptide. Electrophoresis was performed for 90 min at 11 V/cm. (C) Cell cycle regulation of nucleolin DNA binding activity. EMSA with nuclear extracts from biochemically synchronized 444 cells and the labeled HPV18 enhancer oligonucleotide RP3. Lane 1, G0 phase; lane 2, G1 phase; lane 3, S phase; lane 4, G2 phase. Electrophoresis was performed for 90 min at 11 V/cm. (D) Sequence specificity of nucleolin binding to the HPV18 enhancer. For EMSA, a partially purified nucleolin protein fraction and labeled RP3 were used. Partially purified nucleolin (all lanes). The binding complexes were competed by the addition of none (lane 1), 200 ng (lane 2), and 500 ng of RP3 (lane 3), and 200 ng (lane 4) and 500 ng (lane 5) of the RP3 mutant. Electrophoresis was performed for 50 min at 11 V/cm. (E) Specific interaction of nucleolin–GST protein with the HPV18 enhancer. Lanes 1 and 3, affinity-purified nucleolin–GST protein; lanes 2 and 4, GST protein; lanes 1 and 2, EMSA with the labeled RP3 HPV18 enhancer oligonucleotide; lanes 3 and 4, EMSA with an unrelated AP1 recognition site. Electrophoresis was performed for 90 min at 11 V/cm.
Next, we investigated the DNA binding activity of nucleolin during the cell cycle. EMSA experiments were performed with nuclear extracts of 444 cells synchronized to the G0, G1, S, and G2 phase of the cell cycle using the labeled HPV18 enhancer fragment RP3 to detect nucleolin DNA binding. Cell cycle synchronization efficiencies were controlled as previously described (33). Fig. 1 C shows an S phase predominance of nucleolin DNA binding (lane 3). Similar results were obtained using cells synchronized by serum starvation followed by serum induction (not depicted). Furthermore, we could demonstrate the sequence specificity of nucleolin binding to the HPV18 enhancer. A deletion analysis of RP3 revealed that for nucleolin binding the sequence motif 5′-TTGCTTGCATAA is required (33). It was demonstrated by in vivo footprinting that the G residues of this motif are in contact with protein on both the sense and antisense strands in cervical cancer cells (49). To test whether these G residues are required for nucleolin binding, we performed EMSA competition experiments using a RP3 mutant where the G residues of the 5′-TTGCTTGCATAA motif were replaced by A residues in the sense and antisense strands (Fig. 1 D). It was observed that RP3, but not the mutant oligonucleotide, efficiently competed with nucleolin binding to labeled RP3. It is worthy to note that in this experiment partially purified nucleolin was used, which explains the formation of a very intense retarded RP3–protein complex. To additionally demonstrate bona fide nucleolin interaction with the HPV18 enhancer, we used a bacterially expressed, affinity-purified nucleolin–GST fusion protein. This nucleolin–GST protein, but not GST protein itself, bound to the enhancer fragment RP3 (Fig. 1 E, lanes 1 and 2) and not to an unrelated binding site of the transcription factor AP1 (lanes 3 and 4). Thus, nucleolin is a sequence-specific and cell cycle–regulated HPV18 enhancer binding protein.
Activation of HPV18 Oncogene Transcription by Nucleolin.
To directly determine the role of nucleolin in HPV18 oncogene transcription, we used an antisense inhibition approach. We identified an effective antisense oligonucleotide, as monitored by means of immunofluorescence staining (not depicted) and immunoblotting. 444 cells treated by the nucleolin antisense oligonucleotide showed a strong and dose-dependent reduction of concentration of the nucleolin protein in the nuclear extracts (Fig. 2 A, top, lanes 2–4), whereas the nucleolin sense oligonucleotide had no effect (lanes 5–7). 444 untreated cells were used as a reference value (Fig. 2 A, lane 1). It has been suggested that nucleolin could be necessary for the global cellular protein synthesis and the synthesis of ribosomal RNA (27). However, the role of nucleolin in these processes is controversial (50). Therefore, the levels of several ubiquitous and cell cycle regulatory proteins were examined after antisense inhibition of nucleolin expression. We were unable to detect a significant effect on Sp1, Cdk4, and PCNA protein levels (Fig. 2 A, bottom panels). HPV18 oncogene transcription in 444 cells was then analyzed by Northern blotting after antisense inhibition of nucleolin expression (Fig. 2 B, top) with RNA from untreated cells as a reference value (lane 1). The treatment of 444 cells with increasing amounts of the nucleolin antisense oligonucleotide inhibited HPV18 oncogene transcription in a concentration-dependent fashion (Fig. 2 B, lanes 2–4). Transcription of the β actin gene was monitored as control (Fig. 2 B, bottom). The sense oligonucleotide had no effect (Fig. 2 B, top, lanes 5–7). These results demonstrate that nucleolin activates HPV18 oncogene transcription.
Figure 2.


Nucleolin controls HPV18 oncogene transcription. (A) Antisense inhibition of nucleolin expression. 444 cells were treated with phosphorothioate-modified antisense or sense oligonucleotides and then nuclear extracts were prepared. Detection of nucleolin by immunoblotting (top). Lane 1, mock control; lanes 2–4, antisense oligonucleotide: 1 μM, 5 μM, and 9 μM; lanes 5–7, sense oligonucleotide: 1 μM, 5 μM, and 9 μM. The same extracts were analyzed by immunoblotting with an Ab specific for Sp1, Cdk4, and PCNA (bottom). On the side of each immunoblot the positions of detected proteins are indicated by arrows. (B) Nucleolin controls HPV18 oncogene expression. RNA was isolated from aliquots of antisense- or sense-treated cells and analyzed by Northern blotting using an E6-E7 cDNA probe. Lane 1, untreated 444 cells; lanes 2–4, antisense oligonucleotide: 1 μM, 5 μM, and 9 μM; lanes 5–7, sense oligonucleotide: 1 μM, 5 μM, and 9 μM. The differentially spliced HPV18 mRNAs (reference 62) are indicated by arrows. The β actin control is shown at the bottom.
Nucleolin Regulates the Chromatin Structure of the HPV18 Enhancer.
It has previously been shown that nucleolin induces chromatin decondensation in vitro (51). In light of our data, we considered it possible that nucleolin is involved in regulating the chromatin structure of the HPV18 enhancer in vivo. Moreover, a prominent DNase I HS site, indicative of the open chromatin structure, found in transcriptionally active chromatin (52), has been identified within the enhancer of HPV18 in cervical cancer cells (47). When HPV18 enhancer chromatin structure was analyzed as previously described (47) in 444 cells synchronized into an S phase population, this strong DNase I HS site was detectable (Fig. 3 A, lanes 3 and 4), whereas it was just detected in G1 phase (not depicted). Therefore, experiments in 444 cells were performed after antisense inhibition of nucleolin expression. It became evident that the DNase I HS site in the HPV18 enhancer region was not detected in nucleolin antisense oligonucleotide–treated cells (Fig. 3 B, lanes 4–6) compared to sense oligonucleotide–treated cells (Fig. 3 B, lanes 1–3). These results show that nucleolin is involved in opening the chromatin structure of the HPV18 enhancer and suggest that through this mechanism, nucleolin functions as a regulator of HPV18 oncogene transcription.
Figure 3.


Regulation of chromatin structure of the HPV18 enhancer by nucleolin. (A) HPV18 enhancer DNase I HS site mapping was performed as previously described (reference 47). Nuclei from S phase cells were digested with DNase I: 0.25 U, 0.5 U, 1.75 U, and 2.5 U (lanes 1–4). Mock-digested nuclei are not shown. Purified nuclear DNA was then digested with EcoR I and analyzed by Southern blotting using an E6-E7 cDNA probe for indirect end labeling of the 3′ end. The position of a DNase I HS site in the HPV18 enhancer is indicated by an arrow. (B) 444 cells were treated with 9 μM nucleolin sense oligonucleotide and isolated nuclei were digested with DNase I: 0.25 U, 1 U, and 1.75 U (lanes 1–3). In parallel, 444 cells were treated with 9 μM antisense oligonucleotide and isolated nuclei were digested with DNase I: 0.25 U, 1 U, and 1.75 U (lanes 4–6). DNA was purified and analyzed by Southern blotting as described in A. The mock-digested controls are not shown. The HPV18 enhancer DNase I HS site is indicated by an arrow.
Nucleolin as Selective Activator of HPV18 Oncogene Transcription.
To further investigate the role of nucleolin for HPV18 oncogene transcription, we took advantage of a hybrid cell system consisting of the following components: (a) HPV16+ SiHa cells where the contiguous HPV18 URR fused to a CAT is stably integrated. In these cells the HPV18 URR-driven CAT reporter gene is constitutively expressed (Fig. 4 A, lane 1). We refer to these cells as (HPV18 URR-CAT)-SiHa cells throughout the text. (b) A SiHa × HaCaT cell hybrid where the integrated HPV18 URR-CAT reporter gene is not significantly expressed (Fig. 4 A, lane 2). We refer to these cells as (HPV18 URR-CAT)-SiHa × HaCaT hybrids throughout the text. Note that HaCaT cells are normal nontumorigenic human keratinocytes (53). When the DNA binding activity of nucleolin was investigated we found that in (HPV18 URR-CAT)-SiHa × HaCaT hybrids it was just detectable (Fig. 4 B, lane 2) whereas in (HPV18 URR-CAT)-SiHa cells significant amounts were detected (Fig. 4 B, lane 1). At the level of DNA sequence organization the enhancers of the most frequent high risk HPVs, HPV18 and HPV16, are strikingly different (18) and there is no sequence similarity with the HPV18 enhancer fragment RP3 containing the nucleolin binding site (5′-TTGCTTGCATAA) in the HPV16 enhancer. To investigate whether nucleolin controls the activity of the HPV16 URR, we analyzed the expression of HPV16 oncogenes in (HPV18 URR-CAT)-SiHa and (HPV18 URR-CAT)-SiHa × HaCaT hybrid cells by Northern hybridization (Fig. 4 C). The levels of the four HPV16 early gene transcripts (54) in both cell types are similar (Fig. 4 C, lanes 1 and 3), demonstrating that HPV16 oncogene transcription is not affected by nucleolin. To directly test the role of nucleolin in HPV18 URR regulation, we transfected (HPV18 URR-CAT)-SiHa × HaCaT hybrid cells with a nucleolin expression vector (46) and detected a significant induction of HPV18 URR-driven CAT reporter gene expression (Fig. 4 D, lane 1), demonstrating a direct involvement of nucleolin in the regulation of HPV18 oncogene transcription. Thus, in this hybrid cell system nucleolin selectively controls HPV18 oncogene expression.
Figure 4.
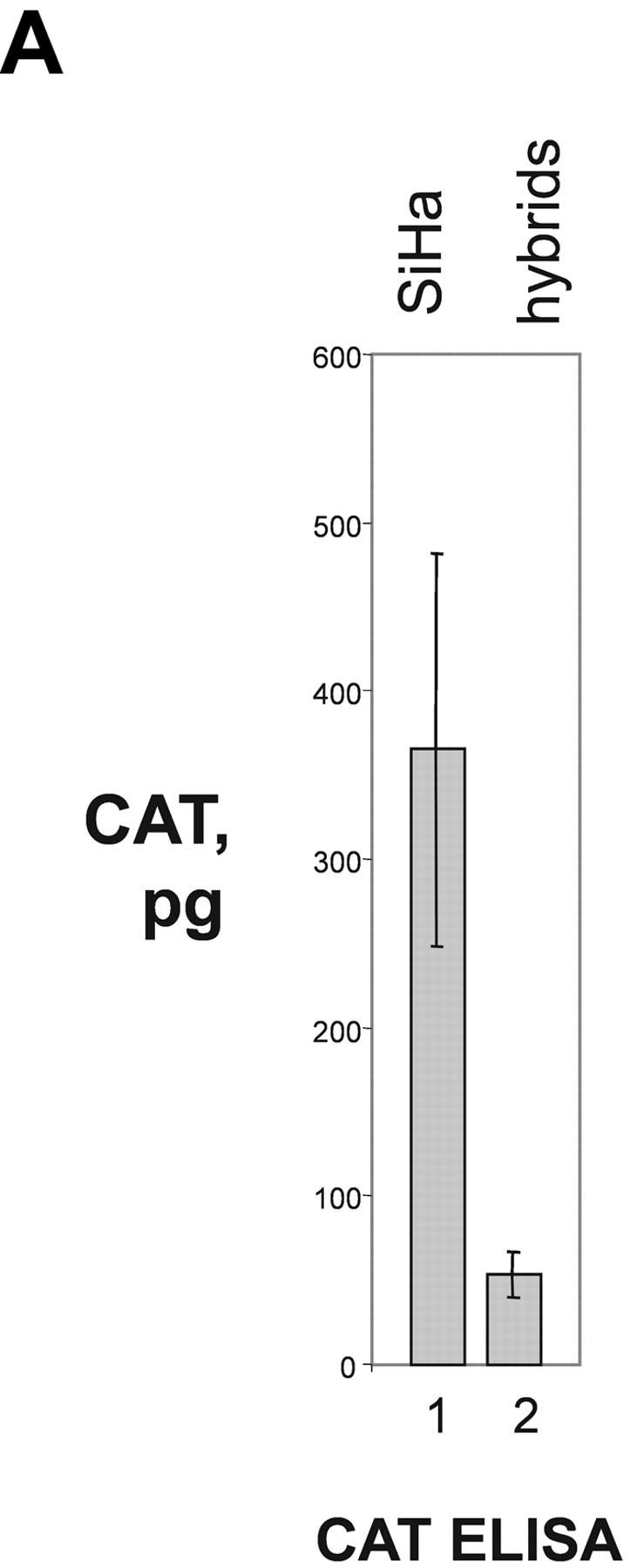
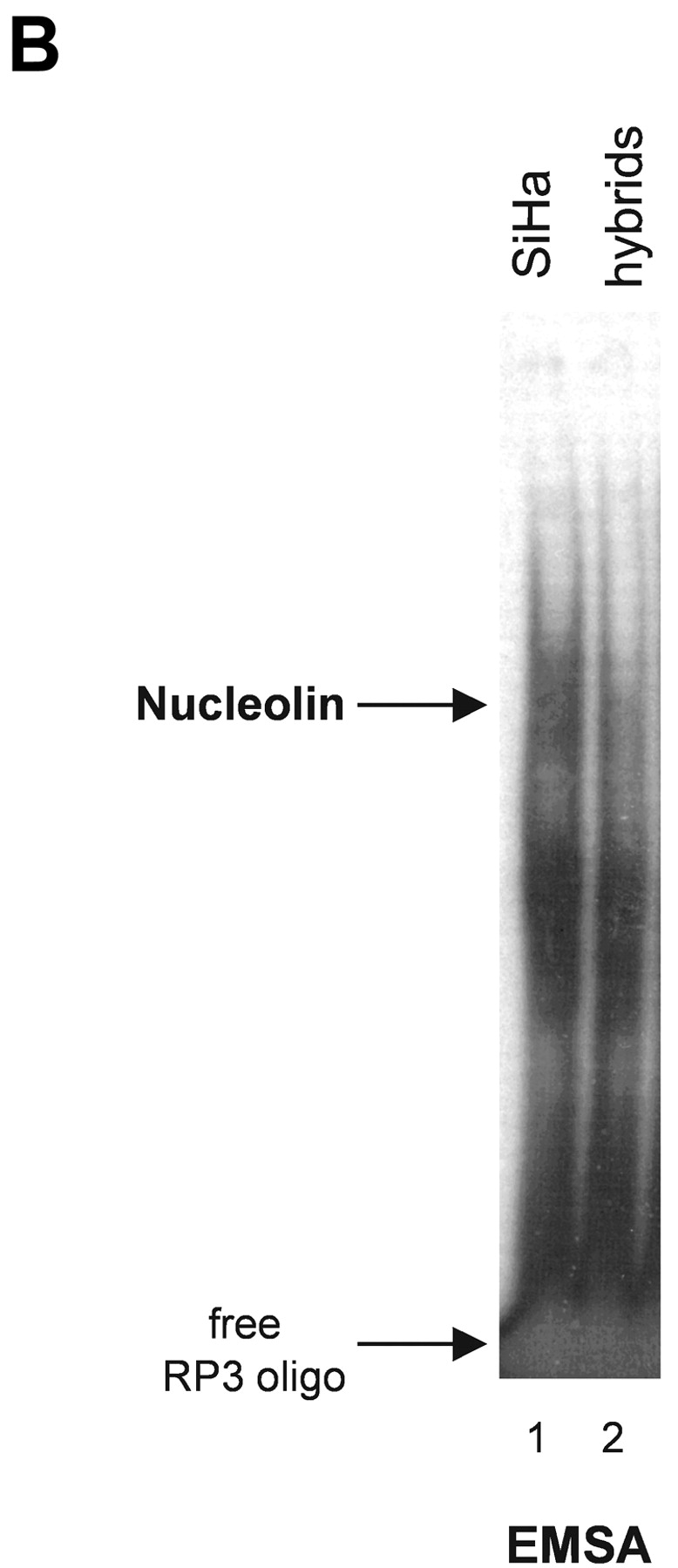

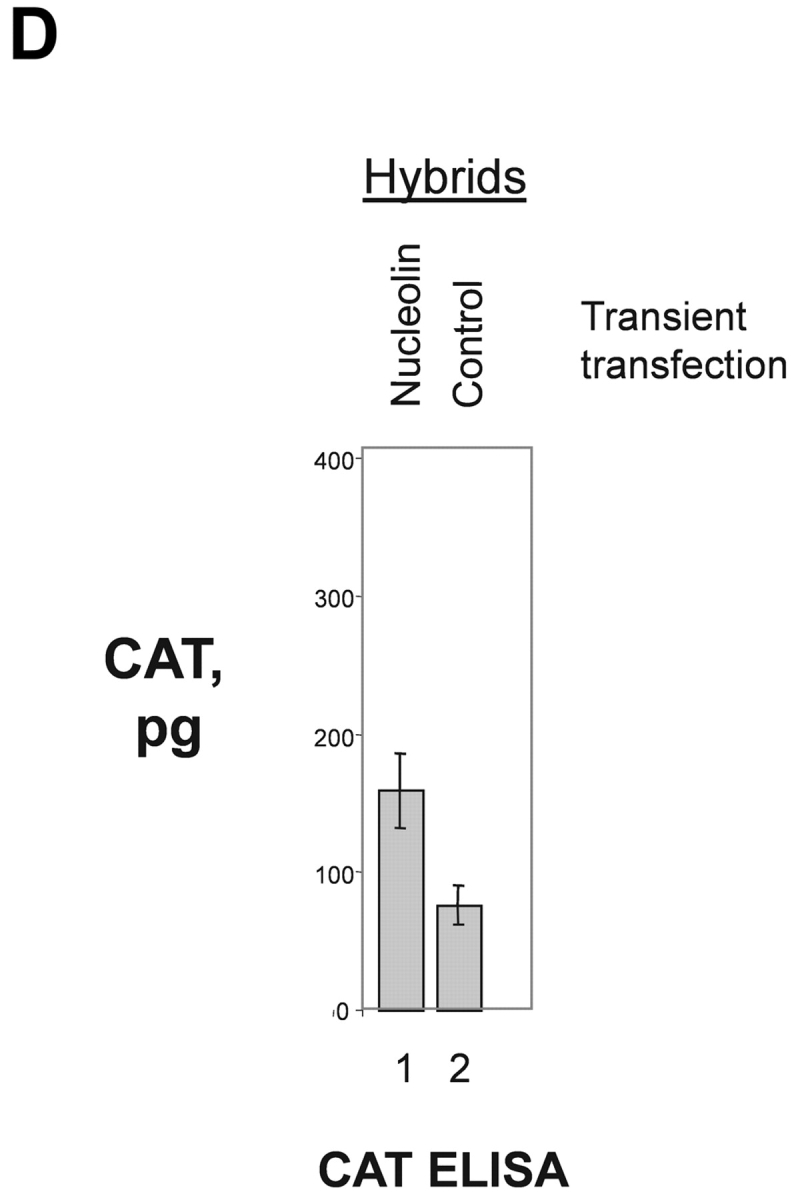
Nucleolin as selective activator of HPV18 oncogene transcription. (A) CAT reporter gene assay. HPV16+ SiHa cervical cancer cells containing an integrated contiguous HPV18 URR fused to a CAT reporter gene (HPV18 URR-CAT)-SiHa (lane 1; reference 39). SiHa × HaCaT hybrid cells containing the HPV18 URR-CAT reporter gene (lane 2). HaCaT cells are normal human keratinocytes (reference 53). CAT reporter gene activity was measured by CAT ELISA. The results were normalized to protein concentration. (B) EMSA with nuclear extracts from (HPV18 URR-CAT)-SiHa cells (lane 1) and SiHa × HaCaT hybrid cells where the HPV18 URR-CAT gene is not significantly expressed (lane 2). The position of the retarded RP3–nucleolin complex is indicated by an arrow. Conditions for EMSA are as described in Fig. 1. (C) HPV16 oncogene transcription in (HPV18 URR-CAT)-SiHa cells (lane 1) and SiHa × HaCaT hybrid cells (lane 3). HaCaT cells were used as a HPV16− control (lane 2). For Northern analysis, 0.5 μg of poly A–selected RNA was used per lane. Filters were consecutively hybridized with probes specific for HPV16 and β actin. The sizes of the corresponding mRNAs are indicated. (D) CAT reporter gene assay. SiHa × HaCaT hybrid cells containing the integrated HPV18 URR-CAT construct were transiently transfected with a nucleolin cDNA expression construct (lane 1; reference 46) or the empty vector (lane 2). CAT activity was measured by CAT ELISA. The results were normalized to protein concentration.
Nucleolin Regulates Proliferation of HPV18+ Cervical Cancer Cells.
It has been reported that the expression of HPV18 oncogenes leads to enhanced proliferation of HPV18+ cervical cancer cells (55). Therefore, we determined whether nucleolin is required for the proliferation of these cells. Three HPV18+ cervical carcinoma cell lines and the HPV16+ carcinoma cell line SiHa were transiently treated with nucleolin sense or antisense oligonucleotide, followed by BrdUrd incorporation measurement. Nucleolin antisense inhibition did not significantly affect the proliferation of HPV16+ SiHa cervical cancer cells (Fig. 5 A, lane SiHa). In contrast, the proliferation rates of the three different HPV18+ cervical cancer cell lines HeLa, 444, and SW756 were strongly inhibited by antisense inactivation of nucleolin (Fig. 5 A). HeLa cells were originally derived from an adenocarcinoma and SW756 cells from a squamous cell carcinoma of the cervix (38, 56). Immunoblotting revealed that nucleolin protein levels were reduced to the same extent in HeLa and SiHa cells by treatment with antisense oligonucleotide (Fig. 5 B top, lanes 2 and 5), whereas the nucleolin sense oligonucleotide had no effect (lanes 3 and 6, respectively). Beta-actin expression was not influenced by the antisense nor the sense oligonucleotide (Fig. 5 B, bottom). Therefore, nucleolin selectively controls the proliferation of HPV18+ cancer cells in vitro.
Figure 5.


Nucleolin controls proliferation of HPV18+ cervical cancer cells. (A) Proliferation rates of SiHa, HeLa, 444, and SW756 cells were measured by means of BrdUrd incorporation after treatment with phosphorothioate-modified nucleolin antisense or sense oligonucleotides, and are displayed relative to mock-treated samples. (B) Nuclear extracts of HeLa and SiHa cells were analyzed by immunoblotting with an Ab specific for nucleolin after treatment with nucleolin sense and antisense oligonucleotides. Lanes 1–3, HeLa cells; lanes 4–6, SiHa cells; lanes 1 and 3, mock control; lanes 2 and 5, nucleolin antisense oligonucleotide; lanes 3 and 6, nucleolin sense oligonucleotide. The same extracts were analyzed by immunoblotting with an Ab specific for β actin (bottom).
Nucleolin Expression in HPV18+ Cervical Neoplasias.
The expression pattern of nucleolin protein in human tissues has not been analyzed so far. Thus, nucleolin expression in four different normal (HPV18-free) and seven HPV18+ cancerous and precancerous tissues of the cervix uteri was examined by immunohistochemistry (Fig. 6) . Nuclear staining was seen in both squamous epithelial and endocervical glandular cells (Fig. 6, A and C). In squamous epithelium of the cervix, nucleolin expression is found throughout except for certain superficial cells within the stratum corneum (Fig. 6 A). In the lower basal and parabasal layers of the squamous epithelium and in the more differentiated cell layers, diffuse nuclear staining was evident (Fig. 6 A). Also, glandular epithelial cells showed diffuse nuclear staining with often more prominent staining of dot-like structures (Fig. 6 C). To evaluate the relevance of our results for HPV18-associated cervical carcinogenesis in vivo, nucleolin expression in HPV18-associated dysplasias, invasive squamous carcinomas, and adenocarcinomas was analyzed by immunohistochemistry. For example, an HPV18+ HSIL is shown in Fig. 6 B, and an HPV18+ adenocarcinoma is shown in Fig. 6 D. It appears that nucleolin is expressed throughout histologic sections of the HSIL and the adenocarcinoma. A similar staining pattern of nucleolin was seen in a total of five different HPV18+ adenocarcinomas and two HPV18-containing squamous cell carcinomas, although the latter ones hardly revealed any nucleolar staining. Upon closer inspection, a difference in the distribution of nucleolin protein expression became apparent between normal and cancerous tissues. Whereas in the nuclei of normal squamous and glandular epithelial cells the intranuclear nucleolin distribution is mostly diffuse, it is speckled in the nuclei of cancer cells and a subset of cells in HSIL (Fig. 6). Thus, the appearance of a speckled nucleolin distribution is associated with HPV18+ cervical carcinomas. HPV DNA was detected in tissue specimens by a published method and typing was performed by reverse line blot hybridization PCR (48).
Figure 6.




Nucleolin expression in normal HPV− cervical tissue and HPV18+ cervical neoplasias. Paraffin-embedded specimens were processed for immunohistochemistry by standard procedures using an Ab specific for nucleolin. Bound antibodies were visualized by avidin-biotin-peroxidase-diaminobenzidine tetrachloride and are stained brown. As a control, the Ab specific for nucleolin was presaturated with the immunizing nucleolin peptide (not depicted). Specimens were counterstained with hematoxylin. The counterstained nuclei are light blue. (A) Squamous epithelium of the human cervix uteri. High power view of lower epithelial cell layers is shown on the bottom right. High power view of more superficial layers is shown on the top right. (B) HPV18+ HSIL. High power view is shown on the right. (C) Endocervical glandular epithelial cells. High power view is shown on the right. (D) HPV18+ adenocarcinoma of the endocervix. High power views of two areas are shown on the right.
Discussion
Our study reveals that the host cell protein nucleolin is a necessary factor for HPV18 oncogene expression in cervical cancer cells and the maintenance of the proliferative state of these cells. Hence, it is essential for the development of HPV18-associated cervical carcinomas. Here, we report that nucleolin binds in a sequence-specific manner to the HPV18 enhancer and activates HPV18 oncogene transcription in S phase of the cell cycle. Thus, we have identified nucleolin as an S phase–specific transcription activator. Moreover, we show that nucleolin controls the formation of a DNase I HS site in the HPV18 enhancer. Nuclease HS regions in chromatin reflect an open chromatin structure that permits the binding of regulatory proteins to cis acting elements in gene control regions thereby facilitating gene transcription (for review see reference 52). A role for nucleolin in chromatin decondensation has been suggested by in vitro experiments with reconstituted chromatin (51). Our results go beyond these in vitro data as we demonstrate that nucleolin acts as a cell cycle stage–specific regulator of chromatin structure in vivo.
We have previously obtained evidence for a cell cycle–regulated protein that binds to the HPV18 enhancer in S phase of the cell cycle and have shown that the DNA binding activity of this protein is controlled by nuclear inhibitory proteins termed I-92, which form noncovalent cell cycle stage–specific complexes (33). Different I-92 activities occur in G1, G2, and G0 phase, whereas no I-92 activity is detectable in S phase. Here, this HPV18 enhancer binding protein was purified, sequenced, and identified as nucleolin. Taken together, these results show how the regulation of nucleolin DNA binding activity during the cell cycle is brought about. Moreover, nucleolin has been identified as a substrate of the cyclin-dependent kinases cdc2, cdk4, and cdk2 (57, 58). This suggests that phosphorylation by these cdks might be involved in controlling the complex formation of nucleolin with stage-specific I-92 inhibitors. Future experiments will clarify this issue.
The results obtained with the (HPV18 URR-CAT)-SiHa × HaCaT hybrid cells indicate that nucleolin does not play a significant role in HPV16 oncogene transcription. It was recently shown by transient transfection assays that the HPV16 contiguous URR is inactive in HeLa cells (59). HeLa cells are derived from an HPV18+ adenocarcinoma (56) and express considerable amounts of nucleolin (unpublished data). Taken together, these findings illustrate that there is a remarkable functional difference of the HPV16 and HPV18 URRs in different cell types. These functional differences can be explained, at least in part, by the diverse sequence organizations of the HPV18 and HPV16 enhancers (18, 25). In this context, it is interesting to note that the URRs of high risk HPVs do not show sequence similarities with the HPV18 enhancer fragment RP3 with the exception of HPV45, which is closely related to HPV18 (unpublished data).
Nucleolin was originally identified as a common phosphoprotein of growing eukaryotic cells (27). Yet the function of nucleolin in growing cells is not completely understood. Here, we have examined the relevance of nucleolin for the proliferation of HPV18-containing cervical cancer cells by antisense inhibition experiments. The data show that nucleolin selectively controls the proliferation of HPV18+ cervical cancer cells by activating E6 and E7 oncogene transcription. As discussed above, the transcription of the HPV16 oncogenes in (HPV18 URR-CAT)-SiHa and (HPV18 URR-CAT)-SiHa × HaCaT hybrid cells is not affected by the level of nucleolin DNA binding activity, and we find that the proliferation rates of these cells are similar. Moreover, antisense inhibition of nucleolin expression in HPV16+ SiHa cells did not significantly affect cell proliferation. Based on previous speculations (28, 29), one could think that nucleolin is essential for the global cellular protein synthesis. Hence, nucleolin antisense inhibition could theoretically block cell proliferation due to an inhibition of global protein synthesis. Yet recent results from the literature (50) and the results we have presented here rule out this possibility. Moreover, a general toxicity of the antisense oligonucleotides can be excluded by controls with the sense oligonucleotide that did not affect HPV18 oncogene expression and had no effect on general protein expression. Transcriptional activation of the E6 and E7 oncogenes is therefore a decisive function of nucleolin required for the proliferation of HPV18-infected cervical carcinoma cells.
Cervical carcinomas develop after high risk HPV infection, although a long period of latency precedes the occurrence of invasive cervical cancer. It is thought that during latency genetic alterations accumulate that ultimately lead to high level transcription of the high risk HPV oncogenes E6 and E7 (for review see reference 10). A continuous expression of E6 and E7 oncoproteins of HPV18 is required for the proliferation of cervical cancer cells in vitro (60) and in vivo (61). HPV oncogene expression is directly linked to the development of progressive squamous epithelial neoplasia (17). To elucidate the role of nucleolin in cervical carcinogenesis, we investigated nucleolin expression in HPV18-free normal cervical tissue and in HPV18+ precancerous and cancerous tissues by immunohistochemistry. This analysis revealed an alteration of the distribution of nucleolin in cervical carcinomas. Nucleolin is homogeneously distributed in the nuclei of normal squamous epithelial cells of the ectocervix and in glandular epithelial cells of the endocervix. In the nuclei of HPV18+ squamous cell carcinomas and adenocarcinomas, however, the nucleolin distribution is speckled. Thus, nucleolin appears to be associated with specific nuclear compartments in cervical cancer. The identity of these nuclear structures is not clear at this stage. One might assume that genetic alterations in the process of tumor development could be involved in causing this deregulation of nucleolin expression.
HPV18 infection is associated with a more aggressive form of cervical neoplasia than HPV16 infection (8). HPV18 has a much stronger transforming activity than HPV16, and it was demonstrated that the URR of HPV18 is responsible for this difference (9). It is tempting to speculate that this difference is due to the fact that the HPV18 URR is sensitive to nucleolin activation, whereas the HPV16 URR is not. Therefore, nucleolin might be directly linked to tumor behavior and clinical outcome. Finally, our data suggest nucleolin as a novel target molecule for the therapy of HPV18+ carcinomas of the cervix uteri.
Acknowledgments
We thank Harald zur Hausen for helpful discussions.
This work was funded by the Deutsche Forschungsgemeinschaft by grants Ro945/2-2, Ro945/2-3, and Ro945/2-4 to H.-D. Royer and by a research grant from the Medical School of the University of Düsseldorf to E. Grinstein.
Footnotes
Abbreviations used in this paper: BrdUrd, bromodeoxuridine; CAT, chloramphenicol acetyltransferase reporter gene; EMSA, electrophoretic mobility shift assay; GST, glutathione S-transferase; HPV, human papillomavirus; HS, hypersensitive; HSIL, high grade squamous intraepithelial lesion; PCNA, proliferating cell nuclear antigen; URR, upstream regulatory region.
References
- 1.zur Hausen, H., and E.-M. de Villiers. 1994. Human papillomaviruses. Annu. Rev. Microbiol. 48:427–447. [DOI] [PubMed] [Google Scholar]
- 2.Shah, K.V. 1997. Human papillomaviruses and anogenital cancers. N. Engl. J. Med. 337:1386–1388. [DOI] [PubMed] [Google Scholar]
- 3.Walboomers, J.M., M.V. Jacobs, M.M. Manos, F.X. Bosch, J.A. Kummler, K.V. Shah, P.J. Snijders, J. Peto, C.J. Meijer, and N. Munoz. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189:12–19. [DOI] [PubMed] [Google Scholar]
- 4.Bosch, F.X., M.M. Manos, N. Munoz, M. Sherman, A.M. Jansen, J. Peto, M.H. Shiffman, V. Moreno, R. Kurman, and K.V. Shah. 1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) study group. J. Natl. Cancer Inst. 88:1361–1365. [DOI] [PubMed] [Google Scholar]
- 5.Barnes, W., G. Delgado, R.J. Kurman, E.S. Petrilli, D.M. Smith, S. Ahmed, A.T. Lorincz, G.F. Temple, A.B. Jenson, and W.D. Lancaster. 1988. Possible prognostic signficance of human papillomavirus type in cervical cancer. Gynecol. Oncol. 29:267–273. [DOI] [PubMed] [Google Scholar]
- 6.Barnes, W., G. Woodworth, S. Waggoner, M. Stoler, A.B. Jenson, G. Delgado, and J. DiPaolo. 1990. Rapid dysplastic transformation of human genital cells by human papillomavirus type 18. Gynecol. Oncol. 38:343–346. [DOI] [PubMed] [Google Scholar]
- 7.Burger, R.A., B.J. Monk, T. Kurosaki, H. Anton-Culver, S.A. Vasilev, M.L. Berman, and S.P. Wilczynski. 1996. Human papillomavirus type 18: association with poor prognosis in early stage cervical cancer. J. Natl. Cancer Inst. 88:1361–1365. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz, S.M., J.R. Darling, K.A. Shera, M.M. Madeleine, B. McKnight, D.A. Galloway, P.L. Porter, and J.K. McDougall. 2001. Human papillomavirus and prognosis of invasive cervical cancer: a population-based study. J. Clin. Oncol. 19:1906–1915. [DOI] [PubMed] [Google Scholar]
- 9.Villa, L.L., and R. Schlegel. 1991. Differences in transcriptional activity between HPV-18 and HPV-16 map to the viral LCR-E6-E7 region. Virology. 181:347–377. [DOI] [PubMed] [Google Scholar]
- 10.zur Hausen, H. 2000. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 92:690–698. [DOI] [PubMed] [Google Scholar]
- 11.Dürst, M., D. Glitz, A. Schneider, and H. zur Hausen. 1992. Human papillomavirus type 16 (HPV16) gene expression and DNA replication in cervical neoplasia: analysis by in situ hybridisation. Virology. 189:132–140. [DOI] [PubMed] [Google Scholar]
- 12.Stoler, M.H., C.R. Rhodes, A. Whitbeck, S.M. Wolinsky, L.T. Chow, and T.R. Broker. 1992. Human papillomavirus type 16 and 18 gene expression in cervical neoplasias. Hum. Pathol. 23:117–128. [DOI] [PubMed] [Google Scholar]
- 13.Wentzensen, N., R. Ridder, R. Klaes, S. Vinokurova, U. Schaefer, and M.K. Doeberitz. 2002. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene. 21:419–426. [DOI] [PubMed] [Google Scholar]
- 14.Dyson, N., P.M. Howley, K. Münger, and E. Harlow. 1989. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 243:934–937. [DOI] [PubMed] [Google Scholar]
- 15.Münger, K., B.A. Werness, N. Dyson, W.C. Phelps, and P.M. Howley. 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 8:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheffner, M., B.A. Werness, J.M. Huibregtse, A.J. Levine, and P.M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 63:1129–1136. [DOI] [PubMed] [Google Scholar]
- 17.Arbeit, J.M., K. Münger, P.M. Howley, and D. Hanahan. 1994. Progressive squamous epithelial neoplasia in K14- human papillomavirus type 16 transgenic mice. J. Virol. 68:4358–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swift, F.V., K. Bhat, H.B. Younghusband, and H. Hamada. 1987. Characterization of a cell type-specific enhancer found in the human papilloma virus type 18 genome. EMBO J. 6:1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Offord, E.A., and P. Beard. 1990. A member of the activator 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. J. Virol. 64:4792–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoppe-Seyler, F., K. Butz, and H. zur Hausen. 1991. Repression of the human papillomavirus type 18 enhancer by the cellular transcription factor Oct-1. J. Virol. 65:5613–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thierry, F., G. Spyrou, M. Yaniv, and P.M. Howley. 1992. Two AP1 sites binding junB are essential for human papillomavirus type 18 transcription in keratinocytes. J. Virol. 66:3740–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauknecht, T., P. Angel, H.-D. Royer, and H. zur Hausen. 1992. Identification of a negative regulatory domain in the human papillomavirus type 18 promoter: interaction with the transcriptional repressor YY1. EMBO J. 11:4607–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yukawa, K., K. Butz, T. Yasui, H. Kikutani, and F. Hoppe-Seyler. 1996. Regulation of papillomavirus transcription by the differentiation-dependent epithelial factor Epoc-1/skn- 1a. J. Virol. 70:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker, J.N., W. Zhao, K.J. Askins, T.R. Broker, and L.T. Chow. 1997. Mutational analyses of differentiation-dependent human papillomavirus type 18 enhancer elements in epithelial raft cultures of neonatal foreskin keratinocytes. Cell Growth Differ. 8:751–762. [PubMed] [Google Scholar]
- 25.Mack, D.H., and L.A. Laimins. 1991. A keratinocyte-specific transcription factor, KRF-1, interacts with AP-1 to activate expression of human papillomavirus type 18 in squamous epithelial cells. Proc. Natl. Acad. Sci. USA. 88:9102–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butz, K., and F. Hoppe-Seyler. 1993. Transcriptional control of human papillomavirus (HPV) oncogene expression: composition of the HPV type 18 upstream regulatory region. J. Virol. 67:6467–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lapeyre, B., H. Bourbon, and F. Amalric. 1987. Nucleolin, the major nucleolar protein of growing eukaryotic cells: an unusual protein structure revealed by the nucleotide sequence. Proc. Natl. Acad. Sci. USA. 84:1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava, M., and H.B. Pollard. 1999. Molecular dissection of nucleolin's role in growth and cell proliferation: new insights. FASEB J. 13:1911–1922. [PubMed] [Google Scholar]
- 29.Ginisty, H., H. Sicard, B. Roger, and P. Bouvet. 1992. Structure and functions of nucleolin. J. Cell Sci. 112:761–772. [DOI] [PubMed] [Google Scholar]
- 30.Bonnet, H., O. Filhol, I. Truchet, P. Brethenou, C. Cochet, F. Amalric, and G. Bouche. 1996. Fibroblast growth factor-2 binds to the regulatory beta subunit of CK2 and directly stimulates CK2 activity toward nucleolin. J. Biol. Chem. 271:24781–24787. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki, N., H. Matsui, and T. Hosoya. 1985. Effects of androgen and polyamines on the phosphorylation of nucleolar proteins from rat ventral prostates with particular reference to 110-kDa phosphoprotein. J. Biol. Chem. 260:8050–8055. [PubMed] [Google Scholar]
- 32.Herblott, S., P. Chastagner, L. Samady, J.L. Moreau, C. Demaison, P. Froussard, X. Liu, J. Bonnet, and J. Theze. 1999. IL-2-dependent expression of genes involved in cytoskeleton organization, oncogene regulation, and transcriptional control. J. Immunol. 162:3280–3288. [PubMed] [Google Scholar]
- 33.Grinstein, E., I. Weinert, B. Droese, M. Pagano, and H.-D. Royer. 1996. Cell cycle regulation of nuclear factor p92 DNA-binding activity by novel phase-specific inhibitors. J. Biol. Chem. 271:9215–9222. [DOI] [PubMed] [Google Scholar]
- 34.Kadonaga, J.T. 1991. Purification of sequence-specific binding proteins by DNA affinity chromatography. Methods Enzymol. 208:10–23. [DOI] [PubMed] [Google Scholar]
- 35.Cole, S.T., and O. Danos. 1987. Nucleotide sequence and comparative analysis of the human papillomavirus type 18 genome. Phylogeny of papillomaviruses and repeated structure of the E6 and E7 gene products. J. Mol. Biol. 193:599–608. [DOI] [PubMed] [Google Scholar]
- 36.Rosenfeld, J., J. Capdevielle, J.C. Guillernot, and P. Ferrara. 1992. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 203:173–179. [DOI] [PubMed] [Google Scholar]
- 37.Stanbridge, E.J., R.R. Flandermeyer, D.W. Daniels, and W.A. Nelson-Rees. 1981. Specific chromosome loss associated with the expression of tumorigenicity in human cell hybrids. Somatic Cell Genet. 7:699–712. [DOI] [PubMed] [Google Scholar]
- 38.Freedman, R.S., J.M. Bowen, A. Leibovitz, S. Pathak, M.J. Siciliano, H.S. Gallager, and B.C. Giovanella. 1982. Characterization of a cell line (SW756) derived from a human squamous carcinoma of the uterine cervix. In Vitro. 18:719–726. [DOI] [PubMed] [Google Scholar]
- 39.Rösl, F., T. Achtstätter, T. Bauknecht, K.-J. Hutter, G. Futtermann, and H. zur Hausen. 1991. Extinction of the HPV18 upstream regulators region in cervical carcinoma cells after fusion with non-tumorigenic human keratinocytes under non-selective conditions. EMBO J. 10:1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava, M., P.J. Fleming, H.B. Pollard, and A.L. Burns. 1989. Cloning and sequencing of human nucleolin cDNA. FEBS Lett. 250:99–105. [DOI] [PubMed] [Google Scholar]
- 41.Chomczynski, P., and N. Sacchi. 1987. Single step method of RNA isolation by acid guanidinium thiocyanate phenol-chloroform extraction. Anal. Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- 42.Holm, P.S., S. Bergmann, K. Jürchott, H. Lage, K. Brand, A. Ladhoff, K. Mantwill, D.T. Curiel, M. Dobbelstein, M. Dietel, et al. 2002. YB-1 relocates to the nucleus in adenovirus-infected cells and facilitates viral replication by inducing E2 gene expression through the late E2 promoter. J. Biol. Chem. 277:10427–10434. [DOI] [PubMed] [Google Scholar]
- 43.Dürst, M., L. Gissmann, H. Ikenberg, and H. zur Hausen. 1983. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA. 80:3812–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khandjian, E.W., and C. Meric. 1986. A procedure for Northern blot analysis of native RNA. Anal. Biochem. 15:227–232. [DOI] [PubMed] [Google Scholar]
- 45.Feinberg, A., and B.A. Vogelstein. 1984. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 137:266–267. [DOI] [PubMed] [Google Scholar]
- 46.Hanakahi, L.A., L.A. Dempsey, M.-J. Li, and N. Maizels. 1997. Nucleolin is one component of the B cell-specific transcription factor and switch region binding protein, LR1. Proc. Natl. Acad. Sci. USA. 94:3605–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rösl., F., E.-M. Westphal, and H. zur Hausen. 1989. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol. Carcinogen. 2:72–80. [DOI] [PubMed] [Google Scholar]
- 48.van den Brule, A.J., R. Pol, N. Fransen-Daalmeijer, L.M. Schouls, C.J. Meijer, and P.J. Snijders. 2002. GP5+/6+ PCR followed by reverse line blot analysis enables rapid and high-throughput identification of human papillomavirus genotypes. J. Clin. Microbiol. 40:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bednarek, P.H., B.J. Lee, S. Gandhi, E. Lee, and B. Phillips. 1998. Novel binding sites for regulatory factors in the human papillomavirus type 18 enhancer and promoter identified by in vivo footprinting. J. Virol. 72:708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roger, B., A. Moisand, F. Amalric, and P. Bouvet. 2002. Repression of RNA polymerase I transcription by nucleolin is independent of the RNA sequence that is transcribed. J. Biol. Chem. 277:10209–10219. [DOI] [PubMed] [Google Scholar]
- 51.Erard, M.S., P. Belenguer, M. Caizergues-Ferrer, A. Pantaloni, and F. Amalric. 1988. A major nucleolar protein, nucleolin, induces chromatin decondensation by binding to histone H1. Eur. J. Biochem. 175:525–530. [DOI] [PubMed] [Google Scholar]
- 52.Gross, D.S., and W.T. Garrard. 1988. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 57:159–197. [DOI] [PubMed] [Google Scholar]
- 53.Boukamp, P., R.T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham, and N. Fusenig. 1988. Normal keratinisation in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker, C.C., W.C. Phelps, V. Lindgren, M.J. Braun, M.A. Gonda, and P.M. Howley. 1987. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 61:962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Knebel Doeberitz, M., T. Oltersdorf, E. Schwarz, and L. Gissmann. 1988. Correlation of modified human papilloma virus early gene expression with altered growth properties in C4-1 cervical carcinoma cells. Cancer Res. 48:3780–3786. [PubMed] [Google Scholar]
- 56.Gey, G.O., W.D. Coffman, and M.T. Kubicek. 1952. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 12:264–265. [Google Scholar]
- 57.Peter, M., J. Nakagawa, M. Doree, J.C. Labbe, and E.A. Nigg. 1990. Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell. 60:791–801. [DOI] [PubMed] [Google Scholar]
- 58.Sarcevic, B., R. Lilischkis, and R.L. Sutherland. 1997. Differential phosphorylation of T-47 human breast cancer cell substrates by D1-, D3-, E-, and A-type cyclin-CDK complexes. J. Biol. Chem. 272:33327–33337. [DOI] [PubMed] [Google Scholar]
- 59.Sailaja, G., R.M. Watts, and H.-U. Bernard. 1999. Many different papillomaviruses have low transcriptional activity in spite of strong epithelial specific enhancers. J. Gen. Virol. 80:1715–1724. [DOI] [PubMed] [Google Scholar]
- 60.Goodwin, E.C., and D. DiMaio. 2000. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA. 97:12513–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bosch, F.X., E. Schwarz, P. Boukamp, N.E. Fusenig, D. Bartscg, and H. zur Hausen. 1990. Suppression in vivo of human papillomavirus type 18 E6-E7 gene expression in nontumorigenic HeLa x fibroblast hybrid cells. J. Virol. 64:4743–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwarz, E., U.K. Freese, L. Gissmann, W. Mayer, B. Roggenbuck, A. Stremlau, and H. zur Hausen. 1985. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 314:111–114. [DOI] [PubMed] [Google Scholar]