

Abstract
Cytomegaloviruses (CMVs) deploy a set of genes for interference with antigen presentation in the major histocompatibility complex (MHC) class I pathway. In murine CMV (MCMV), three genes were identified so far: m04/gp34, m06/gp48, and m152/gp40. While their function as immunoevasins was originally defined after their selective expression, this may not necessarily reflect their biological role during infection. The three immunoevasins might act synergistically, but they might also compete for their common substrate, the MHC class I complexes. To approach this question in a systematic manner, we have generated a complete set of mutant viruses with deletions of the three genes in all seven possible combinations. Surface expression of a set of MHC class I molecules specified by haplotypes H-2d (Kd, Dd, and Ld) and H-2b (Kb and Db) was the parameter for evaluation of the interference with class I trafficking. The data show the following: first, there exists no additional MCMV gene of major influence on MHC class I surface expression; second, the strength of the inhibitory effect of immunoevasins shows an allele-specific hierarchy; and third, the immunoevasins act not only synergistically but can, in certain combinations, interact antagonistically. In essence, this work highlights the importance of studying the immunosubversive mechanisms of cytomegaloviruses in the context of gene expression during the viral replicative cycle in infected cells.
Keywords: murine cytomegalovirus, BAC, immune evasion, MHC class II, allele
Introduction
The capacity of a virus to actively modulate the host immune response defines the biological balance between a persistent virus and its host. A number of viral functions are instrumental for maintaining and tuning this balance. The highly species-specific CMVs are a representative example. Human CMV (HCMV), a herpesvirus that defines the β-herpesvirus subfamily, is an important pathogen only in the immunologically immature or immunocompromised host, whereas in the normal population the infection occurs without clinical symptoms. CMVs interfere with innate and adaptive cellular and humoral immune effector mechanisms by affecting cytokine networks, by activation and silencing of natural killer (NK)* cells, by downmodulating antigen presentation in the MHC class I and II pathways, and by the regulation of apoptosis. Some viral genes that affect host immune functions are homologues pirated from the vertebrate genome, whereas others show no apparent homology to host genes (1, 2).
Several HCMV and murine CMV (MCMV) gene products, usually glycoproteins, have been shown to interfere with the MHC class I pathway of antigen processing and presentation. With one exception, all these proteins interfere with antigen presentation by downmodulation of MHC class I cell surface expression. Individually, none of those is essential for virus growth in cell culture. They lack significant homologies to other herpesviral or cellular genes and are dispersed over the left and right genome termini (3, 4). The HCMV proteins US2 and US11 cause proteasomal degradation of MHC class I molecules by retrograde transport from the ER to the cytosol (5, 6). The US3-encoded protein binds to MHC class I molecules and arrests them in the ER (7, 8). Finally, US6 binds to the peptide transporter associated with processing to prevent peptide loading of MHC class I molecules (9–11).
MCMV is comparable to HCMV with regard to a variety of functions by which it can modulate the host response. The infection of the mouse with MCMV offers the advantage that defined mutants can be tested in the natural host. In MCMV, three genes encoding glycoproteins that interact with MHC class I molecules, here referred to as immunoevasins, have been identified: the immunoevasin m04/gp34 binds to MHC class I molecules without affecting cell surface expression, which is exceptional for MHC class I interfering CMV proteins (12). Nevertheless, m04/gp34 is involved in the prevention of antigen presentation to selected CTL clones by a so far unknown mechanism (13). The second immunoevasin m06/gp48 reduces cell surface expression of MHC class I molecules by binding them and by retargeting the complexes to lysosomes for degradation (14). The third immunoevasin m152/gp40 also reduces cell surface expression of MHC class I molecules, but achieves this by a different mechanism, namely by retention of the peptide-loaded complexes in the ER-cis-Golgi intermediate compartment (15). The m152/gp40 immunoevasin was the first and is still the only MHC class I interacting CMV glycoprotein shown to be relevant for CMV replication in vivo in the face of functional immunity (16).
Previous studies on CMV-encoded MHC class I reactive proteins have had the aim to elucidate the specific mode of function of defined viral proteins. With the exception of m152/gp40 of MCMV, there exist no data on the relevance of the individual immunoevasins in the course of CMV infection in vitro and in vivo. It is unknown whether, in the context of the CMV genome, the effects of these genes are additive, cooperative, redundant, or even competitive. Likewise, little is known about possible MHC class I allele-specific differences in the action of the different immunoevasins. This limited knowledge is because of a lack of appropriate virus mutants that would allow to perform a systematic analysis of immunoevasin interactions in the context of virus infection. The generation of CMVs with targeted gene deletions by recombination in cells is laborious (17) and the generation of mutants with multiple targeted mutations dispersed over the genome is almost not achievable. Cloning of CMVs as infectious bacterial artificial chromosomes (BACs; reference 18) has opened perspectives for novel site–directed and random mutagenesis techniques in Escherichia coli (for a review, see reference 19). Further work from several laboratories on the generation and mutagenesis of BAC-cloned CMV genomes (20–24) has confirmed the attractivity and general applicability of CMV mutagenesis in bacteria. Until now, targeted mutagenesis usually involved subcloning of viral DNA and usage of available restriction sites, which is unfavorable for the introduction of independent mutations in close proximity (a distance of <1.5 kbp; reference 18), as it is the case for genes m04 and m06.
Here, we systematically investigated the influence on MHC class I molecule surface expression of the immunoevasins m04/gp34, m06/gp48, and m152/gp40 during MCMV infection. We generated a complete set of MCMV mutants which lack or express these three genes in all possible combinations. To this end, we applied to the viral MCMV-BAC a fast mutagenesis procedure for targeted mutation via a double crossing-over event between PCR-generated DNA fragments and the viral DNA. We tested the effect of this set of seven MCMV immunoevasin deletion mutants on cell surface expression of H-2d and H-2b MHC class I alleles and conclude from the data the following: (i) there exists no additional MCMV gene product that would significantly interfere with MHC class I molecule export; (ii) individual MHC class I alleles are differentially affected; (iii) the isolated deletion of gene m06 has the strongest effect on MHC class I, indicating a dominant role of the immunoevasin m06/gp48; and (iv) the immunoevasins m06/gp48 and m152/gp40 cooperate in their function. In contrast, m04/gp34 and m152/gp40 compete for MHC class I molecules, whereby m04/gp34 antagonizes the function of m152/gp40. In summary, the viral MHC class I–modulating proteins interact in their effects on MHC class I molecules, and the outcome of this interaction is allele specific.
Materials and Methods
Cells, Viruses, and Plaque Assay.
BALB/c mouse embryonic fibroblasts (MEFs) were cultured in DMEM supplemented with 10% (vol/vol) FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. These cells were used for virus reconstitution from recombinant BAC plasmids as described previously (25). NIH3T3 cells (CRL1658; American Type Culture Collection) were cultured in DMEM supplemented with 10% (vol/vol) newborn calf serum and antibiotics (see above). These cells were used for propagation of recombinant viruses, for determination of viral growth, and for Western blot analysis. For the detection of MHC class I molecules on the cell surface after infection with the respective viruses, the following fibroblast cell lines were used: B12, which is a BALB/c (haplotype H-2d)-derived SV40-transformed fibroblast cell line (26); and C57SV, which is a C57BL/6 (haplotype H-2b)-derived SV40-transformed fibroblast cell line (27). The MCMV wild-type (wt) and wt-like strains used in these studies were strain Smith (VR-194; American Type Culture Collection) and the BAC-derived recombinant MW97.01 (20). Virus stocks were prepared following published protocols (28). In vitro growth kinetics of the MCMV mutants and of parental wt MCMV were determined on NIH3T3 cells as follows: NIH3T3 cells were infected with a multiplicity of infection of 0.1 PFU per cell (without centrifugal enhancement) and supernatants of infected cells were harvested every 24 h from day 0 (input virus titer) through day 5 after infection. Virus titers in supernatants were measured in triplicate by a plaque assay performed on second-passage MEF monolayers in 48-well plates as described previously (29).
Plasmid Construction.
Plasmid cloning was performed by standard methods (30). The parental shuttle plasmid pST76A-SacB is a derivative of plasmid pST76A (31) into which the Bacillus amyloliquefaciens sacB gene (32) was introduced. The sacB gene was used for selection of clones with resolved cointegrates and lost shuttle plasmids by adding 5% (wt/vol) sucrose to the agar plates as described previously (21). The shuttle plasmid pSTm06, used for generation of the BAC plasmid pΔm06, was cloned as follows: the MCMV sequence from nt positions 2701–9760 was excised from plasmid pHindIII A (33) by HindIII and KpnI digestion, and was inserted into plasmid pBluescriptII (KS+) (Stratagene). Within this plasmid, the MCMV sequence from nt positions 5392–6235, containing open reading frame m06, was deleted by NheI treatment and replaced by the kanamycin resistance gene (kn) from plasmid pCP15 (34). The MCMV sequence containing the kn instead of m06 was then excised by BstEII and EcoRI treatment and inserted into the SmaI site of plasmid pST76A-SacB, hereby generating shuttle plasmid pSTm06. Plasmid pSTm06 now contains the kn flanked by MCMV sequences upstream (nt 2701–5392) and downstream (nt 6235–9760) of m06.
Mutagenesis of Viral BAC Plasmids Using Linear PCR Fragments.
A PCR-based mutagenesis procedure was used for the generation of the recombinant MCMV BAC plasmids pΔm04, pΔm152, pΔm04+m06, pΔm04+m152, pΔm06+m152, and pΔm04+m06+m152 that contain exact single-, double- or triple-deletions of the MCMV open reading frames m04, m06, and m152, respectively. The technique was based on homologous recombination mediated by the recombination functions redα (exo) and redβ (bet), and the exonuclease inhibitor redγ (gam) of bacteriophage λ expressed from the plasmid pBADαβγ (35) (for the strategy, see Fig. 1 A). First, linear DNA recombination fragments were generated. These fragments contained the prokaryotic kn from plasmid pACYC177 (New England BioLabs) or the resistance gene zeocin (zeo) from plasmid pZero1 (Invitrogen), flanked by sequences of 40 to 60 nts in length, homologous to the up and downstream regions of the target gene. For the generation of these linear fragments by PCR, contiguous pairs of synthetic primers were used, which all bind to the resistance gene with their 3′-ends and which carry sequences homologous to the MCMV genomic sequence at their 5′-ends. For deletion of gene m152 (nt 210244–211377), the PCR fragment m152-PCR, containing zeo flanked by 34-bp minimal FRT sites (5′-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC-3′), was generated by using plasmid pZero1 as template DNA and the contiguous primers m152-FRT-zeo-5 (5′GCTCGAGCGAGAGCACCCGACGATCTGACATTGTCCAGTGTGCCGGT-CGCACGAACATCAGAAGTTCCTATTCTCTAGAAAGT-ATAGGAACTTCAACGTTTACAATTTCGCCTGATGCG-3′) and m152-FRT-zeo-3 (5′TCACAAGCCGTGTCACCGC-TCCACGTTTCACCGTCGTCGGTCTCCCGATCGCTA-GCCTGAACAGAAGTTCCTATACTTTCTAGAGAATAG-GAACTTCTGAAGTTTTAGCACGTGTCAGTCCT-3′). For deletion of gene m04 (nt 3270 to 4067), the PCR fragment m04-PCR, containing kn, was generated using plasmid pACYC177 as template DNA and contiguous primers m04–5 (5′-TAATGATCTAGACGGCAATTTCTGTCTCATTCGTTGTTCC- AGAGCGACGGATGGTACAAG-3′) and m04–3 (5′-TACTCAGAACACCGGAAAATGGTT TACTCAAGGGGATTTTTATTTAGGGGGTTAGTTACT-3′). With the exception of the single m06 deletion BAC plasmid pΔm06 (see below), deletion of the m06 gene (nt 5300–6334) in the MCMV BAC plasmids was performed by using PCR fragment m06-PCR, which was generated using plasmid pZero1 as template DNA and the contiguous primers m06-zeo-5 (5′CACGCCCAAAATCACGCAATCATATATAAATGGACAATGAAGCCAATCTAA- CGTTTACAATTTCGCCTGATGCG-3′) and m06-zeo-3 (5′GCAGTCTAGCTAGCGGCTCGCGGCCGTAGGTGTA-GAACAACATAGCACCTGAAGTTTTAGCACGTGTCAGTCCT-3′). The PCR products were purified using the QIAGEN PCR purification kit (QIAGEN). For removal of residual methylated template plasmid DNA, products were digested with 20 U of restriction enzyme DpnI for 1.5 h. The PCR products were then immediately precipitated with 100% ethanol, washed with 70% ethanol, and resuspended in 10 μl of sterile water. 5 μl of each linear fragment was electroporated into electrocompetent E. coli (strain DH10B), containing the respective MCMV BAC plasmid and the recombination proteins (see below), using a Bio-Rad Gene Pulser (2.5 kV, 200 Ω, and 25 μF). After addition of 1 ml of LB medium followed by shaking for 2 h at 37°C, the bacteria were grown on LB agar plates containing 25 μg/ml chloramphenicol and 25 μg/ml kanamycin or zeocin and incubated overnight at 37°C.
Figure 1.

Strategy for construction of the MCMV BACs and genomic organization of the resulting deletion mutants. (A) Mutagenesis steps for single-, double-, and triple-deletion of the MHC class I interacting genes m04, m06, and m152 by PCR-based BAC mutagenesis. By using primers that contain 40–60 nucleotides homologous to the up- and downstream regions of the viral target sequence at their 5′-ends (black boxes), FRT sites (dotted boxes), and primer binding sites to a resistance gene, a linear DNA recombination fragment was amplified by PCR. This fragment was electroporated into DH10B containing the MCMV BAC with the chloramphenicol resistance gene (Cm) and containing the recombination functions redα, -β, and -γ expressed from plasmid pBADαβγ. Incubation under selection conditions results in recombinant MCMV BACs. These BACs were used for the second round of mutagenesis using a different resistance marker (marker 2). Alternatively, the resistance marker could be excised by using FLP-mediated recombination, leaving one residual FRT site in the genome. This allowed the usage of the same resistance marker for a further round of mutagenesis. (B) Genomic organization of immune evasion gene deletion mutants. Shown are the HindIII fragments of the MCMV wt (strain Smith) genome. Genes m04 and m06, coding for the glycoproteins gp34 and gp48, respectively, are located on fragment HindIII A. Gene m152, coding for gp40, is located on fragment HindIII E. (B1) In the first round of mutagenesis, the kn was inserted into the BAC-derived wt MCMV genome MW97.01, thereby generating the mutant genome Δm04-MCMV. Into this recombinant genome, the zeo gene was additionally introduced, thereby deleting gene m152 and generating mutant Δm04+152-MCMV. Since zeo was flanked by 34-bp FRT sites (crosshatched boxes), it could be excised by FLP-mediated recombination. This allowed again insertion of the zeo gene, this time for deletion of gene m06, generating mutant Δm04+06+152-MCMV. (B2) For the generation of mutant Δm152-MCMV, gene m152 was replaced by a zeo gene that was flanked by FRT sites. After FLP-mediated excision of zeo in position of m152, zeo was inserted once again for deletion of m06, generating mutant Δm06+152-MCMV. (B3) Mutant Δm04+06-MCMV was generated by two rounds of recombination inserting the zeo in the place of gene m06 and the kn in the place of gene m04. (B4) Mutant Δm06-MCMV was generated by the two step replacement procedure using the shuttle plasmid pSTm06.
Electrocompetent DH10B were prepared as follows: 2 ng DNA of plasmid pBADαβγ (45) was introduced into DH10B, which contain the parental MCMV BAC plasmid, by chemical transformation using standard protocols (30). 2 ml of an overnight culture of these transformed DH10B were then added to 200 ml of LB growth medium containing 25 μg/ml chloramphenicol and 25 μg/ml kanamycin. 2 ml of 10% (wt/vol) l-arabinose in LB medium was added at an OD600 of 0.2 in order to induce the expression of the exo protein, which is controlled by the inducible BAD-promoter (Invitrogen). After 30 min of incubation at 37°C, bacteria were put on ice for 15 min. After three washing steps with ice-cold 10% (vol/vol) glycerol, bacteria were resuspended in a total volume of 800 μl of 10% glycerol, subdivided into 60 μl aliquots, and frozen at −80°C.
Excision of the zeo from the MCMV BAC plasmid pΔm152-zeo via the flanking FRT sites by FLP-mediated site specific recombination was performed by using plasmid pCP20 as described previously (34).
Mutagenesis of BAC Plasmids Using Shuttle Plasmids.
For deletion of 82% of the m06 gene sequence (nt 5392 to 6235) within the wt MCMV BAC plasmid pSM3fr, a two-step recombination strategy based on RecA-mediated homologous recombination in the E. coli strain CBTS (36) was applied as described previously (18). Shuttle plasmid pSTm06 was used for the generation of the recombinant MCMV BAC plasmid pΔm06 (Fig.1).
Reconstitution of Recombinant Viruses by Transfection.
For reconstitution of viral progeny, 80% confluent MEF in a 6-cm culture dish were transfected with 1.5 μg of MCMV BAC plasmid DNA by the calcium phosphate precipitation technique as described previously (30). 5 h after transfection, the MEF were treated with 15% glycerol (vol/vol) in Hepes-buffered saline for 2.5 min as described previously (30). After 3–5 d of cultivation, virus plaques appeared. The recombinant viruses Δm04-MCMV, Δm04+152-MCMV, and Δm152-MCMV were already used in a previous study and published under the names Δm4-MW99.03, Δm4+m152-MW99.04, and Δm152-MW99.05, respectively (13).
Isolation and Analysis of DNA.
Isolation of plasmid DNA from E. coli cultures was performed using an alkaline lysis procedure as described previously (30). Isolation of MCMV BAC DNA from E. coli was performed using the Nucleobond kit from Machery & Nagel. Viral DNA from infected cells was isolated as published previously (33).
Western Blot Analysis.
For protein detection in infected cells, Western blot analysis was performed at 16 h after infection. For the detection of m04/gp34, the specific peptide antiserum m04–3 (13) was used. The m152/gp40 protein was detected with mAb gpM3D10 (37) and protein m06/gp48 was detected with polyclonal antiserum m06–2 (38). The lysates of virus-infected cells were suspended in Laemmli sample buffer containing 5% β-mercaptoethanol, sonified for 10 s, and incubated at 95°C for 5 min. After electrophoresis on a 10% SDS-PAGE, the proteins were transferred to Hybond-P membranes (Amersham Biosciences) in the presence of blotting buffer (25 mM Tris, 192 mM glycine, 20% [vol/vol] methanol, pH 8.3). After blocking of the membranes for 30 min at room temperature with TBS-T (Tris-buffered saline/0,05% Tween-20) containing 5% (wt/vol) dry milk, the primary antibody was added in a dilution (in TBS-T) of 1:5,000. After washing with TBS-T, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibody. Proteins were visualized using the ECL system (Amersham Biosciences).
Flow Cytometry.
For the detection of MHC class I molecules on the surface of infected cells, the following antibodies were used (see also Table I in Results): mAb 28–14–8s (HB27; American Type Culture Collection), specific for the α3 domain of H-2Ld and Lq molecules, served for the detection of the Ld molecule on cells of H-2d haplotype. mAb SF1.1.1 (HB-159; American Type Culture Collection) served for the detection of H-2Kd, mAb 34–2-12s (HB-87; American Type Culture Collection) for H-2Dd, mAb Y-3 (39) for H-2Kb, and mAb B22.249 (40) for H-2Db. Subconfluent cell-layers in a 6-cm culture dish were infected with recombinant or wt MCMVs with a virus dose of 0.3 PFU per cell using centrifugal enhancement of infectivity, which results in an effective MOI of 6 (41). At 4, 7, 12 and 16 h after infection, the cells were washed with PBS, trypsinized, and rinsed with FACS®-buffer (PBS supplemented with 2% FCS and 0.03% [wt/vol] NaN3), followed by labeling for 60 min at 37°C with the indicated primary antibodies. After washing with FACS®-buffer, antibodies bound to MHC class I molecules were stained by adding fluorescein-conjugated goat anti–mouse IgG (Sigma-Aldrich).
Table I.
Compilation of Cell Lines and Antibodies Used for the Cytofluorometric Analysis of MHC Class I Cell Surface Expression
| Cell line | MHC haplotype | Class I allele | Antibody |
|---|---|---|---|
| B12 | H-2d | Kd | SF1.1.1 |
| B12 | H-2d | Dd | 34-2-12s |
| B12 | H-2d | Ld | 28-14-8s |
| C57SV | H-2d | Kb | Y-3 |
| C57SV | H-2d | Db | B22.249 |
A comparable infection rate for all wt and mutant viruses (>90% of the cells infected) was confirmed for a separate aliquot of the infected cells by labeling of the intranuclear viral IE1 protein pp89 with mouse mAb (IgG1) CROMA101 (provided by S. Jonjic, University of Rijeka, Rijeka, Croatia). IE1 is expressed during all kinetic phases of viral gene expression (42). For permeabilization of the cells, which is required for detection of intracellular pp89, saponin was added to the buffers in a final concentration of 0.1% (vol/vol). Fluorescein-staining was performed as specified above. Labeling of the cell surface marker CD44 with rat mAb IM7.8.1 (TIB235; American Type Culture Collection) was used to control for the expression of cell surface molecules not affected by MCMV gene products in order to exclude downregulation of cell surface molecules due to a general cytopathogenic effect of the infection. For detection of antibodies bound to CD44, fluorescein-conjugated goat anti–rat IgG (Sigma-Aldrich) was used.
Throughout, cells incubated with the second antibody alone were used as negative controls and the resulting fluorescence signals were subtracted from the values gained with the first and second antibody for the same probes. For each expression profile, 3 × 106 or 5 × 106 cells were labeled and analyzed on a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson) for data processing.
Assay for Competitive Virus Fitness In Vivo.
Tissue tropism and in vivo growth characteristics of wt MCMV and of the immune evasion gene triple deletion mutant Δm04+06+152-MCMV were compared by coinfection of immunocompromised mice followed by two-color in situ DNA hybridization (2C-ISH) of viral genomes in infected tissues. In infected tissue cells, viral DNA is accumulated during the late phase in an intranuclear inclusion body, which is the site of DNA packaging and nucleocapsid assembly.
Conditions of Infection.
Female BALB/c mice (8-wk-old) were immunocompromised by hematoablative total-body γ-irradiation with a single dose of 7 Gy delivered by a 137Cs source. At 24 h after the irradiation, recipients were coinfected intravenously in the tail vein with a 1:1 mixture (5 × 104 PFU each) of the two MEF-propagated, sucrose-gradient purified viruses (41) contained in 0.5 ml of physiological saline. Infection of host tissues was determined by 2C-ISH on day 8 after infection, that is, at a prefinal stage of multiple-organ CMV disease.
Hybridization Probes for 2C-ISH.
The hybridization probe M55-P spans 1541 bps from nucleotide positions 84250–85790 of the MCMV strain Smith genome (4) (GenBank/EMBL/DDBJ accession no. MCU68299[complete sequence]) within the gB-coding gene M55. It was synthesized by PCR using plasmid pACYC-gB (43) as the template, and oligonucleotides 5′-GCACGTCGTAGGTAAATTGC-3′ and 5′-TCGCTCCTCGAGCTGGTACG-3′ as primers. M55-P was tagged during the PCR by incorporation of fluorescein-conjugated dUTP (fluorescein-12-dUTP) (no. 1373242; Roche Laboratories). Probe m152-P spans 1115 bps from nucleotide positions 210254 to 211368 within gene m152. It was synthesized by PCR using plasmid pGEM-4Z-m152 as the template, and oligonucleotides 5′-CAGTTGATGTAGACCAGGCGATAC-3′ and 5′-GCTATCACCTACTTGCTCCTCTCG-3′ as primers. For the construction of pGEM-4Z-m152, the desired sequence within gene m152 was amplified from virion DNA by using the primers mentioned above with their 5′ ends extended by HindIII and BamHI cleavage sites, respectively. The amplificate was then ligated into cloning vector pGEM-4Z (GenBank/EMBL/DDBJ accession no. X65305) (no. P2161; Promega). Probe m152-P was tagged during the PCR by incorporation of digoxigenin-11-dUTP (no. 1093088; Roche Laboratories).
2C-ISH.
A red label is used to visualize gene M55, which is shared by the two viruses, whereas a black label is assigned to gene m152 contained in the genome of wt MCMV only (for the design, see Fig. 4 A). The hybridization of viral DNA in deparaffinized 2-μm tissue sections was performed with procedures described in greater detail by Grzimek et al. (44). In a first step, gene M55 was stained red by using alkaline phosphatase-conjugated anti-fluorescein antibody (no. 1426338; Roche Laboratories) with fuchsin as the chromogenic substrate. In the second step, gene m152 was stained black by using peroxidase-conjugated antidigoxigenin antibody (no. 1207733; Roche Laboratories) with diaminobenzidine tetrahydrochloride as the substrate, followed by color enhancement with ammonium nickel sulfate hexahydrate. Accordingly, cells infected by mutant Δm04+ 06+152-MCMV are marked by a clean red color, whereas in wt MCMV the red staining of gene M55 is superimposed by the black staining of gene m152. Quantitative microscopic analysis as well as the photodocumentation of the tissue sections were performed as described previously (44).
Figure 4.

Equivalent tissue tropism and comparable in vivo replication efficacy of wt MCMV and mutant Δm04+06+152-MCMV in the absence of immune control. (A) Maps (not drawn to scale) illustrating the design of probes for two-color in situ hybridization (2C-ISH), discriminating between wt (BAC-derived MW97.01) MCMV (mixed black & red staining) and mutant Δm04+06+152-MCMV (clean-red staining). (B) Comparative morphometric quantitation of infection in liver, spleen, and adrenal/suprarenal glands (per mouse, only one of the paired glands was included in the analysis) of immunodepleted (7Gy γ-irradiated) BALB/c recipients on day 8 after intravenous coinfection with wt MCMV and mutant MCMV. Note that some adrenal glands were colonized clonally by either wt or mutant MCMV. Shown are linked data pairs for individual mice no. 1–5. Infected cells were counted for representative 10-mm2 tissue areas, except for the adrenal glands for which the results from 2-mm2 areas were extrapolated to 10 mm2. (C1–C3) 2C-IHC–stained sections of the indicated organs (derived from individual mouse no. 5), showing distinct black-red or clean-red foci of infection by wt and mutant MCMV, respectively. Counterstaining was performed with hematoxylin. Bar marker, 100 μm.
Results
Construction of Recombinant MCMV Genomes by PCR-based BAC Mutagenesis in E. coli.
To revisit the roles of the known MHC class I interacting genes m04/gp34, m06/gp48, and m152/gp40 in the course of infection, we intended to generate a complete set of seven mutants with single-, double-, and triple-deletions of these three genes in all possible combinations. To this end, we applied to the MCMV-BAC a PCR-based one-step recombination procedure (45) using conditional expression of the proteins redα, -β, and -γ from bacteriophage λ (Materials and Methods and Fig. 1 for the mutagenesis strategy).
Using this procedure, six recombinant MCMV BACs with deletions of the known immunoevasin genes m04, m06, and m152 in all combinations were generated: pΔm04; pΔm152; pΔm04+06; pΔm04+m152; pΔm06+m152; and pΔm04+m06+m152 (Fig. 1, B1–B3). BAC plasmid pΔm06 was constructed by using the established two-step recombination method (18) (Fig. 1 B4). Correct deletions of the respective genes and the integrity of the whole genomes were confirmed by restriction pattern analysis using appropriate restriction enzymes followed by Southern blot analysis (unpublished data).
Expression of Immunoevasins m04/gp34, m06/gp48, and m152/gp40 by MCMV Recombinants.
The MCMV BAC plasmids pΔm04, pΔm06, pΔm152, pΔm04+m06, pΔm04+ m152, pΔm06+m152, and pΔm04+m06+m152 were transfected into permissive MEF for the reconstitution of viruses. The correct expression of gp34, gp48, and gp40 by the MCMV mutants Δm04-MCMV, Δm06-MCMV, Δm152-MCMV, Δm04+06-MCMV, Δm04+152-MCMV, Δm06+152-MCMV, and Δm04+06+152-MCMV, respectively, was analyzed by Western blot analysis of lysates from NIH3T3 cells infected for 16 h. All mutants showed the expected pattern of glycoprotein expression (Fig. 2) .
Figure 2.
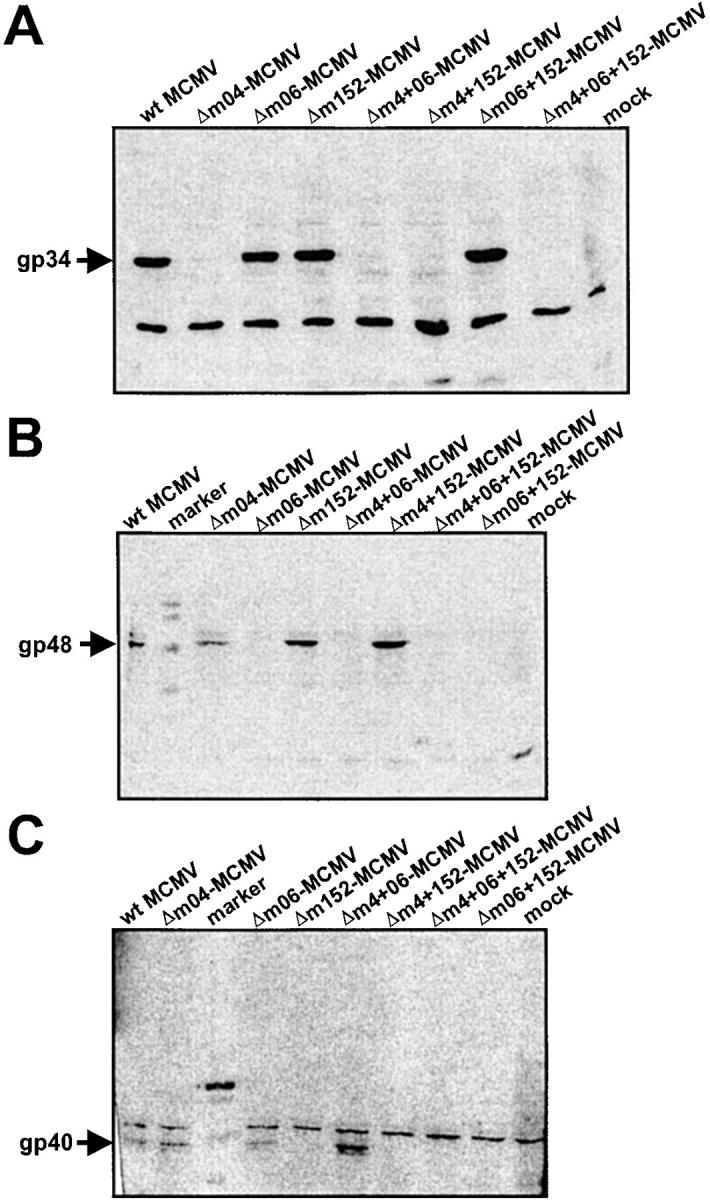
Expression of gp34, gp40, and gp48. NIH3T3 cells were infected with the indicated viruses for 16 h or were left uninfected (mock). The immunoevasin molecules were detected by Western blot analysis. (A) Detection of gp48 with a polyclonal antiserum confirms lack of expression in all mutants with m06 gene deletion. (B) Detection of gp40 with a mAb confirms lack of expression in all mutants with m152 gene deletion, and (C) detection of gp34 with a polyclonal antibody confirms lack of expression in all mutants with m04 gene deletion. In mutants with no deletion of m04, m06, and m152, respectively, expression of the corresponding glycoprotein was confirmed.
Immunoevasins m04/gp34, m06/gp48, and m152/gp40 Are Collectively Dispensable for Virus Replication in Fibroblasts.
Previous work has already shown that each of the immune evasion genes m04, m06, and m152 can be deleted without affecting virus growth in fibroblast cell cultures. However, since these genes could possibly substitute each other in an essential function, deletion of all three or of combinations of two might still abolish virus growth. In addition, an impaired viral replication could result from undesired spontaneous mutations that may have accumulated elsewhere in the viral genomes during the repeated mutagenesis procedures. To explore these possibilities, we have taken multistep growth curves for wt MCMV and the complete set of mutants (Fig. 3) . Specifically, NIH3T3 cells were infected at an MOI of 0.1 PFU per cell (without centrifugal enhancement), and virus titers in the supernatants were determined daily until day 5. All mutants, including the triple-deletion mutant Δm04+06+152-MCMV, were found to grow like wt MCMV. This finding excluded adverse effects of undesired mutations and confirmed that the three immune evasion genes are not essential for virus replication in fibroblasts in cell culture.
Figure 3.

Multistep virus growth curves in cell culture. NIH3T3 cells were infected with the indicated wt and mutant viruses at an MOI of 0.1 PFU per cell and virus titers in the supernatants were detected every 24 h after infection until day 5. All seven mutants show a virus productivity comparable to wt MCMV (MW97.01).
The Triple Immunoevasin Deletion Mutant Δm04+06+ 152-MCMV Has No Growth Deficiency Phenotype in Immunocompromised Mice.
So far, the data have shown that the three immune evasion genes, individually or in any possible combination, are dispensable for virus replication in fibroblast cell cultures. Yet, in vivo, MCMV replicates in a wide variety of cell types, including macrophages (46, 47), dendritic cells (48), endothelial cells, and different types of epithelial cells such as hepatocytes, pneumocytes, enterocytes, and glandular epithelial cells (49). Therefore, normal virus growth in fibroblast cell cultures did not exclude an altered cell type tropism and/or a modulated replication efficacy in host organs. If interference with antigen presentation is the only biological function of the immunoevasins, their deletion in mutant Δm04+06+152-MCMV should not affect in vivo replication in absence of host immune functions. For testing this prediction, we used here coinfection of severely immunocompromised mice followed by quantitative 2C-ISH analysis of virus infection of host organs to directly compare the in vivo fitnesses of wt MCMV and mutant Δm04+06+152-MCMV (Fig. 4). In essence, the mutant was found to replicate at organ sites relevant to CMV disease, including liver (Fig. 4, B and C1), spleen (Fig. 4, B and C2), adrenal glands (Fig. 4, B and C3), and lungs (unpublished data). Foci of infection in tissues, each representing the clonal progeny of a single hit infection event, were either mixed black and red (representing wt MCMV) or clean-red (representing the mutant). The existence of clean-red foci proves that the mutant can replicate in the respective tissues independent of wt MCMV. The histomorphometric quantitation of infected tissue cells indicated an identical efficacy of wt and mutant virus spread in the liver and the adrenal glands, whereas a minor disadvantage of the mutant in the spleen may be attributed to some residual immunity in this lymphoid organ after the 7-Gy γ-irradiation. In conclusion, the data gave no indication for a nonimmunological role of the immunoevasin genes.
Evidence Against the Expression of a Further MHC Class I Downmodulating Immunoevasin during the E Phase.
The three currently known immune evasion proteins of MCMV were identified by their interaction with MHC class I molecules. In principle, there could have existed further viral proteins performing such a function. The triple mutant Δm04+06+152-MCMV gave us the opportunity to address that question for the first time.
Cell surface expression of a set of MHC class I molecules specified by haplotypes H-2d and H-2b was tested by cytofluorometric analysis using the cell lines and MHC allele-specific antibodies compiled in Table I. As time point for the analysis, we chose 12 h after infection, because this is an E phase time at which all known immune evasion genes are simultaneously expressed and at which expression of other cell surface molecules, as verified for CD44 as a marker, is not yet affected by cytopathic effects of the infection (tested were 4, 7, 12, and 16 h after infection, unpublished data). An infection rate of >90% of the cells was assured by the detection of the intranuclear IE1 protein.
As it is shown in Fig. 5 A, cell surface expression of the set of MHC class I molecules of haplotypes H-2d and H-2b was essentially identical for uninfected cells and cells infected with Δm04+06+152-MCMV. This finding allows the conclusion that there exists no further viral E gene that would significantly contribute to the downmodulation of MHC class I cell surface expression in the two MHC haplotypes tested herein. Yet, it should be emphasized that the existence of unknown functional analogs of m04/gp34, which interact with MHC class I molecules but do not downmodulate their cell surface expression, remains to be considered.
Figure 5.

Cell surface expression of MHC class I molecules. (A) Comparison between cells infected with mutant Δm04+06+152-MCMV and uninfected cells. (B) Comparison between cells infected with wt MCMV (MW97.01) and cells infected with mutant Δm04+06+152-MCMV. The expression of MHC class I molecules of haplotypes H-2d (Kd, Dd, and Ld) and H-2b (Kb and Db) was measured by cytofluorometry at 12 h after infection (multiplicity of infection of 6) of B12 and C57SV cells, respectively. Left panels show original fluorescence histograms for one selected experiment. Right panels show data compiled from 3 to 5 independent experiments. The columns represent the mean values (standard deviations indicated by bars) of the percentage of expression relative to uninfected cells (in A) and relative to cells infected with Δm04+06+152-MCMV (in B).
The Efficacy of MHC Class I Downmodulation by the Concerted Action of All Three Immunoevasins Shows Allele-specific Differences.
With the rationale in mind that presence and absence of all three immunoevasins would reveal the maximal possible difference, we compared MHC class I cell surface expression after infection with wt MCMV and mutant Δm04+06+152-MCMV (Fig. 5 B). With the notable exception of Kb, the tested MHC class I molecules were significantly downregulated by the three immunoevasins in wt MCMV. The degree of downregulation was similar for Kd, Dd, and Db, and a bit less for Ld. The finding that certain MHC class I molecules, Kb in the specific case, are apparently not efficiently targeted by the immunoevasins, predicts functional, allele-specific differences in the immune control of infection.
Combinatorial Gene Deletions Reveal Synergistic As Well As Antagonistic Interactions between the Immunoevasins.
It is the beauty of our immunoevasin gene deletion library of virus mutants that immune evasion proteins can now be expressed in the context of infection in any possible combination (Table II).
Table II.
Overview on the Expression of Immunoevasins by wt MCMV and the Set of Deletion Mutants
| Virus | Immunoevasin molecule expressed
|
||
|---|---|---|---|
| m04/gp34 | m06/gp48 | m152/gp40 | |
| Δm04+06+152-MCMV | − | − | − |
| Δm06+152-MCMV | + | − | − |
| Δm04+152-MCMV | − | + | + |
| Δm04+06-MCMV | − | − | + |
| Δm152-MCMV | + | + | − |
| Δm06-MCMV | + | − | + |
| Δm04-MCMV | − | + | + |
| wt MCMV | + | + | + |
Fig. 6 gives a comprehensive view on the effects of immunoevasins on the cell surface expression of all MHC class I molecules specified by haplotypes H-2d (Fig. 6 A) and H-2b (Fig. 6 B). Cytofluorometric analysis was again performed at 12 h after infection, since, at that time point, it was guaranteed that cells infected with the different viruses all expressed the corresponding immunoevasins. The level of MHC class I surface expression observed was normalized to the expression obtained after infection with mutant Δm04+06+152-MCMV. As shown above (Fig. 5 A), the expression was not significantly different in uninfected cells and cells infected with Δm04+06+152-MCMV, but we preferred the mutant as the standard for the data normalization to account for putative general effects of the infection on MHC class I gene expression, trafficking, and metabolism.
Figure 6.


Comprehensive patterns of MHC class I surface expression for all possible combinations of immunoevasins expressed by the set of immunoevasin gene deletion mutants. (A) Pattern for haplotype H-2d. (B) Pattern for haplotype H-2b. B12 and C57SV cells, respectively, were infected at an MOI of 6, and the expression of MHC class I molecules at the cell surface was determined by cytofluorometry at 12 h after infection. The columns represent the mean values (SDs indicated by bars) of the percentage of expression relative to the expression in cells infected with mutant Δm04+06+152-MCMV (set as 100%) from 3 to 5 independent experiments. The head legends indicate the combinations of immunoevasins expressed by the various mutants (see Table II) and (farthest right column) by BAC-cloned wt MCMV MW97.01.
Let us view the data in Fig. 6 from the left to the right. At a glance, isolated expression of immunoevasin m04/gp34 by mutant Δm06+152-MCMV had no downmodulating effect on the cell surface expression of any of the tested MHC class I molecules. Rather, there appears to be some upregulation in the case of Ld. Immunoevasins m06/gp48 and m152/gp40, expressed by mutants Δm04+152-MCMV and Δm04+06-MCMV, respectively, showed comparable efficacy regarding the downmodulation of Dd, Ld, and Db, whereas m06/gp48 was superior in downmodulating K locus alleles, namely Kd and Kb. In addition, allele-specific differences became apparent in that, for instance, Dd was less affected than Db. Immunoevasin m04/gp34, when coexpressed with m06/gp48 in mutant Δm152-MCMV, showed an indifferent role. It had no significant influence on the expression of Kd, Dd and Db, but instead of supporting downmodulation it rather appeared to slightly antagonize the effect of m06/gp48 on Ld and Kb. The most striking result was obtained after the coexpression of m04/gp34 and m152/gp40 in mutant Δm06-MCMV: m04/gp34 clearly antagonized the downmodulating effect of m152/gp40. Remarkably, this was true for all five MHC class I molecules tested, including Kd, even though gp34 was found not to form complexes with Kd at the cell surface (12). In contrast, m06/gp48 and m152/gp40, coexpressed in mutant Δm04-MCMV, showed a cooperative function. This cooperation was particulary significant when single expression of either one was inefficient, as it was the case for Dd. Finally, expression of all three molecules in wt MCMV did not further enhance the cooperative downmodulation effected by m06/gp48 and m152/gp40. Instead, in the case of Kb, the antagonizing function of m04/gp34 was so strong that it partially rescued the surface expression. Thus, our previous assumption that the maximal downmodulation should be seen for wt MCMV has found an exception in Kb.
In conclusion, this study has unraveled the effects of the three MCMV-encoded immunoevasins on MHC class I cell surface expression for two important, frequently studied haplotypes. The most important new finding is that the immunoevasins interact not only in a cooperative but also in a competitive fashion.
Discussion
Both, MCMV and HCMV deploy a set of genes to alter stability and export of MHC class I molecules. These immune evasion genes are unique for each virus and have different functional properties (50). Studies on the function of individually expressed proteins have identified for each of the seven MHC reactive CMV immunoevasins the principle of the interaction with the MHC class I molecule, the intracellular compartment of activity, and the relevance of viral protein domains (for a review, see reference 51).
The fact that each of the two viruses possesses multiple genes for similar functions forces us to explain this apparent redundance. Rapid MHC molecule degradation is the dominant phenotype of HCMV infection, whereas destabilization of nascent MHC class I complexes in the ER is definitely not the hallmark of the MCMV gene functions. The MCMV genes all affect the MHC complex export to the plasma membrane. Therefore, in MCMV-infected cells, but less so in HCMV-infected cells, the nascent MHC class I complexes could interact with more than one viral protein simultaneously or subsequently. Since the viral proteins may have allelic preferences, this could result in a complex pattern of allele-specific cooperative and competitive effects. This might lead to results completely different from what is seen when a viral protein is expressed and tested in isolation.
Experimentally, this question can only be addressed by construction and direct comparison of appropriate mutants as a set. Until recently, the ability to mutate virus genomes depended for most viruses on recombination techniques in mammalian cells, a procedure which is difficult to control. The advent of functional genomics has led to the development of cloning vectors, suitable for the maintenance of large inserts. These F-factor derived BACs fit perfectly to the insert size requirements of herpesvirus genomes (120–240 kb), and after pioneering MCMV BAC cloning by our group (18), several herpesvirus genomes have been successfully cloned as BACs. Since the large DNA inserts in BACs preclude conventional enzymatic cleavage and ligation techniques, targeted BAC mutation in E. coli requires sophisticated recombination procedures (for a review, see reference 52). Here we adapted a recently developed mutagenesis approach for manipulation of conventional bacterial plasmids (45) to the viral BAC. This method uses double crossing over of a linear DNA fragment with the viral BAC mediated by the recombinases redα and -β derived from bacteriophage λ (53). Compared with the conventional two-step shuttle mutagenesis, the homologies required for efficient recombination are reduced to only 25–50 nts. These short sequences can be supplied by synthetic primers. This new strategy allows repeated mutagenesis of herpesvirus genomes without any cloning and thus provided the basis for the construction of the first systematic and comprehensive set of mutants for multiple immunoevasins in the genomic context, the major intention of this study.
By testing the growth of the MCMV mutants in cell culture and in immunodepleted mice, we confirmed the immunological nature of the attenuation phenotype and the fidelity of the mutagenesis method. The in vivo replication is an important control, as it can reveal even subtle differences in virus fitness. According to our results, mutants that have undergone repeated mutagenesis procedures are not unspecifically attenuated and therefore can reliably be used in future in vivo studies on immune control and immune evasion.
Here, we focused on the hallmark of MHC class-I–interfering functions, namely the surface expression of MHC class I molecules. We used cytofluorometric analysis, because this method is sufficiently standardized and reproducible to identify consistent patterns between large scale experiments performed with different cell lines. Three major findings are emerging: first, only the previously identified immunoevasins m06/gp48 and m152/gp40 downmodulate surface expression of MHC class I molecules after infection of cells with MCMV. Our data exclude the existence of additional unknown MCMV genes that would indirectly affect MHC molecule stability, as it is the case in HCMV for gene US6 which inhibits transporter associated with processing–dependent peptide translocation into the ER and loading of MHC molecules (9–11).
Second, the third known immunoevasin, namely m04/gp34, operates in an essentially different fashion in that it does not downmodulate MHC class I cell surface expression, although it binds to MHC class I molecules in the ER as well as at the cell surface (54). This is consistent with our original hypothesis that m04 contributes only indirectly to immune evasion, for instance through NK control (12). Since cell surface expression was the parameter tested in our study, the data cannot rule out the existence of further immunoevasins that function like m04/gp34. Data presented here indicate that gp34 destines MHC complexes for export to the plasma membrane. This does not yet address the functionality of the MHC molecule complexed with m04/gp34. One consequence of m04 expression in cells of H-2d haplotype is the presentation of an antigenic peptide of m04/gp34 itself (55). Studying the recognition of MCMV-infected cells by CTL clones in the H-2b haplotype, we observed that m04/gp34 compromises Kb- but not Db-restricted CTL clones by an as yet unknown mechanism (13). Thus, although the surface export of MHC molecules is not negatively affected, the binding site for the TCR or the cargo of peptides might be different for MHC molecules escorted by m04/gp34.
Third, and most striking, the immunoevasins have cooperative as well as antagonistic effects with regard to surface expression of MHC molecules. Whether or not there is a hierarchy in the strength of MHC downregulation of m06 over m152 must await further MHC molecule testing. Clearly, m06/gp48 and m152/gp40 do cooperate to reduce MHC class I expression. This becomes detectable when molecules such as Dd are analyzed, which are not strongly regulated by either of these two immunoevasins when expressed in isolation. Therefore, the redundance may be helpful to effectively downregulate MHC alleles in cases in which each individual immunoevasin has only a limited effect. The moderate effect of the isolated m152 in the genomic context comes as a surprise, given the fact that deletion of m152 has strong functional effects on CTL recognition of antigenic peptides presented by MHC molecules such as Ld (16), Kb, and Db (13). The finding that m152 does have effects on CTL recognition which do not completely parallel with its effects on the modulation of MHC expression at the plasma membrane suggests that this gene may affect CTL function through more than one mechanism. Our recent finding that m152/gp40 has dual effects in that it, besides MHC class I downregulation, also downmodulates H-60, thereby downregulating NK cell function and possibly also accessory CTL functions, provides an explanation (56). Therefore, we have to be careful with regard to functional predictions based on the amount of MHC molecules at the cell surface. In fact, the role of m06 is less stringently corroborated by functional testing, and future analysis of the mutants will have to show whether m06 can control CTL recognition to the extent predicted by its effect on MHC export (14).
Remarkably, m04/gp34 antagonizes and overrides the downmodulating effect of m152/gp40 for all MHC class I molecules tested. As this effect is seen only after deletion of gene m06, a situation which is unlikely to exist in a natural MCMV genome, one apparent role of immunoevasin m06/gp48 is to prevent an m04/gp34-mediated restoration of class I surface expression.
In contrast, m04/gp34 did not antagonize the downmodulating effect m06/gp48. There must exist a structural basis for this difference: in accordance with the gene expression kinetics, m06 gets the first chance to interact with MHC class I, since even for an MHC molecule such as Dd, for which the downmodulation by m06/gp48 is incomplete, the subsequent expression of the m04 gene cannot revert the inhibition (Fig. 6). One should consider that there is stable complex formation between viral immunoevasins and MHC class I molecules only for m04/gp34 (12) and m06/gp48 (14) but not for m152/gp40. m152/gp40 is the earliest immunoevasin to be expressed during the viral replication cycle (15) but its interaction with MHC class I molecules is only transient. Nevertheless, after interaction with m152/gp40 expressed in isolation, MHC molecules lose the intrinsic capacity to migrate to the plasma membrane (37). Most probably, the nascent molecules get their irreversible destination by the interaction with either m06/gp48 or m04/gp34, resulting in lysosome or plasma membrane targeting, respectively, whereas the interaction with m152/gp40 does not determine the final fate of the MHC complexes. Therefore, it is not unreasonable to speculate that in the absence of m06/gp48 at least some of the MHC complexes that were originally retained in the ER by m152/gp40 are eventually escorted to the cell surface by m04/gp34, whereas in the presence of m06/gp48 the MHC class I traffic is preferentially directed toward lysosomal degradation.
In summary, the testing of a set of immune evasion gene mutants has disclosed functional interactions between immunoevasins. With the new mutagenesis methods, virus constructs can be generated in the near future in which by promoter exchange the expression of immunoevasins is altered from synchronous to sequential or even conditional expression patterns in order to study the temporal regulation of immunoevasin interactions. This study has already revealed that, with respect to MHC class I surface expression, the function of one immunoevasin, namely m04/gp34, is strongly antagonistic to m152/gp40, another immunoevasin encoded in a distant region of the viral genome.
Acknowledgments
We thank Sabine Linke for excellent technical assistance.
U.H. Koszinowski and A. Gutermann were supported by grants from the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 455, individual project A7, and SPP Neue Vakzinierungsstrategien. M.J. Reddehase and J. Podlech were supported by a grant from the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 490, individual project B1.
Footnotes
Abbreviations used in this paper: BAC, bacterial artificial chromosome; MEF, mouse embryonic fibroblast; NK, natural killer; wt, wild-type.
References
- 1.Tortorella, D., B.E. Gewurz, M.H. Furman, D.J. Schust, and H.L. Ploegh. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926. [DOI] [PubMed] [Google Scholar]
- 2.Alcami, A., and U.H. Koszinowski. 2000. Viral mechanisms of immune evasion. Trends Microbiol. 8:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chee, M.S., A.T. Bankier, S. Beck, R. Bohni, C.M. Brown, R. Cerny, T. Horsnell, C.A. Hutchison, T. Kouzarides, J.A. Martignetti, et al. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125–169. [DOI] [PubMed] [Google Scholar]
- 4.Rawlinson, W.D., H.E. Farrell, and B.G. Barrell. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiertz, E.J., D. Tortorella, M. Bogyo, J. Yu, W. Mothes, T.R. Jones, T.A. Rapoport, and H.L. Ploegh. 1996. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 384:432–438. [DOI] [PubMed] [Google Scholar]
- 6.Wiertz, E.J., T.R. Jones, L. Sun, M. Bogyo, H.J. Geuze, and H.L. Ploegh. 1996. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 84:769–779. [DOI] [PubMed] [Google Scholar]
- 7.Ahn, K., A. Angulo, P. Ghazal, P.A. Peterson, Y. Yang, and K. Fruh. 1996. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc. Natl. Acad. Sci. USA. 93:10990–10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones, T.R., E.J. Wiertz, L. Sun, K.N. Fish, J.A. Nelson, and H.L. Ploegh. 1996. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. USA. 93:11327–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn, K., A. Gruhler, B. Galocha, T.R. Jones, E.J. Wiertz, H.L. Ploegh, P.A. Peterson, Y. Yang, and K. Fruh. 1997. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity. 6:613–621. [DOI] [PubMed] [Google Scholar]
- 10.Hengel, H., J.O. Koopmann, T. Flohr, W. Muranyi, E. Goulmy, G.J. Hammerling, U.H. Koszinowski, and F. Momburg. 1997. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity. 6:623–632. [DOI] [PubMed] [Google Scholar]
- 11.Lehner, P.J., J.T. Karttunen, G.W. Wilkinson, and P. Cresswell. 1997. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA. 94:6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleijnen, M.F., J.B. Huppa, P. Lucin, S. Mukherjee, H. Farrell, A.E. Campbell, U.H. Koszinowski, A.B. Hill, and H.L. Ploegh. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kavanagh, D.G., M.C. Gold, M. Wagner, U.H. Koszinowski, and A.B. Hill. 2001. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194:967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reusch, U., W. Muranyi, P. Lucin, H.G. Burgert, H. Hengel, and U.H. Koszinowski. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziegler, H., R. Thaele, P. Lucin, W. Muranyi, T. Flohr, H. Hengel, H. Farrell, W. Rawlinson, and U.H. Koszinowski. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 6:57–66. [DOI] [PubMed] [Google Scholar]
- 16.Krmpotic, A., M. Messerle, M.I. Crnkovic, B. Polic, S. Jonjic, and U.H. Koszinowski. 1999. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 190:1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mocarski, E.S., Jr., and G.W. Kemble. 1996. Recombinant cytomegaloviruses for study of replication and pathogenesis. Intervirology. 39:320–330. [DOI] [PubMed] [Google Scholar]
- 18.Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U.H. Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA. 94:14759–14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brune, W., M. Messerle, and U.H. Koszinowski. 2000. Forward with BACs: new tools for herpesvirus genomics. Trends Genet. 16:254–259. [DOI] [PubMed] [Google Scholar]
- 20.Wagner, M., S. Jonjic, U.H. Koszinowski, and M. Messerle. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borst, E.M., G. Hahn, U.H. Koszinowski, and M. Messerle. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 73:8320–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hobom, U., W. Brune, M. Messerle, G. Hahn, and U.H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGregor, A., and M.R. Schleiss. 2001. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol. Genet. Metab. 72:15–26. [DOI] [PubMed] [Google Scholar]
- 24.Yu, D., G.A. Smith, L.W. Enquist, and T. Shenk. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner, M., D. Michel, P. Schaarschmidt, B. Vaida, S. Jonjic, M. Messerle, T. Mertens, and U. Koszinowski. 2000. Comparison between human cytomegalovirus pUL97 and murine cytomegalovirus (MCMV) pM97 expressed by MCMV and vaccinia virus: pM97 does not confer ganciclovir sensitivity. J. Virol. 74:10729–10736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.del Val, M., H.J. Schlicht, T. Ruppert, M.J. Reddehase, and U.H. Koszinowski. 1991. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 66:1145–1153. [DOI] [PubMed] [Google Scholar]
- 27.Trinchieri, G., D.P. Aden, and B.B. Knowles. 1976. Cell-mediated cytotoxicity to SV40-specific tumour-associated antigens. Nature. 261:312–314. [DOI] [PubMed] [Google Scholar]
- 28.Brune, W., H. Hengel, and U. Koszinowski. 1999. A mouse model for cytomegalovirus infection. Curr. Protocols Immunol. 1971–1973. [DOI] [PubMed]
- 29.Reddehase, M.J., F. Weiland, K. Muench, S. Jonjic, A. Lueske, and U.H. Koszinowski. 1985. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 55:264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook, J.T., and D. Russel. 2001. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Press, Cold Spring, NY. 1836 pp.
- 31.Posfai, G., M.D. Koob, H.A. Kirkpatrick, and F.R. Blattner. 1997. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J. Bacteriol. 179:4426–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gay, P., D. Le Coq, M. Steinmetz, E. Ferrari, and J.A. Hoch. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J. Bacteriol. 153:1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebeling, A., G.M. Keil, E. Knust, and U.H. Koszinowski. 1983. Molecular cloning and physical mapping of murine cytomegalovirus DNA. J. Virol. 47:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cherepanov, P.P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 158:9–14. [DOI] [PubMed] [Google Scholar]
- 35.Muyrers, J.P., Y. Zhang, G. Testa, and A.F. Stewart. 1999. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 27:1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kempkes, B., D. Pich, R. Zeidler, B. Sugden, and W. Hammerschmidt. 1995. Immortalization of human B lymphocytes by a plasmid containing 71 kilobase pairs of Epstein-Barr virus DNA. J. Virol. 69:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziegler, H., W. Muranyi, H.G. Burgert, E. Kremmer, and U.H. Koszinowski. 2000. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 19:870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bubeck, A., U. Reusch, M. Wagner, T. Ruppert, W. Muranyi, P.M. Kloetzel, and U.H. Koszinowski. 2002. The glycoprotein gp48 of murine cytomegalovirus. Proteasome-dependent cytosolic dislocation and degradation. J. Biol. Chem. 277:2216–2224. [DOI] [PubMed] [Google Scholar]
- 39.Jones, B., and C.A. Janeway, Jr. 1981. Cooperative interaction of B lymphocytes with antigen-specific helper T lymphocytes is MHC restricted. Nature. 292:547–549. [DOI] [PubMed] [Google Scholar]
- 40.Lemke, H., G.J. Hammerling, and U. Hammerling. 1979. Fine specificity analysis with monoclonal antibodies of antigens controlled by the major histocompatibility complex and by the Qa/TL region in mice. Immunol. Rev. 47:175–206. [DOI] [PubMed] [Google Scholar]
- 41.Kurz, S., H.P. Steffens, A. Mayer, J.R. Harris, and M.J. Reddehase. 1997. Latency versus persistence or intermittent recurrences: evidence for a latent state of murine cytomegalovirus in the lungs. J. Virol. 71:2980–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keil, G.M., M.R. Fibi, and U.H. Koszinowski. 1985. Characterization of the major immediate-early polypeptides encoded by murine cytomegalovirus. J. Virol. 54:422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapp, M., M. Messerle, B. Buehler, M. Tannheimer, G.M. Keil, and U.H. Koszinowski. 1992. Identification of the murine cytomegalovirus glycoprotein B gene and its expression by recombinant vaccinia virus. J. Virol. 66:4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grzimek, N.K., J. Podlech, H.P. Steffens, R. Holtappels, S. Schmalz, and M.J. Reddehase. 1999. In vivo replication of recombinant murine cytomegalovirus driven by the paralogous major immediate-early promoter-enhancer of human cytomegalovirus. J. Virol. 73:5043–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang, Y., F. Buchholz, J.P. Muyrers, and A.F. Stewart. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20:123–128. [DOI] [PubMed] [Google Scholar]
- 46.Hanson, L.K., J.S. Slater, Z. Karabekian, H.W. Virgin, C.A. Biron, M.C. Ruzek, N. van Rooijen, R.P. Ciavarra, R.M. Stenberg, and A.E. Campbell. 1999. Replication of murine cytomegalovirus in differentiated macrophages as a determinant of viral pathogenesis. J. Virol. 73:5970–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoddart, C.A., R.D. Cardin, J.M. Boname, W.C. Manning, G.B. Abenes, and E.S. Mocarski. 1994. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol. 68:6243–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrews, D.M., C.E. Andoniou, F. Granucci, P. Ricciardi-Castagnoli, and M.A. Degli-Esposti. 2001. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat. Immunol. 2:1077–1084. [DOI] [PubMed] [Google Scholar]
- 49.Podlech, J., R. Holtappels, N. Wirtz, H.P. Steffens, and M.J. Reddehase. 1998. Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J. Gen. Virol. 79:2099–2104. [DOI] [PubMed] [Google Scholar]
- 50.Hengel, H., U. Reusch, A. Gutermann, H. Ziegler, S. Jonjic, P. Lucin, and U.H. Koszinowski. 1999. Cytomegaloviral control of MHC class I function in the mouse. Immunol. Rev. 168:167–176. [DOI] [PubMed] [Google Scholar]
- 51.Reddehase, M.J. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. In press. [DOI] [PubMed] [Google Scholar]
- 52.Wagner, M., Z. Ruzsics, and U.H. Koszinowski. 2002. Herpesvirus genetics has come of age. Trends Microbiol. 10:318–324. [DOI] [PubMed] [Google Scholar]
- 53.Muyrers, J.P., Y. Zhang, and A.F. Stewart. 2001. Techniques: recombinogenic engineering–new options for cloning and manipulating DNA. Trends Biochem. Sci. 26:325–331. [DOI] [PubMed] [Google Scholar]
- 54.Kavanagh, D.G., U.H. Koszinowski, and A.B. Hill. 2001. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 167:3894–3902. [DOI] [PubMed] [Google Scholar]
- 55.Holtappels, R., D. Thomas, J. Podlech, G. Geginat, H.P. Steffens, and M.J. Reddehase. 2000. The putative natural killer decoy early gene m04 (gp34) of murine cytomegalovirus encodes an antigenic peptide recognized by protective antiviral CD8 T cells. J. Virol. 74:1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krmpotic, A., D. Busch, I. Bubic, F. Gerhard, H. Hengel, M. Hasan, A.A. Scalzo, U. Koszinowski, and S. Jonjic. 2002. Dual function of MCMV immunoevasion gene. m152/gp40 glycoprotein confers virus resistance to CD8+ T lymphocytes and NK cells in vivo. Nat. Immunol. 3:529–535. [DOI] [PubMed] [Google Scholar]